
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defect-mediated selective hydrogenation of nitroarenes on nanostructured WS2†
Yifan
Sun‡
 ae,
Albert J.
Darling‡
a,
Yawei
Li
b,
Kazunori
Fujisawa
ce,
Cameron F.
Holder
a,
He
Liu
a,
Michael J.
Janik
*b,
Mauricio
Terrones
*acde and
Raymond E.
Schaak
*ab
ae,
Albert J.
Darling‡
a,
Yawei
Li
b,
Kazunori
Fujisawa
ce,
Cameron F.
Holder
a,
He
Liu
a,
Michael J.
Janik
*b,
Mauricio
Terrones
*acde and
Raymond E.
Schaak
*ab
aDepartment of Chemistry and Materials Research Institute, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: res20@psu.edu
bDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: mjj13@psu.edu
cDepartment of Physics, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: mut11@psu.edu
dDepartment of Materials and Science Engineering, The Pennsylvania State University, University Park, PA 16802, USA
eCenter for 2-Dimensional and Layered Materials, The Pennsylvania State University, University Park, PA 16802, USA
First published on 19th September 2019
Abstract
Transition metal dichalcogenides (TMDs) are well known catalysts as both bulk and nanoscale materials. Two-dimensional (2-D) TMDs, which contain single- and few-layer nanosheets, are increasingly studied as catalytic materials because of their unique thickness-dependent properties and high surface areas. Here, colloidal 2H-WS2 nanostructures are used as a model 2-D TMD system to understand how high catalytic activity and selectivity can be achieved for useful organic transformations. Free-standing, colloidal 2H-WS2 nanostructures containing few-layer nanosheets are shown to catalyze the selective hydrogenation of a broad scope of substituted nitroarenes to their corresponding aniline derivatives in the presence of other reducible functional groups. Microscopic and computational studies reveal the important roles of sulfur vacancy-rich basal planes and tungsten-terminated edges, which are more abundant in nanostructured 2-D materials than in their bulk counterparts, in enabling the functional group selectivity. At tungsten-terminated edges and on regions of the basal planes having high concentrations of sulfur vacancies, vertical adsorption of the nitroarene is favored, thus facilitating hydrogen transfer exclusively to the nitro group due to geometric effects. At lower sulfur vacancy concentrations on the basal planes, parallel adsorption of the nitroarene is favored, and the nitro group is selectively hydrogenated due to a lower kinetic barrier. These mechanistic insights reveal how the various defect structures and configurations on 2-D TMD nanostructures facilitate functional group selectivity through distinct mechanisms that depend upon the adsorption geometry, which may have important implications for the design of new and enhanced 2-D catalytic materials across a potentially broad scope of reactions.
Introduction
Two-dimensional transition metal dichalcogenide (TMD) materials are active catalysts for a broad range of chemical transformations, including the hydrogen evolution reaction,1 the electrochemical reduction of CO2,2 and various hydrotreating and hydrogenation reactions.3–5 The near-zero free energy of adsorption of molecular hydrogen on edge and defect sites makes TMDs ideal candidates as active hydrogenation catalysts.6 Hydrogenation catalysts that can target one reducible group on a molecule when others are present and accessible are especially important. For example, the selective hydrogenation of substituted nitroarenes is important for accessing aniline compounds that are useful intermediates in pharmaceutical and agrochemical production.7Catalysts that facilitate selective hydrogenation reactions using H2 are desirable based on atom economy considerations and the formation of water as the only reduction byproduct.8–10 Among heterogeneous catalysts, chemically modified noble metals can achieve selective hydrogenation,11–13 but recent interest in new and low-cost materials has resulted in a growing number of non-platinum group (NPG) transition metal catalysts for such reactions.14–18 TMD materials are well known NPG hydrogenation catalysts.19 The more recent ability to synthesize two-dimensional (2-D) TMDs containing single- and few-layer nanosheets has expanded the catalytic scope of these materials and enabled new capabilities in achieving functional group selectivity. For example, nanostructured MoS2 catalyzes the selective hydrogenation of substituted nitroarenes using H2 to form their corresponding anilines, but requires transition metal promotors to facilitate the formation of active sites that arise from a significantly lowered metal–sulfur bond energy.9,10,19 Understanding how functional group selectivity emerges in 2-D TMDs catalysts is important, but insights remain limited. Here, we show that colloidal WS2 nanostructures containing single- and few-layer nanosheets, and without transition metal promotors, catalyze the selective hydrogenation of substituted nitroarenes to their corresponding aniline derivatives. Complementary microscopic and computational studies provide important insights into the origin of the catalytic selectivity on the TMD nanostructures, pointing to the important roles of atomic sulfur vacancies on the basal planes and tungsten-terminated edges. These WS2 nanostructures provide a well-defined platform to study how catalytic activity and selectivity can be achieved in nanostructured 2-D TMD materials.20
Results and discussion
Nanostructured WS2 was synthesized in solution by reacting WCl6, hexamethyldisilazane, and CS2 in oleylamine and oleic acid.21 TEM (Fig. 1a) and HAADF-STEM (Fig. 1b) images of the resulting ∼100 nm colloidal nanostructures show that they contain nanosheets in a flower-like morphology.22 The crystal structure of 2H-WS2, shown in Fig. 1c and d, consists of alternating stacked monolayers that contain tungsten atoms coordinated by sulfur atoms in a trigonal prismatic geometry. The Raman spectrum in Fig. 1e shows the in-plane (E12g) and out-of-plane (A1g) vibration modes, as well as higher-order modes [LA(M) and 2LA(M)],23,24 which are characteristic of 2H-WS2 nanostructures and indicate the presence of defects.25 The XRD pattern in Fig. 1f shows further evidence for the formation of crystalline 2H-WS2. Polycrystallinity and strain arising from the curled nanosheets and small domain sizes result in a broad, yet characteristic and identifiable diffraction pattern that is consistent with 2H-WS2.26,27 STEM-EDS data reveal that the 2H-WS2 nanostructures are chalcogen deficient (Fig. S1†).28 XPS, which is sensitive to the chemical environment around each atom and can distinguish between the two WS2 polymorphs, confirms that the majority (∼90%) of the as-synthesized sample contains 2H-WS2, with the remainder being 1T-WS2 (Fig. S2 and Table S1†). The surface area of the flower-like 2H-WS2 nanostructures is estimated by BET to be 62.7 m2 g−1, which is much higher than that of the bulk powder sample (7.6 m2 g−1, Fig. S3†).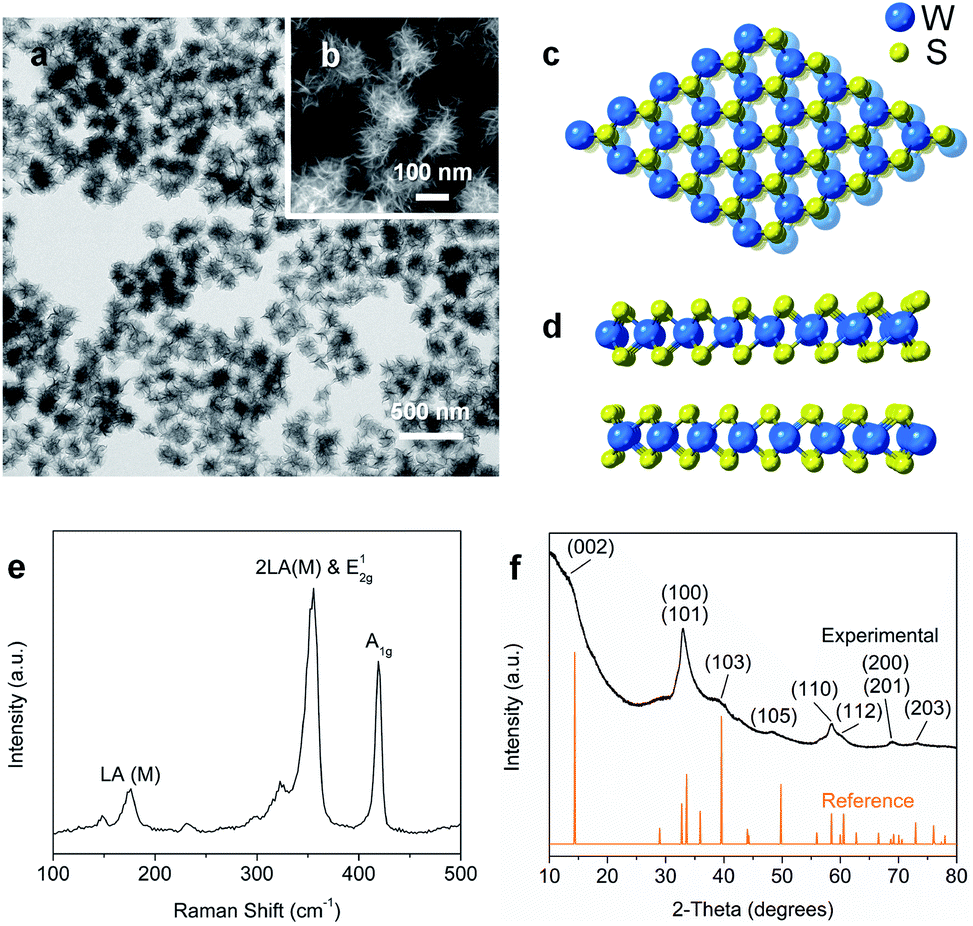 | ||
| Fig. 1 (a) TEM and (b) HAADF-STEM images of 2H-WS2 nanoflowers. (c) Top (basal plane) and (d) side (edge) views of the hexagonal structure for 2H-WS2. (e) Raman spectrum and (f) powder XRD data for the as-prepared 2H-WS2 nanostructures. | ||
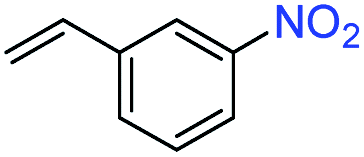
In a typical catalytic experiment, the 2H-WS2 nanostructures were added to a solution mixture containing THF, deionized water, and a substituted nitroarene. Hydrogenation was carried out for 8 h at 120 °C with 50 bar of H2, which are conditions comparable to previous studies of transition metal-based catalysts for analogous reactions.15,16,29 The model substrate was 3-nitrostyrene, which contains reducible nitro and vinyl groups. Using the aforementioned conditions, the 2H-WS2 nanoflowers facilitate >99% conversion of the nitro group to an amine and 94% retention of the vinyl group. In contrast, commercially available Pt/C unselectively hydrogenates both nitro and vinyl groups under the same conditions, while no detectable products were obtained with bulk WS2 powders (Table S2†). At lower H2 pressures (20 bar), 3-nitrostyrene can also be selectively transformed to 3-vinylaniline, though a longer reaction time (12 h) is required to achieve similar conversion (Fig. S4†).
Aliquots taken during the reaction indicate that the conversion of 3-nitrostyrene to 3-vinylaniline at 120 °C with 50 bar of H2 is nearly linear with time up to 8 h, reaching >99% conversion and 98% selectivity (Fig. 2a). After 8 h, reduction of the vinyl group results in increasing formation of the 3-ethylaniline byproduct, and consequently a decrease in selectivity. Therefore, reduction of the nitro group takes place prior to that of the vinyl group, and adjusting experimental parameters can optimize both activity and selectivity. As shown in Fig. 2b, the 2H-WS2 catalysts remain highly active and selective over five consecutive hydrogenation reactions of 3-nitrostyrene, achieving 98% conversion with >99% selectivity on the fifth cycle and indicating a high degree of recyclability. TEM, XRD, EDS, and Raman data for 2H-WS2 nanostructures before catalysis and after one and five catalytic cycles are indistinguishable (Fig. S5 and S6†). XPS shows no change in the 2H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1T phase ratio after 5 cycles. However, the W 4f and S 2p peaks exhibit a small shift (∼0.5 eV) towards lower binding energies after five cycles, suggesting that the surface of the nanostructured 2H-WS2 catalyst becomes slightly reduced after long-term reaction with hydrogen (Fig. S7†).
:1T phase ratio after 5 cycles. However, the W 4f and S 2p peaks exhibit a small shift (∼0.5 eV) towards lower binding energies after five cycles, suggesting that the surface of the nanostructured 2H-WS2 catalyst becomes slightly reduced after long-term reaction with hydrogen (Fig. S7†).
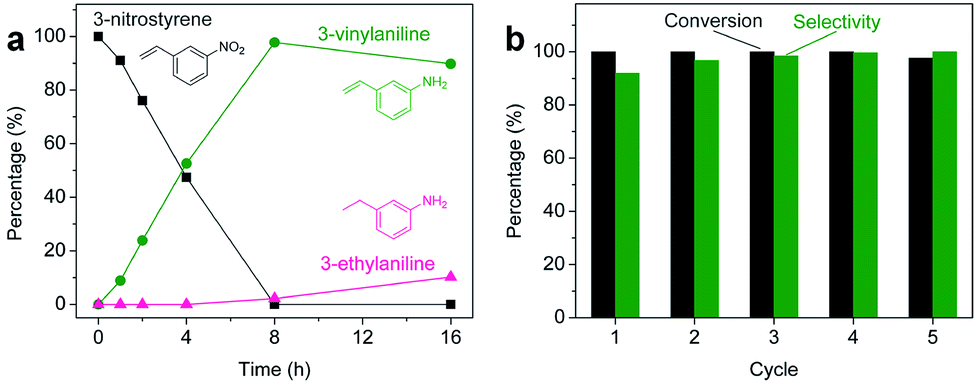 | ||
| Fig. 2 (a) Aliquot study for the selective hydrogenation of 3-nitrostyrene catalyzed by 2H-WS2 nanostructures at 50 bar H2 and 120 °C, showing the percentage of 3-nitrostyrene (grey), 3-vinylaniline (green), and 3-ethylaniline (pink) at different reaction times. (b) Percent conversion (grey) and selectivity (green) for the hydrogenation of 3-nitrostyrene to 3-vinylaniline using the same nanostructured 2H-WS2 catalyst over five successive cycles. | ||
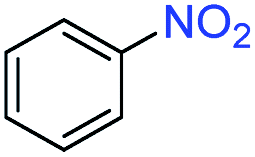
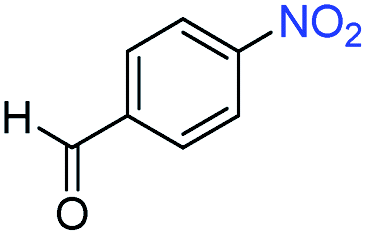
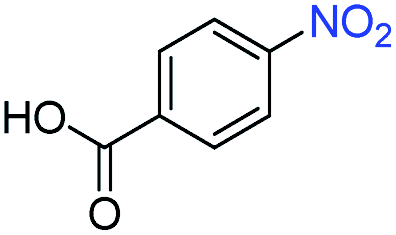
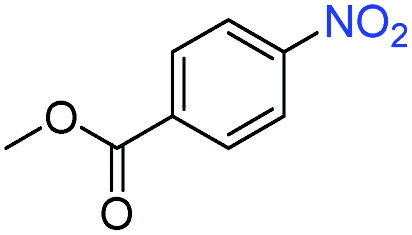
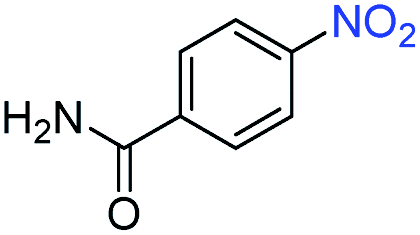
A broad scope of nitroarenes with diverse reducible groups including aldehydes, carboxylic acids, esters, amides, sulfonamides, nitriles, thioethers, ketones, halogens, and pyridines, as well as combinations of groups, were also tested for conversion to their corresponding anilines. As shown in Table 1, most substrates achieved complete conversion and >99% selectivity towards hydrogenation of the nitro group, leaving the other functional groups unaltered. Though most halide-containing anilines were obtained with >99% selectivity through hydrogenation of the corresponding nitro compounds under standard reaction conditions, a lower reaction temperature (100 °C) was used for 4-iodobenzene to avoid dehalogenation. Hydrogenation of 4-nitrobenzaldehyde to 4-aminobenzaldehyde could only be achieved with 78% selectivity, as hydrogenation of the aldehyde led to the formation of 4-methylaniline. In addition, nitrobenzene, nitrosobenzene, and N-phenylhydroxylamine were exclusively converted to aniline under standard reaction conditions, supporting the Haber mechanism where the nitro groups are converted to amines with nitroso and hydroxylamine intermediates.30
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Substrate | Conv. (%) | Selec. (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Unless otherwise noted, hydrogenations were performed under the following conditions: WS2 (5 mg), THF (1 mL), H2O (125 μL), substrate (0.125 mmol), 8 h, 120 °C. b Run at 100 °C. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
|
>99 | 94 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
|
>99 | 78 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |
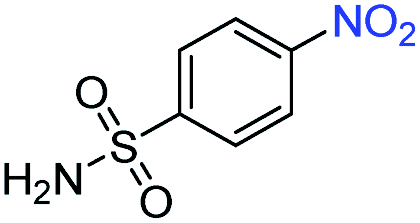
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 |
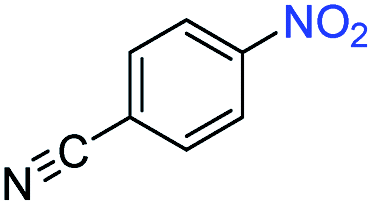
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 |
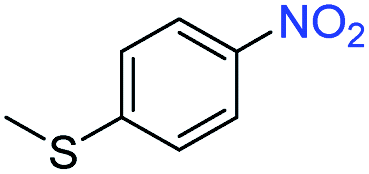
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 |
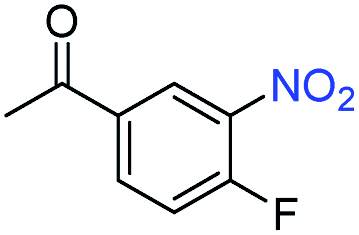
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13b |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 |
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 |
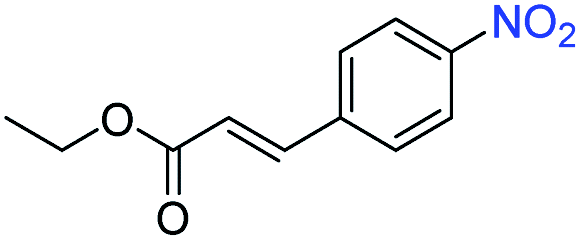
|
>99 | >99 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Flower-like nanostructured WS2 is an active catalyst for selective hydrogenation while bulk WS2 shows no hydrogenation activity under analogous conditions (Table S2†). To understand how nanostructuring of this model 2-D TMD system enables catalysis, ADF-STEM was used to gain atomic-level insights into the structures of the as-prepared nanosheet catalysts. As shown in Fig. 3a, monolayer and bi-layer WS2 nanosheets were observed near the edges of the 2H-WS2 nanoflowers, with visible holes on the nanosheets indicating the presence of atomic vacancies. ADF-STEM can differentiate atoms based on Z-contrast (Z = atomic number), thus a line scan at high magnification can be analyzed to determine which atoms are in a particular location.31Fig. 3b–d show three line scans across the basal plane of a WS2 monolayer, highlighting a region with no vacancies, a region with an individual monosulfur vacancy (VS), and a region with a tungsten vacancy (VW) associated with a monosulfur vacancy (VS). While both tungsten and sulfur vacancies are present, STEM-EDS quantification reveals that the 2H-WS2 nanostructures are chalcogen-deficient, suggesting that sulfur vacancies dominate. Additional microscopic data also suggest that both tungsten- and sulfur-terminated edges are abundantly present in the flower-like nanostructures (Fig. S8†). Although not observed, disulfur vacancies (VS2) likely also exist, considering the sulfur vacancy concentration and previous reports.23
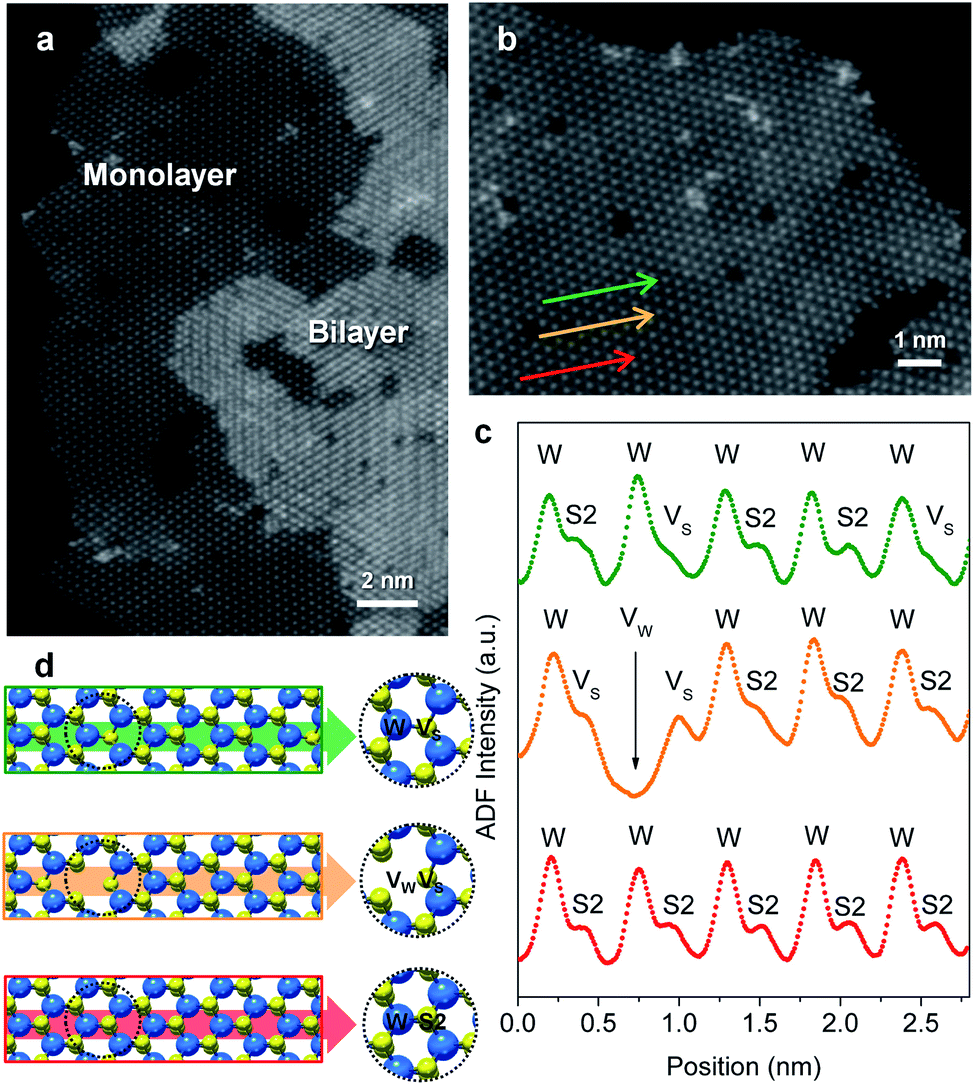 | ||
| Fig. 3 (a) High-resolution ADF-STEM image showing bilayer and monolayer domains with atomic vacancies on the surface (basal plane) of the 2H-WS2 nanostructures. (b) Atomically resolved ADF-STEM image for a region of monolayer 1H-WS2. (c) Experimental ADF intensity curves and (d) structural models corresponding to the three line scans indicated by the green, orange, and red arrows in (b), showing the alternation of tungsten (W) and sulfur (S2) configurations, as well as existence of tungsten (VW) and monosulfur (VS) vacancies. | ||
Surface engineering of nanostructured TMD catalysts has been demonstrated as an effective approach to generate active edge and vacancy sites for hydrogen activation and production.32–34 Similar insights are important for selective hydrogenation reactions on TMDs in order to understand how selectivity emerges and therefore how it can be optimized, manipulated, and controlled. DFT calculations were performed to identify the optimized adsorption geometry of 3-nitrostyrene and the hydrogen transfer process on 2-D WS2 with different sulfur vacancy configurations. Monolayers of 1H-WS2 with a monosulfur vacancy (1VS), a disulfur vacancy (1VS2), and four evenly-distributed monosulfur vacancies (4VS) on a basal plane were constructed in a 4 × 4 × 1 supercell, corresponding to 3%, 6% and 12% sulfur vacancy concentrations, respectively (Fig. 4).
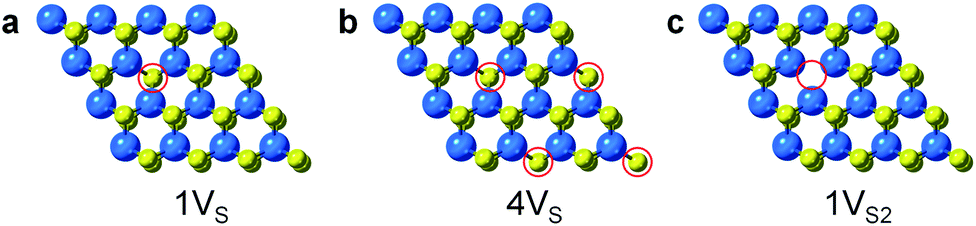 | ||
| Fig. 4 Simulated structures for different types of S-vacancies on the basal planes of 1H-WS2: (a) 1VS, (b) 4VS, and (c) 1VS2. The vacancy sites are highlighted by red circles. | ||
During hydrogenation on a transition metal disulfide (MS2) catalyst, molecular hydrogen is adsorbed on the surface and is activated by either homolytic dissociation on the sulfur atoms to form two –S–H bonds or heterolytic dissociation to yield –S–H and –M–H species.35 The activated hydrogen atoms are then transferred to the adsorbed nitroarene molecule through active sites (i.e. vacancies and edge sites of nanostructured WS2), where the adsorption geometry of the nitroarene molecule plays a key role in determining the activity and selectivity. We thus first investigated the adsorption of 3-nitrostyrene on the 1VS, 1VS2, and 4VS surfaces of WS2. Three adsorption geometries, including two vertical modes where the molecule stands up with the nitro group bound to the WS2 surface and one parallel mode where the molecule lies flat, parallel to the WS2 plane, were investigated (Fig. S9†).36,37 Calculation results suggest that the weakly adsorbed parallel configuration is preferable in the 1VS and 1VS2 models, while the quasi-vertical configuration with two oxygen atoms from the nitro group bound to W atoms in the sulfur vacancy site is slightly preferred in the case of 4VS (Fig. 5 and Table S3†). The positive adsorption energies in the 1VS and 1VS2 models indicate that the vertical adsorbed states are unstable local minima. A high concentration of sulfur vacancies significantly promotes the vertical adsorption of 3-nitrostyrene on WS2. Favorable adsorption with a high concentration of vacancies could also lead to higher adsorbate coverages that motivate a vertical orientation due to surface crowding. This vertical orientation facilitates selective hydrogenation of the nitro group since it is positioned directly on the surface of the WS2 catalyst while the vinyl group does not interact with the surface.
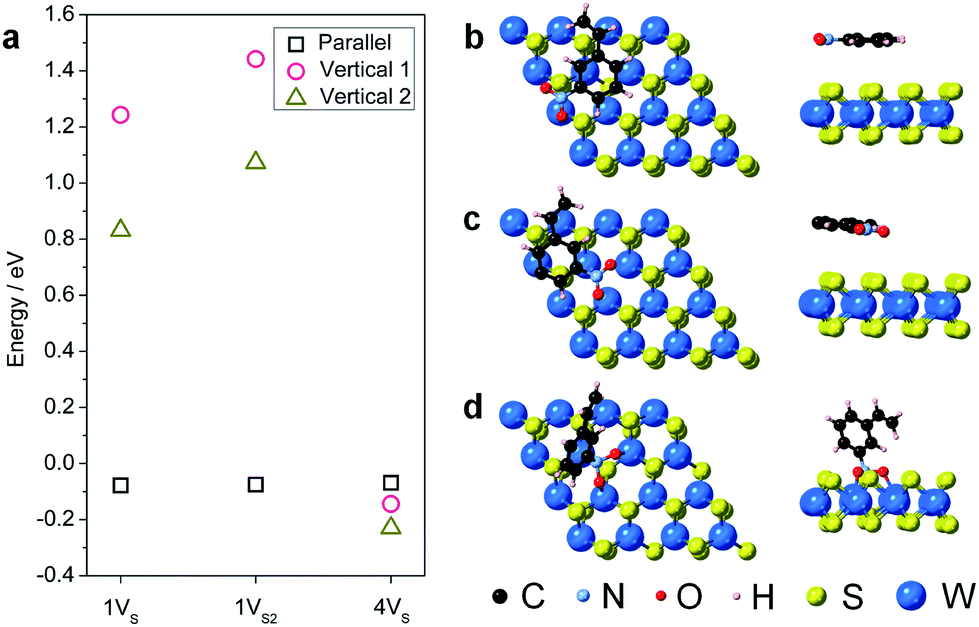 | ||
| Fig. 5 (a) Calculated adsorption energies of the three adsorption geometries (vertical 1, vertical 2, and parallel) on the three sulfur vacancy models (1VS, 1VS2, and 4VS) for 1H-WS2 basal planes. Lowest energy adsorbed structures of 3-nitrostyrene absorbed on the surface of a 1H-WS2 monolayer are shown for the (b) 1VS (parallel); (c) 1VS2 (parallel); and (d) 4VS (vertical) orientations on the different vacancy models. | ||
For the 1VS and 1VS2 models where 3-nitrostyrene preferentially adsorbs parallel to the WS2 surface and both the nitro and vinyl groups are oriented so that they could undergo catalytic hydrogenation, we carried out additional kinetic calculations involving stepwise hydrogenation of both the nitro and vinyl groups. We calculated the activation barriers (Ea) and reaction energies (ΔE) for the hydrogenation of the nitro and vinyl groups, using the 1VS model as a representative parallel-adsorption system. As summarized in Fig. S10 and S11,† the rate-determining step for hydrogenation of the nitro group is the transformation from R–NO2 to R–NOOH, which has a reaction barrier of 0.97 eV that is noticeably lower than that of the first step of the vinyl hydrogenation pathway (1.07 eV). Thus, selective hydrogenation of the nitro group is kinetically favored when there is a low concentration of sulfur vacancies.
In addition to point defects involving sulfur vacancies on the basal planes, the WS2 nanoflowers also provide abundant edge sites that could also serve as active sites for selective hydrogenation of 3-nitrostyrene. We therefore also calculated the adsorption energy of 3-nitrostyrene on the tungsten- and sulfur-terminated edges, which were constructed using 4 × 1 × 1 nanoribbons. Previous DFT calculations have demonstrated that, at high temperatures and partial pressures of H2, partial edge site coverage of sulfur and hydrogen adatoms is likely based on the previous ab initio thermodynamics methodology.38 Given the high pressures and temperatures utilized in our catalytic experiments, modeling of the tungsten-terminated edge sites included a coverage of θS = 0.5 and θH = 0.5, and the sulfur-terminated edge sites were modeled with θS = 1 and θH = 1 (Fig. S12†). As shown in Fig. 6, S13 and S14,† the nitro adsorption energies were calculated for five distinct configurations. Three adsorption geometries for the tungsten-terminated edge sites were considered, including head-on adsorption (W-1), the nitro group adsorbed to the tungsten atoms modified with one sulfur atom (W-2), and the nitro group adsorbed to the tungsten atoms modified with one sulfur atom and one hydrogen atom (W-3). Two geometries for the sulfur-terminated sites were also considered: head-on adsorption (S-1) and staggered head-on adsorption (S-2). Based on the optimized absorption energies, only the W-2 and W-3 configurations, involving the tungsten-terminated edge sites, revealed favorable adsorption of 3-nitrostyrene via a quasi-vertical geometry, where the two oxygen atoms from the nitro group are separately bonded to the two exposed tungsten atoms of WS2 (Fig. 6). For the other three geometries, either weak or unfavorable interactions were observed (Table S4†). For both the W-2 and W-3 orientations, the nitro group directly bonds to the tungsten atoms at the edges and results in selective hydrogenation, whereas the vinyl group does not directly interact with the WS2 nanostructures.
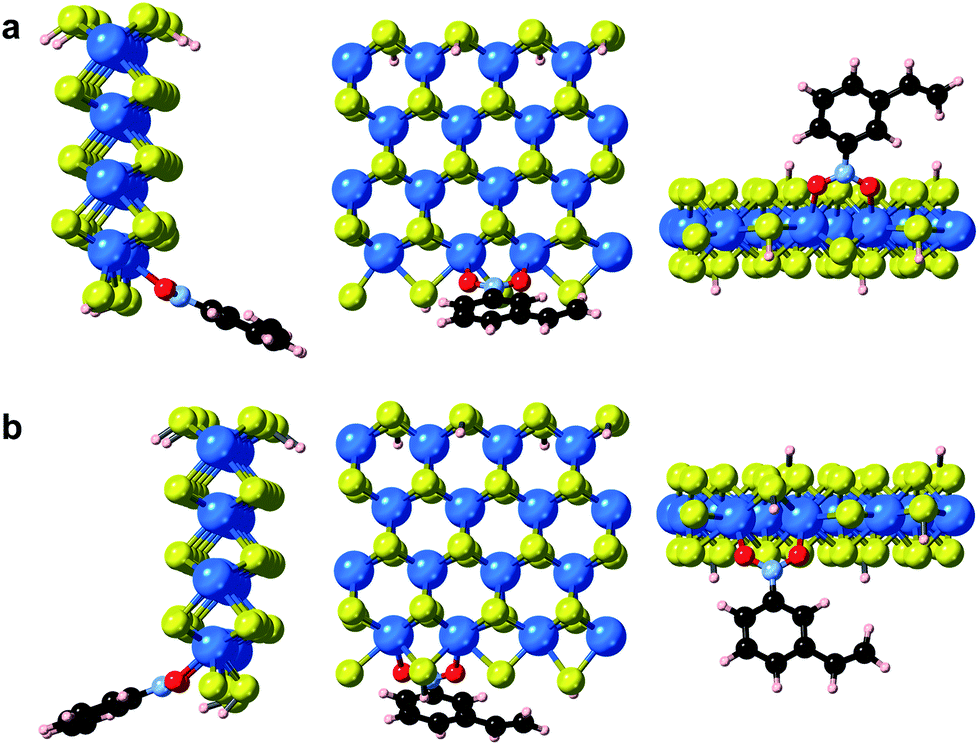 | ||
| Fig. 6 Optimized geometries of 3-nitrostyrene absorbed on the tungsten-terminated edges of 1H-WS2 monolayer are shown for the (a) W-2 and (b) W-3 models from three viewing angles. | ||
The mechanisms that result in selective hydrogenation of 3-nitrostyrene to 3-vinylaniline are different for regions of the nanostructured surface that have high vs. low sulfur vacancy concentrations and different edge terminations. However, the end result is the same – the nitro group is selectively hydrogenated relative to the vinyl group, and the selectivity arises from interactions between the substrate molecule and sulfur defects on the WS2 surface.
Conclusions
Colloidally synthesized 2H-WS2 nanostructures, which have high surface areas and contain single- and few-layer nanosheets with high vacancy concentrations, serve as a model 2-D TMD system for understanding the origin of selectivity during hydrogenation reactions. The 2H-WS2 nanostructures catalyze the selective hydrogenation of substituted nitroarenes with molecular hydrogen, transforming them to the corresponding anilines in the presence of a broad scope of reducible functionalities. Microscopic and computational studies indicate that sulfur vacancies on the basal planes and tungsten-terminated edges facilitate chemoselectivity, where hydrogen preferentially transfers to the nitro group due either to the preferential vertical adsorption geometry at high basal-plane vacancy concentrations and tungsten-terminated edge sites, or to smaller kinetic barriers at lower basal-plane vacancy concentrations where parallel adsorption is favored. Such insights are important for guiding nanostructuring efforts in 2-D TMD catalyst systems, as well as other applications including responsive gas sensors, where vacancy-rich surfaces are common and defect engineering could tune adsorption behavior.39 Future efforts to synthesize more complex and precisely-tailored TMD nanostructures, including composition-tunable alloys and noble metal-anchored heterostructures,28,40–42 will provide a broader scope of systems to enable more comprehensive studies on the nature of the active sites, the various mechanism by which selectivity can be achieved and modified, and the role of interactions between active species and underlying substrates.Experimental
Materials
Oleylamine (technical grade, 70%), tungsten(VI) chloride (WCl6, ≥99.9%, trace metal basis), hexamethyldisilazane (HMDS, reagent grade, ≥99%), carbon disulfide (CS2, ≥99.9%, anhydrous), tetrahydrofuran (THF, anhydrous, contains 250 ppm BHT as inhibitor, ≥99%), azobenzene (98%), 4-nitrobenzamide (98%), 4-nitrobenzenesulfonamide (97%), 4′-fluoro-3′-nitroacetophenone (97%), 3-nitrostyrene (96%), hydrazobenzene, 1-bromo-4-nitrobenzene (99%), ethyl 4-nitrocinnamate (predominately trans, 99%), 4′-aminoacetophenone (99%), 3-vinylaniline (contains KOH as inhibitor, 97%), 4-nitrothioanisole (96%), 1-chloro-4-nitrobenzene (99%), 1-iodo-4-nitrobenzene (98%), tetradecane (≥99%), nitrosobenzene (≥97%), 3-ethylaniline (98%), p-toluidine (99%), 4-(4-nitrobenzyl)pyridine (98%), 4-nitrobenzonitrile (97%), hydroxylamine hydrochloride (99%), azoxybenzene, and nitrobenzene (99%) were purchased from Sigma Aldrich. Oleic acid (technical grade, 90%) and 4-nitrobenzoic acid (99%) were purchased from Alfa Aesar. Methyl 4-nitrobenzoate (≥98.0%) and 4-nitrobenzaldehyde (≥98.0%) were purchased from TCI. 10% Pt on activated carbon powder was purchased from Premetek. Solvents, including hexane and isopropanol, were of analytical grade. All chemicals were used as received without further purification.Synthesis of 2H-WS2 nanostructures
The colloidal synthesis of the 2H-WS2 nanostructures was carried out according to the previously reported approach.21 15 mL of oleylamine was added to a 100 mL three-neck flask and degassed for 30 min under vacuum at 120 °C. HMDS (0.5 mL) was then injected into the flask after cooling to 100 °C under argon and the mixture was heated to 320 °C. Meanwhile, WCl6 (50 mg, 0.125 mmol) was dissolved in 0.3 mL of oleic acid (0.95 mmol) and mixed with 5 mL of oleylamine (15.2 mmol) in an argon-flushed septum-capped vial. Upon injection, 0.24 mL of CS2 was introduced to the vial, forming a homogeneous solution that became hot due to the exothermicity of the reaction. The solution in the vial was subsequently injected dropwise into the three-neck flask at a rate of 10 mL h−1 using a syringe pump. After 30 min, the injection was stopped and the heating mantle was removed. After cooling down to room temperature, the products were washed three times with a 1:1 toluene/ethanol mixture, collected with centrifugation, and kept as a powder under argon.
Hydrogenation reactions
In a typical reaction, 20 mg of the 2H-WS2 nanostructures in powder form was added to a septum-capped glass vial along with a Teflon-coated magnetic stir bar, 4 mL of THF, 0.5 mL of deionized water, and 0.5 mmol of substrate molecules. Sonication was carried out to make sure the dispersion was fully mixed. A 300 mL Parr stainless steel autoclave (4766-T-SS-3000-VGR) was used for all hydrogenation reactions. The septum cap of every sample was punctured with a disposable 18 gauge needle to allow the partitioning of H2 into the vial. Once prepared and sealed, the autoclave was flushed twice with H2 gas at 25 bar to remove ambient air, and then filled to the final pressure (50 bar H2). Once pressurized, the autoclave was heated to 120 °C and held at that temperature for 8 h. The reaction was cooled to room temperature, the H2 atmosphere was discharged, and the samples were prepared for GC-MS as described below. For the hydrogenation of substituted nitroarenes, a smaller scale with 5 mg of 2H-WS2 nanostructures in powder form, 1 mL of THF, 0.125 mL of deionized water, and 0.125 mmol of substrate molecules were utilized.GC-MS analysis
Typically, tetradecane (100 μL for 0.5 mmol scale reactions, 25 μL for 0.125 mmol scale reactions) was injected into the reaction mixture post-reaction as an internal standard. The catalyst from the reaction mixture was then removed via centrifugation at 13500 rpm and the resulting solution was diluted with THF and submitted for GC-MS analysis. Product yields were determined via calibration curves made using commercially available reaction products. If no substrate signal was observed via GC-MS, it was assumed that the reaction had proceeded to >99% conversion. Similarly, if the desired aniline was observed as the only product, it was assumed that the reaction had proceeded with >99% selectivity.
Recycling experiments
The hydrogenation of 0.5 mmol 3-nitrostyrene using 20 mg WS2 nanostructures as the catalyst was performed as described above. Following the completion of the reaction, 100 μL tetradecane was added to the reaction mixture. The WS2 nanostructures were separated from the reaction mixture via centrifugation, and the resulting supernatant was diluted with THF and submitted for GC-MS analysis to determine reaction yields, as described above. The remaining WS2 nanostructures were washed and centrifuged twice with THF, and then used as the catalyst for another hydrogenation of 3-nitrostyrene (0.5 mmol scale). This process was repeated for a total of five times to evaluate the catalyst recyclability.Characterization
Powder X-ray diffraction (XRD) patterns were collected using a Bruker-AXS D8 Advance diffractometer equipped with Cu Kα radiation and a LynxEye 1-D detector. Simulated XRD patterns of 2H-WS2 were generated using the CrystalMaker/CrystalDiffract software package. Transmission electron microscopy (TEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images, and energy dispersive X-ray spectroscopy (EDS) data with element maps were acquired using a FEI Talos F200X operating at 200 kV. High-resolution ADF-STEM images were obtained using a FEI Titan3 G2 60/300 TEM with a spherical aberration corrector on both the probe- and the image-forming lens at an accelerating voltage of 80 kV. ADF line scan was obtained using ImageJ software. Bruker ESPRIT 2 software was applied for EDS data interpretation. Micro-Raman measurements were performed in a Renishaw inVia confocal microscope-based Raman spectrometer with 514.5 nm laser. The laser spot size using the 100× objective lens was approximately 1 μm. X-ray photoelectron spectroscopy (XPS) experiments were performed using a Physical Electronics VersaProbe II instrument equipped with a monochromatic Al Kα X-ray source (hν = 1486.7 eV) and a concentric hemispherical analyzer. Charge neutralization was performed using both low energy electrons (<5 eV) and argon ions. The binding energy axis was calibrated using sputter cleaned Cu foil (Cu 2p3/2 = 932.7 eV, Cu 3p3/2 = 75.1 eV). Peaks were charge referenced to the CHx peak in the carbon 1s spectra at 284.8 eV. Measurements were made at a takeoff angle of 45° with respect to the sample surface plane. This resulted in a typical sampling depth of 3–6 nm (95% of the signal originated from this depth or shallower). Quantification was carried out using instrumental relative sensitivity factors (RSFs) that account for the X-ray cross section and inelastic mean free path of the electrons. Nitrogen adsorption analysis was carried at 77 K with an Accelerated Surface Area and Porosimetry Analyzer (ASAP 2020; Micromeritics Instrument Corp.). Prior to the measurement, the sample was degassed at 333 K for 24 h under 4 μm Hg vacuum. Surface area was estimated using the Brunauer, Emmett and Teller (BET) equation.43Calculations
Density functional theory (DFT) calculations were performed using Vienna ab initio simulation package (VASP) 5.4.4.44 The projector-augmented-wave (PAW) approach was utilized to treat the core electrons,45,46 and the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional of the generalized gradient approximation (GGA) was used to describe electron interactions.47 Kinetic cut-off energy for the plane-wave basis set was set as 450 eV. Gaussian smearing method was applied with smearing width of 0.01 eV.Monolayer WS2 with trigonal prismatic coordination (1H-WS2) was selected as the surface slab model for the calculation of molecule adsorption and reaction pathways. Considering the large steric hinderance of 3-nitrostyrene, we applied a 4 × 4 × 1 supercell to prevent the potential intermolecular repulsion effect induced by coverage. Three distinct types of sulfur vacancies were modeled: only one sulfur atom removed from the lattice (denoted as 1VS); both top and bottom sulfur atoms removed (denoted as 1VS2); and four sulfur vacancies uniformly distributed on one side of the monolayer structure (denoted as 4VS). The vacuum space between periodic images is at least 12 Å to minimize the interactions between adjacent images. For this supercell, a 1 × 1 × 1 Γ-centered k-point mesh was applied. We tested the convergence with 3 × 3 × 1 k-point mesh and found that for selected adsorption and reaction, the energy difference between the results obtained with two meshes was less than 0.05 eV. In the case of 3-nitrostyrene adsorbed on tungsten- and sulfur-terminated edges, calculations were performed with a 3 × 1 × 1 gamma-centered k-point due to the one-dimensional structure. The electronic convergence criteria was set as 1 × 10−6 eV. Structural optimization was considered complete when the magnitude of the forces on the atoms was less than 0.01 eV Å−1, while force convergence was set as 0.05 eV Å−1 for the search of the transition states. The transition states located using the climbing image nudged elastic band (CI-NEB) method were further refined using the dimer method with a force criteria of 0.01 eV Å−1.48,49 Transition states were confirmed by an imaginary frequency corresponding to the reactive mode. In some cases, there was another small imaginary frequency corresponding to the rocking vibration mode of the benzene group. However, due to the tight force convergence, further energy gains to eliminate the small imaginary frequency for the transition states would be low.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. S. and R. E. S. were supported by the U.S. National Science Foundation Grant No. DMR-1607135. A. J. D. and C. F. H. were supported by the ACS Petroleum Research Fund Grant No. 58373ND5. Y. L. and M. J. J. recognize support from the US Department of Energy, Office of Science, Basic Energy Sciences, Catalysis Science Program under award # DE-SC0016529. Computational work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. H. L., K. F. and M. T. acknowledge support from NSF EFRI 2-DARE grant 1433311 and the Air Force Office of Scientific Research (AFOSR) grant 17RT0244. M. T. also acknowledges the Center for 2-Dimensional and Layered Materials at the Pennsylvania State University. Electron microscopy was performed at the Electron Microscopy Facility at the Materials Characterization Lab of the Penn State Materials Research Institute. We thank Jeff Shallenberger for the help with XPS acquisition, Phillip Smith for his assistance with collection of GC-MS data, and Alexey Silakov for helpful discussions.References
- R. Lv, J. A. Robinson, R. E. Schaak, D. Sun, Y. Sun, T. E. Mallouk and M. Terrones, Acc. Chem. Res., 2015, 48, 56–64 CrossRef CAS PubMed.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- S. Kamiguchi, K. Arai, K. Okumura, H. Iida, S. Nagashima and T. Chihara, Appl. Catal., A, 2015, 505, 417–421 CrossRef CAS.
- E. J. M. Hensen, P. J. Kooyman, Y. van der Meer, A. M. van der Kraan, V. H. J. de Beer, J. A. R. van Veen and R. A. van Santen, J. Catal., 2001, 199, 224–235 CrossRef CAS.
- T. A. Pecoraro and R. R. Chianelli, J. Catal., 1981, 67, 430–445 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- J. I. Paredes, J. M. Munuera, S. Villar-Rodil, L. Guardia, M. Ayán-Varela, A. Pagán, S. D. Aznar-Cervantes, J. L. Cenis, A. Martínez-Alonso and J. M. D. Tascón, ACS Appl. Mater. Interfaces, 2016, 8, 27974–27986 CrossRef CAS PubMed.
- I. Sorribes, L. Liu and A. Corma, ACS Catal., 2017, 7, 2698–2708 CrossRef CAS.
- L. Huang, P. Luo, M. Xiong, R. Chen, Y. Wang, W. Xing and J. Huang, Chin. J. Chem., 2013, 31, 987–991 CrossRef CAS.
- P. Serna and A. Corma, ACS Catal., 2015, 5, 7114–7121 CrossRef CAS.
- S. Furukawa, K. Takahashi and T. Komatsu, Chem. Sci., 2016, 7, 4476–4484 RSC.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- J. R. Morse, J. F. Callejas, A. J. Darling and R. E. Schaak, Chem. Commun., 2017, 53, 4807–4810 RSC.
- F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M.-M. Pohl, J. Radnik, A.-E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Nat. Chem., 2013, 5, 537–543 CrossRef CAS PubMed.
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schunemann, A. Bruckner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- M. Shen, H. Liu, C. Yu, Z. Yin, M. Muzzio, J. Li, Z. Xi, Y. Yu and S. Sun, J. Am. Chem. Soc., 2018, 140, 16460–16463 CrossRef CAS PubMed.
- D. Cantillo, M. Baghbanzadeh and C. O. Kappe, Angew. Chem., Int. Ed., 2012, 51, 10190–10193 CrossRef CAS PubMed.
- R. J. Voorhoeve, J. Catal., 1971, 23, 236–242 CrossRef CAS.
- M. Cargnello, Chem. Mater., 2019, 31, 576–596 CrossRef CAS.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- Y. Sun, Y. Wang, D. Sun, B. R. Carvalho, C. G. Read, C.-H. Lee, Z. Lin, K. Fujisawa, J. A. Robinson, V. H. Crespi, M. Terrones and R. E. Schaak, Angew. Chem., Int. Ed., 2016, 55, 2830–2834 CrossRef CAS PubMed.
- Z. Lin, B. R. Carvalho, E. Kahn, R. Lv, R. Rao, H. Terrones, M. A. Pimenta and M. Terrones, 2D Mater., 2016, 3, 022002 CrossRef.
- D. Sun, S. Feng, M. Terrones and R. E. Schaak, Chem. Mater., 2015, 27, 3167–3175 CrossRef CAS.
- W. Shi, M.-L. Lin, Q.-H. Tan, X.-F. Qiao, J. Zhang and P.-H. Tan, 2D Mater., 2016, 3, 025016 CrossRef.
- Y. Sun, K. Fujisawa, M. Terrones and R. E. Schaak, J. Mater. Chem. C, 2017, 5, 11317–11323 RSC.
- W. J. Schutte, J. L. De Boer and F. Jellinek, J. Solid State Chem., 1987, 70, 207–209 CrossRef CAS.
- Y. Sun, K. Fujisawa, Z. Lin, Y. Lei, J. S. Mondschein, M. Terrones and R. E. Schaak, J. Am. Chem. Soc., 2017, 139, 11096–11105 CrossRef CAS PubMed.
- R. V. Jagadeesh, T. Stemmler, A.-E. Surkus, H. Junge, K. Junge and M. Beller, Nat. Protoc., 2015, 10, 548–557 CrossRef CAS PubMed.
- J. Song, Z. F. Huang, L. Pan, K. Li, X. Zhang, L. Wang and J. J. Zou, Appl. Catal., B, 2018, 227, 386–408 CrossRef CAS.
- X. Zhao, S. Ning, W. Fu, S. J. Pennycook and K. P. Loh, Adv. Mater., 2018, 30, 1802397 CrossRef PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- C. Tsai, H. Li, S. Park, J. Park, H. S. Han, J. K. Nørskov, X. Zheng and F. Abild-Pedersen, Nat. Commun., 2017, 8, 15113 CrossRef PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- A. Travert, H. Nakamura, R. A. van Santen, S. Cristol, J.-F. Paul and E. Payen, J. Am. Chem. Soc., 2002, 124, 7084–7095 CrossRef CAS PubMed.
- L. Zhang, X.-M. Cao and P. Hu, Appl. Surf. Sci., 2017, 392, 456–471 CrossRef CAS.
- L. Zhang, J. Jiang, W. Shi, S. Xia, Z. Ni and X. Xiao, RSC Adv., 2015, 5, 34319–34326 RSC.
- A. S. Rosen, J. M. Notestein and R. Q. Snurr, J. Phys. Chem. C, 2018, 122, 15318–15329 CrossRef CAS.
- J. H. Han, H. K. Kim, B. Baek, J. Han, H. S. Ahn, M.-H. Baik and J. Cheon, J. Am. Chem. Soc., 2018, 140, 13663–13671 CrossRef CAS PubMed.
- G. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS PubMed.
- Y. Shi, Y. Zhou, D.-R. Yang, W.-X. Xu, C. Wang, F.-B. Wang, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2017, 139, 15479–15485 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional material characterization and DFT calculation data. See DOI: 10.1039/c9sc03337h |
| ‡ These authors have contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2019 |