
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversal of reaction type selectivity by Lewis acid coordination: the ortho photocycloaddition of 1- and 2-naphthaldehyde†
Simone
Stegbauer
,
Noah
Jeremias
,
Christian
Jandl
and
Thorsten
Bach
 *
*
Department Chemie and Catalysis Research Center (CRC), Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; Fax: +49 89 28913315; Tel: +49 89 28913330
First published on 29th July 2019
Abstract
The value of a specific substrate class for synthetic applications is greatly enhanced if different types of reactions can be performed selectively upon a judicious choice of reaction conditions. In the present study it was shown that the typical photochemical behaviour of naphthaldehydes is completely altered in the presence of Lewis acids. Without Lewis acids, reactions at the carbonyl group are exclusively observed while Lewis acids facilitate a visible light-mediated cycloaddition at the arene core providing access to products of aromatic C–H functionalization via cyclobutane intermediates.
Introduction
Upon direct excitation, 1-naphthaldehyde (1) and 2-naphthaldehyde (2) undergo photochemical transformations at the aldehyde carbonyl group. Like for many other aromatic aldehydes, the typical reaction products are oxetanes (Paternò–Büchi reaction1,2),3 alcohols (photoreduction)4 or carbonyl addition products (photoaddition).5 The reaction of 1-naphthaldehyde (1) in 2,3-dihydrofuran for example has been reported to deliver oxetane 3 in a yield of 55% (Scheme 1).3d The reactivity pattern is determined by the nπ* character of the reactive excited states (singlet or triplet)6 or it is due to electron transfer pathways which generate a radical anion by reduction of the carbonyl group.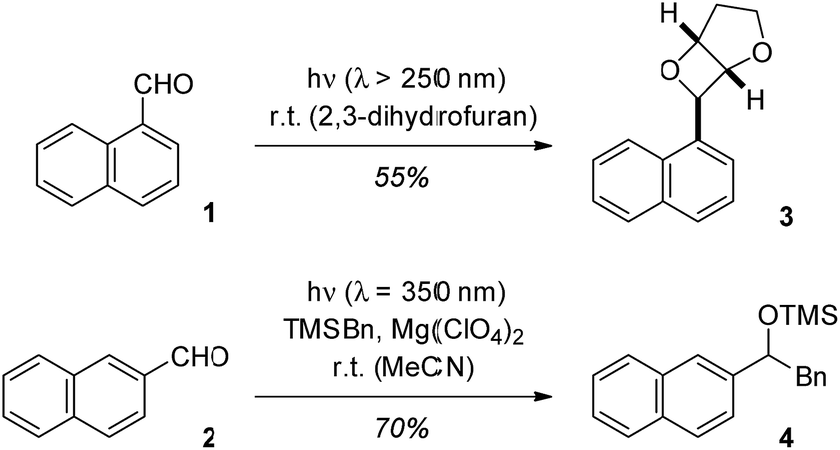 | ||
| Scheme 1 Paternò–Büchi reaction of 1-naphthaldehyde (1) to oxetane 3 (top) and Mg(ClO4)2-mediated photoaddition of benzyltrimethylsilane to 2-naphthaldehyde (2). | ||
In recent work, the influence of Lewis acids on photochemical reactions has been studied with a focus on enantioselective transformations.7 However, it was also noted that the Lewis acid modifies other selectivity parameters.8 We hypothesized that by Lewis acid coordination to the carbonyl group of naphthaldehydes their typical photochemical behaviour may change because the lowest excited states are of ππ* character. This speculation was supported by previous experiments on photoinduced electron transfer (PET) reactions of silanes to naphthaldehydes.9 In the absence of a Lewis acid the aldehydes are not sufficiently reactive to undergo a PET with silanes such as benzyltrimethylsilane (TMSBn). The addition of magnesium perchlorate (4 eq. relative to 2) allows for the desired transformation and product 4 was obtained in the reaction of 2-naphthaldehyde (2) and TMSBn (2 eq.).
In the context of these reactions, Fukuzumi and co-workers studied the photophysical properties of Lewis acid [Mg(ClO4)2 or Sc(OTf)3] complexes with naphthaldehydes 1 and 2 in great detail.9b While the enhanced reactivity in the reaction with silanes was ascribed to the higher redox potential of the photoexcited Lewis acid complex, it was also concluded that “the π,π* excited state becomes the lowest excited state in the Mg(ClO4)2 complex as compared with the lowest n,π* triplet excited state in the uncomplexed carbonyl compound”.
In the present study, we have compared the photochemical reactivity of 1- and 2-naphthaldehyde towards a typical olefin component (2,3-dimethyl-2-butene) in the absence and in the presence of catalytic amounts of strong Lewis acids. The Lewis acids induced a complete reversal of the type selectivity from carbonyl reactions in the absence of a Lewis acid to reactions at the C1/C2 double bond of the aromatic core. To the best of our knowledge, a complete type selectivity reversal in the photochemistry of aldehydes has not yet been reported. Previous studies in photochemistry have mainly focussed on the choice of irradiation wavelength10 or the addition of a triplet sensitizer. One of the most notable type selectivity changes is observed with β,γ-unsaturated enones which undergo an oxadi-π-methane rearrangement in the presence and a 1,3-acyl shift in the absence of a triplet sensitizer.11
Results and discussion
Although 1- and 2-naphthaldehyde had been previously shown to undergo Paternò–Büchi and photocycloaddition reactions with olefins3 we closely examined the yet unreported reaction with 2,3-dimethylbutene in order to rule out any involvement of the aromatic π system. Upon irradiation at λ = 366 nm product formation was observed and three major products could be isolated and identified for both aldehydes (Fig. 1). 1-Naphthaldehyde (1) delivered oxetane 5a and the two carbonyl addition products 6a and 7a in a combined yield of 63%. 2-Naphthaldehyde (2) reacted similarly and furnished oxetane 5b and the alcohols 6b and 7b in a combined yield of 79%. The reactions were notoriously slow and proceeded only with an acceptable conversion if performed in the olefin as the solvent.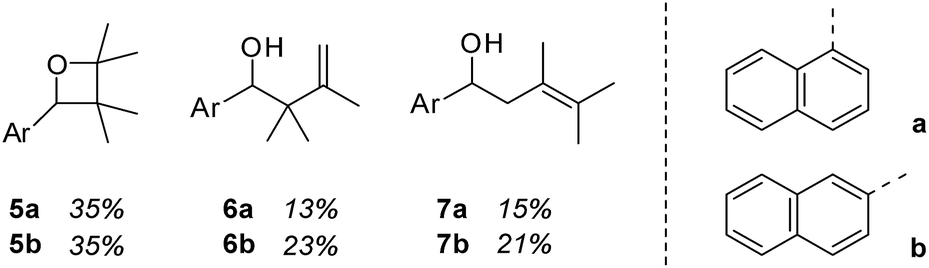 | ||
| Fig. 1 Products 5–7 obtained by irradiation (λ = 366 nm) of 1-naphthaldehyde (1) and 2-naphthaldehyde (2) in a solution of excess (100 eq.) 2,3-dimethyl-2-butene (t = 20 h). | ||
There was no photochemical reaction of 1 and 2 upon irradiation at a longer wavelength (λ ≥ 400 nm). This observation is in line with the fact that the two naphthaldehydes are colourless with no absorption bands in the visible part of the spectrum. The spectrum of 1-naphthaldehyde (1) is depicted in Fig. 2 (for the spectrum of 2, see the ESI†) that also displays the spectral changes upon gradual addition of EtAlCl2 as the Lewis acid. Isosbestic points at 255 nm, 288 nm, and 340 nm indicate that there are only two UV active species present in solution. The absorption of the Lewis acid complex stretches into the visible region and the solution of the complex is yellow-coloured. Likewise 2-naphthaldehyde (2) showed a less pronounced but clearly identifiable absorption maximum at λ = 398 nm.
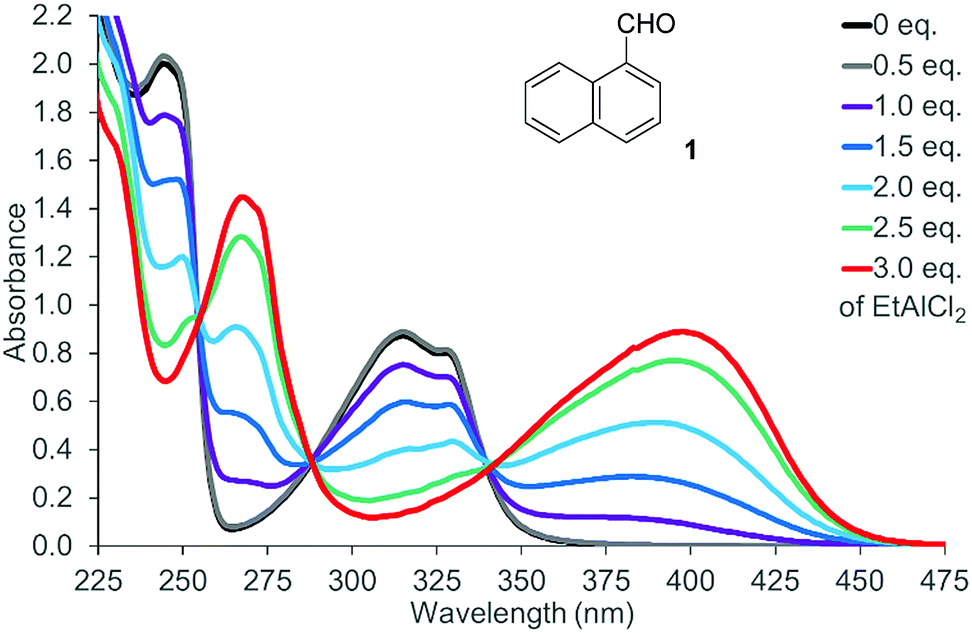 | ||
| Fig. 2 UV/Vis spectrum of 1-naphthaldehyde (1) (c = 1.0 mM in CH2Cl2) in the presence of variable equivalents (eq.) of EtAlCl2. | ||
Based on its UV/Vis spectra it was attempted to photochemically convert 1-naphthaldehyde (1) and 2,3-dimethylbutene in the presence of Lewis acids. At λ = 405 nm there was a Lewis acid catalysed reaction with EtAlCl2 and AlBr3 showing similar activity. The cleanest reaction was recorded with 25 mol% AlBr3 at −78 °C in dichloromethane solution (Scheme 2). In none of these reactions was there an indication for the formation of carbonyl addition products 5a–7a. Surprisingly, the obtained product was aldehyde 8 with a rearranged carbon skeleton. Deuterium labelling experiments revealed that the aldehyde proton remains at its position and that the proton at C2 migrates to C1 in the course of the reaction. Final proof for the structure was obtained upon reduction of aldehyde 8 to alcohol 9 which in turned delivered crystals suitable for single crystal X-ray crystallography.
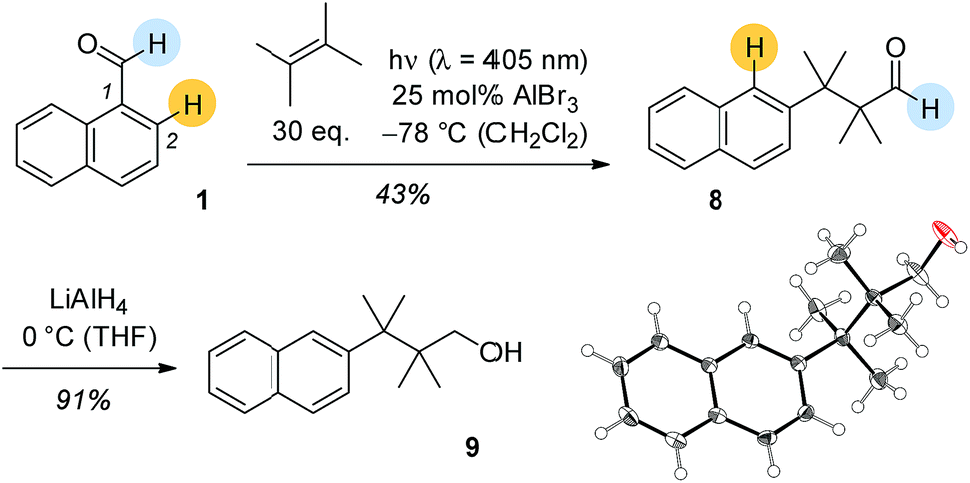 | ||
| Scheme 2 AlBr3-catalysed photochemical reaction of 1-naphthaldehyde (1) and 2,3-dimethyl-2-butene and consecutive reaction of aldehyde 8 to alcohol 9 the constitution of which was established by single crystal X-ray crystallography. The marked positions indicate the positions of hydrogen atoms before and after the reaction as determined by deuterium labelling. | ||
Since we speculated that aldehyde 8 was derived from an ortho [2 + 2] photocycloaddition12–14 to the naphthalene C1/C2 bond, cyclobutane 10 was prepared by an independent route (see ESI†) and subjected to Lewis acidic conditions (Scheme 3). The formation of aldehyde 8 supported our hypothesis and suggests a reaction pathway that commences with Lewis acid coordination to the aldehyde carbonyl group of 10. The reaction cascade is subsequently initiated by a 1,2-shift to the cationic centre15,16 generating benzylic cation 11. An intramolecular hydride shift delivers cation 12 which eventually undergoes ring opening to the final product.
 | ||
| Scheme 3 Suggested reaction course for the formation of aldehyde 8 from ortho photocycloaddition product 10via intermediates 11 and 12. | ||
Once known from independent synthesis,17 minor amounts of cyclobutane 10 were also identified and isolated from the product mixture of some Lewis acid catalysed reactions lending further evidence towards its temporary existence in the formation of aldehyde 8.
In the case of 2-naphthaldehyde (2), the cyclobutane reaction product was stable and could be isolated. A low Lewis acid loading of either EtAlCl2 or AlBr3 was sufficient to alter the type selectivity of the photochemical reaction with 2,3-dimethylbutene (Scheme 4). As for 1-naphthaldehyde (1) there was no reaction observed at the aldehyde carbonyl atom and products 5b–7b were not detected in the Lewis acid catalysed reactions. The optimal irradiation source turned out to be a light emitting diode (LED) with an emission maximum at λ = 457 nm (Scheme 4). When the photocycloaddition was performed with 5 mol% of AlBr3 in dichloromethane at −78 °C 56% of product 13 were isolated. With other Lewis acids [BF3, AlCl3, Sc(OTf)3] there was no reaction under these conditions.
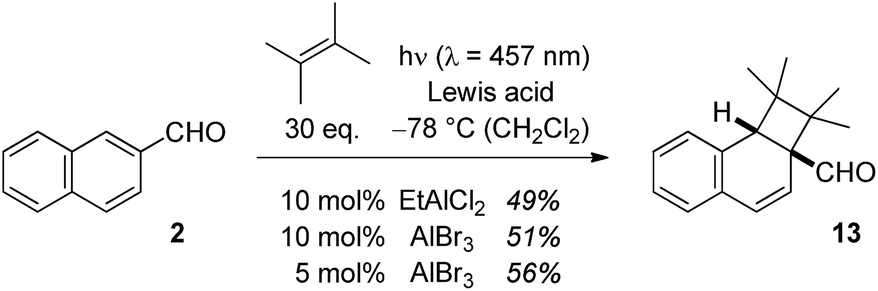 | ||
| Scheme 4 Intermolecular Lewis acid catalysed ortho photocycloaddition of 2-naphthaldehyde (2) and 2,3-dimethylbutene to cyclobutane 13. | ||
In the reaction with AlBr3 as catalyst a side product was obtained in varying amounts and there were hints that the product was a consecutive product of cyclobutane 13. If the reaction mixture was warmed to 0 °C after complete photochemical conversion, a further transformation to this product continued which was complete after another 12 hours. The product was identified as alcohol 14 which could be isolated in 74% yield (Scheme 5). The reaction cascade thus offers access to a formal [3 + 2] cycloaddition product of 2-naphthaldehyde with C–C bond formation at the aldehyde carbon atom and at carbon atom C1.
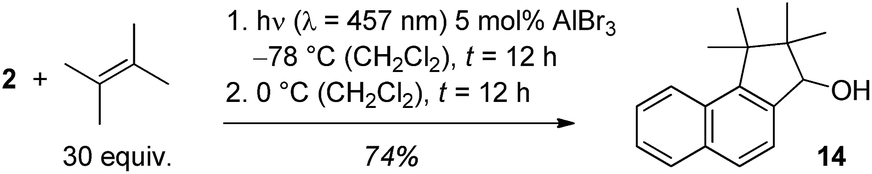 | ||
| Scheme 5 Optimised conditions for the generation of secondary alcohol 14 by a sequence of AlBr3-catalysed ortho photocycloaddition and rearrangement. | ||
Despite the fact that the synthetic scope of the ortho photocycloaddition was not in the focus of our current experiments we performed a related formal [3 + 2] cycloaddition sequence with 2-naphthaldehyde (2) and cyclopentene. After the photocycloaddition/rearrangement cascade (λ = 457 nm, 5 mol% AlBr3, CH2Cl2) the expected alcohol could not be isolated in pure form. It was therefore oxidised (Dess–Martin periodinane) to the respective ketone 15 (Fig. 3) which was isolated in an overall yield of 41% as a single diastereoisomer.
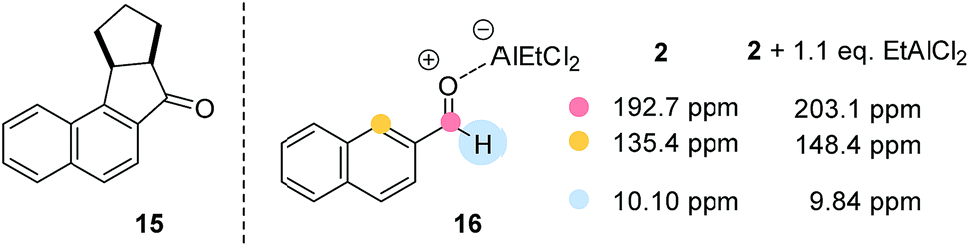 | ||
| Fig. 3 Structure of the photocycloaddition/rearrangement cascade product 15 (left) and selected NMR shift changes (right) in the Lewis acid complex 16 derived from 2-naphthaldehyde (2) and EtAlCl2 (−60 °C, CD2Cl2). | ||
Some preliminary mechanistic experiments18 were performed to shed light on the underlying principles of the observed type selectivity reversal. NMR spectra of 2-naphthaldehyde (2) were recorded at −60 °C in CD2Cl2 solution (see the ESI† for details). Upon addition of EtAlCl2 (1.1 eq.) as Lewis acid there were marked changes19 in the NMR shift data which are in full agreement with a coordination of the Lewis acid to the oxygen atom of the carbonyl group. Most notable are the shift changes to lower field for the 13C NMR signals of the carbonyl carbon atom and the carbon atom C1 (Fig. 3). In the 1H NMR spectrum the shielding of the aldehyde proton is most prominent. Together with the UV/Vis spectra (isosbestic point) it seems reasonable to assume that upon addition of Lewis acid a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex 16 is formed. Attempts to detect this species by luminescence were successful and luminescence spectra could be obtained for both aldehydes 1 and 2 in the presence of EtAlCl2 (3 eq.) as the Lewis acid in dichloromethane solution. The lifetimes of the luminescent state as determined by time-dependent spectra are below 10 μs (detection limit). Given the fact that the emission was observed in solution and given the short lifetime of the luminescent state it appears reasonable to assume that the luminescence is due to emission from the singlet state S1 (fluorescence). A delayed luminescence (phosphorescence) could be detected neither in solution nor in a matrix. The fluorescence maxima of the EtAlCl2 complexes were determined to be at λ = 468 nm for 1 (λexc = 400 nm) and at λ = 478 nm for 2 (λexc = 390 nm, Fig. 4).
:1 complex 16 is formed. Attempts to detect this species by luminescence were successful and luminescence spectra could be obtained for both aldehydes 1 and 2 in the presence of EtAlCl2 (3 eq.) as the Lewis acid in dichloromethane solution. The lifetimes of the luminescent state as determined by time-dependent spectra are below 10 μs (detection limit). Given the fact that the emission was observed in solution and given the short lifetime of the luminescent state it appears reasonable to assume that the luminescence is due to emission from the singlet state S1 (fluorescence). A delayed luminescence (phosphorescence) could be detected neither in solution nor in a matrix. The fluorescence maxima of the EtAlCl2 complexes were determined to be at λ = 468 nm for 1 (λexc = 400 nm) and at λ = 478 nm for 2 (λexc = 390 nm, Fig. 4).
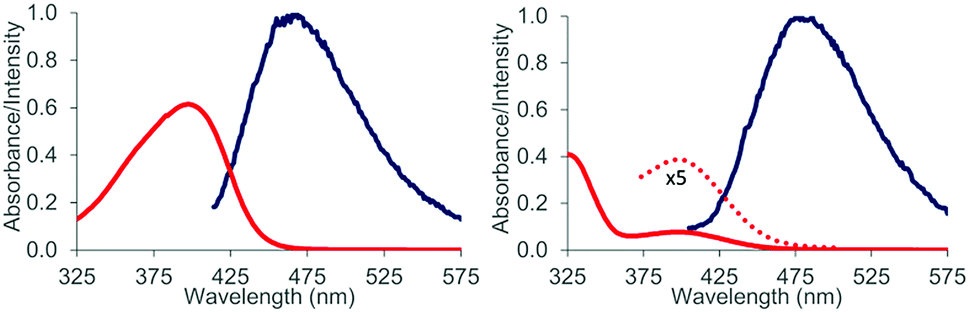 | ||
| Fig. 4 Normalized absorption and emission spectra as measured for a CH2Cl2 solution (0.1 mM) of 1-naphthaldehyde (1, left) and 2-naphthaldehyde (2, right) in the presence of three equivalents EtAlCl2. For clarity only the region between λ = 325–575 nm is displayed and the weak long wavelength absorption obtained for complex 16 (2·AlEtCl2) is also expanded by a factor of five (dotted line). | ||
The emission behaviour of the EtAlCl2 complexes is in stark contrast to the uncomplexed naphthaldehydes which are non-fluorescent in solution but show a strong phosphorescence in a dichloromethane matrix at 77 K (see the ESI† for more details). Our results are in agreement with previous work of Fukuzumi et al. (vide supra) who had reported for the complexes of Mg(ClO4)2 and naphthaldehydes 1 and 2 a fluorescence in acetonitrile solution with emission maxima at λ = 437 nm and at λ = 440 nm.9b With Sc(OTf)3 the emission maxima were red-shifted to λ = 487 nm and λ = 510 nm. In the Fukuzumi study, the fluorescence was quenched upon addition of silanes and products of a PET were obtained (cf.Scheme 1). Surprisingly, in our case, the fluorescence of the naphthaldehyde-EtAlCl2 complexes was not quenched upon addition of up to 1000 equivalents of 2,3-dimethylbutene. The fluorescence intensity remained identical and there was no change in shape of the fluorescence signal. The result makes it unlikely that the fluorescent states are involved in the observed ortho photocycloaddition. Further evidence that the photocycloaddition does not occur from a singlet state intermediate was obtained from the reaction of 3-hexene (17) with 2-naphthaldehyde (2). The reaction was not stereospecific as both (E)-17 and (Z)-17 delivered mainly a single product 18. The reaction time was kept identical (12 hours) and starting material was recovered in both experiments (20% and 38%). The yields given in Scheme 6 are not corrected for conversion. When calculated relative to conversion the yield for 18 was 78% from (E)-17 and 63% from (Z)-17.
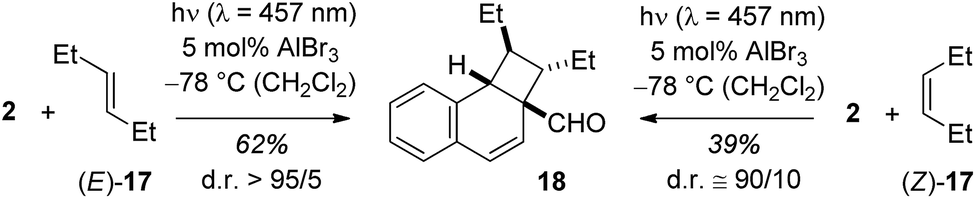 | ||
| Scheme 6 Stereoconvergent reaction course in the Lewis acid catalysed ortho photocycloaddition of 2-naphthaldehyde (2) and 3-hexene (17). | ||
Based on NOESY data for 18 and for the alcohol obtained from 18 by reduction (LiAlH4 in THF, 88% yield, see ESI† for further details), the relative configuration of the two ethyl groups within the cyclobutane ring is trans. Formation of this product from (Z)-17 requires the involvement of an intermediate in which free rotation is possible, potentially a triplet 1,4-diradical.20
Despite this circumstantial evidence, any conclusions regarding the mechanism of the Lewis acid catalysed ortho photocycloaddition reactions are premature at this point in time. When the UV/Vis spectra of 1- and 2-naphthaldehyde were recorded in the presence of a large (>3 eq.) excess of EtAlCl2 (see ESI†) they indicated the formation of new species which could potentially also be photochemically active. It is well established that a minor component can be responsible for the reactivity in a catalytic reaction.21 The exact assignment of these species needs to be performed and it will also be required to learn more about the reactivity features of the 1:1 Lewis acid complexes. The sheer number of various ππ* transition accessible to naphthaldehydes and other aromatic aldehydes complicates the analysis which is much more facile for α,β-unsaturated carbonyl compounds where apart from the nπ* transition only a single ππ* transition is responsible for the photochemical reactivity.
Conclusion
In summary, we have found that naphthaldehydes change their photochemical reactivity pattern dramatically in the presence of EtAlCl2 or AlBr3 as Lewis acids. The site of reactivity is shifted from the carbonyl group to the C1/C2 arene double bond where an initial ortho photocycloaddition with olefins occurs. Consecutive Lewis acid promoted reactions lead to an opening of the cyclobutane ring and to the formation of a C2-alkylated naphthalene in the case of 1-naphthaldehyde (1) and to a formal [3 + 2] cycloaddition product for 2-naphthaldehyde (2). Both reaction pathways create selectively C–C bonds at the naphthalene skeleton at the expense of existing C–H bonds. In combination with the facile execution of the reaction which requires only catalytic amounts (5–25 mol%) of the Lewis acid and visible light as energy source (λ = 405 or 457 nm) this feature should make the reactions a valuable supplement to the portfolio of C–C bond formation reactions at the naphthalene core.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 665951 – ELICOS) and by the Fonds der Chemischen Industrie (PhD fellowship to S. S.) is gratefully acknowledged. We thank Dr J. D. Jolliffe for his help with crystal structure analysis and Dr T. S. Chung for the fluorescence measurements.Notes and references
- (a) E. Paternò and G. Chieffi, Gazz. Chim. Ital., 1909, 39, 341 Search PubMed; (b) G. Büchi, C. G. Inman and E. S. Lipinsky, J. Am. Chem. Soc., 1954, 76, 4327 CrossRef.
- For reviews on the Paternò–Büchi reaction: (a) M. D'Auria, Photochem. Photobiol. Sci., 2019 10.1039/c9pp00148d; (b) M. Abe, in Handbook of Synthetic Photochemistry, ed. A. Albini and M. Fagnoni, Wiley-VCH, Weinheim, 2009, p. 217 Search PubMed; (c) A. G. Griesbeck and S. Bondock, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. Horspool and F. Lenci, CRC Press, Boca Raton, 2nd edn, 2004, pp. 60–1 Search PubMed; (d) T. Bach, Synthesis, 1998, 683 CrossRef CAS.
- (a) N. C. Yang, Pure Appl. Chem., 1964, 9, 591 Search PubMed; (b) N. C. Yang, M. Nussim, M. J. Jorgenson and S. Murov, Tetrahedron Lett., 1964, 3657 CrossRef CAS; (c) N. C. Yang, M. Kimura and W. Eisenhardt, J. Am. Chem. Soc., 1973, 95, 5058 CrossRef CAS; (d) A. G. Griesbeck, H. Mauder, K. Peters, E.-M. Peters and H. G. von Schnering, Chem. Ber., 1991, 124, 407 CrossRef CAS; (e) M. Abe, M. Ikeda, Y. Shirodai and M. Nojima, Tetrahedron Lett., 1996, 37, 5901 CrossRef CAS; (f) M. Abe, M. Ikeda and M. Nojima, J. Chem. Soc., Perkin Trans. 1, 1998, 3261 RSC; (g) A. G. Griesbeck, S. Bondock and J. Lex, J. Org. Chem., 2003, 68, 9899 CrossRef CAS PubMed.
- (a) J. Khan and S. G. Cohen, J. Org. Chem., 1991, 56, 938 CrossRef CAS; (b) S. Kohtani, E. Yoshioka, K. Saito, A. Kudo and H. Miyabe, Catal. Commun., 2010, 11, 1049 CrossRef CAS; (c) D. W. Manley, L. Buzzetti, A. MacKessack-Leitch and J. C. Walton, Molecules, 2014, 19, 15324 CrossRef PubMed.
- (a) A. Takuwa, H. Tagawa, H. Iwamoto, O. Soga and K. Maruyama, Chem. Lett., 1987, 1091 CrossRef CAS; (b) S. V. Kessar, A. K. S. Mankotia, J. C. Scaiano, M. Barra, J. Gebicki and K. Huben, J. Am. Chem. Soc., 1996, 118, 4361 CrossRef CAS; (c) S. S. Kim, Y. J. Mah and A. R. Kim, Tetrahedron Lett., 2001, 42, 8315 CrossRef CAS; (d) S. S. Kim, J. A. Chang and Y. J. Mah, J. Photosci., 2002, 9, 5 CAS; (e) J. Sun, X. Peng and H. Guo, Tetrahedron Lett., 2015, 56, 797 CrossRef CAS.
- (a) N. C. Yang, S. L. Murov and T.-C. Shieh, Chem. Phys. Lett., 1969, 3, 6 CrossRef CAS; (b) D. W. Boldridge, B. L. Justus and G. W. Scott, J. Chem. Phys., 1984, 80, 3179 CrossRef CAS; (c) A. M. J. van Eijk, G. B. Ekelmans, W. Klinkenberg, A. H. Huizer and C. A. G. O. Varma, J. Chem. Soc., Faraday Trans., 1990, 86, 2083 RSC.
- (a) R. Brimioulle and T. Bach, Science, 2013, 342, 840 CrossRef CAS PubMed; (b) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef CAS PubMed; (c) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391 CrossRef CAS PubMed; (d) X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120 CrossRef CAS PubMed; (e) S. Poplata and T. Bach, J. Am. Chem. Soc., 2018, 140, 3228 CrossRef CAS PubMed; (f) S. Poplata, A. Bauer, G. Storch and T. Bach, Chem.–Eur. J., 2019, 25, 8135 CAS.
- Review: C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338 CrossRef CAS PubMed.
- (a) S. Fukuzumi, T. Okamoto and J. Otera, J. Am. Chem. Soc., 1994, 116, 5503 CrossRef CAS; (b) S. Fukuzumi, N. Satoh, T. Okamoto, K. Yasui, T. Suenobu, Y. Seko, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 7756 CrossRef CAS PubMed.
- For reviews on the wavelength selectivity of photochemical reactions: (a) S. Protti, D. Ravelli and M. Fagnoni, Photochem. Photobiol. Sci., 10.1039/c8pp00512e; (b) N. J. Turro, V. Ramamurthy, W. Cherry and W. Farneth, Chem. Rev., 1978, 78, 125 CrossRef CAS.
- For reviews on the photochemistry of β,γ-unsaturated enones: (a) M. G. Banwell and D. J.-Y. D. Bon, in Molecular Rearrangements in Organic Synthesis, ed. C. M. Rojas, Wiley, Hoboken, NJ, 2015, ch. 9, p. 261 Search PubMed; (b) T. Tsuno, in Handbook of Synthetic Photochemistry, ed. A. Albiniand M. Fagnoni, Wiley-VCH, Weinheim, 2010, p. 95 Search PubMed; (c) D. Armesto, M. J. Ortiz and A. R. Agarrabeitia, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. Horspool and F. Lenci, CRC Press, Boca Raton, 2nd edn, 2004, pp. 77–1 Search PubMed; (d) H. E. Zimmerman and D. Armesto, Chem. Rev., 1996, 96, 3065 CrossRef CAS PubMed; (e) K. N. Houk, Chem. Rev., 1976, 76, 1 CrossRef CAS.
- Reviews: (a) R. Remy and C. G. Bochet, Chem. Rev., 2016, 116, 9816 CrossRef CAS PubMed; (b) N. Hoffmann, Photochem. Photobiol. Sci., 2012, 11, 1613 RSC; (c) U. Streit and C. G. Bochet, Beilstein J. Org. Chem., 2011, 7, 525 CrossRef CAS PubMed; (d) U. Streit and C. G. Bochet, Chimia, 2008, 62, 962 CrossRef CAS; (e) J. Mattay, Angew. Chem., Int. Ed., 2007, 46, 663 CrossRef CAS PubMed; (f) N. Hoffmann, Synthesis, 2004, 481 CrossRef CAS; (g) J. Cornelisse and R. de Haan, in Molecular and Supramolecular Photochemistry, ed. V. Ramamurthy and K. S. Schanze, Dekker, New York, 2001, vol. 8, p. 1 Search PubMed; (h) P. J. Wagner, Acc. Chem. Res., 2001, 34, 1 CrossRef CAS PubMed.
- For some recent contributions to dearomatization by arene photocycloaddition, see: (a) U. Streit, F. Birbaum, A. Quattropani and C. G. Bochet, J. Org. Chem., 2013, 78, 6890 CrossRef CAS PubMed; (b) A. Zech and T. Bach, J. Org. Chem., 2018, 83, 3069 CrossRef CAS PubMed; (c) M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624 CrossRef CAS PubMed; (d) T. W. Bingham, L. W. Hernandez, D. G. Olson, R. L. Svec, P. J. Hergenrother and D. Sarlah, J. Am. Chem. Soc., 2019, 141, 657 CrossRef CAS PubMed.
- For a para photocycloadditon to 1-naphthaldehyde (1), see: D. Döpp and H. R. Memarian, Chem. Ber., 1990, 123, 315 CrossRef.
- (a) M. Yamashita, J. Onozuka, G.-i. Tsuchihashi and K. Ogura, Tetrahedron Lett., 1983, 24, 79 CrossRef CAS; (b) K. Ishihara and K. Nakano, J. Am. Chem. Soc., 2007, 129, 8930 CrossRef CAS PubMed; (c) À. Gutiérrez-Bonet, A. Flores-Gaspar and R. Martin, J. Am. Chem. Soc., 2013, 135, 12576 CrossRef PubMed.
- For reviews on rearrangement of carbocations by a 1,2-shift see: (a) X.-M. Zhang, Y.-Q. Tu, F.-M. Zhang, Z.-H. Chen and S.-H. Wang, Chem. Soc. Rev., 2017, 46, 2272 RSC; (b) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef CAS PubMed; (c) V. G. Shubin, Top. Curr. Chem., 1984, 117, 267 Search PubMed.
- J. J. McCullough, R. C. Miller and W.-S. Wu, Can. J. Chem., 1977, 55, 2909 CrossRef CAS.
- For seminal studies on the influence of Lewis acids on the photochemistry of carbonyl compounds, see: (a) F. D. Lewis and S. V. Barancyk, J. Am. Chem. Soc., 1989, 111, 8653 CrossRef CAS; (b) F. D. Lewis, S. V. Barancyk and E. L. Burch, J. Am. Chem. Soc., 1992, 114, 3866 CrossRef CAS.
- (a) R. F. Childs, D. L. Mulholland and A. Nixon, Can. J. Chem., 1982, 60, 801 CrossRef CAS; (b) P. Laszlo and M. Teston, J. Am. Chem. Soc., 1990, 112, 8750 CrossRef CAS.
- (a) W. L. Dilling, T. E. Tabor, F. P. Boer and P. P. North, J. Am. Chem. Soc., 1970, 92, 1399 CrossRef CAS; (b) N. P. Peet, R. L. Cargill and D. F. Bushey, J. Org. Chem., 1973, 38, 1218 CrossRef CAS; (c) N. C. Yang, M. Kimura and W. Eisenhardt, J. Am. Chem. Soc., 1973, 95, 5058 CrossRef CAS; (d) J. F. D. Kelly, J. M. Kelly and T. B. H. McMurry, J. Chem. Soc., Perkin Trans. 2, 1999, 1933 RSC; (e) R. Brimioulle, A. Bauer and T. Bach, J. Am. Chem. Soc., 2015, 137, 5170 CrossRef CAS PubMed.
- (a) J. Halpern, Science, 1982, 217, 401 CrossRef CAS PubMed; (b) J. M. Brown, Chem. Soc. Rev., 1993, 22, 25 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization for all starting materials and products (5–10, 13–15, 18), spectroscopic data, low temperature NMR spectra. CCDC 1937926. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03315g |
| This journal is © The Royal Society of Chemistry 2019 |