
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTertiary amine-directed and involved carbonylative cyclizations through Pd/Cu-cocatalyzed multiple C–X (X = H or N) bond cleavage†
Qiu-Chao
Mu‡
ab,
Yi-Xue
Nie‡
b,
Xing-Feng
Bai
ab,
Jing
Chen
*a,
Lei
Yang
ab,
Zheng
Xu
*b,
Li
Li
b,
Chun-Gu
Xia
a and
Li-Wen
Xu
 *ab
*ab
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute (SRI), Lanzhou Institute of Chemical Physics (LICP), University of the Chinese Academy of Sciences (UCAS), Lanzhou 730000, P. R. China. E-mail: chenj@licp.cas.cn; liwenxu@hznu.edu.cn
bKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Key Laboratory of Organosilicon Material Technology of Zhejiang Province, Hangzhou Normal University, Hangzhou 311121, P. R. China. E-mail: zhengxu@hznu.edu.cn
First published on 19th August 2019
Abstract
A novel Pd/Cu-cocatalyzed carbonylative cyclization by C–H activation and N-dealkylative C–N bond activation has been developed for the chemoselective construction of synthetically useful heterocycles. The N,N-dimethylamine group on o-indolyl-N,N-dimethylarylamines was found to act as both the directing group and reactive component in this C–H carbonylative cyclization reaction. Furthermore, a unique C–H oxidation/carbonylative lactonization of diarylmethylamines is firstly demonstrated under modified reaction conditions, which could be easily applicable to the one-step synthesis of multi-substituted phthalides bearing an N,O-ketal skeleton that is difficult to access by previously reported methods. Mechanistic studies implicate that Pd/Cu-cocatalyzed C–H oxidation/carbonylative lactonization is a sequential reaction system via Cu-catalyzed C(sp3)–H oxidation and Pd-catalyzed oxidative carbonylation of the C(sp2)–H bond. It was found that trace amounts of water are essential to promote the Cu-catalyzed C(sp3)–H oxidation of diarylmethylamine for the formation of the hydroxyl group, which could act as an in situ-formed directing group in the intramolecular carbonylative lactonization step.
Introduction
The concept of transition-metal-catalyzed C–H activation/functionalization has re-captured the imagination of chemists and become an exciting field of research in chemistry in the last 20 years.1 After two decades of effort, dramatic achievements in the field of C–H activation have been achieved and applications of C–H functionalization have permeated and transformed versatile fields including polymer chemistry, materials science, chemical biology and medicinal chemistry.2 In this regard, by introducing a directing group, numerous catalyst systems have been established to offer direct synthetic methodologies to transform C–H bonds into various valuable C–Z bonds (e.g., C–C, C–O, C–N, C–S, C–P, and C–halides).3 For instance, Daugulis and Corey demonstrated N-heterocycle-directed palladium-catalyzed β-C(sp3)–H arylation of amide derivatives, which realized the synthetic potential of double C(sp3)–H activation to give homo-diarylated α-amino acid derivatives.4a,b Yu4c and Martin4d independently reported carboxyl-directed Pd- and Cu-catalyzed C–H bond activation/C–O bond formation to give lactones and coumarins. The Shi4e and Cheng4f groups reported hydroxy-directed transition-metal-catalyzed C–H bond activation/carbonylation of 2-arylphenols to synthesize benzopyranones.As a simple yet useful tertiary amine directing group, the N,N-dimethylamine group has been widely utilized in C–H activation/functionalization.5 In this context, nitrogen-directed Pd(II)-catalyzed ortho-carbonylation, alkoxycarbonylation of arenes with CO and alcohols, and functionalization of substituted toluenes through ortho-olefination of N,N-dimethylbenzylamines have been well established in the past years (Scheme 1b).5 Very recently, You5a and Sebesta5b independently reported the cross-coupling reaction of ferrocenylmethanamines through Pd-catalyzed C(sp2)–H functionalization using N,N-dimethylamine as a directing group (Scheme 1a). Notably, Xu5c reported a new approach for the synthesis of optically active diarylmethylamines via Ir-catalyzed N,N-dimethylamine-directed C–H activation/C–B bond formation. Despite these advances, the N,N-dimethylamine group was the only used directing group in most of the documented examples. While the N,N-dimethylamine group can occasionally become removable in some cases to access further transformation of the C–H activation products,5d its removal reduced the atom-economy and potential derivative diversity of the whole C–H activation process. To overcome these limitations, the combinational use of C–H and C–N activation in N,N-dimethylamine-directed C–H functionalization to ensure that the N,N-dimethylamine group could work beyond simple directing effects appeared and some rare examples have also been reported.6 However, in the research area of N,N-dimethylamine-directed C–H carbonylation with CO, to date, only the Lei group reported a straightforward oxidative C–H bond activation/N-dealkylative carbonylation of tertiary [1,10-biphenyl]-2-anilines, which provided an alternative process to the synthesis of various biologically important lactams and phenanthridin-6(5H)-ones.6d,e
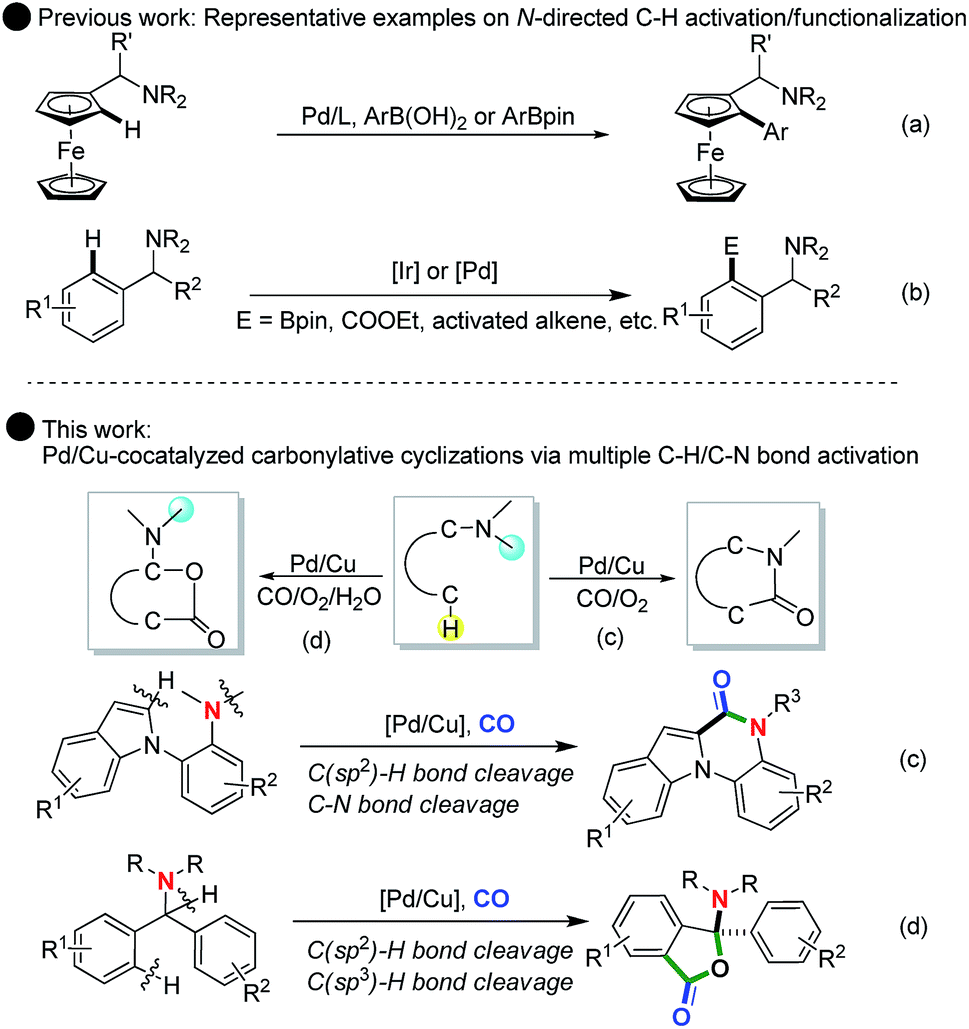 | ||
| Scheme 1 Tertiary amine-directed C–H functionalization: single role as the directing group (previous work) and dual role as the directing group and reactive component (this work). | ||
Notably, the direct transition-metal-catalyzed group-directed C–H activation-initiated carbonylation of diarylmethylamines could provide a convenient and powerful alternative for the construction of new backbones of heterocyclic compounds, such as lactam and lactone skeletons, that have been well identified as potentially useful and bioactive molecules. Herein, we reported a Pd/Cu-cocatalyzed unprecedented and multiple C–H activation/N-dealkylative C–N bond activation/carbonylation sequence by using the N,N-dimethylamine group as both the directing group and reactive component, allowing for a facile synthesis of biologically important N-heterocycles, such as indolo[1,2-a]quinoxalin-6-ones. Further studies also offered the first example of Pd/Cu co-controlled hybrid and chemoselective C–H oxidation/carbonylation to access the functionalized N,O-ketal-containing phthalides, in which the copper plays a dual role in the first step of C(sp3)–H oxidation with the aid of water by single electron transfer (SET) and in the second step of Pd(II)-catalyzed C(sp2)–H carbonylation by oxidation of Pd(0) to Pd(II) respectively. These findings provide a new access to combine the attractive features of C–H functionalization and C–N bond activation/cleavage with catalytic carbonylation for atom-economic synthesis of novel heterocycles.
Results and discussion
To validate our hypothesis that the construction of a novel indolo[1,2-a]quinoxalin-6-one moiety could be completed by Pd/Cu-cocatalyzed carbonylation through C–H activation/N-dealkylative C–N bond activation, o-indolyl-N,N-dimethylarylamine (2a) was chosen as a model substrate. It should be noted that, among these lactam-containing heterocycles, indolo[1,2-a]quinoxalin-6-ones rank as a class of important tetracyclic moieties that represent a diverse range of pharmacological properties, and their synthesis has attracted the interest of many scientists in the past decades.7,8 Meanwhile, the key motifs of phthalides that bear a lactone scaffold have been found in many potentially bioactive products.7 Despite the long-standing efforts in the synthesis of the indolo[1,2-a]quinoxalin-6-ones8 and N,O-ketal-containing phthalides,9 most of the reported methods to afford these compounds usually suffer from inherent limitations, including poor reaction efficiency, pre-functionalized substrates, and limited scope or operational simplicity. Nevertheless, to access the indolo[1,2-a]quinoxalin-6-ones and N,O-ketal-containing phthalides from easily available compounds with the N,N-dimethylamine group, the development of practical and one-step processes still remains highly desirable. Thus to overcome the aforementioned challenges, initially we sought to develop a C–H activation/carbonylation process by merging the C–N bond activation and C–H oxidative carbonylation (Scheme 1c).To establish the Pd/Cu-cocatalyzed carbonylative cyclization of o-indolyl-N,N-dimethylarylamine (2a) by optimizing various reaction parameters, we found that the combination of PdCl2 (10 mol%), Cu(TFA)2·xH2O (30 mol%) and PivOH (1.0 equiv.) in a dry solvent of dioxane/DMA (3/1) under a mixture of CO/O2 (1/3, 1 atm) gave the best result (Table 1, entry 1). The reaction did not work in the absence of either PdCl2 or Cu(TFA)2·xH2O (Table 1, entries 2 and 3). Pd(OAc)2 and Pd(TFA)2 were found to be obviously inferior to PdCl2 in catalytic activity and resulted in trace yields (Table 1, entries 5 and 6). Copper salts also proved to be essential for the reactivity, for example, CuCl2, Cu(OAc)2, and Cu(acac)2 gave decreased yields of 3a (Table 1, entries 7–9). The yield also dropped dramatically, when using AcOH, AdOH, or Boc–Val–OH instead of PivOH (Table 1, entries 10–12). The effect of solvents on the reaction was then investigated, and it was found that protic solvents, such as i-PrOH (Table 1, entry 17), impeded the reaction and the polar aprotic solvents proved to work well in this reaction. The mixture of dioxane/DMA appears to be the optimal choice (Table 1, entries 13–16). In addition, a remarkable decrease of the yield of 3a was observed when adjusting the ratio of CO and O2 (Table 1, entries 18 and 19). Moreover, we proved that the O2 is essential for this reaction (Table 1, entries 20–22). Notably, reducing the catalyst loading would result in lower yields of 3a (Table 1, entries 23–25).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
a
2a (0.1 mmol), PdCl2 (10 mol%), Cu(TFA)2·xH2O (30 mol%), PivOH (1.0 equiv.), dioxane/DMA (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2.0 mL), V(CO)/V(O2) (1:3), 100 °C, 48 h.
b Determined by GC-MS, isolated yield is given in the parenthesis. The trace by-products are detected as the various demethylated products coming from 2a and 3a. And the similar polarity of these products makes the purification by flash column chromatography really difficult, which resulted in a loss of isolated yield.
c Toluene/dioxane (3:1, 2.0 mL), 110 °C.
d 110 °C.
e The amount of 2a is 0.5 mmol in this case.
f ND = not determined.
g The amount of Pd or Cu catalyst is reduced and the others are not changed.
h 1.0 mmol scale of 2a. :1, 2.0 mL), V(CO)/V(O2) (1:3), 100 °C, 48 h.
b Determined by GC-MS, isolated yield is given in the parenthesis. The trace by-products are detected as the various demethylated products coming from 2a and 3a. And the similar polarity of these products makes the purification by flash column chromatography really difficult, which resulted in a loss of isolated yield.
c Toluene/dioxane (3:1, 2.0 mL), 110 °C.
d 110 °C.
e The amount of 2a is 0.5 mmol in this case.
f ND = not determined.
g The amount of Pd or Cu catalyst is reduced and the others are not changed.
h 1.0 mmol scale of 2a.
|
||
| 1 | None | 88(60)e |
| 2c | Without PdCl2 | NDf |
| 3c | Without Cu(TFA)2·xH2O | ND |
| 4c | Without PivOH | 60 |
| 5c | Pd(OAc)2 instead of PdCl2 | Trace |
| 6c | Pd(TFA)2 instead of PdCl2 | Trace |
| 7c | CuCl2 instead of Cu(TFA)2·xH2O | Trace |
| 8c | Cu(OAc)2 instead of Cu(TFA)2·xH2O | Trace |
| 9c | Cu(acac)2 instead of Cu(TFA)2·xH2O | Trace |
| 10c | AcOH instead of PivOH | Trace |
| 11c | AdOH instead of PivOH | Trace |
| 12c | Boc–Val–OH instead of PivOH | Trace |
| 13d | Toluene/dioxane = 3:1 |
69 |
| 14d | Toluene/DMF = 3:1 |
69 |
| 15d | Toluene/NMP = 3:1 |
29 |
| 16d | Toluene/DMA = 3:1 |
82 |
| 17d | i-PrOH instead of the mixture solvent | Trace |
| 18c | CO/O2 = 1:2 |
55 |
| 19c | CO/O2 = 1:4 |
57 |
| 20c | AgOAc instead of O2 | Trace |
| 21c | K2S2O8 instead of O2 | Trace |
| 22c | TEMPO instead of O2 | Trace |
| 23 | 5 mol% PdCl2g | 55 |
| 24 | 10 mol% Cu(TFA)2·xH2Og | Trace |
| 25 | 20 mol% Cu(TFA)2·xH2Og | 25 |
| 26 | Noneh | 46 |
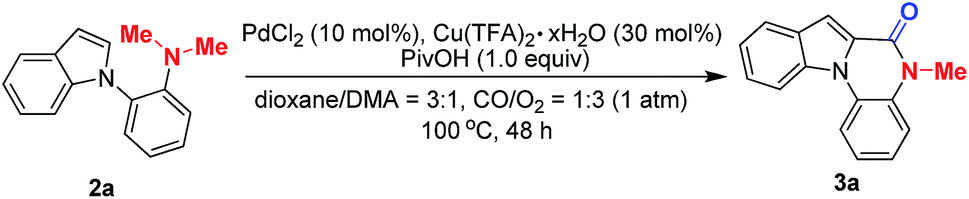
The substrate scope was next explored under the optimized reaction conditions. As shown in Scheme 2, o-indolyl-N,N-dimethylarylamines bearing various substituents at the 3-, 4-, 5-, 6-, and 7-positions of the indole ring were amenable under the optimal conditions and enabled formation of corresponding products in moderate to good yields (3a–3r). It is important to note that the functional groups, such as 5-Br (3d) and 5-CN (3g), could provide handles for further derivation. 3h and 3i were also produced in 38% and 43% yields respectively from the reaction of C3-substituted indole partners, suggesting that steric hindrance did not dramatically hamper the transformation. Gratifyingly, introduction of a 7-Me group was also tolerated (3o). Switching to disubstituted substrates still furnished the desired products (3p). Interestingly, when 2q is used in the Pd/Cu-cocatalyzed carbonylative cyclization, the major product is 3r but not 3q, in which double C–N bond cleavage occurs under the optimized reaction conditions (Scheme 2, eqn (2)).
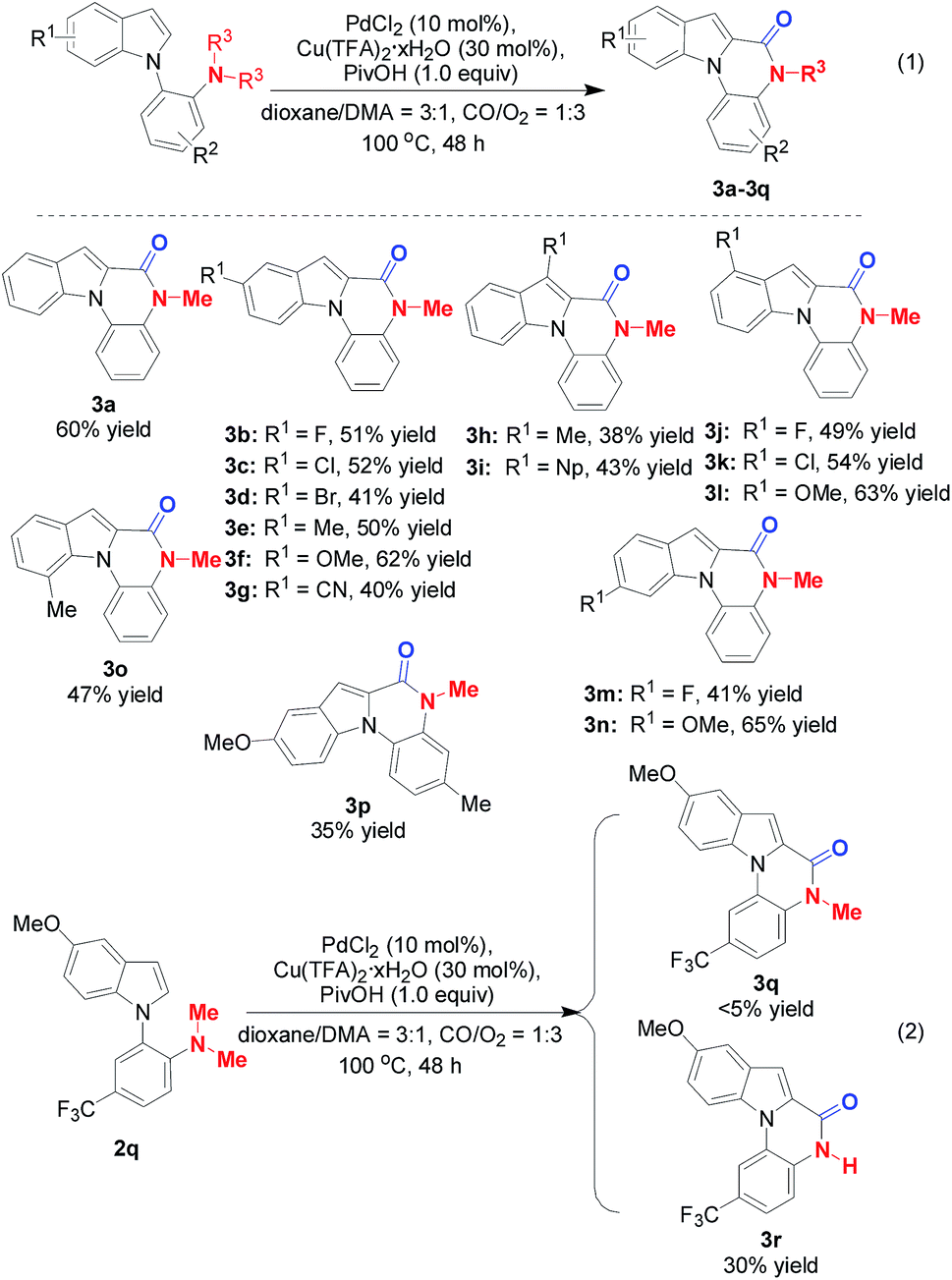 | ||
| Scheme 2 Reaction scope. The reactions were run on a 0.5 mmol scale. | ||
Considering the importance of the isoindolinone and phthalide scaffolds in many pharmaceuticals and bioactive natural products (see Fig. 1),7 we extended the practicality of this synthetic strategy with diarylmethylamines (4a) as the substrate. Surprisingly, the unexpected N,O-ketal-containing phthalide 5a was detected. After judicious optimization of various reaction parameters (see Table 2 and ESI, Tables S1–S9†), the N,O-ketal-containing phthalide 5a was obtained in 90% GC-MS yield upon heating 4a with a combination of PdCl2 (10 mol%), Cu(OAc)2 (30 mol%), Boc–Val–OH (30 mol%) and PivOH (1.0 equiv.) in a dry solvent of toluene/dioxane (5/1) under a mixture of CO/O2 (2/1, 1 atm).
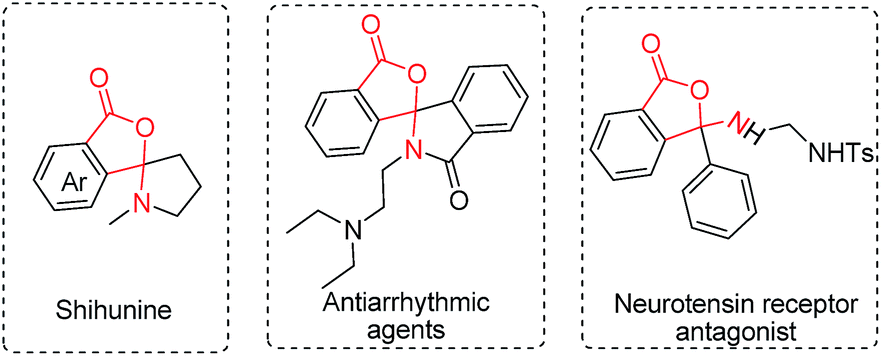 | ||
| Fig. 1 Representative examples of biologically active molecules based on the N,O-ketal-containing phthalide core. | ||
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
|
a
4a (0.1 mmol), PdCl2 (10 mol%), Cu(OAc)2 (30 mol%), Boc–Val–OH (30 mol%), PivOH (1.0 equiv.), toluene/dioxane (5:1, 1.2 mL), V(CO)/V(O2) (2:1), 100 °C, 60 h.
b Determined by GC-MS, isolated yield is given in the parenthesis.
c Toluene/DMF (5:1, 1.2 mL), 120 °C.
d 100 °C, 48 h.
e ND = not determined.
|
||
| 1 | None | 90(65) |
| 2c | Without PdCl2 | NDe |
| 3c | Without Cu(OAc)2 | ND |
| 4c | Without PivOH | 42 |
| 5c | AcOH instead of PivOH | 55 |
| 6c | Cs2CO3 instead of PivOH | ND |
| 7c | Pd(OA)2 instead of PdCl2 | Trace |
| 8c | Pd(TFA)2 instead of PdCl2 | Trace |
| 9c | CuCl2 instead of Cu(OAc)2 | Trace |
| 10c | Cu(acace)2 instead of Cu(OAc)2 | 45 |
| 11c | AgOAc as oxidant | Trace |
| 12c | K2S2O8 as oxidant | Trace |
| 13c | Toluene instead of mixed solvent | Trace |
| 14c | i-PrOH instead of mixed solvent | Trace |
| 15c | DMA instead of DMF | 35 |
| 16 | 110 °C instead of 100 °C | 82 |
| 17 | 90 °C instead of 100 °C | 55 |
| 18 | Boc–Phe–OH | 80 |
| 19 | Fomc–Val–OH | 72 |
| 20 | CO/O2 = 4:1 |
35 |
| 21d | Add H2O (10 equiv.) | 17 |
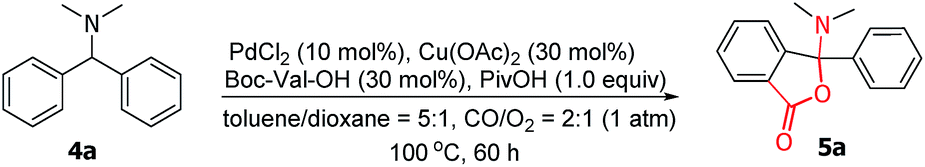
Representative results to support the optimized reaction conditions are shown in Table 2. For example, no product was detected in the absence of either PdCl2 or Cu(OAc)2 (Table 1, entries 2 and 3). The yield also dropped dramatically in the absence of PivOH or using AcOH instead of PivOH (Table 1, entries 4 and 5). Although various palladium salts, copper salts and oxidants were screened, no positive results were obtained (Table 1, entries 7–12). Solvent effects on the reaction were also investigated. It was found that both non-polar (i.e. toluene) and protic solvents (i.e. i-PrOH) impeded the reaction, thus resulting in trace amounts of target product 5a. Only polar aprotic solvents proved to work well in this reaction, and the mixture of toluene/dioxane gave better results (Table 1, entries 1, 13–15). The higher temperature (110 °C) slightly decreased the yield to 80% and the lower temperature led to a lower yield (Table 1, entries 16 and 17). Acid-derived ligands were also screened in this work, and Boc–Val–OH was proved to be the best one (Table 1, entries 1, 18, and 19). In addition, a remarkable change of this transformation was observed by increasing or decreasing the concentrate of CO (Table 1, entries 1 and 20). In addition, adding extra water has no benefits but harms the reaction (Table 1, entry 21).
We next evaluated the scope and generality of this novel lactonization by Pd/Cu-cocatalyzed C–H carbonylation. As shown in Scheme 3, both substrates bearing electron-rich and electron-poor substituents on the aryl ring could be activated and a C–H carbonylation process occurred to give corresponding N,O-ketal-containing phthalides in moderate to good yields. The steric hindrance of substituents on the phenyl rings exhibited an important effect on the yields. For instance, while the substrate bearing a meta- or para-methyl substituent gave yields greater than 50% (5c, 5e), ortho-methyl substituted substrates decreased the yields of the corresponding products to 36–45% (5b, 5m). Substrates bearing either an electron-donating or electron-withdrawing substituent on the para-position of the aryl rings were well-tolerated under the current conditions (5e–5k). A wide range of functional groups, including fluoride (5f), chloride (5g), methoxy (5h), and trifluoromethoxy (5k), were well compatible with the reaction. Furthermore, the gram-scale reaction of 4a has been pursued and the desired product 5a was obtained in 55% isolated yield (Scheme 4).
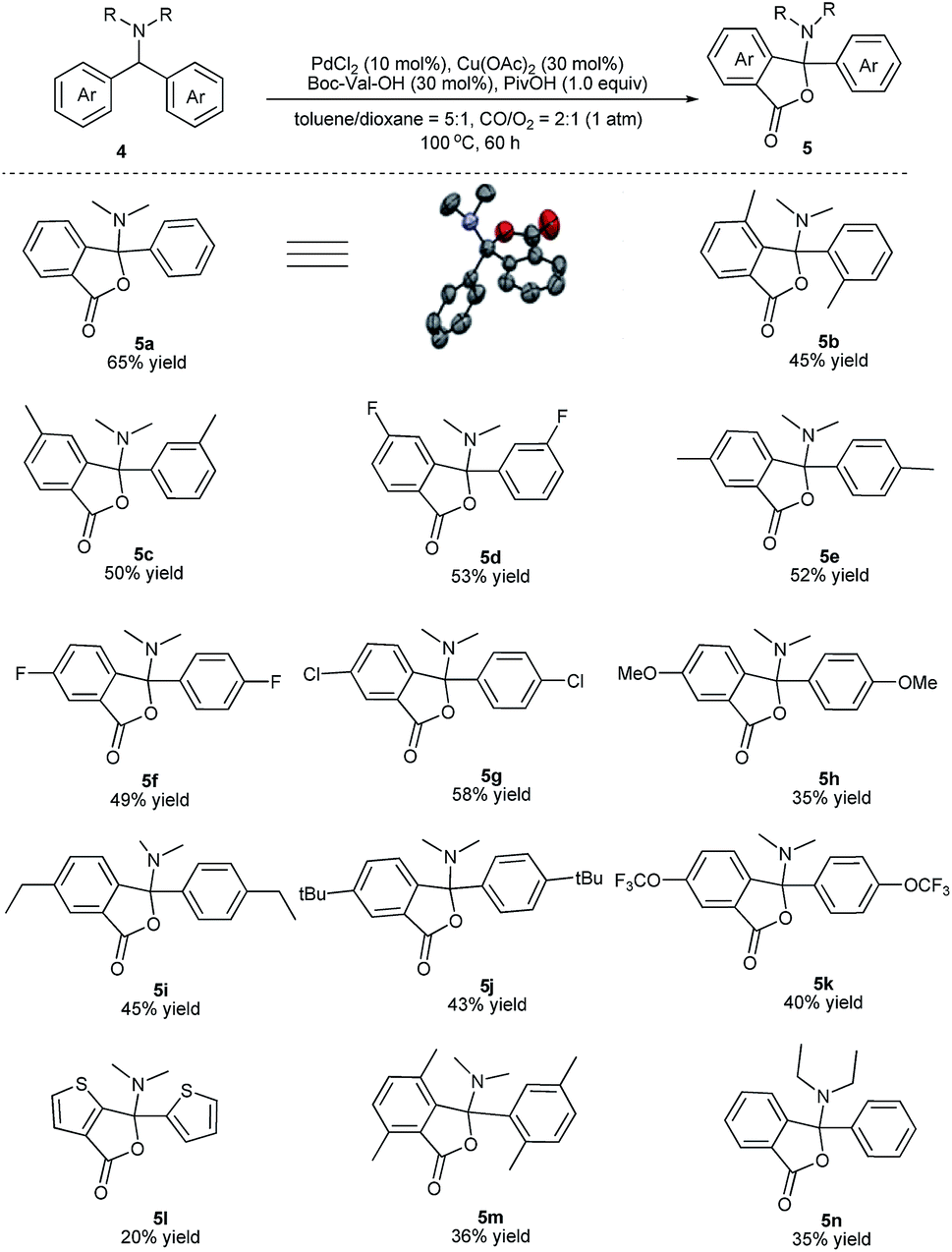 | ||
| Scheme 3 Reaction scope. The reactions were run on a 0.5 mmol scale. | ||
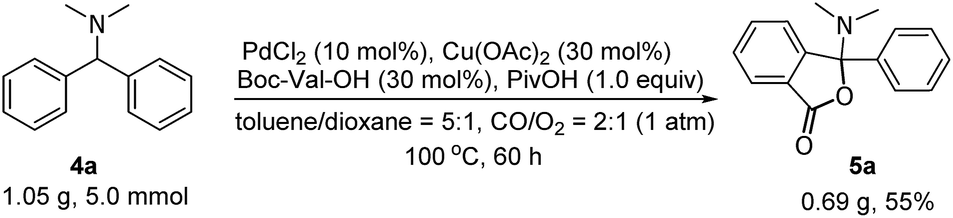 | ||
| Scheme 4 Gram-scale reaction. | ||
Mechanistic studies
To gain more insight into the N-dealkylative C–N bond activation/carbonylation, various control experiments and kinetic isotope effect (KIE) studies were conducted. Firstly, compound 3a was detected by GC-MS using 2a-H as the substrate under the standard conditions, which clearly indicated that 2a-H was the key intermediate during the reaction (Scheme 5, eqn (1)). As shown in eqn (2) and (3), KH/KD = 1.3 and 1.2 were observed respectively for the intermolecular KIE experiments, suggesting that C–N bond cleavage might not be involved in the rate-determining step, while the C–H bond cleavage step did (KH/KD = 1.9, eqn (4)). All these experiments suggested C–N bond cleavage as the key step, which led to the intermediate 2a-H in the initial step, and the resulting –NHMe group of 2a-H worked as the directing group in the following C–H activation/carbonylation sequence.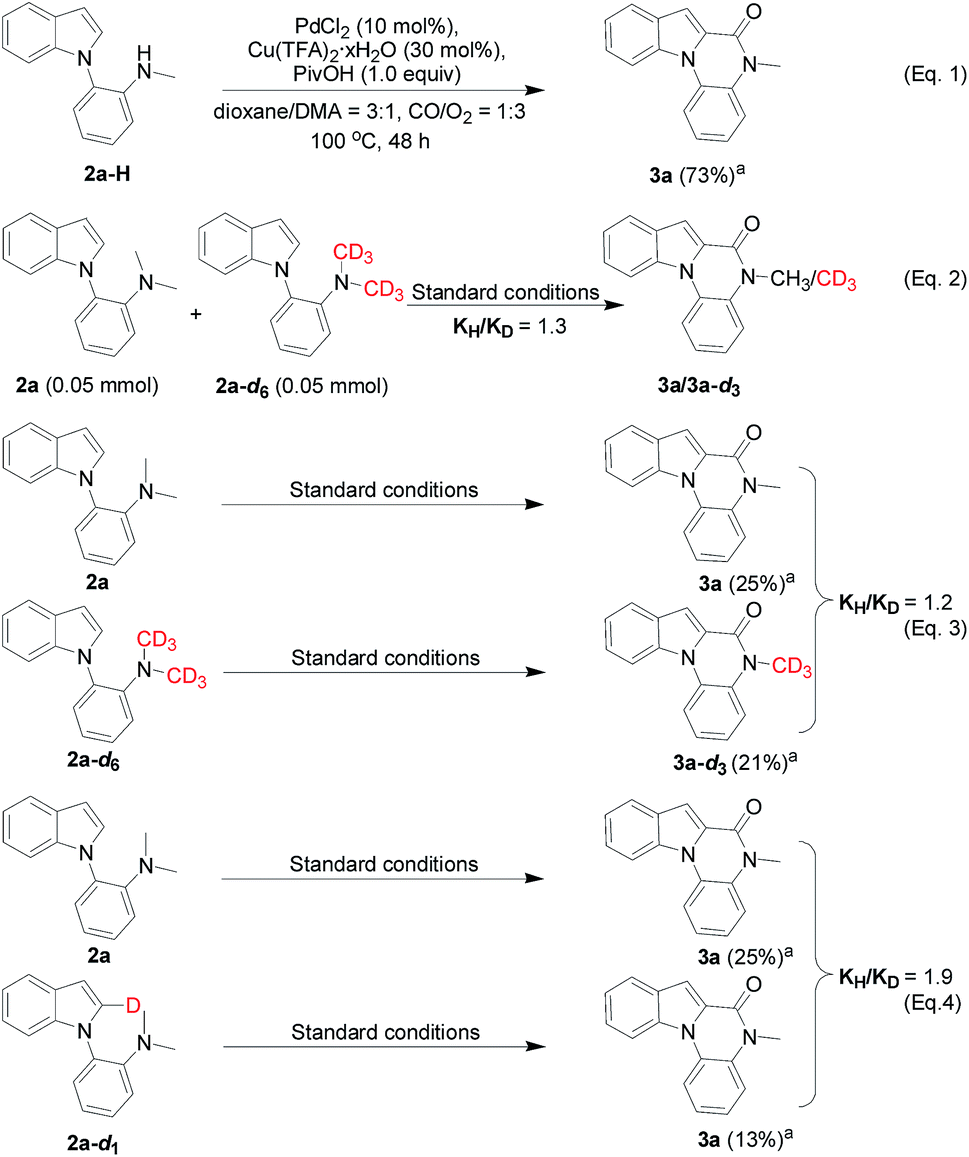 | ||
| Scheme 5 Control experiments and KIE experiments. aDetermined by GC-MS. | ||
Furthermore, to clarify the highly attractive protocol for the unexpected synthesis of phthalides bearing an N,O-ketal skeleton, two plausible mechanisms of Pd/Cu-cocatalyzed C–H functionalization are proposed in Scheme 6. Palladium-catalyzed C–H activation has been well studied and normally needs the presence of directing groups. For instance, the C(sp2)–H bond activation with the N,N-dimethylamine group as the directing group has been successfully used in catalytic carbonylation, olefination, and cross-coupling reactions. According to the literature precedence, it is supposed that the reaction may proceed through pathway a shown in Scheme 6, which involves two sequential C(sp2)–H and C(sp3)–H activation via two independent catalytic cycles by Pd/Cu-based cooperative catalysis directed by the N,N-dimethylamine group and carboxyl group respectively.5a,d,6a The carbonylation process was considered to occur in the first C(sp2)–H activation cycle (left), which is followed by anion exchange and reductive elimination to form intermediate 6a. In this pathway, 6a is proposed to be a key intermediate and the final product 5a is released by the reductive elimination in the subsequent cycle of the C(sp3)–H activation (pathway a, right cycle).10a However, we did not detect 6a or a related intermediate during the reaction process. In addition, the desired product 5a was not formed when 6a was used as a starting material under the standard reaction conditions (Scheme 6, eqn (1)). Thus on the basis of the above experimental results, the proposed pathway is not a major process and could be excluded.
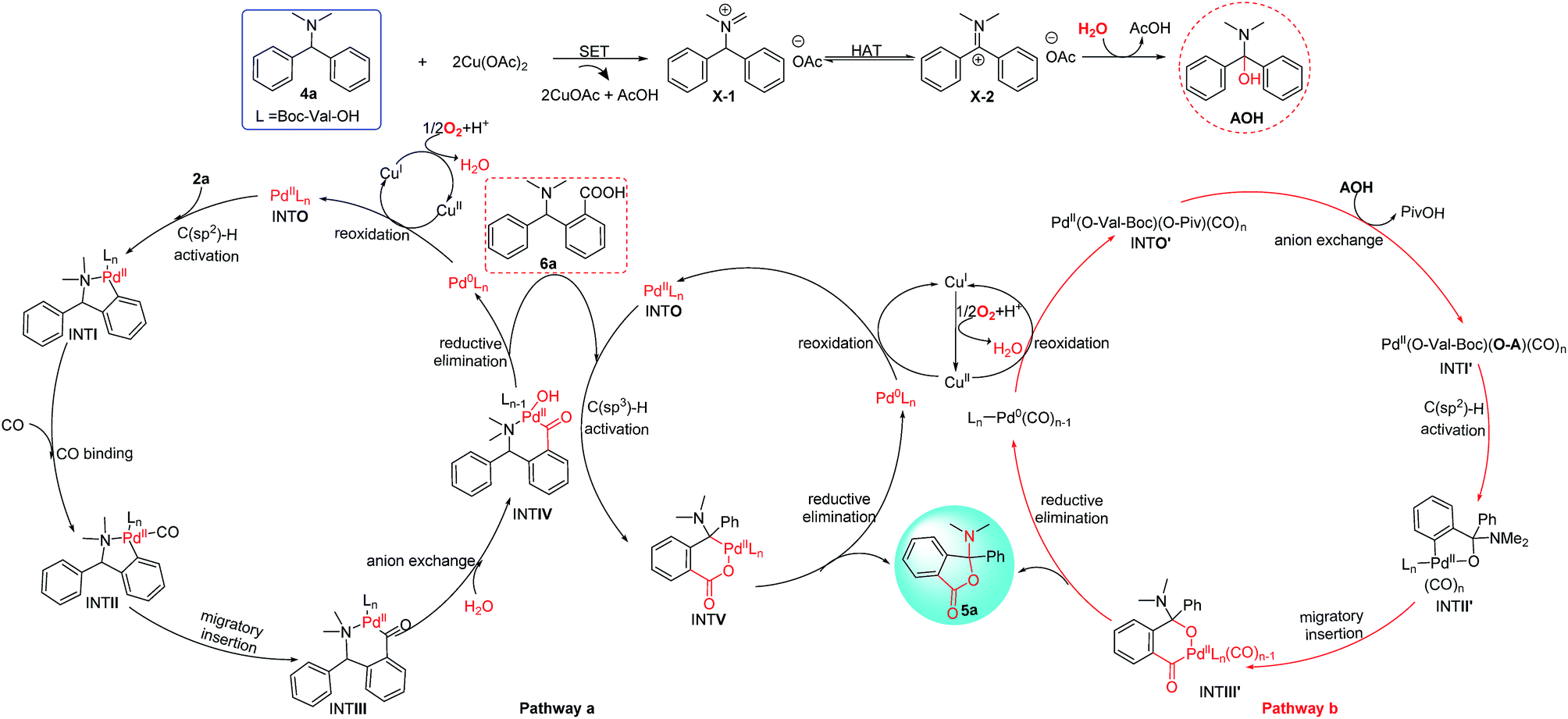 | ||
| Scheme 6 Two proposed mechanisms of the double C–H functionalization for Pd/Cu-cocatalyzed oxidative carbonylation/lactonization of diarylmethylamines. | ||
Then we proposed an alternative mechanism (pathway b) that is shown in Scheme 6 (the third cycle). In this case, the C(sp3)–H bond of N,N-dimethylphenylamine is activated via Cu-catalyzed a single electron transfer (SET) together with a hydrogen-migration process.11 Substrate 4a was initially considered to be converted into intermediate X-1 with the assistance of Cu(OAc)2 through the SET pathway.11a–c And it could be transformed into X-2 through a proton transfer process because intermediate X-1 lies in an equilibrium with intermediate X-2.11d Hydrolysis of X-2 would help shift the equilibrium to form AOH by the nucleophilic attack of H2O to X-2. Then the active CO-ligated Pd(II) intermediate (INTO′) is allowed to react with AOH by anion exchange that induced by chelating of the hydroxyl group to Pd center, which can make C(sp2)–H bond activation more easily by the Pd(II) center of INTI′. The resulting five-membered palladacycle (INTII′) becomes a key intermediate in the subsequent carbonylation in this pathway. Then the migratory insertion of one coordinated CO into the aryl–Pd bond forms the six-membered palladacycle (INTIII′). Reductive elimination from INTIII′ leads to 5a and generates the Pd(0) species, which will be reoxidized to form the active palladium(II) complex by Cu(OAc)2 in the presence of O2.
To gain more insights into the reaction mechanism that 5a was produced through C(sp3)–H oxidation and the subsequent C(sp2)–H activation, oxidative carbonylation of AOH, followed by reductive elimination (Scheme 5, pathway b),10b–d various control experiments, kinetic isotope effect (KIE) studies and density functional theory (DFT) investigations were conducted. According to the above proposed mechanism (pathway b), the following speculations may be deduced: (1) the starting material 4a cannot be activated through the SET process without the Cu catalyst; (2) in the absence of CO, the whole reaction may stop at intermediate AOH; (3) trace water is important for the successful hydrolysis of X-2 to AOH, whose hydroxyl group is derived from water or oxygen in the reaction system. To verify these assumptions, several control experiments were carefully designed and carried out. As shown in Scheme 7, all controlled experiments were performed under standard conditions unless notified.
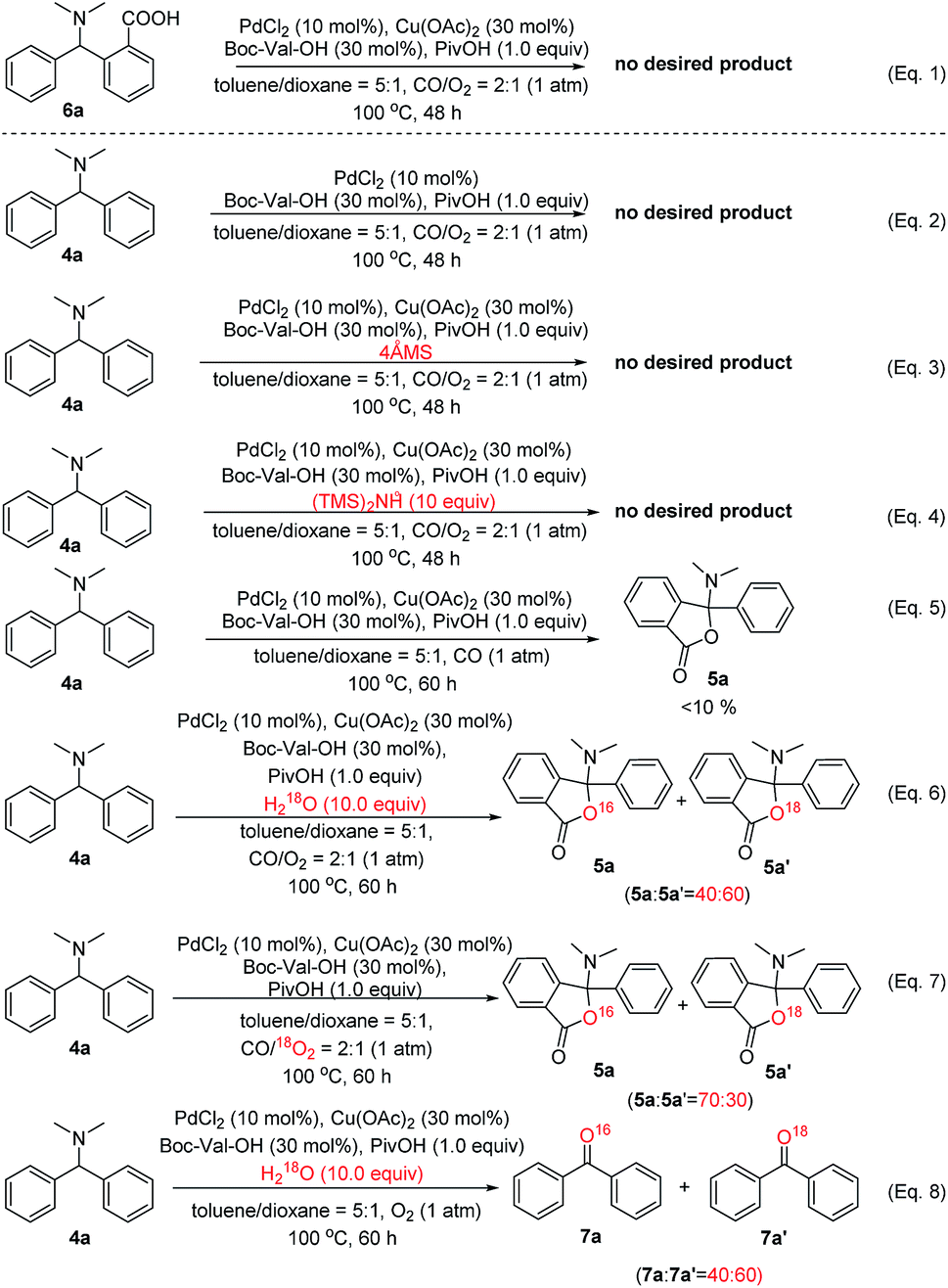 | ||
| Scheme 7 Mechanistic studies with control experiments. | ||
Firstly, it was found that no lactonization reaction was observed in the absence of Cu(OAc)2 (Scheme 7, eqn (2)), suggesting that Cu(II) species indeed act as the initiator involved in the transformation of substrate 4a. Secondly, we have tried to completely remove the trace amount of water in the reaction system with a 4 Å molecular sieve or hexamethyldisilazane (Scheme 7, eqn (3) and (4)). Interestingly, almost no desired product was observed in these two cases. On the other hand, if an excessive amount of water (up to 15 equiv.) is added to the reaction system (see Table S9 in the ESI†), the yield of product 3a decreased from 72% to lower than 10%, suggesting that water plays an important role in the whole catalytic cycle. As such, we postulate that the catalytic generation of the AOH intermediate in this system is facilitated by the copper catalyst and water in the presence of O2 and can undergo rapid C(sp3)–H oxidation of 4a. It was noteworthy that, in pathway b of Scheme 3, the O2 was initially introduced into the reaction system via a Cu-promoted redox reaction in the presence of carboxylic acid (PivOH). In accord with this expectation, the yield of 5a was lower than 10% in the absence of O2, indicating that the oxygen atom in O2 exists in the catalytic cycle in the form of water (Scheme 7, eqn (5)). To support the importance of water in the formation of product 5avia intermediate AOH, an experiment was carried out in the presence of trace amounts of H218O under the standard reaction conditions (Scheme 7, eqn (6)). As expected, both the 5a and O18-containing 5a′ were obtained in the ratio of 40:60 (5a/5a′). Another 18O labelled experiment was conducted with the 18O2 as the oxidant to give the different ratio of 5a and 5a′ (Scheme 7, eqn (7)). Additionally, in the absence of CO, the isolation of diphenylketone, a byproduct that formed via the C–N bond cleavage approach, is surprising, as its existence would normally be thought as potent evidence for hydrolysis of starting material 4a. Therefore, these control experiments strongly support the unprecedented potential of ppm levels of water-driven Pd/Cu catalysis, which will be an innovative process to understand the mechanism of catalytic C–H oxidation/carbonylation reaction featuring trace water as the carrier of oxygen (O2) in this reaction.
Furthermore, a complicated mixture of diphenylketone O16-7a and O18-7a′ (40:60) can be detected when H218O was added to the system (Scheme 7, eqn (8)). These results seem to further suggest that diphenylketone is mostly likely generated by removing one dimethylamine from intermediate AOH and water may participate in the reaction through the hydrolysis process of intermediate X-2. These experimental observations clearly indicated that both the trace amounts of H2O and O2 were the source of the oxygen atom for the formation of 5a, and O2 was involved in the reaction in the form of H2O which was transformed into intermediate AOH by nucleophilic addition to intermediate X-2. The subsequent reaction of this AOH would then occur smoothly in the presence of palladium and copper catalysts. These control experiments suggest the possibility of pathway b as the mechanism for the catalytic C–H functionalization/carbonylation reaction.
To further clarify the mechanism of the Pd-catalyzed C(sp2)–H carbonylation process, the catalytic cycle starting from intermediate AOH to product 5a has been explored with density functional theory (DFT) calculations. Our calculation results show that the reaction may proceed through the catalytic cycle shown in Fig. 2. The corresponding free energy profiles are shown in Fig. 3. The Pd(II) carboxylic acid salt (O) coordinated with one molecule of CO, which can be formed by deprotonation and ligand exchange of PdCl2 in the presence of Boc–Val–OH, PivOH and CO, was considered as the active catalyst in this reaction. It is set to a relative zero value. O was firstly transformed into the Pd(II) complex (Int1′) by the ligand exchange of AOH with PivO-. In Int1′, the ortho-C(sp2)–H bond of the AO-fragment was slightly activated via the weak π interactions between the phenyl group and Pd center. This can be seen from the elongation of the C(sp2)–H bond. Int1′ then undergoes C(sp2)–H activation via a six-membered transition state TS1 with a free energy barrier of 19.5 kcal mol−1, generating the Pd(II) Int2. In this step, a new Pd–C(sp2) bond is formed and the proton is transferred to the carbonyl of the Boc fragment. If the carbonyl group on the carboxylate fragment acts as the base, the calculated free energy barrier increases to 28.8 kcal mol−1 (see Fig. S9 in the ESI†), indicating that the Boc-based carbonyl group is a better base to activate the C(sp2)–H. The proton on the carbonyl oxygen of Int2 will rapidly transfer to the carboxylate oxygen of Boc–Val–O–, yielding energetically more stable intermediate Int3. Interestingly, progressing from Int3, a second CO molecule will coordinate to Int3 leading to the reactive intermediate Int4, which is exergonic by only 8.7 kcal mol−1. After that, the carbonyl insertion into the Pd–C(sp2) bond can occur easily via the transition state TS3 with a free energy barrier of 32.0 kcal mol−1 to form the intermediate Int5, while the carbonyl insertion into the Pd–O bond was found to be unfavourable by 7.4 kcal mol−1. Subsequently, Int5 rapidly undergoes a rapid reductive elimination viaTS6 with an energy barrier of 2.1 kcal mol−1 to give the final product 3a and release the Pd(0) catalyst (Int7). This process is exergonic by 57.1 kcal mol−1. Finally, Pd(0) species could be re-oxidized to release the active center Pd(II) with the aid of copper as well as the presence of O2 to close the catalytic cycle.
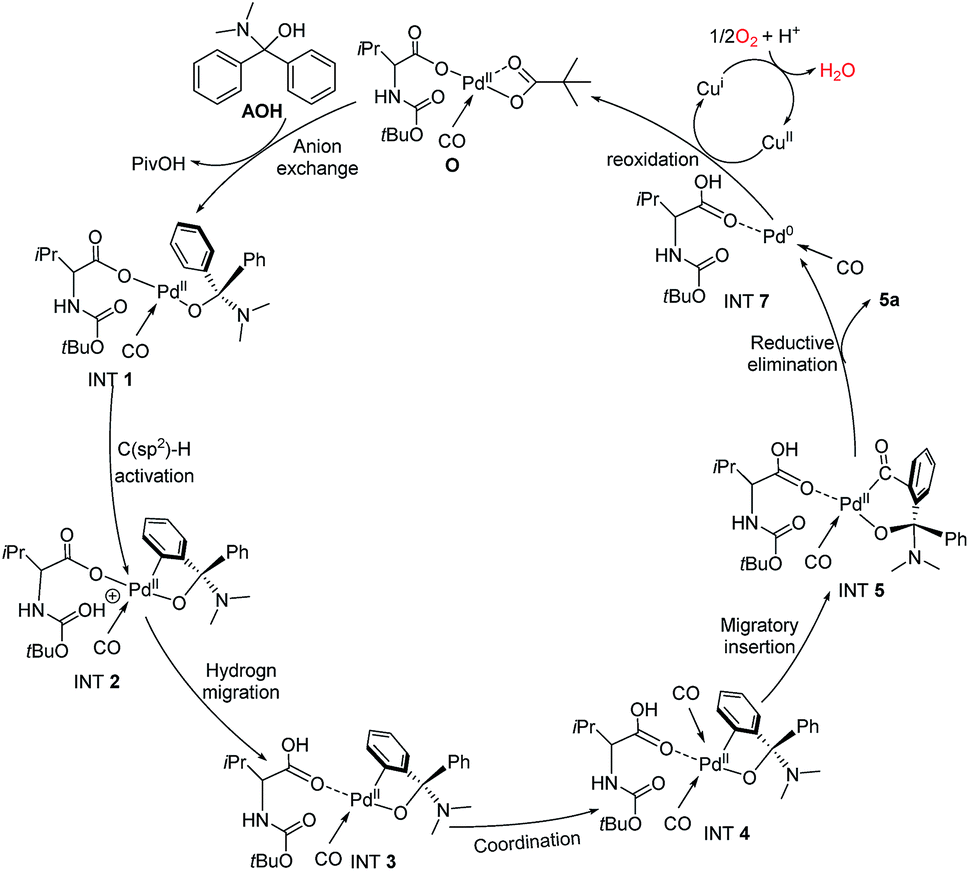 | ||
| Fig. 2 Calculated catalytic cycle for the double C–H functionalization of diarylmethylamines (4a) catalyzed by the Pd(II) catalyst. | ||
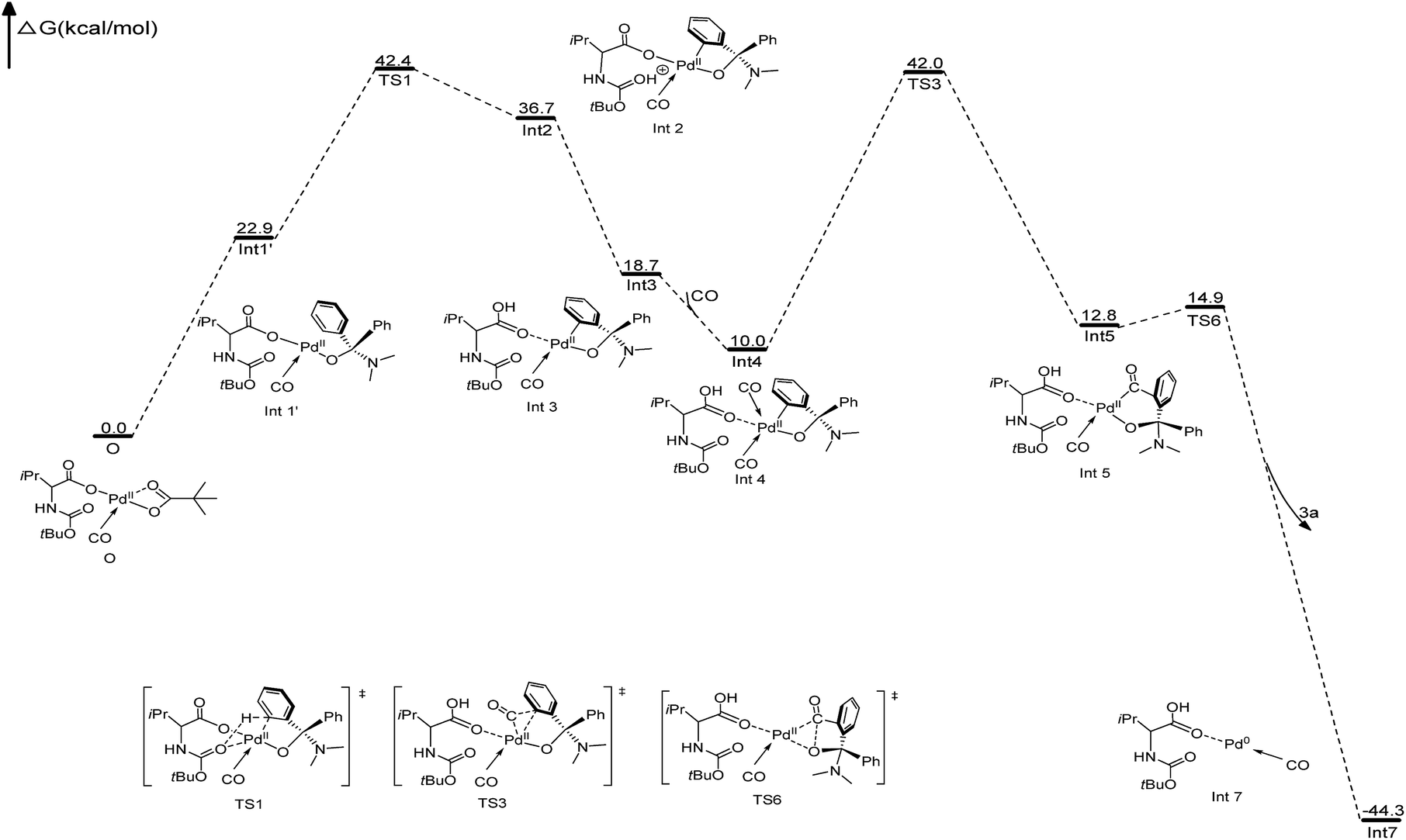 | ||
| Fig. 3 The free energy profiles for the Pd/Cu-cocatalyzed double C–H functionalization/carbonylation. The free energies (in kcal mol−1) are calculated at the B3lyp and BS (BS = 6–31 g (d,p) for main group elements and LanL2DZ for Pd) level of theory. | ||
In order to identify the rate-determining step of the whole reaction cycle, we carried out two parallel experiments to compare the kinetic isotope effect for the C(sp3)–H and C(sp2)–H functionalization, respectively (Scheme 8). The results clearly show that the migratory insertion step of the C(sp2)–H bond is the rate-limiting step and the cleavage of C(sp2)–H is more difficult than that of C(sp3)–H oxidation, which is in agreement with the DFT calculations (Fig. 2, the free energy barrier of TS3 is 12.5 kcal mol−1, and it is higher than that of TS1).
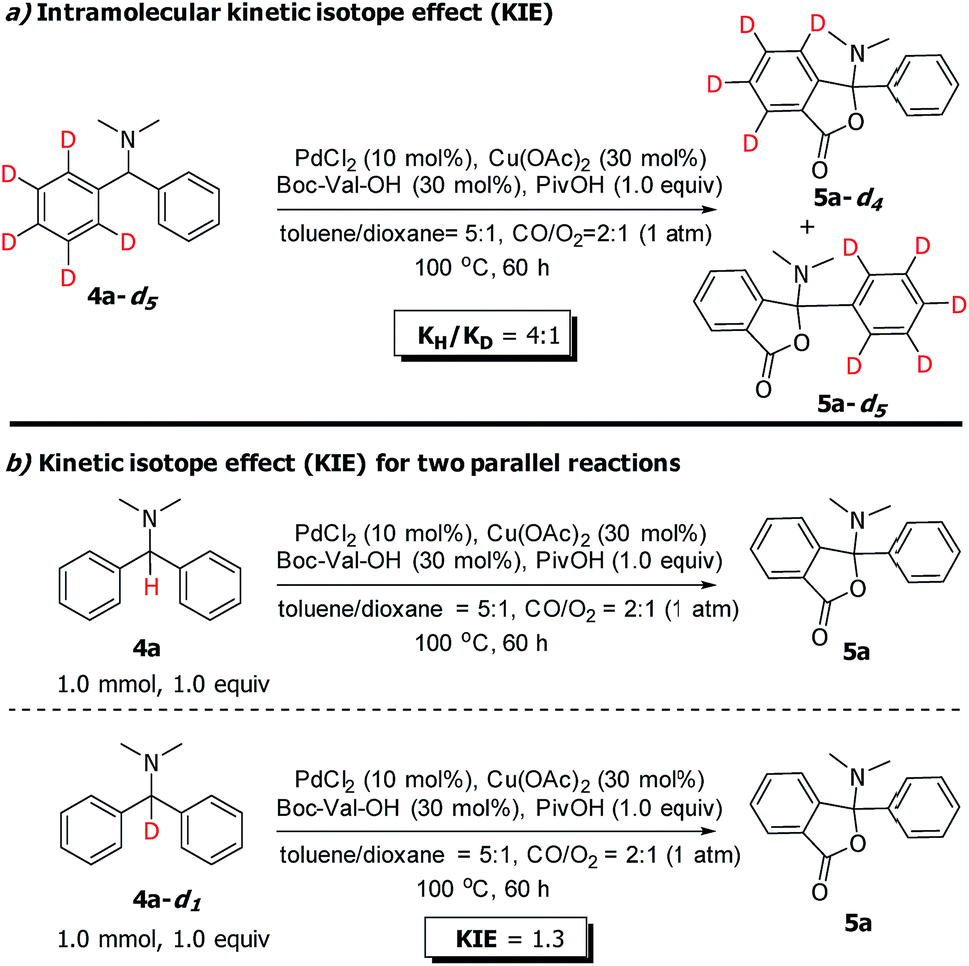 | ||
| Scheme 8 KIE experiments. | ||
Conclusion
In summary, we reported an unprecedented and multiple C–H activation/N-dealkylative C–N bond activation/carbonylative cyclization sequence by Pd/Cu-based bimetallic catalysis, allowing for the direct and facile preparation of a wide variety of novel heterocycles, such as indolo[1,2-a]quinoxalin-6-ones and phthalides, in good chemoselectivity. This novel protocol showed good functional group tolerance and broad substrate scope and will contribute substantially to the preparation of heterocyclic compounds bearing the lactam skeleton. On the other hand, we have also developed a unique palladium/copper-cocatalyzed double C(sp3)–H/C(sp2)–H functionalization/carbonylation/lactonization of diarylmethylamines to form N,O-ketal-containing phthalide derivatives in promising yields with good substrate scope. This serendipitously discovered reaction is found to be the combined sequential C(sp3)–H/C(sp2)–H cleavage and oxidative carbonylation. This protocol could be easily applicable to the one-step synthesis of multi-substituted phthalides bearing an N,O-ketal skeleton, which is difficult to access by previously reported methods. The experimental results suggested that the novel Pd/Cu-cocatalyzed C–H carbonylative cyclization is a sequential reaction system by Cu-catalyzed C(sp3)–H oxidation and Pd-catalyzed oxidative carbonylation of the C(sp2)–H bond. Mechanistic insights also implicate that ppm levels of water are essential to promote the Cu-catalyzed C(sp3)–H oxidation of diarylmethylamine for the formation of the hydroxyl group during the construction of the key N,O-ketal intermediate. Thus, this C(sp3)–H oxidation process is proved to be a key step for the catalytic cycle of C(sp2)–H carbonylation, which further supported the mutual importance of sequential and hybrid C(sp3)–H oxidation and C(sp2)–H carbonylation in the hydroxyl-directed intramolecular oxidative carbonylative lactonization. We believe that this work strongly supports the unprecedented and new potential of trace water as the carrier of oxygen (O2) in transition-metal catalysis, which will be an innovative process to understand the mechanism of the catalytic C–H oxidation/carbonylation reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (NSFC 21702211, 21773051, 21703051, and 21801056), Natural Science Foundation of Jiangsu Province (BK20170421), and Zhejiang Provincial Natural Science Foundation of China (LZ18B020001 and LY17B030005) for financial support of this work. We also thank Dr K. Z. Jiang, Dr K. F. Yang, Dr C. Q. Sheng, Dr X. Q. Xiao, Dr Z. J. Zheng, Dr J. Cao, and Dr G. W. Yin (all at HZNU) for their technical and analytical support.Notes and references
- For recent examples on the C–H functionalization, see: (a) F. Berger, M. B. Plutschack, J. Riegger, M. W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS PubMed; (b) X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Sheng, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540 CrossRef CAS PubMed; (c) R. G. Kinney, J. Tjutrins, G. M. Torres, N. J. Liu, O. Kulkarni and B. A. Arndtsen, Nat. Chem., 2018, 10, 193 CrossRef PubMed; (d) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489 CrossRef CAS PubMed; (e) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. Davies, Nature, 2016, 533, 230 CrossRef CAS PubMed; (f) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef; (g) D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851 CrossRef CAS; (h) F. L. Zhang, K. Hong, T. J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS; (i) M. H. Shaw, V. M. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. Macmillan, Science, 2016, 352, 1304 CrossRef CAS; (j) A. K. Cook, S. D. Schimler, A. J. Matzger and M. S. Sanford, Science, 2016, 351, 1421 CrossRef CAS; (k) B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403 CrossRef CAS PubMed; (l) X. Li, X. Li and N. Jiao, J. Am. Chem. Soc., 2015, 137, 9246 CrossRef CAS PubMed; (m) B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250 CrossRef CAS PubMed.
- For selected reviews on C–H functionalization, see: (a) J. B. Williamson, S. E. Lewis, R. R. Johnson III, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654 CrossRef CAS PubMed; (b) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925 RSC; (c) H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936 CrossRef CAS PubMed; (d) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302 CrossRef CAS PubMed; (e) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS; (f) M. P. Doyle and K. I. Goldberg, Acc. Chem. Res., 2012, 45, 777 CrossRef CAS PubMed; (g) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (h) H. M. L. Davies, J. D. Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855 RSC; (i) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (j) X.-F. Wu, H. Neumann and M. Beller, ChemSusChem, 2013, 6, 229 CrossRef CAS PubMed; (k) B. Gabriele, R. Mancuso and G. Salerno, Eur. J. Org. Chem., 2012, 6825 CrossRef CAS.
- For selected reviews, see: (a) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (b) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS; (c) W. Ye, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864 CrossRef; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (e) J. A. Labinger, Chem. Rev., 2017, 117, 8483 CrossRef CAS PubMed; (f) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (g) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMed; (h) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (i) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (j) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (k) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (l) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (m) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (n) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (o) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC; (p) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (q) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (r) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (s) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (t) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (u) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (v) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (w) C. Sambiagio, D. Schonbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnurch, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed; (c) X.-F. Cheng, Y. Li, Y. M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X. S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed; (d) J. Gallardo-Donaire and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350 CrossRef CAS PubMed; (e) S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598 CrossRef CAS; (f) T.-H. Lee, J. Jayakumar, C.-H. Cheng and S.-C. Chuang, Chem. Commun., 2013, 49, 11797 RSC.
- For selected reports, see: (a) Z.-J. Cai, C.-X. Liu, Q. Gu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 2149 CrossRef CAS PubMed; (b) K. Plevova, B. Mudrakova, E. Rakovsky and R. Sebesta, J. Org. Chem., 2019, 84, 7312 CrossRef CAS PubMed; (c) X. Zou, H. Zhao, Y. Li, Q. Gao, Z. Ke and S. Xu, J. Am. Chem. Soc., 2019, 141, 5334 CrossRef CAS PubMed; (d) G. Cai, Y. Fu, Y. Li, X. Wan and Z.-J. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS PubMed; (e) H. Li, G.-X. Cai and Z.-J. Shi, Dalton Trans., 2010, 39, 10442 RSC; (f) B. Liu and B. F. Shi, Synlett, 2013, 24, 2274 CrossRef CAS; (g) Z. Wang, Y. Li, F. Zhu and X.-F. Wu, Adv. Synth. Catal., 2016, 358, 2855 CrossRef CAS.
- (a) H. Li, G.-X. Cai and Z.-J. Shi, Dalton Trans., 2010, 39, 10442 RSC; (b) R. Shi, L. Lu, H. Zhang, B. Chen, Y. Sha, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 10582 CrossRef CAS PubMed; (c) X. Li, J. Pan, H. Wu and N. Jiao, Chem. Sci., 2017, 8, 6266 RSC; (d) R. Shi, H. Niu, L. Lu and A. Lei, Chem. Commun., 2017, 53, 1908 RSC; (e) R. Shi, L. Lu, H. Xie, J. Yan, T. Xu, H. Zhang, X. Qi, Y. Lan and A. Lei, Chem. Commun., 2016, 52, 13307 RSC; (f) S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589 CrossRef CAS PubMed.
- For selected reports, see: (a) H. Iqbal and F. Xua, WO 2016/179505 A1, 2016; (b) O. Key, O. Etsuo, O. Hiroyuki, O. Yoshimasa, N. Massaki and K. Kazuhiro, Eur. Pat. Appl., EP 273401 A1, 1988; (c) M. Snider, D. A. Pereira, K. P. Longo, R. E. Davidson, F. J. Vinick, K. Laitinen, K. Genc-Sehitoglu and J. N. Crawley, Bioorg. Med. Chem. Lett., 1992, 2, 1535 CrossRef; (d) E. Breuer and S. Zbaida, Tetrahedron, 1975, 31, 499 CrossRef CAS; (e) A. Brossi, W. Kloetzed, S. Teitel and J. F. Blount, J. Am. Chem. Soc., 1971, 93, 4321 CrossRef CAS PubMed; (f) G. Campiani, S. Butini, F. Trotta, C. Fattorusso, B. Catalanotti, F. Aiello, S. Gemma, V. Nacci, E. Novellino, J. A. Stark, A. Cagnotto, E. Fumagalli, F. Carnovali, L. Cervo and T. Mennini, J. Med. Chem., 2003, 46, 3822 CrossRef CAS PubMed.
- For selected reports, see: (a) A. Gogoi, P. Sau, W. Ali, S. Guin and B. K. Patel, Eur. J. Org. Chem., 2016, 1449 CrossRef CAS; (b) Q. Yuan and D. Ma, J. Org. Chem., 2008, 73, 5159 CrossRef CAS; (c) A. Huang, F. Liu, C. Zhan, Y. Liu and C. Ma, Org. Biomol. Chem., 2011, 9, 7351 RSC; (d) M. J. Beach, R. Hope, D. H. Klaubert and R. K. Russell, Synth. Commun., 1995, 25, 2165 CrossRef CAS; (e) W. Liu, J. Bang, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 14137 CrossRef CAS PubMed; (f) L. Kong, Y. Sun, Z. Zheng, R. Tang, M. Wang and Y. Li, Org. Lett., 2018, 20, 5251 CrossRef CAS PubMed.
- For selected reports, see: (a) K. Mitobe, T. Kawasaki-Takasuka, T. Agou and T. Kubota, J. Fluorine Chem., 2019, 218, 3647 CrossRef; (b) K. Kasamatsu, T. Yoshimura, A. Mandi, T. Taniguchi, K. Monde, T. Furuta and T. Kawabata, Org. Lett., 2017, 19, 352 CrossRef CAS PubMed; (c) C. Engelbert, Organic Reactions, Hoboken, NJ, United States, 2008, vol. 72, pp. 1–366 Search PubMed; (d) Y. Nagao, M. Tachikawa and K. Kozawa, Shikizai Kyokaishi, 2001, 74, 339 CAS; (e) M. Sakamoto, N. Sekine, H. Miyoshi, T. Mino and T. Fujita, J. Am. Chem. Soc., 2000, 122, 10210 CrossRef CAS; (f) K. Bowden, S. P. Hiscocks and A. Perjess, J. Chem. Soc., Perkin Trans. 2, 1998, 291 RSC; (g) K. Unverferth, R. Dorre, B. Korner, H. Scheibe and E. Morgenstern, Arch. Pharm., 1991, 324, 809 CrossRef CAS; (h) T. Hisato and E. Shoji, J. Chem. Soc., Perkin Trans. 1, 1988, 2149 Search PubMed; (i) K. Akiba, Y. Negishi and N. Inamoto, Bull. Chem. Soc. Jpn., 1984, 57, 2188 CrossRef CAS; (j) P. L. Faye, K. Vaughan and D. L. Hooper, Can. J. Chem., 1983, 61, 179 CrossRef CAS; (k) M. V. Bhatt, K. M. Kamath and M. Ravindranathan, J. Chem. Soc. C, 1971, 1772 RSC.
- (a) X.-F. Cheng, Y. Li, Y. M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X. S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed; (b) S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 10598 CrossRef CAS PubMed; (c) T.-H. Lee, J. Jayakumar, C.-H. Cheng and S.-C. Chuang, Chem. Commun., 2013, 49, 11797 RSC; (d) Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2015, 54, 2255 CrossRef CAS PubMed; (e) J. Zhang, Y. Hou, Y. Ma and M. Szostak, J. Org. Chem., 2019, 84, 338 CrossRef CAS PubMed; (f) L. Ren, X. Li and N. Jiao, Org. Lett., 2016, 18, 5852 CrossRef CAS PubMed.
- For selected reports, see: (a) J. Shearer, C. X. Zhang, L. Q. Hatcher and K. D. Karlin, J. Am. Chem. Soc., 2003, 125, 12670 CrossRef CAS PubMed; (b) X. Li, J. Pan, H. Wu and N. Jiao, Chem. Sci., 2017, 8, 6266 RSC; (c) R. K. Kawade, D. B. Huple, R. J. Lin and R. S. Liu, Chem. Commun., 2015, 51, 6625 RSC; (d) M. L. Deb, C. D. Pegu, P. J. Borpatra and P. K. Baruah, RSC Adv., 2016, 6, 40552 RSC; (e) S. Guo, B. Qian, Y. Xie, C. Xia and H. Huang, Org. Lett., 2011, 13, 522 CrossRef CAS PubMed; (f) J.-S. Tian and T.-P. Loh, Angew. Chem., Int. Ed., 2010, 49, 8417 CrossRef CAS PubMed; (g) L. Huang, X. Zhang and Y. Zhang, Org. Lett., 2009, 11, 3730 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 1944894. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03081f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |