
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silicon clusters with six and seven unsubstituted vertices via a two-step reaction from elemental silicon†‡
Lorenz J.
Schiegerl
ab,
Antti J.
Karttunen
 c,
Wilhelm
Klein
a and
Thomas F.
Fässler
*ab
c,
Wilhelm
Klein
a and
Thomas F.
Fässler
*ab
aDepartment of Chemistry, Technische Universität München, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: thomas.faessler@lrz.tum.de
bWACKER Institute of Silicon Chemistry, Technische Universität München, Lichtenbergstraße 4, 85748 Garching, Germany
cDepartment of Chemistry and Materials Science, Aalto University, 00076 Aalto, Finland
First published on 15th August 2019
Abstract
Unsaturated silicon clusters with only partial substitution, and thus, “naked” Si atoms are well studied species as they are proposed intermediates in gas-phase deposition processes. Although a remarkable number of stable molecular clusters has been reported, they are typically still obtained by multi-step syntheses. Herein we introduce a newly developed synthetic approach which led to the formation of the anionic species {Si(TMS)3}3Si9− (1a) and {Si(TMS)3}2Si92− (1b), and an extension of this synthetic protocol resulted in the first covalent attachment of ligands through metal atoms to these clusters, (SnCy3)3Si9− (2a) and (SnCy3)2Si92− (2b). The influence of the substituents on the electron localization in the central Si9 unit is analyzed by means of intrinsic bond orbital (IBO) analysis and partial atomic charge distribution. The IBO analyses reveal a new type of delocalization including 5-center-6-electron besides 3-center-2-electron bonds. The Raman spectra of 1b and 2b allow an assignment of the Si–Si intra-cluster vibrations by comparison to calculated (DFT-PBE0) spectra. The anions are formed in a one-step synthesis from binary K12Si17 which can easily be obtained by fusing the elements K and Si. The anions are characterized by ESI mass spectrometry and comprehensive NMR studies (1H, 13C, 29Si, 119Sn). Attempts to crystallize 1a and 2a as their (K–222crypt)+ salts yielded after the loss of one of the substituents single crystals containing 1b and 2b. The single crystal X-ray structure analyses reveal the presence of anionic siliconoids with surfaces of seven unsubstituted silicon atoms.
Introduction
The call for new sources of silicon-based materials is steadily increasing due to applications in numerous daily-life products as e.g. batteries, photovoltaics and electronic devices.1–8 The wide range of applications is promoted by the abundancy, low costs, non-toxicity and semiconducting properties of silicon. After the first reports on Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si double bonds9,10 a new field for the exploration of tailor-made low-valent silicon compounds has been established that undergoes constant expansion, mirrored e.g. by the synthesis of stable silaethenes (SiMe3)2SiC(OSiMe3)R (R = adamantyl, CEt3, CMe),11,12 compounds with silicon–silicon triple bonds as in (iPrR′2)Si–Si
Si double bonds9,10 a new field for the exploration of tailor-made low-valent silicon compounds has been established that undergoes constant expansion, mirrored e.g. by the synthesis of stable silaethenes (SiMe3)2SiC(OSiMe3)R (R = adamantyl, CEt3, CMe),11,12 compounds with silicon–silicon triple bonds as in (iPrR′2)Si–Si![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–Si(R′2iPr) (R′ = CH(SiMe3)2),13 a stable silylene (CH)2(NC(CH3)3)2Si,14 an aromatic hexasilabenzene isomer (Tip)6Si6 (Tip = 2,4,6-triisopropylphenyl),15 the triatomic Si(0) unit (CAAC)3Si3 (CAAC = cyclic (alkyl)amino carbene),16 and of so-called siliconoid clusters.17–21 Siliconoids are best described as partially substituted silicon clusters with ligand-free silicon atoms (Fig. 1), and further modifications of such silicon compounds have frequently been achieved, which underlines their versatile synthetic potential for the formation of silicon-based materials.19–27
Si–Si(R′2iPr) (R′ = CH(SiMe3)2),13 a stable silylene (CH)2(NC(CH3)3)2Si,14 an aromatic hexasilabenzene isomer (Tip)6Si6 (Tip = 2,4,6-triisopropylphenyl),15 the triatomic Si(0) unit (CAAC)3Si3 (CAAC = cyclic (alkyl)amino carbene),16 and of so-called siliconoid clusters.17–21 Siliconoids are best described as partially substituted silicon clusters with ligand-free silicon atoms (Fig. 1), and further modifications of such silicon compounds have frequently been achieved, which underlines their versatile synthetic potential for the formation of silicon-based materials.19–27
 | ||
| Fig. 1 Selected examples of known molecular silicon cluster species with naked cluster atoms: (a) (SiMeDis2)3Si4− (Dis = CH(SiMe3)2);28 (b) {Si(SiMetBu2)3}4Si5;29 (c) the siliconoid (Tip)5Si6− (Tip = 2,4,6-triisopropylphenyl);18 (d) (SitBu3)6Si8;21 (e) (tBu)4{C4(SiMe3)4}2Si8;20 (f) the siliconoid (SiHtBu2)2Si92− (3b).30 Silicon clusters are shown as yellow polyhedra; naked and substituted Si cluster atoms are shown as red and black spheres, respectively, and the cluster substituents are drawn in the wire-and-stick mode. | ||
Their synthesis has made major progress in recent years, and just lately a step-wise, atomically precise expansion of the anionic siliconoid (Tip)5Si6− (Fig. 1b) was reported using (Cp*)2Si.31 The formation of siliconoids of higher nuclearity from molecular precursors, however, generally affords several synthetic steps. In 1993, Wiberg et al. already suggested in their report on the synthesis of (SitBu3)4Si4 that such substituted Si4 cluster compounds should probably be accessible in a more straightforward manner through the reaction of alkyl halides with tetrahedral Si44− polyanions that occur in binary alkali metal alloys of silicon.32 This idea was promoted by subsequent reports on (SiMeDis2)3Si4− (Dis = CH(SiMe3)2)28 (Fig. 1a) and (SitBu3)3Si4− (ref. 33) in which – again via molecular precursors – tri-substituted tetrahedral Si4 clusters have been obtained. The idea of using Zintl anions as precursors for Si-rich molecules has constantly been pursued for more than two decades, nevertheless, many approaches were repeatedly discarded due to the high reducing properties of such silicides.19,32
Examples of Zintl phases with deltahedral clusters are A4Si4 (ref. 34–37) (A = Li–Cs) and A12Si17 (ref. 38 and 39) (A = K–Cs), which contain solely Si44− units and Si44− alongside Si94− in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, respectively. The Si44− and Si94− clusters are unsaturated species with interesting properties firstly due to their nucleophilic character (multiple negative charge). Secondly, they are electrophilic in character at the same time due to an electron-deficient bonding situation of the cluster skeleton.40,41 Since the A4Si4 phases are rather insoluble in any solvent, the focus was set on the A12Si17 phases which are soluble in liquid ammonia, from which solvates containing Si44− (ref. 42 and 43) and Si94− (ref. 43–45) units could be obtained, and it has been shown that the Si44−Zintl clusters from such an A12Si17 phase are receptive to chemical conversion. (CuMes)2Si44− (ref. 46) was obtained by the reaction with CuMes (Mes = 1,3,5-trimethylbenzene). But also the Si94− clusters could be derivatized e.g. by the addition of transition metal fragments yielding (PhZn)Si93−,47 ({Ni(CO)2}2Si9)28− (ref. 48) and (NHCDippCu)Si93− (NHCDipp = 1,3-bis-(2,6-di-iso-propylphenyl)imidazole-2-ylidene).49
:1 ratio, respectively. The Si44− and Si94− clusters are unsaturated species with interesting properties firstly due to their nucleophilic character (multiple negative charge). Secondly, they are electrophilic in character at the same time due to an electron-deficient bonding situation of the cluster skeleton.40,41 Since the A4Si4 phases are rather insoluble in any solvent, the focus was set on the A12Si17 phases which are soluble in liquid ammonia, from which solvates containing Si44− (ref. 42 and 43) and Si94− (ref. 43–45) units could be obtained, and it has been shown that the Si44−Zintl clusters from such an A12Si17 phase are receptive to chemical conversion. (CuMes)2Si44− (ref. 46) was obtained by the reaction with CuMes (Mes = 1,3,5-trimethylbenzene). But also the Si94− clusters could be derivatized e.g. by the addition of transition metal fragments yielding (PhZn)Si93−,47 ({Ni(CO)2}2Si9)28− (ref. 48) and (NHCDippCu)Si93− (NHCDipp = 1,3-bis-(2,6-di-iso-propylphenyl)imidazole-2-ylidene).49
However, a synthetic approach including Si44− or Si94− units to form covalent bonds to ligands is still missing, although such reactions of the corresponding Ge94− clusters (from the precursor Zintl phase K4Ge9)50–53 are very well known. Investigations on the solubility of the silicon clusters in A12Si17 revealed that in liquid ammonia solution the mono-protonated species HSi93− (ref. 45 and 54) is present, and that a subsequent transfer to pyridine yields even the doubly-protonated species H2Si92−.55 Furthermore, theoretical studies suggest the formation of siliconoids with Si94− units by the attachment of sp3-Si linkers.56 Just recently, we used Si9 clusters from the precursor K12Si17 for the production of the anionic siliconoids (SiHtBu2)3Si9− (3a) and (SiHtBu2)2Si92− (3b, Fig. 1d) via direct ligand attachment.30 The silylation of Si9 clusters by the reaction of K12Si17 with SiHtBu2Cl yields species with covalently bonded SiHtBu2 groups at the cluster vertex atoms. These siliconoids contain the so far highest number of unsubstituted silicon atoms (six in 3a and seven in 3b) and are more easily accessible than those siliconoids obtained via the established “molecular multi-step” approaches. By a recent definition of siliconoids,19 the unsubstituted Si atoms only display homoatomic bonds with a hemispheroidal coordination sphere and are free of ligands. Herein we report on the reactivity of K12Si17 towards SiTMS3Cl and SnCy3Cl (TMS = trimethylsilane, Cy = cyclohexyl).
Results and discussion
The recently introduced synthetic route for the formation of anionic siliconoids by substitution of Si9 clusters from K12Si17 was further explored employing SiTMS3Cl and SnCy3Cl as reagents. Reactions of Ge9 clusters from the K4Ge9 precursor with such reactants led to an attachment of silyl51,57–61 and stannyl62,63 groups at the cluster cores, and the products were characterized as tri-substituted cluster species by ESI-MS and NMR investigations in solution as well as by X-ray analysis in solid-state. Regarding the corresponding reaction of Si9 clusters from K12Si17 in solution, an “activation” of the precursor by liquid NH3/222crypt30,55 has been found to be a key-step. The silylation of Si9 was accomplished by the reaction of K12Si17 after pretreatment with liquid NH3/222crypt30 with Si(TMS)3Cl and SnCy3Cl in thf and pyridine, respectively, and yielded deep brownish filtrates which were dried in vacuo. Digesting the residue with fluorobenzene, decanting of the solutions, and removal of all volatile ingredients yielded bulk materials which were further characterized by ESI-MS and NMR spectroscopy (1H, 13C, 119Sn, 29Si).The ESI-MS spectrum of the reaction product with SiTMS3Cl shows the mass peak of the tri-silylated species {Si(TMS)3}3Si9− (1a) as single species (Fig. 2a). Fragmentation of the isolated mass peak leads to the corresponding di- and mono-silylated cluster species (Fig. 2b) and confirms the composition of 1a. The isotope distribution of the mass peaks comprises a unit of 21 silicon atoms in accordance with the presence of three Si(TMS)3 substituents at the Si9 cluster. In analogy, the ESI-MS spectrum of the reaction product with SnCy3Cl shows the mass peak for the tri-stannylated species (SnCy3)3Si9− (2a) (Fig. 2c). However, the mass peak of the di-stannylated species (SnCy3)2Si9− (Fig. 2d) was also present, although with lower intensity. A fragmentation mass experiment of 2a confirmed the composition by the loss of SnCy3 in analogy to 1a and 3a.30
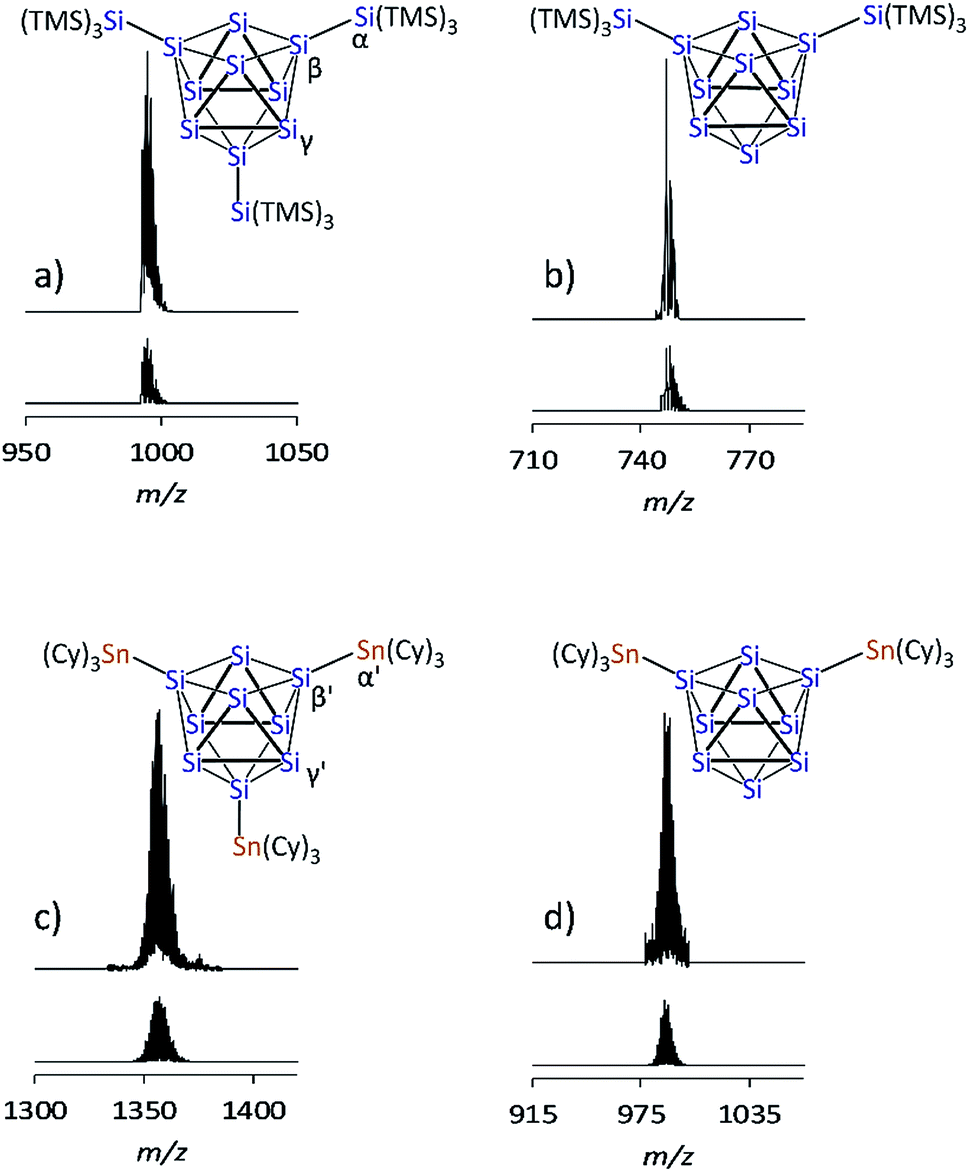 | ||
| Fig. 2 ESI-MS mass peaks of the Si9 species (top: measured spectrum, bottom: simulated isotope pattern): (a) {Si(TMS)3}3Si9 (1a), m/z = 996; (b) {Si(TMS)3}2Si9 from mass fragmentation (1b), m/z = 748; (c) (SnCy3)3Si9 (2a), m/z = 1357; (d) (SnCy3)2Si9 (2b), m/z = 989. For details of the measurement, see ESI.‡ | ||
1H and 13C NMR spectra of 1a (NMR spectra in the ESI‡) exclusively show one type of TMS group (1H: 0.25 ppm; 13C: 3.51 ppm) and one set of signals for 222crypt (1H: 3.64, 3.59, 2.61 ppm; 13C: 71.47, 67.44, 55.01 ppm). The 1H NMR integral ratio of TMS:222crypt = 3:1 confirms the composition of a triply silylated species for 1a (Fig. 2a). The 1H and 13C NMR spectra of 2a show the expected signals for the cyclohexyl groups. The Cy signals appear superimposed due to their signal splitting, but the 13C NMR spectrum reveals four peaks (33.96, 31.10, 25.86, 21.54 ppm) for the SnCy3 groups of 2a. A 119Sn NMR measurement reveals one single peak at −70.50 ppm, indicative of one sort of SnCy3 groups in the reaction product, although the integral ratio (SnCy3:222crypt) in the 1H NMR measurement does not perfectly match a ratio of 3:1 of 2a. Most likely, small amounts of side-products containing (K–222crypt)+ units are responsible for this observation.
The 29Si NMR spectrum of a solution of the bulk material containing 1a reveals four signals at −8.70, −129.94, −175.29, and −360.72 ppm, indicative of a tri-silylated D3h symmetric Si9 core. The signals at −8.70 ppm (TMS) and −129.94 ppm (α, Fig. 2a) originate from the attached silyl groups and conform well to reported shifts of the corresponding Ge9 species {Si(TMS)3}3Ge9−.51,60 The signals at −175.29 (β, Fig. 2a) and −360.72 ppm (γ, Fig. 2a) match well with the signals reported for the Si9 cluster atoms of (SiHtBu2)3Si9− (3a) (−175.16 and −358.81 ppm, which were confirmed in computational studies).30 The 29Si NMR spectrum of 2a reveals signals at −100.01 (β′, Fig. 2c) and −335.50 ppm (γ′, Fig. 2c). The signals of the substituted cluster atoms β/β′ are shifted more downfield if compared to the one of the ligand-free cluster atoms of the prismatic faces γ/γ′, which bear the highest negative ppm values for known siliconoids.19 The shift range of γ/γ′ is comparable to that of the protonated species H2Si92− (−346 ppm)55 and HSi93− (−359 ppm).54
Yellow block-shaped crystals suitable for single crystal X-ray diffraction were obtained from fluorobenzene/hexane solutions of bulk materials 1a and 2a. However, the structure determinations show the presence of the di-anionic siliconoid species {Si(TMS)3}2Si92− (1b) and (SnCy3)2Si92− (2b). The occurrence of the di-substituted species in the single crystals is traced back to substituent cleave from the cluster cores during crystallization. Ligand scrambling has been observed before in anionic Si4 clusters.28 However, a disproportionation according to “2{Si(TMS)3}3Si9− → {Si(TMS)3}2Si92− + {Si(TMS)3}4Si9” is excluded by to calculations due to the strongly endoenergetic nature of +92 kJ mol−1 (further information in ESI‡). EDX analyses confirmed the corresponding K:Si (1b) and K:Si:Sn (2b) ratios in the single crystals. The molecular structures of the siliconoid di-anions 1b and 2b are shown in Fig. 3a and b, respectively. The structures can be described as di-substituted Si9 clusters with the shape of a C2v-distorted mono-capped square anti-prism. The substituents are attached at two opposing silicon vertex atoms of the open square of the cluster.
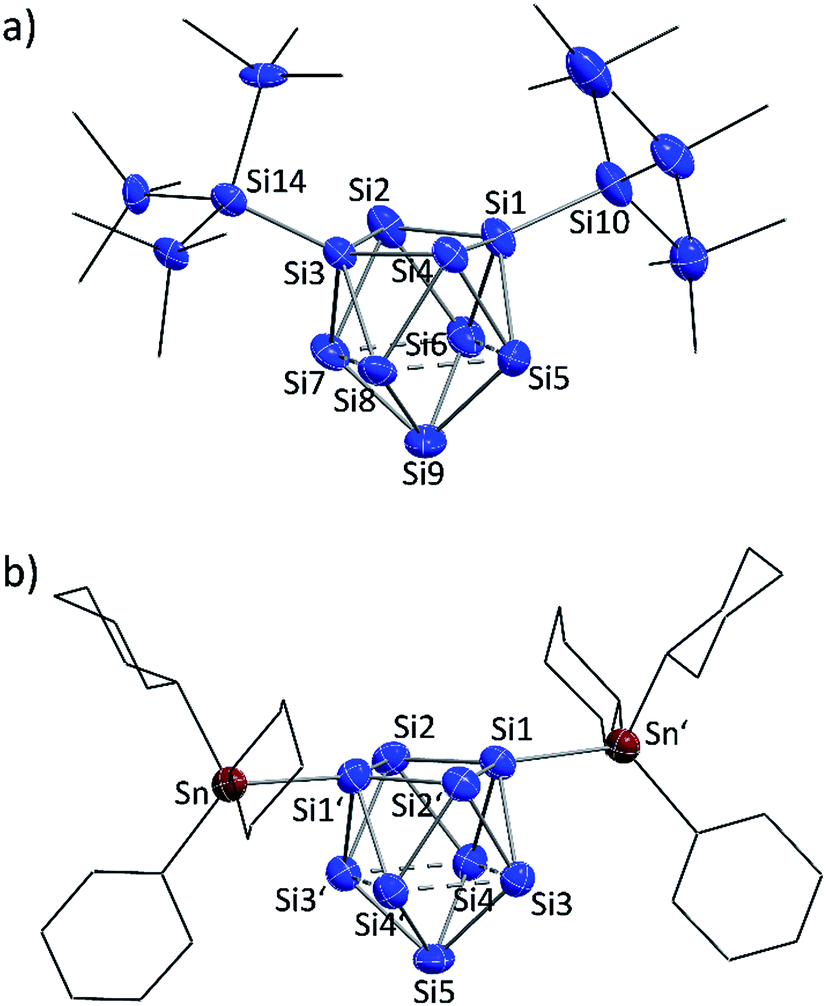 | ||
| Fig. 3 Single crystal structures of di-anionic siliconoids: (a) {Si(TMS)3}2Si92− (1b); (b) (SnCy3)2Si92− (2b); symmetry operation: (′) = −x, y, 0.5 − z; Si and Sn atoms (blue and red-brown, respectively) are shown as ellipsoids at 50% probability level; C atoms are drawn in the wire-and-stick mode; H atoms are omitted. | ||
Beside the Si–Si and Si–Sn exo-bonds, the nine Si atoms of the central units in 1b and 2b display two groups of Si–Si bond lengths, which are listed and compared to the ones in the known anion 3b in Table 1 (type 1 and type 2). The shorter bonds (type 1) in the range between 2.40 and 2.48 Å are slightly longer than typical single bonds. Interestingly, the elongations are quite small considering that Si1/Si3 in 1a and Si1/Si1′ in 2b show a coordination number of 5. A comparison to the bond lengths within the non-capped square in the cluster HSi93− shows a significant influence of the hydrogen substituent on the Si–Si distances. The bonds within the square with the substituted Si atom (coordination number 5) of 2.34 Å are clearly shorter if compared to the ones between the unsubstituted atoms (2.55 Å).45,54 By contrast, the atoms Si9 (1a) and Si5 (2b) with coordination number 4 form an umbrella-type coordination to the neighboring Si atoms. Most intriguingly, the atoms Si5 to Si8 in 1b and Si3 to Si4 in 2b each show, beside three shorter contacts of type 1, two longer contacts to atoms of the same kind (type 2). The corresponding Si–Si distances between 2.569(6)–2.738(5) Å in 1b and 2.565(2)–2.664(2) Å in 2b are in the typical region of unsubstituted Si atoms with an umbrella-type coordination sphere. Distances between such silicon atoms in known siliconoids as e.g. (Tip)5Si6− (2.5506(9) Å, Fig. 1b),18 (Tip)6Si6 (2.7076(8) Å)22 and (Mes)6Si5 (2.636(1) Å)64 are comparable to the type 2 bonds.
| Bond type | {Si(TMS)3}2Si92− (1b) | (SiHtBu2)2Si92− (3b)30 | (SnCy3)2Si92− (2b) |
|---|---|---|---|
| Exo-bonds | Si1–Si10: 2.357(5) | Si1–Si10: 2.349(2) | Si1–Sn: 2.578(1) |
| Si3–Si14: 2.339(5) | Si3–Si11: 2.379(2) | Si1′–Sn′: 2.578(1) | |
| Type 1 | Si1–Si2: 2.396(5) | Si1–Si2: 2.405(2) | Si1–Si2: 2.430(2) |
| Si1–Si4: 2.427(5) | Si1–Si4: 2.423(2) | Si1–Si2′: 2.433(2) | |
| Si2–Si3: 2.395(4) | Si2–Si3: 2.418(2) | Si1′–Si2: 2.433(2) | |
| Si3–Si4: 2.398(5) | Si3–Si4: 2.414(2) | Si1′–Si2′: 2.430(2) | |
| Si5–Si9: 2.468(6) | Si5–Si9: 2.429(2) | Si4–Si5: 2.436(2) | |
| Si6–Si9: 2.470(6) | Si6–Si9: 2.441(2) | Si3–Si5: 2.432(2) | |
| Si7–Si9: 2.427(6) | Si7–Si9: 2.450(2) | Si4′–Si5: 2.436(2) | |
| Si8–Si9: 2.475(7) | Si8–Si9: 2.447(2) | Si3′–Si5: 2.432(2) | |
| Type 2 | Si5–Si6: 2.772(6) | Si5–Si6: 2.782(2) | Si3–Si4: 2.664(2) |
| Si7–Si8: 2.738(5) | Si7–Si8: 2.764(2) | Si3′–Si4′: 2.664(2) | |
| Si6–Si7: 2.569(6) | Si6–Si7: 2.534(2) | Si3–Si4′: 2.565(2) | |
| Si5–Si8: 2.585(6) | Si5–Si8: 2.534(2) | Si3′–Si4: 2.565(2) |
A comparison of the molecular structures of the siliconoid di-anions 1b, 2b and 3b (Fig. 3 and 4) shows specific differences for the arrangement of the two substituents at the respective Si9 cluster core. As expected, the longest Si9 cluster exo-bonds are detected for the stannyl derivative 3b with Si9–Sn bond lengths of 2.578(1) Å, while the Si9–Si cluster exo-bonds in 1b and 2b are significantly shorter with values of 2.357(5)/2.339(5) Å (1b) and 2.349(2)/2.379(2) Å (3b) (Table 1). Si–Sn bonds in low-valent Si-compounds are scarce, but the Si9–Sn bond lengths in 2b compare well to the reported Si–Sn distance in the disilene (Tip)3(SnMe3)Si2 (2.5675(6) Å).65 The anion 2b is located on a twofold symmetry axis, and the two equivalent Sn–Si1–Si1′ and Si1–Si1′–Sn′ angles are 176.92(6)°. In 2b, the Sn atoms of the SnCy3 ligands are almost in plane with the non-capped cluster square formed by the atoms Si1/Si1′/Si2/Si2. In the silyl derivatives 1b and 3b, the corresponding angles are smaller, and the exo-bonds are oriented towards the open face of the clusters [1b: Si10–Si1–Si3 with 161.1(2)°, Si1–Si3–Si14 with 150.3(2)°; 3b: Si10–Si1–Si3 with 158.54(6)°, Si1–Si3–Si11 with 175.58(6)°]. The arrangement of the stannyl substituents in 2b is consistent with the corresponding angles in the di-substituted Ge9 derivative (SnPh3)2Ge92− [corresponding angles: 171.49(5)° and 172.22(5)°] in which one exo-bond is orientated towards the capped face of the cluster.66
 | ||
| Fig. 4 Molecular structures: (a) {Si(TMS)3}2Si92− (1b); (b) (SiHtBu2)2Si92− (3b);30 (c) (SnCy3)2Si92− (2b, symmetry operation: (′) = −x, y, 0.5 − z); partial atomic charge distributions (DFT-PBE0/TZVP level of theory, all values in e−): (d) {Si(TMS)3}2Si92− (1b); (e) (SiHtBu2)2Si92− (3b); (f) (SnCy3)2Si92− (2b); (g) {Si(TMS)3}3Si9− (1a); (h) (SiHtBu2)3Si9− (3a); (i) (SnCy3)3Si9− (2a). Molecular structures: Si atoms in blue and Sn atoms in red-brown are shown as ellipsoids at 50% probability level; organic groups are shown as black sticks; HSi atoms in 3b are omitted. | ||
This indicates a certain degree of interaction of the tin atoms with next-nearest neighbor silicon cluster atoms in 2b as it was also indicated in the NMR experiments for the tin substituents in the disilene (Tip)3(SntBu2Cl)Si2.65 Such an interaction is further supported by the formation of (SnPh3)Ge93− in which the stannyl ligand is bonded to two Ge9 cluster atoms.66
The highly dispersed 29Si NMR signals in solution in a range from −8.70 to −360.72 ppm for 1a and from −100.01 to −335.50 ppm for 2a hint for an inhomogeneous electron distribution due to the different oxidation numbers. Such a broad range was observed before in (Tip)6Si6 (Tip = 2,4,6-triisopropylphenyl) with a tricyclic structure featuring silicon atoms with two, one, and no substituents outside the ring framework. Consequently, (Tip)6Si6 can be regarded as a tricyclic aromatic isomer of hexasilabenzene.15 In contrast to (Tip)6Si6 with two Si atoms not attached to Tip substituents, bare Si94− clusters reveal nine such atoms. Si94− possesses a fully delocalized electronic system which fits the superatom model of a 40-electron cluster.67–70 Ligand attachment to Si94− allows for a step-wise transition to molecules with partially delocalized bonds.
In order to investigate the bond properties of the di- and tri-substituted Si9 atom clusters we calculated the partial atomic charges [e−] of the silicon cluster atoms of 1a/1b, 2a/2b and 3a/3b (Fig. 4).30 Comparison of the overall charge distributions shows that lower partial charges are located at substituted silicon atoms including substituent-specific charge differences underlining the electronic influence of the respective substituent on the cluster atoms (partial charges: SnCy3 > SiHtBu2 > Si(TMS)3). Interestingly, the partial charges at the unsubstituted Si atoms (prism faces) in the tri-substituted species 1a/2a/3a are all identical with a value of 0.14e− and show a homogeneous distribution of the extra negative charge at the cluster prism faces of the D3h symmetric cluster. By contrast, the two extra negative charges in the di-substituted C2v symmetric clusters 1b, 2b and 3b are distributed more versatilely on the cluster surfaces consisting of ligand-free Si atoms. The partial atomic charges of the silicon atoms in the capped cluster squares [1b/3b: Si5 to Si8; 2b: Si3(′) and Si4(′)] are equal for all species with a value of 0.23e− for each atom, whereas the highest partial charges at the square-capping silicon atoms, from which the substituents are detached during crystallization, is found in 1b (0.40e−, Si9 atom).
Further insight into the bonding situation within the cluster units is provided by an IBO analysis of 1a and 1b (Fig. 5) that manifests an influence of the third substituent on the bonding situation within the Si9 cluster cores. The analysis shows a delocalization of the cluster valence electrons (total: 40e−) that is in accordance with a previous report for related tris-substituted nona-germanium clusters71 stronger for D3h symmetric 1a than for C2v symmetric 1b. Delocalization in 1a occurs by three 5c-6e (5 center-6 electron) bonds (18e−) within the three capped square faces and by two 3c-2e bonds (4e−) in the two prism faces. A comparison to the bonding situation in 1b shows that only one delocalized 5c-6e bond (6e−) is present here which is located within the capped square of the cluster from which cap the substituent is released. Most interestingly, causes the attachment of two ligands a high degree of bond localization in form of four covalent 2c-2e bonds (8e−) in 1b in the non-capped square of the cluster. Furthermore, two 3c-2e bonds (total 8e−) are present in the triangular faces of 1b. The remaining 18 cluster valence electrons of 1a and 1b are located in the covalent cluster exo-bonds (1a: 6e−; 1b: 4e−) and in six lone pairs which are distributed over the naked Si cluster atoms (1a: 12e−, six lone pairs: 1b: 14e−, seven lone pairs). Similar bond delocalization was also reported for hexasilabenzene15 in which theoretical analysis revealed the cyclic delocalization of six mobile electrons of the p-, s- and non-bonding type across the central four-membered ring and which was described as dismutational aromatic. The herein presented study to the charge distributions within substituted Si9 clusters adds these cluster species as delocalized species to the known silicon molecules in the literature.
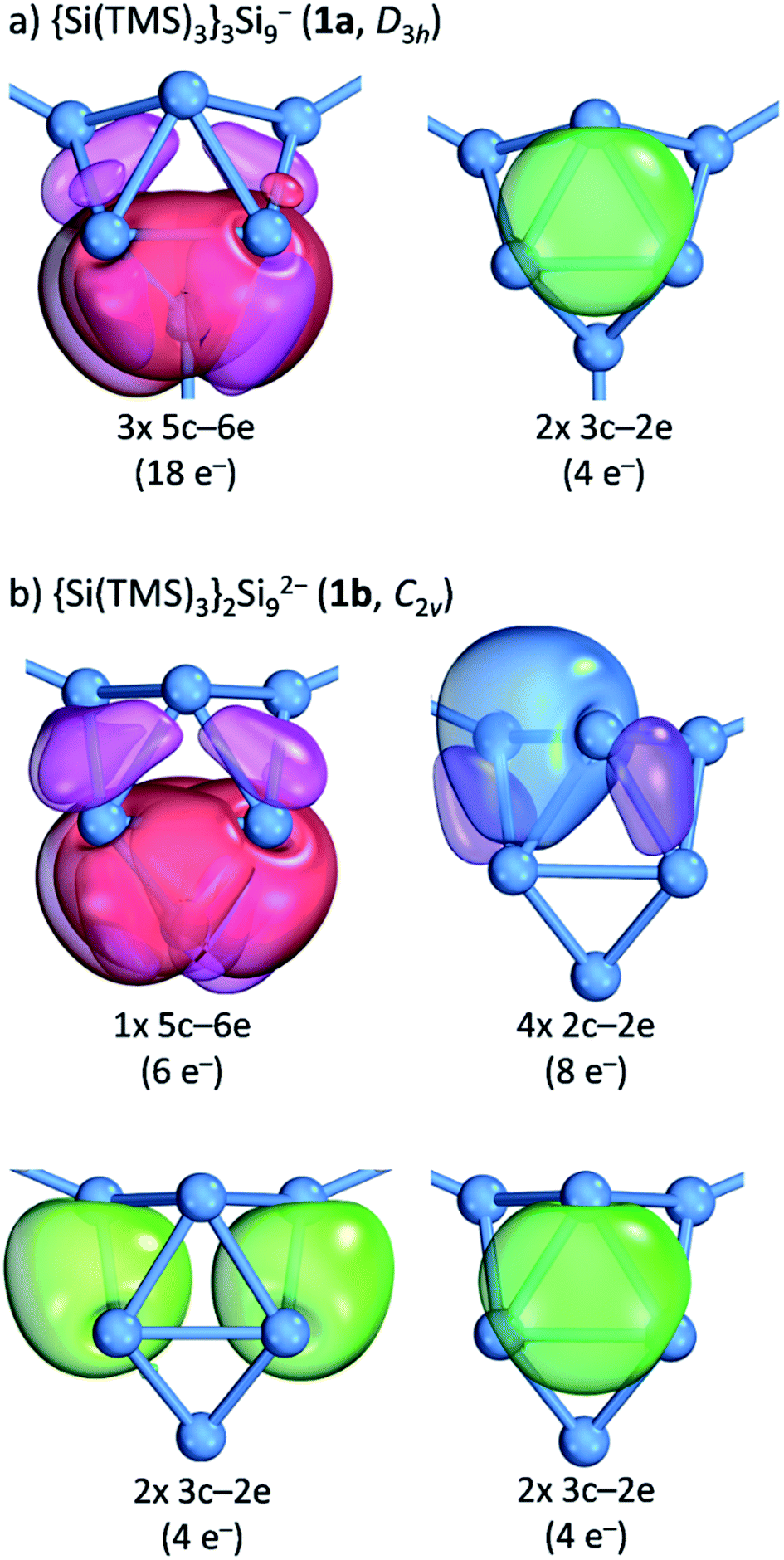 | ||
| Fig. 5 Intrinsic bond orbital (IBO) analysis: (a) the D3h symmetric cluster in {Si(TMS)3}2Si92− (1a); (b) the C2v symmetric cluster in {Si(TMS)3}2Si92− (1b). The plotted IBO isosurfaces enclose 80% of the total electron density of the IBO (DFT-PBE0/def2-TZVP level of theory). Details to cluster atom distributions to the respective bonds as well as to the 5c-6e bond in 1b are shown in the ESI.‡ | ||
Furthermore, the crystals were characterized by Raman spectroscopy, and the measured vibrations are assigned by comparison to calculated Raman spectra of 1b and 2b (Fig. 6). Changes of intensities might be due to specific packing effects (calculations were performed on discrete anionic species) and to orientation effects of the single crystal. In both cases characteristic ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) (Si–Si) stretching vibrations of the Si9 cluster cores were detected which agree well with the calculated values. Interestingly, the peaks and the distribution range of the cluster vibrations are different for the two siliconoids (1b: 294, 346, 371, 447 cm−1; 2b: 273, 295, 352, 405 cm−1), which indicates an influence of the respective substituent on the Si9 intra-bonds. All vibrations for the stannyl derivative 2b (range: 273–405 cm−1) are found at lower wavenumbers than those of 1b (range: 294–512 cm−1). For 1b, an Si9–Si(TMS)3 vibration for the cluster exo-bond was found at 512 cm−1, whereas the Si9–SnCy3 exo-bonds are not detectable due to laser absorption effects in the spectrum below 200 cm−1. Moreover, [Si–C] and [C–H] vibrations were observed for 1b and [Sn–C], [C–C] and [C–H] vibrations for 2b, which also include the vibrations for the (K–222crypt) units in the single crystals.
(Si–Si) stretching vibrations of the Si9 cluster cores were detected which agree well with the calculated values. Interestingly, the peaks and the distribution range of the cluster vibrations are different for the two siliconoids (1b: 294, 346, 371, 447 cm−1; 2b: 273, 295, 352, 405 cm−1), which indicates an influence of the respective substituent on the Si9 intra-bonds. All vibrations for the stannyl derivative 2b (range: 273–405 cm−1) are found at lower wavenumbers than those of 1b (range: 294–512 cm−1). For 1b, an Si9–Si(TMS)3 vibration for the cluster exo-bond was found at 512 cm−1, whereas the Si9–SnCy3 exo-bonds are not detectable due to laser absorption effects in the spectrum below 200 cm−1. Moreover, [Si–C] and [C–H] vibrations were observed for 1b and [Sn–C], [C–C] and [C–H] vibrations for 2b, which also include the vibrations for the (K–222crypt) units in the single crystals.
 | ||
| Fig. 6 Raman spectra of single crystals: (a) measured spectrum of a (K–222crypt)21b single crystal (blue, top), calculated spectrum of {Si(TMS)3}2Si92− (1b) by DFT-PBE0/TZVP (blue, bottom); (b) measured spectrum of a (K–222crypt)22b single crystal (red-brown, top), calculated spectrum of (SnCy3)2Si92− (2b) by DFT-PBE0/TZVP (red-brown, bottom); details of the calculations can be found in the ESI.‡ | ||
Up to this study, the Raman spectra of Si9 cluster compounds comprised the protonated cluster species H2Si92− and Si94− that show less Raman vibrations. The spectrum of H2Si92− displays a prominent resonance at 386 cm−1 and a resonance of Si94− as part of the solid phase K12Si17 at 390 cm−1.55 In contrast, multiple Si9 vibrations are confirmed for the ligand-stabilized clusters 1b/2b, and a possible correlation with the delocalized bonding situation from the IBO analysis is of interest for future reports.
Conclusions
The synthetic approach for silicon-rich molecules comprising silicon atoms with low oxidation state from the binary intermetallic precursor K12Si17 is established. For the first time a metal atom is attached to a Si9 cluster core pathway allowing for the structural characterization of Cy3Sn–(Si92−)–SnCy3 (2b). In addition the anionic siliconoid {Si(TMS)3}2Si92− (1b) is structurally characterized. Both anions form via the tris-substituted derivatives (SnCy3)3Si9− (1a) and {Si(TMS)3}3Si9− (2a) that have been spectroscopically characterized. In contrast to known “molecular multi-step” approaches, the syntheses became feasible via a two-step reaction from elemental silicon. The molecular anions possess high numbers of unsubstituted Si cluster atoms and add as novel delocalized representatives to known silicon compounds in the literature. Moreover, the influence of the ligands on bond localization at the Si9 cluster cores was pointed out.Experimental section
General
All reactions and manipulations were performed under a purified argon atmosphere using standard Schlenk and glove box techniques. Thf was dried using the solvent purificator MBraun MB-SPS, and fluorobenzene was dried over CaH2 prior to use. 222crypt was dried in vacuo prior to use. Liquid ammonia was dried and stored over sodium metal, and all other solvents (including deuterated solvents) were stored over molecular sieves (3 Å). All other chemicals were received commercially and used without further purification.K12Si17 (activated)
The Zintl compound K12Si17 was synthesized by heating (heating rate: 2 °C min−1) a stoichiometric mixture of 0.55 g K (14.0 mmol, 1 eq.) and 0.59 g Si (21.1 mmol, 1.5 eq.) in a sealed tantalum ampoule to 800 °C for 15 h and subsequent cooling (cooling rate: 0.5 °C min−1) to room temperature. The ampoule was opened in a glove box, and the product was finely ground yielding 1.04 g K12Si17 (1.10 mmol, 94%) as a black solid. The solid was characterized by powder X-ray diffraction showing high purity for the solid phase (ESI‡). For the activation, 0.20 g K12Si17 (0.21 mmol, 1 eq.) and 0.15 g 222crypt (0.39 mmol, 1.9 eq.; 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosan) were weighed into a Schlenk tube, and liquid ammonia was added at −78 °C (iPrOH/CO2) to give a dark red solution. The solution was stirred at −78 °C for 2 h, and K12Si17 (activated) was obtained as a brown solid after the removal of liquid ammonia.(K–222crypt)+ salts of 1a and 1b
K12Si17activated (batch with 200 mg K12Si17) was cooled to 0 °C (ice bath), and thf (12 mL) was added. 0.36 g Si(TMS)3Cl (1.26 mmol, 6 eq.) was added under continuous stirring as a pre-cooled thf solution (3 mL). The reaction mixture was stirred overnight and allowed to warm to room temperature. A brownish filtrate was obtained after filtration, and all volatiles were removed in vacuo. The residue was extracted with fluorobenzene (5 mL), and a red filtrate was obtained. After washing with hexane and vacuum drying, a light brown solid containing the (K–222crypt)+ salt of 1a was obtained (yield: 0.12 g, 40% based on K12Si17). The solid was characterized by ESI-MS in thf and NMR spectroscopy (1H, 13C, 29Si) in solution (thf-d8). For crystallization, a fluorobenzene solution of the solid (3 mL) was layered with hexane. Yellow block-shaped crystals of the (K–222crypt)+ salt of 1b suitable for single crystal X-ray diffraction were obtained after 10 d (10 mg, 10% based on (K–222crypt)1a). The crystals were investigated by Raman spectroscopy, and the K:Si ratio was confirmed by EDX analyses on single crystals.
:Si = 6.30%:93.7% (calcd: 6.22%:93.8%); Raman (532 nm): 294 [(Si–Si) calcd 304], 346 [(Si–Si) calcd 341], 371 [(Si–Si) calcd 373], 447 [(Si–Si) calcd 445], 512 [(Si–Si) calcd 505], 627 [(Si–C) calcd 622], 690 [(Si–C) calcd 687], 748 [(C–H) calcd 745], 838 [(C–H) 849], 866 [(C–H) calcd 872] cm−1.
(K–222crypt)+ salts of 2a and 2b
K12Si17activated (batch with 200 mg K12Si17) was cooled to 0 °C (ice bath), and pyridine (12 mL) was added. 0.51 g (1.26 mol, 6 eq.) SnCy3Cl was added under continuous stirring as pre-cooled pyridine solution (3 mL). The reaction mixture was stirred overnight and filtered under continuous cooling yielding a colored filtrate which was dried in vacuo and extracted with fluorobenzene (5 mL). The red filtrate was dried in vacuo, and a brown solid containing the (K–222crypt)+ salt of 2a was obtained (yield: 0.10 g, 27% based on K12Si17). The solid was investigated by ESI-MS in thf and NMR spectroscopy (1H, 13C, 29Si, 119Sn) in solution (thf-d8). For crystallization, a fluorobenzene solution of the solid (3 mL) was layered with hexane. Yellow block-shaped crystals of the (K–222crypt)+ salt of 2b suitable for single crystal X-ray diffraction were obtained after 12 d (7 mg, 7% based on (K–222crypt)2a). The crystals were investigated by Raman spectroscopy, and the K:Si ratio was confirmed by EDX analyses on single crystals.
:Si:Sn = 15.5%:44.1%:40.4% (calcd: 13.8%:44.5%:41.8%); Raman (785 nm):, 273 [(Si–Si) calcd 271], 295 [(Si–Si) calcd 298], 352 [(Si–Si) calcd 348], 405 [(Si–Si) calcd 407], 638 [(Sn–C) calcd 641], 804 [(C–C) calcd 808], 839 [(C–H) calcd 842], 881 [(C–C) calcd 891], 1015 [(C–H) calcd 1022], 1062 [(C–H) calcd 1070], 1162 [(C–H) calcd 1173] cm−1.
Computational details
Quantum-chemical calculations at the DFT-PBE0/TZVP level of theory were carried out using the TURBOMOLE program package.72–76 Intrinsic atomic orbitals (IAOs) and intrinsic bond orbitals were used to analyze the partial charges and bonding of the clusters, respectively.77 Full computational details are available in the ESI.‡Single crystal structure determination
For single crystal data collection, the crystals were fixed on a glass capillary and positioned in a cold stream of N2 gas. Single crystal data collection was performed with a STOE StadiVari (Mo Kα radiation) diffractometer equipped with a DECTRIS PILATUS 300K detector. Structures were solved by Direct Methods (SHELXS-2014) and refined by full-matrix least-squares calculations against F2 (SHELXL-2014).78 The positions of the hydrogen atoms were calculated and refined using a riding model. Unless otherwise stated, all non-hydrogen atoms were treated with anisotropic displacement parameters. In 1b some hypersilyl groups show rotational disorder and were refined at two split positions. In 2b, one cyclohexyl group is found in two different orientations and was refined at two split positions. For visualization, the crystal structures have been plotted with Diamond.‡79Electron dispersive X-ray (EDX) analysis
Single crystals of all compounds were analyzed with a SWIFT-ED-TM (Oxford Instruments) and a Hitachi TM-1000 Tabletop microscope (Hitachi High-Technologies) with the INCA system software.NMR spectroscopy
1H, 13C and 29Si NMR spectra were recorded on a Bruker AVIII Ultrashield 400 MHz or a Bruker AVIII 500 MHz Cryo system, 119Sn NMR spectra were measured on a Bruker AVIII 300 MHz (Bruker Inc) instrument. The signals of the 1H and 13C spectra were calibrated on the rest proton signal of the used deuterated solvent thf-d8. Chemical shift values are given in δ values by parts per million (ppm). The coupling constants J are stated in Hz. Signal multiplicities are abbreviated as follows: s – singlet, d – doublet, t – triplet, m – triplet. The spectra were evaluated with MestReNova.80Electrospray ionization mass spectrometry (ESI-MS)
The preparation of the samples was done in a glove box. The spectra were measured on an HCT instrument (Bruker Inc). The data were analyzed using the program Bruker Compass Data Analysis 4.0 SP 5. The dry gas temperature was adjusted to 300 °C and the injection speed to 240 μL s−1. For fragmentation experiments, the respective mass peaks were isolated (width: 40) and fragmented (amplitude: 2.0). Visualization of the spectra was carried out with the programs OriginPro (Origin Lab Inc) or Microsoft Excel (Microsoft Inc).Raman spectroscopy
Raman spectra were recorded with an inVia Raman Microscope RE04 with a CCD detector and 500 mW maximal power (Renishaw PLC; Software: WiRE 4.2 build 5037) at λ = 532 nm. For the measurements the samples were sealed in glass capillaries in a glove box.Powder X-ray diffraction (PXRD)
The data were collected at room temperature on a STOE Stadi P diffractometer (Ge(111) monochromator, Cu Kα1 radiation, λ = 1.54056 Å) with a Dectris MYTHEN 1K detector in Debye–Scherrer geometry. For the measurements the samples were sealed in glass capillaries (Ø 0.5 mm). The raw data were processed with WinX-POW,81OriginPro (Origin Lab Inc) was used for the visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial support by WACKER Chemie AG and Deutsche Forschungsgemeinschaft (DFG, FA 198/14-1). They thank Sebastian Geier and Christoph Wallach for the Raman measurements, Maria Müller for the EDX analyses, Brigita Bratic for supporting the syntheses and Dr Annette Schier for proofreading. A. J. K. thanks the Academy of Finland for funding (grant 308089) and the Finnish IT Center for Science (CSC) for computational resources.References
- M. Ashuri, Q. He and L. L. Shaw, Nanoscale, 2016, 8, 74–103 RSC.
- M. G. Kanatzidis, Adv. Mater., 2007, 19, 1165–1181 CrossRef CAS.
- R. A. Bley and S. M. Kauzlarich, J. Am. Chem. Soc., 1996, 118, 12461–12462 CrossRef CAS.
- C. Eun-Chel, P. Sangwook, H. Xiaojing, S. Dengyuan, C. Gavin, P. Sang-Cheol and A. G. Martin, Nanotechnology, 2008, 19, 245201 CrossRef PubMed.
- L. T. Canham, Appl. Phys. Lett., 1990, 57, 1046–1048 CrossRef CAS.
- J. K. Rath, B. Stannowski, P. A. T. T. van Veenendaal, M. K. van Veen and R. E. I. Schropp, Thin Solid Films, 2001, 395, 320–329 CrossRef CAS.
- M. L. Snedaker, Y. Zhang, C. S. Birkel, H. Wang, T. Day, Y. Shi, X. Ji, S. Kraemer, C. E. Mills, A. Moosazadeh, M. Moskovits, G. J. Snyder and G. D. Stucky, Chem. Mater., 2013, 25, 4867–4873 CrossRef CAS.
- L. Venema, Nature, 2011, 479, 309 CrossRef CAS PubMed.
- D. N. Roark and G. J. D. Peddle, J. Am. Chem. Soc., 1972, 94, 5837–5841 CrossRef CAS.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst and R. K. Kallury, Chem. Commun., 1981, 191–192 RSC.
- A. G. Brook, S. C. Nyburg, F. Abdesaken, B. Gutekunst, G. Gutekunst, R. Krishna, M. R. Kallury, Y. C. Poon, Y. M. Chang and W. N. Winnie, J. Am. Chem. Soc., 1982, 104, 5667–5672 CrossRef CAS.
- A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755–1757 CrossRef CAS PubMed.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- K. Abersfelder, A. J. P. White, H. S. Rzepa and D. Scheschkewitz, Science, 2010, 327, 564–566 CrossRef CAS PubMed.
- K. C. Mondal, S. Roy, B. Dittrich, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2016, 55, 3158–3161 CrossRef CAS PubMed.
- D. Scheschkewitz, Angew. Chem., Int. Ed., 2005, 44, 2954–2956 CrossRef CAS PubMed.
- P. Willmes, K. Leszczyńska, Y. Heider, K. Abersfelder, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2016, 55, 2907–2910 CrossRef CAS PubMed.
- Y. Heider and D. Scheschkewitz, Dalton Trans., 2018, 47, 7104–7112 RSC.
- T. Iwamoto, N. Akasaka and S. Ishida, Nat. Commun., 2014, 5, 5353 CrossRef CAS PubMed.
- G. Fischer, V. Huch, P. Mayer, S. K. Vasisht, M. Veith and N. Wiberg, Angew. Chem., Int. Ed., 2005, 44, 7884–7887 CrossRef CAS PubMed.
- K. Abersfelder, A. J. P. White, R. J. F. Berger, H. S. Rzepa and D. Scheschkewitz, Angew. Chem., Int. Ed., 2011, 50, 7936–7939 CrossRef CAS PubMed.
- K. Abersfelder, A. Russell, H. S. Rzepa, A. J. P. White, P. R. Haycock and D. Scheschkewitz, J. Am. Chem. Soc., 2012, 134, 16008–16016 CrossRef CAS PubMed.
- N. Akasaka, S. Ishida and T. Iwamoto, Inorganics, 2018, 6, 107 CrossRef CAS.
- S. David, Chem. Lett., 2011, 40, 2–11 CrossRef.
- Y. Ohmori, M. Ichinohe, A. Sekiguchi, M. J. Cowley, V. Huch and D. Scheschkewitz, Organometallics, 2013, 32, 1591–1594 CrossRef CAS.
- K. Takeuchi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2008, 130, 16848–16849 CrossRef CAS PubMed.
- M. Ichinohe, M. Toyoshima, R. Kinjo and A. Sekiguchi, J. Am. Chem. Soc., 2003, 125, 13328–13329 CrossRef CAS PubMed.
- T. Iwamoto, M. Tamura, C. Kabuto and M. Kira, Science, 2000, 290, 504–506 CrossRef CAS PubMed.
- L. J. Schiegerl, A. J. Karttunen, W. Klein and T. F. Fässler, Chem.–Eur. J., 2018, 24, 19171–19174 CrossRef CAS PubMed.
- K. I. Leszczyńska, V. Huch, C. Präsang, J. Schwabedissen, R. J. F. Berger and D. Scheschkewitz, Angew. Chem., Int. Ed., 2019, 58, 5124–5128 CrossRef PubMed.
- N. Wiberg, C. M. M. Finger and K. Polborn, Angew. Chem., Int. Ed., 1993, 32, 1054–1056 CrossRef.
- T. M. Klapötke, S. K. Vasisht, G. Fischer and P. Mayer, J. Organomet. Chem., 2010, 695, 667–672 CrossRef.
- T. Goebel, A. Ormeci, O. Pecher and F. Haarmann, Z. Anorg. Allg. Chem., 2012, 638, 1437–1445 CrossRef CAS.
- H. G. von Schnering, M. Schwarz, J. H. Chang, K. Peters, E. M. Peters and R. Nesper, Z. Kristallogr. – New Cryst. Struct., 2005, 220, 525 CAS.
- L. A. Stearns, J. Gryko, J. Diefenbacher, G. K. Ramachandran and P. F. McMillan, J. Solid State Chem., 2003, 173, 251–258 CrossRef CAS.
- J. He, D. D. Klug, K. Uehara, K. F. Preston, C. I. Ratcliffe and J. S. Tse, J. Phys. Chem. B, 2001, 105, 3475–3485 CrossRef CAS.
- V. Quéneau, E. Todorov and S. C. Sevov, J. Am. Chem. Soc., 1998, 120, 3263–3264 CrossRef.
- C. Hoch, M. Wendorff and C. Röhr, J. Alloys Compd., 2003, 361, 206–221 CrossRef CAS.
- K. Wade, Inorg. Nucl. Chem. Lett., 1972, 8, 559–562 CrossRef CAS.
- K. Wade, Chem. Commun., 1971, 792–793 RSC.
- C. Lorenz, S. Gärtner and N. Korber, Z. Anorg. Allg. Chem., 2017, 643, 141–145 CrossRef CAS.
- C. B. Benda, T. Henneberger, W. Klein and T. F. Fässler, Z. Anorg. Allg. Chem., 2017, 643, 146–148 CrossRef CAS.
- S. Joseph, C. Suchentrunk, F. Kraus and N. Korber, Eur. J. Inorg. Chem., 2009, 2009, 4641–4647 CrossRef.
- T. Henneberger, W. Klein and T. F. Fässler, Z. Anorg. Allg. Chem., 2018, 644, 1018–1027 CrossRef CAS.
- M. Waibel, F. Kraus, S. Scharfe, B. Wahl and T. F. Fässler, Angew. Chem., Int. Ed., 2010, 49, 6611–6615 CrossRef CAS PubMed.
- J. M. Goicoechea and S. C. Sevov, Organometallics, 2006, 25, 4530–4536 CrossRef CAS.
- S. Joseph, M. Hamberger, F. Mutzbauer, O. Härtl, M. Meier and N. Korber, Angew. Chem., Int. Ed., 2009, 48, 8770–8772 CrossRef CAS PubMed.
- F. S. Geitner and T. F. Fässler, Chem. Commun., 2017, 53, 12974–12977 RSC.
- M. W. Hull and S. C. Sevov, Angew. Chem., Int. Ed., 2007, 46, 6695–6698 CrossRef CAS PubMed.
- F. Li and S. C. Sevov, Inorg. Chem., 2012, 51, 2706–2708 CrossRef CAS PubMed.
- F. S. Geitner, J. V. Dums and T. F. Fässler, J. Am. Chem. Soc., 2017, 139, 11933–11940 CrossRef CAS PubMed.
- F. S. Geitner, W. Klein and T. F. Fässler, Angew. Chem., Int. Ed., 2018, 57, 14509–14513 CrossRef CAS PubMed.
- C. Lorenz, F. Hastreiter, J. Hioe, L. Nanjundappa, S. Gärtner, N. Korber and R. M. Gschwind, Angew. Chem., Int. Ed., 2018, 57, 12956–12960 CrossRef CAS PubMed.
- L. J. Schiegerl, A. J. Karttunen, J. Tillmann, S. Geier, G. Raudaschl-Sieber, M. Waibel and T. F. Fässler, Angew. Chem., Int. Ed., 2018, 57, 12950–12955 CrossRef CAS PubMed.
- L.-A. Jantke and T. Fässler, Inorganics, 2018, 6, 31 CrossRef.
- O. Kysliak, C. Schrenk and A. Schnepf, Inorg. Chem., 2015, 54, 7083–7088 CrossRef CAS PubMed.
- L. J. Schiegerl, F. S. Geitner, C. Fischer, W. Klein and T. F. Fässler, Z. Anorg. Allg. Chem., 2016, 642, 1419–1426 CrossRef CAS.
- K. Mayer, L. J. Schiegerl, T. Kratky, S. Günther and T. F. Fässler, Chem. Commun., 2017, 53, 11798–11801 RSC.
- K. Mayer, L. J. Schiegerl and T. F. Fässler, Chem.–Eur. J., 2016, 22, 18794–18800 CrossRef CAS PubMed.
- O. Kysliak, T. Kunz and A. Schnepf, Eur. J. Inorg. Chem., 2017, 2017, 805–810 CrossRef CAS.
- L. G. Perla and S. C. Sevov, J. Am. Chem. Soc., 2016, 138, 9795–9798 CrossRef CAS PubMed.
- L. G. Perla, A. Muñoz-Castro and S. C. Sevov, J. Am. Chem. Soc., 2017, 139, 15176–15181 CrossRef CAS PubMed.
- D. Nied, R. Köppe, W. Klopper, H. Schnöckel and F. Breher, J. Am. Chem. Soc., 2010, 132, 10264–10265 CrossRef CAS PubMed.
- K. Abersfelder, T.-l. Nguyen and D. Scheschkewitz, Z. Anorg. Allg. Chem., 2009, 635, 2093–2098 CrossRef CAS.
- A. Ugrinov and S. C. Sevov, Chem.–Eur. J., 2004, 10, 3727–3733 CrossRef CAS PubMed.
- A. J. Stone, Inorg. Chem., 1981, 20, 563–571 CrossRef CAS.
- A. J. Stone and M. J. Alderton, Inorg. Chem., 1982, 21, 2297–2302 CrossRef CAS.
- W. A. de Heer, Rev. Mod. Phys., 1993, 65, 611–676 CrossRef CAS.
- P. Andre Clayborne and H. Häkkinen, Phys. Chem. Chem. Phys., 2012, 14, 9311–9316 RSC.
- N. V. Tkachenko and A. I. Boldyrev, Chem. Sci., 2019, 10, 5761–5765 RSC.
- TURBOMOLE V7.3 2018, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, 2007 Search PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- C. Adamo and V. J. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- Diamond Version 3.2k, Crystal Impact GbR, 1997–2014 Search PubMed.
- MestReNova v9.1.0, Mestrelab Research S.L., 2014 Search PubMed.
- WinXPOW v3.0.2.1, STOE & Cie GmbH, 2011 Search PubMed.
Footnotes |
| † Dedicated to Professor Reinhold Tacke on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1896557 and 1896556. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03324f |
| This journal is © The Royal Society of Chemistry 2019 |