
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective catecholamine detection in living cells by a copper-mediated oxidative bond cleavage†
Ka Yan
Tong‡
 a,
Jia
Zhao‡
b,
Chun-Wai
Tse
a,
Pui-Ki
Wan
a,
Jianhui
Rong
b and
Ho Yu
Au-Yeung
*a
a,
Jia
Zhao‡
b,
Chun-Wai
Tse
a,
Pui-Ki
Wan
a,
Jianhui
Rong
b and
Ho Yu
Au-Yeung
*a
aThe University of Hong Kong, State Key Laboratory for Synthetic Chemistry, Department of Chemistry, Pokfulam Road, P. R. China. E-mail: hoyuay@hku.hk
bSchool of Chinese Medicine, The University of Hong Kong, 10 Sassoon Road, Pokfulam, Hong Kong, P. R. China
First published on 14th August 2019
Abstract
The development of a new triggered-release system for selective detection of catecholamines in biological samples including living cells is reported. Catecholamines are a class of tightly regulated hormones and neurotransmitters in the human body and their dysregulation is implicated in various neurodegenerative diseases. It is highly challenging to selectively sense and detect catecholamines in a complex biological environment due to their small size, non-specific molecular shape and trivial chemical properties. In this study, a copper-based, catecholamine-triggered oxidation that releases a fluorescent reporter is described. The probe is highly sensitive and selective for detecting changes in catecholamine levels in aqueous buffer, human plasma, and cellular models of neuronal differentiation and Parkinson's disease. This new catecholamine sensing strategy features chemical reactivity as part of small molecule recognition as opposed to the conventional use of a well-designed host for reversible binding.
Introduction
Catecholamines (e.g. dopamine, adrenaline and noradrenaline) are important small molecule neurotransmitters involved in vital physiological processes such as learning and memory, blood pressure regulation, motor control and physical responses.1,2 Abnormal catecholamine levels are associated with various neurological disorders such as schizophrenia,3 psychosis,4 and attention deficit hyperactivity disorder.5 At the cellular level, catecholamine dynamics is tightly regulated to maintain proper cellular functions. In neurons, for example, dopamine is stored in synaptic vesicles and released into the synaptic cleft when an action potential is reached, and the released dopamine could be taken up again and repackaged for further release.6 In particular, dopamine depletion in dopaminergic neurons in the mid-brain is attributed to motor deficits which are the main symptoms of Parkinson's disease.7 Recognition and detection of catecholamine in cells are therefore critical to track their dynamics for a further understanding of their roles in neuronal development and related diseases, as well as developing diagnostic and therapeutic strategies for catecholamine-related disorders.8Development of selective recognition and detection for catecholamines in biological cells or other biological samples is far from trivial because catecholamines have only minimal structural features and diol/amine functional groups are widely present in other biomolecules. The small size and neutral charge of catecholamines also add extra complexity in designing recognition systems in aqueous environments. Although there are a few sophisticated synthetic hosts that show promising catecholamine binding, they are not biocompatible, sensitive or selective for biological applications.9–14 Methods to engineer an easily detectable response from these hosts are also not straightforward. Very recently, several (bio)macromolecular hosts that sense catecholamines by a conformational change of the host upon catecholamine binding have been reported.15–17 For example, protein-based sensors have been constructed by fusing a known catecholamine-binding protein with a fluorescent protein.15,16 The genetically engineered proteins offer a high selectivity and strong binding to catecholamines but their use may require cell transfection or implantation of genetically modified cells. A few systems based on DNA-wrapped carbon nanotubes or boronic acid-functionalized conjugated polymers have also been reported.18–21 Compared with macromolecular sensors, small molecule systems for fluorescent catecholamine detection are operationally simple and applicable in different types of biological samples with minimum preparative procedures. Rational chemical modifications on small molecule probes also offer a facile tuning of photophysical, localization, cell-permeation and other properties to suit the specific need of the experiment. Examples of small molecule probes for direct catecholamine sensing are however scarce, with one example featuring reversible iminium and/or boronate ester formation between dopamine/noradrenaline and an aldehyde- and/or a boronic acid-containing probe.22–24 The small association constant (K ∼ 102 to 104 M−1) of iminium and boronic acid catechol ester may also limit its application to samples with a high catecholamine concentration. Other bioanalytical methods such as liquid chromatography,25–27 electrochemical methods28,29 and the use of fluorescent false neurotransmitters30–34 are either not applicable in living cells or only indirectly studying neuronal activity.
In view of the distinctive difficulty and importance of catecholamine detection, we sought to develop a new strategy for the selective recognition and fluorescence detection of the small molecules, which not only will provide new optical tools for studying the neurotransmitters in different samples and conditions, but also could be versatilely applied to other catecholamine-responsive systems for potential controlled-release, triggered response and other applications. Because catecholamine binding via reversible boronic acid catechol ester and/or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N exchange is inherently weak,35,36 we turned our attention to include chemical reactivity as part of the recognition.37,38 In particular, we are inspired by the oxidative reactivity of dopamine β-hydroxylase (DβH), which is a copper oxygenase featuring a His/His/Met coordination, and reason that a similar catecholamine-triggered, copper-based oxidation can be harnessed for the selective release of a caged fluorescent reporter (Scheme 1).39–43 Here we report the design and development of a prototypical catecholamine probe (CAP) that is based on a copper-based oxidative reaction and demonstrate that it can successfully detect changes in catecholamine levels in PC12 models of neuronal differentiation and Parkinson's disease. Combined with morphological assessment of the cells in the same imaging experiment, an integrative study of important neuronal processes by morphology and molecular imaging is demonstrated for the first time. On the other hand, connecting the chemical reactivity of a small molecule to that of metal-based oxidation is demonstrated to be an effective strategy for the selective detection of small organic molecules that are challenging recognition targets using conventional host–guest chemistry.
N exchange is inherently weak,35,36 we turned our attention to include chemical reactivity as part of the recognition.37,38 In particular, we are inspired by the oxidative reactivity of dopamine β-hydroxylase (DβH), which is a copper oxygenase featuring a His/His/Met coordination, and reason that a similar catecholamine-triggered, copper-based oxidation can be harnessed for the selective release of a caged fluorescent reporter (Scheme 1).39–43 Here we report the design and development of a prototypical catecholamine probe (CAP) that is based on a copper-based oxidative reaction and demonstrate that it can successfully detect changes in catecholamine levels in PC12 models of neuronal differentiation and Parkinson's disease. Combined with morphological assessment of the cells in the same imaging experiment, an integrative study of important neuronal processes by morphology and molecular imaging is demonstrated for the first time. On the other hand, connecting the chemical reactivity of a small molecule to that of metal-based oxidation is demonstrated to be an effective strategy for the selective detection of small organic molecules that are challenging recognition targets using conventional host–guest chemistry.
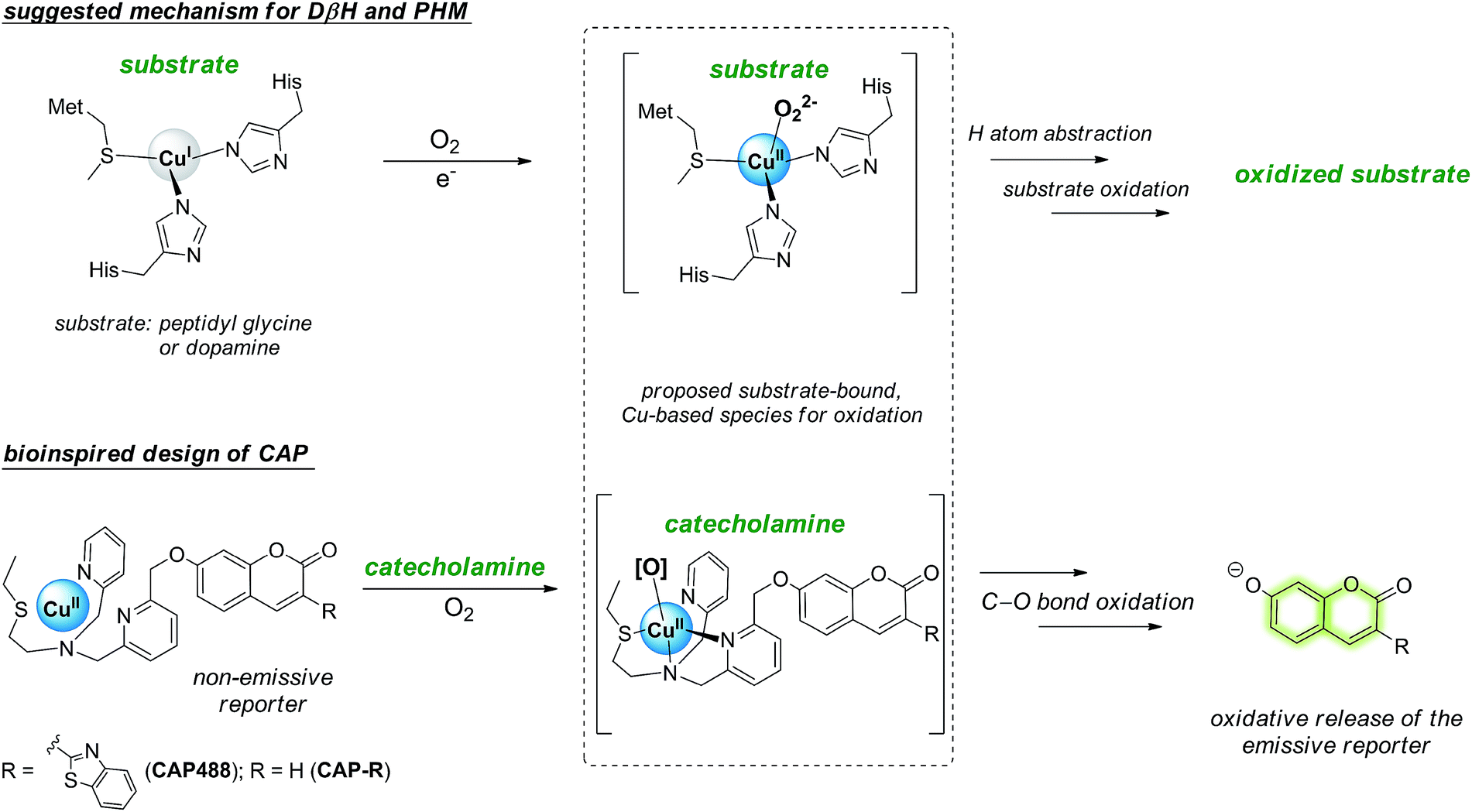 | ||
| Scheme 1 The His–His–Met copper coordination in the mono-copper active site of dopamine β-hydroxylase (DβH) and related enzymes featuring a proposed substrate-bound, CuII-peroxo species for substrate oxidation inspires the design of the catecholamine probe (CAP) that consists of a non-luminescent reporter caged by an N3S–CuII complex, in which dopamine (and catecholamine) binding will trigger O2 activation at the copper for the oxidative release of the reporter in its luminescent form for catecholamine sensing. | ||
Results and discussion
Design, synthesis and initial evaluation of CAP
Our strategy to develop a catecholamine-triggered oxidative release of a caged reporter was inspired by the oxidation (hydroxylation) of dopamine to noradrenaline by dopamine β-hydroxylase (DβH). DβH is a copper monooxygenase with a copper co-factor coordinated by two histidine residues and one methionine residue.44,45 Detailed structural, spectroscopic and kinetic studies on DβH and the related peptidylglycine α-hydroxylating monooxygenase (PHM) suggest that substrate oxidation involves hydrogen atom abstraction from the substrate by a copper-peroxo species generated from dioxygenation of the copper and substrate-mediated electron transfer (Scheme S1†).46 If a synthetic copper complex in a similar coordination environment could trigger an oxidation upon reaction with catecholamines, a caged reporter could be released to produce a luminescence response. A copper(II) complex supported by two sp2 nitrogen and one thioether donor that mimics the His/His/Met coordination in DβH was therefore conjugated to 3-(2-benzothiazolyl)-7-hydroxycoumarin (λem = 488 nm) to give CAP488. When caged as an ether, the coumarin is only weakly fluorescent, but a C–O bond cleavage will release the fluorophore in its phenol/phenolate form to produce a fluorescence turn-on (Scheme 1 and Fig. S1†).43,47 It is worth noting that related O2 dependent oxidations based on iron, cobalt and copper have been developed as selective fluorescent probes for the metals and reactive oxygen species, and have been shown to be useful imaging tools for many different biological samples including live cells, tissues and animals.48–51 The use of an organic molecule to trigger such a metal-based oxidation for sensing is however not known.CAP488 was obtained from coordinating a Cu2+ ion to the corresponding ligand–fluorophore conjugate that was synthesized in three steps from commercially available materials (Scheme S2†). Due to the modular probe design, variants of CAP488 of other properties, for example the ratiometric CAP-R, can be easily obtained by simply linking the ligand to other reporters. An X-ray structure of CAP-R prepared from CuCl2 shows the expected N3S–CuII coordination with a fifth chloride ligand (Fig. S83†).52 Initial evaluation of CAP488 in PBS buffer (pH 7.4) showed a significant 70-fold fluorescence enhancement after treating with 1 eq. of dopamine for 1 h. A ca. 15-fold enhancement of the intensity ratio I455/I385 was also observed when CAP-R was treated under the same conditions (Fig. 2b). When a pyridine donor in CAP488 is replaced by a phenyl ring (N2S-cyan) or a quinoline (N3qS-cyan), or the thioether is replaced by a pyridine (N4-cyan), the fluorescence response towards dopamine was significantly reduced or became negligible (Fig. 1), showing that the observed dopamine reactivity of CAP488 is specific to the N3S-coordinated Cu2+.53 In addition, similar fluorescence responses were observed when CAP488 and CAP-R were treated with adrenaline and noradrenaline (Fig. S2†), showing that the N3S–CuII complex could serve as a general catecholamine-responsive trigger.
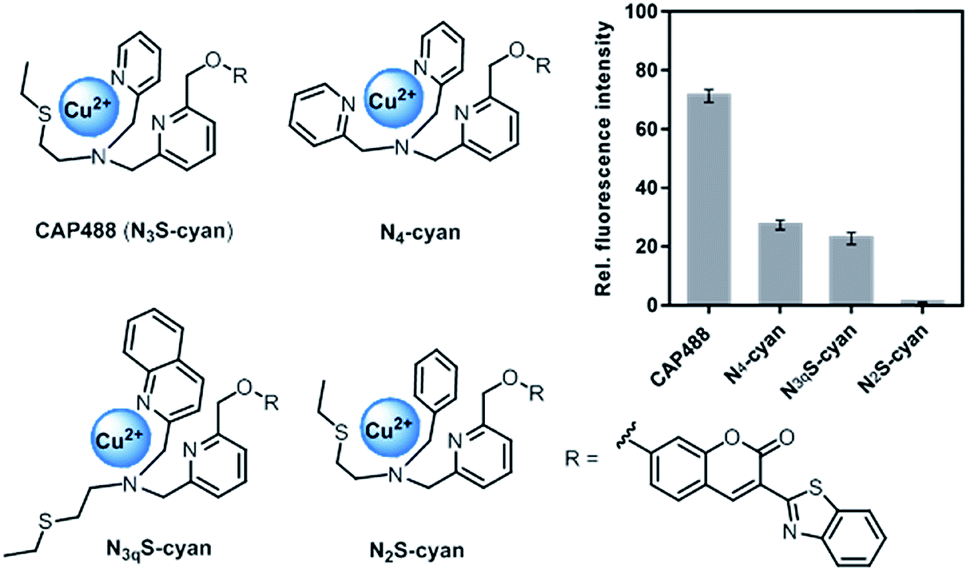 | ||
| Fig. 1 Structures and fluorescence response towards 1 eq. of dopamine of Cu(II) complex–fluorophore conjugates with combinations of N,S-donors. | ||
Catecholamine reactivity, fluorescence response and selectivity
The fluorescence response of CAP488 was evaluated in more detail. As discussed above, CAP488 (10 μM, PBS, pH 7.4) showed a 70-fold emission enhancement centered at 488 nm after a 1 h reaction with dopamine (Fig. 2). Adrenaline (84-fold) and noradrenaline (72-fold) also gave comparable fluorescence turn-on. The counter anion, which probably is dissociated from the Cu2+ in solution, does not affect the catecholamine response and similar fluorescence enhancements were observed when CAP488 was prepared using different copper(II) salts (Fig. S4†).54 The observed fluorescence turn-on was due to the proposed C–O bond cleavage and the uncaged fluorophore, ligand fragments and other oxidized products could be identified by LCMS and ESI-MS analysis of the solution mixtures after the reactions (Fig. S17–S19†). The yield of the released coumarin from CAP488 by dopamine was ca. 5% as determined by the LC analysis. Albeit being a seemingly low cleavage yield, the emission intensity at 488 nm of 0.05 μM 3-(2-benzothiazolyl)-7-hydroxycoumarin is indeed found to be about 70-fold stronger than that of a 10 μM CAP488 solution, consistent with the observed 70-fold fluorescence enhancement for dopamine (Fig. S5†).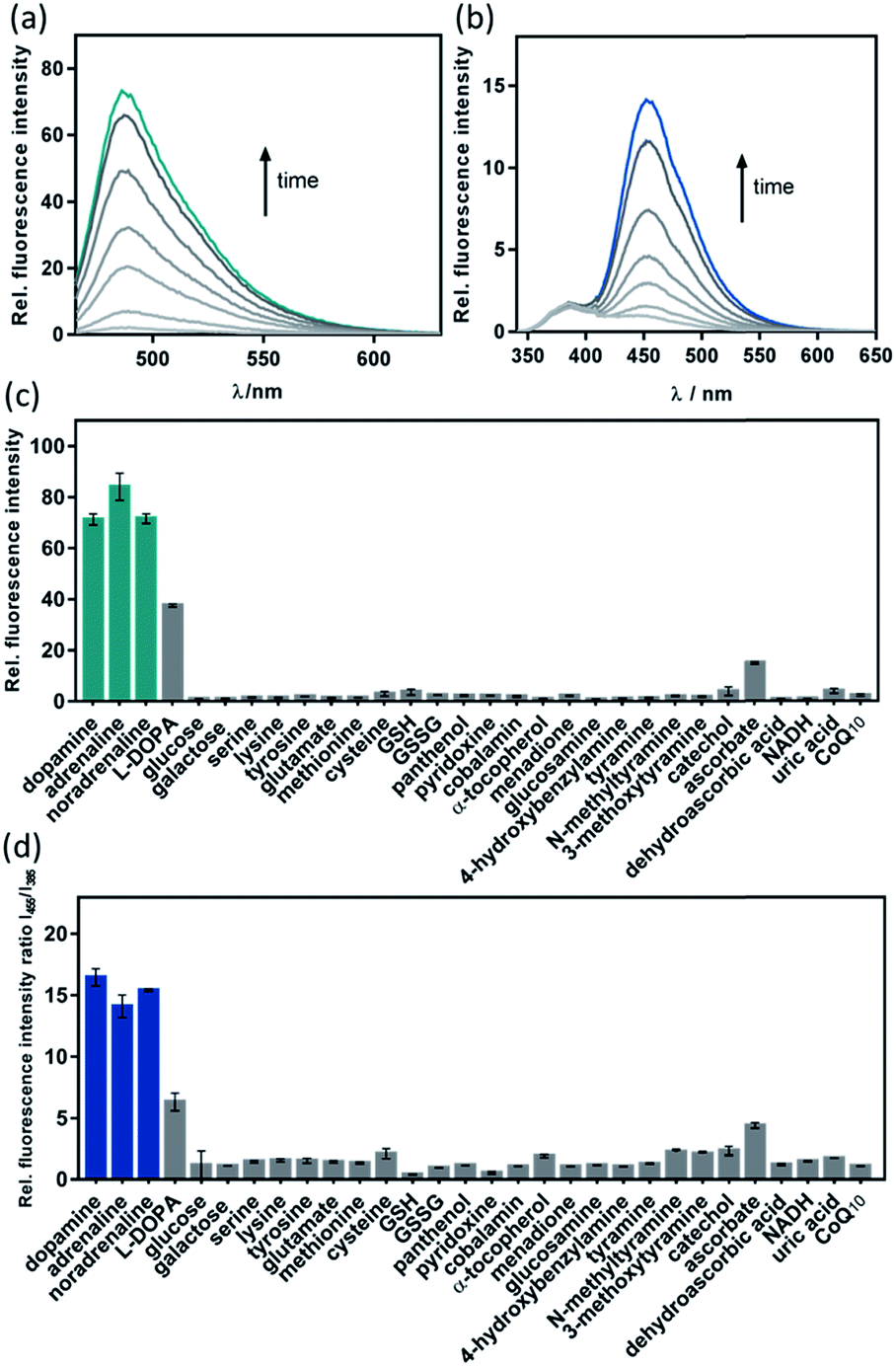 | ||
| Fig. 2 Time-dependent fluorescence response (0, 1, 3, 5, 10, 30 and 60 min) of 10 μM (a) CAP488 and (b) CAP-R towards 1 eq. of dopamine; and selectivity of (c) CAP488 and (d) CAP-R against other biomolecules. Error bars are ±SD (n = 3). | ||
The fluorescence response of CAP488 is selective to catecholamines. In addition to dopamine, adrenaline and noradrenaline, L-DOPA, a precursor to dopamine, also showed a turn-on response. On the other hand, other biologically relevant diols (e.g. sugars), amines (e.g. amino acids), hydroquinones/quinones (e.g. α-tocopherol, menadione or CoQ10) and glucosamine did not result in significant fluorescence responses (Fig. 2c). Reactions of CAP488 with catechol, 4-hydroxybenzylamine, tyramine, N-methyltyramine and 3-methoxytyramine or dopamine derivatives that are masked as silyl ethers and/or trimethylammonium also resulted in a low to negligible fluorescence response, suggesting that both the catechol and amine functional groups in catecholamines are essential for the reaction (Fig. 2c and S6†). Of note, developing an activity-based recognition that targets both amine and diol while avoiding cross reactivity is non-trivial because these functional groups are also widely present in a biological matrix. Because of the necessity of the C–O bond cleavage for the fluorescence response, CAP differentiates catecholamines from other competitive species that react with the N3S–CuII complex differently.55–59 In fact, synthetic copper complexes supported by ligands with primary N3S donors have been intensively studied as models of DβH and PHM.52,60,61 Dioxygenation of the corresponding CuI complexes has been shown to generate a copper-oxygen species (e.g. superoxo and peroxo) that is reactive towards the oxidation of model substrates. It has also been demonstrated that the weak thioether donor can raise the CuII/I reduction potential to result in a more electrophilic copper-superoxo species for better hydrogen atom abstraction.52b On the other hand, other studies have demonstrated that the stability and reactivity of a copper-oxygen species are also affected by interactions in the secondary coordination sphere.52c A correct combination of the ligand donors (in both the first and second coordination sphere), terminal oxidant and substrate is therefore critical for useful copper-based oxidations. Different from the bioinorganic model studies or previous ROS/metal sensing in which the metal-oxygen oxidant is generated directly from oxygenation or ROS coordination,39,41–43CAP488 is not reactive towards O2 until being activated by catecholamines, therefore representing a new way to activate copper-based, O2-dependent oxidation.
With the strong catecholamine-dependent fluorescence enhancement, CAP488 was further studied in a real biological matrix. Linear fluorescence responses to the three catecholamines in both aqueous buffer and human plasma solutions were observed, showing that the probe could be applied in real biological samples (Fig. S7–S9†). Detection limits of CAP488 for the three catecholamines are around 0.2–0.5 μM as determined by the 3σ method. The fluorescence response has a reasonable kinetics in which the fluorescence signal is saturated under 30 min, which is of a timescale similar to those of other activity-based probes that have been successfully applied in cell and animal imaging.62,63 Some cellular processes with a change in intracellular catecholamine levels such as neuron differentiation also take place in a much longer time frame,64,65 suggesting that the probe could be applied in these biological processes.
Characterization of the copper-based oxidation
Before further evaluation in biological systems, the oxidative bond cleavage was studied in more detail. First, O2 was found to be the terminal oxidant, but not other ROS, for the oxidative cleavage of CAP488. No fluorescence enhancement was observed when CAP488 was treated with the catecholamines under anaerobic conditions (Fig. S10†). In addition, reaction of CAP488 with H2O2, O2− and other ROS also gave no significant fluorescence turn-on (Fig. S11†), and that control reactions of CAP488 and the catecholamines in the presence of catalase or superoxide dismutase showed that the fluorescence responses were not affected (Fig. S12†). The reaction of CAP488 with the catecholamines also produced no detectable amount of ROS as revealed by LCMS analysis of the CAP488/catecholamine reaction mixtures in the presence of a H2O2 probe,66 an O2− probe39 or a general ROS probe (Fig. S20–S22†).67 The ROS probes were found to remain unreacted along with the release of the coumarin and other oxidation products. Indeed, a computational study has shown that ROS generation from a Fenton-like reaction involving copper is significantly inhibited in the presence of catecholamines.68 The presence of common transition metal ions like Fe2+, Fe3+ and Zn2+ also showed no effect on the catecholamine-induced fluorescence response (Fig. S13†). All these observations are consistent with a copper-based oxidative bond cleavage. Furthermore, HR-ESI-MS analysis of an immediate mixture of CAP488 and dopamine showed two new peaks at m/z = 881.1278 and 657.1074 that are assignable to a CAP488-dopamine adduct and the one-electron reduced form of the probe respectively (Fig. S23–S27†). Similar species were also observed when adrenaline or noradrenaline was used (Fig. S28–S31†).69 These results suggest that catecholamine binding may lead to a reduction of CAP488, and subsequent dioxygenation would lead to the formation of a copper-based oxidant for the bond cleavage. Indeed, aqueous Cu2+ is known to oxidize adrenaline to adrenochrome with itself being reduced,70 and adrenochrome can also be identified from the reaction of CAP488 and adrenaline (Fig. S18†). This oxidative cyclization is also consistent with the observation that both the catechol and amine groups in the catecholamines are necessary for inducing the bond cleavage. Interestingly, dioxygenation of the Cu(I) analogue of CAP488 did not lead to a significant fluorescence turn-on (Fig. S14†). One possible reason could be the further participation of the catecholamine after the initial electron transfer. In fact, CV studies showed that the direct CAP488 reduction by dopamine may not be thermodynamically feasible (Fig. S34†). Preliminary kinetic studies also showed a fractional reaction order for the catecholamines (Fig. S35, and Tables S1 and S2†).71 Since the catecholamine-induced cleavage showed a strong solvent dependence (Fig. S15 and S33†), stabilization and characterization of the reactive copper-based oxidant at low temperature are not trivial.52 Nevertheless, it is clear that the C–O bond cleavage in CAP488 involves a copper-based oxidation that is triggered by catecholamines and further mechanistic studies are warranted.Imaging of dopamine accumulation in a neuronal differentiation model using CAP488
Given the promising analytical performance of CAP488, it was further employed in live cell fluorescence imaging. First, only negligible emission was observed when a lysate solution of HeLa cells was treated with CAP488, showing that common intracellular components do not have a significant effect on the bond cleavage (Fig. S15†). A cellular model of neuron differentiation using adrenal pheochromocytoma PC12 cells was then studied.72–74 Upon stimulation with nerve growth factor (NGF), PC12 differentiates and resembles the phenotype of sympathetic ganglion neurons with an increase in tyrosine hydroxylase (TH) activity and dopamine biosynthesis.75–79 While the differentiation process is conventionally assessed by monitoring the cell morphology (e.g. length and number of neurites),64,65,80 changes in the intracellular molecular content have only been separately determined by HPLC or electrochemical methods on cell lysates.73,78,81,82 Molecular imaging of PC12 cells using CAP488 will therefore establish a direct correlation of the dopamine dynamics with the NGF-stimulated differentiation process. As expected, significant neurite outgrowth was observed upon stimulating the cells with NGF, which is a characteristic morphological change of neuron differentiation of PC12 (Fig. 3g).78 By staining the cells with CAP488, a significant increase in the intracellular fluorescence was also observed, which is consistent with the previously reported dopamine accumulation due to an increased TH activity (Fig. 3c). In addition, blocking the NGF receptor TrkA with the inhibitor K252a, or treating the undifferentiated cells with only K252a showed no significant fluorescence enhancement,83,84 further confirming that the observed fluorescence enhancement in NGF-treated cells was due to an increase of intracellular catecholamines (Fig. 3b). Furthermore, LC/MS/MS analysis of cell lysates from PC12 cells that were stimulated with NGF under the same conditions also showed a significant increase in the dopamine level and no detectable amount of adrenaline or noradrenaline, further confirming that the fluorescence increase in NGF-stimulated PC12 cells observed using CAP488 is due to dopamine accumulation in the cells (Fig. S36 and S37†). Altogether, these results demonstrate that CAP488 is a new imaging tool for the direct catecholamine imaging in cells. While matching cell morphology to different stages of neuronal development may require extensive technical experience, fluorescence imaging by measuring emission intensity from a fluorescent probe could be more straightforward that provides additional information at the molecular level.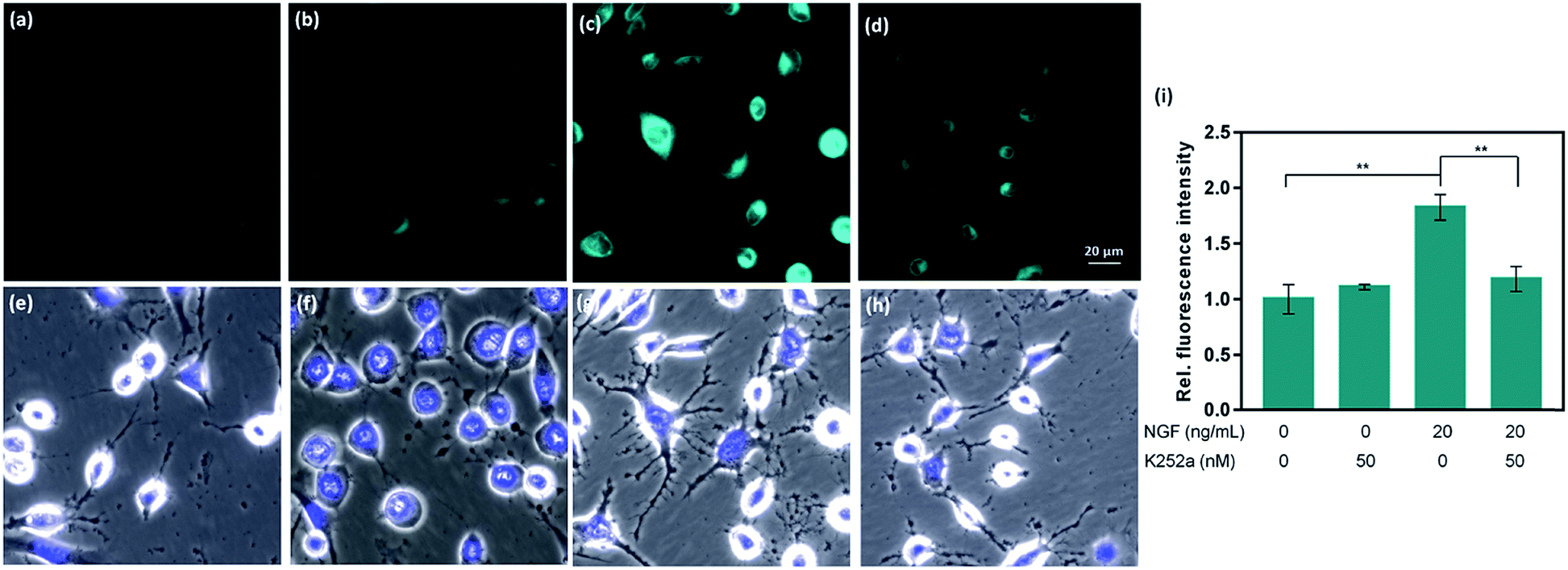 | ||
| Fig. 3 Representative fluorescent images of PC12 cells stimulated with NGF and stained with CAP488. (a) Control cells; and cells treated with (b) 50 nM K252a; (c) 20 ng ml−1 NGF; (d) 20 ng ml−1 NGF and 50 nM K252a; (e–h) overlays of bright field and Hoechst 33342 in (a–d); (i) average fluorescence intensity of PC12 cells treated under conditions in (a)–(d) measured across the entire field from triplicate experiments. Scale bar = 20 μm. Error bars are ±SD (n = 3). Statistical analysis was performed with one-way ANOVA followed by the Tukey test. **: p < 0.01. | ||
CAP488 is non-cytotoxic under the imaging conditions. Nuclear staining with Hoechst 33342 and MTT assays showed that the cells were viable throughout the experiment (Fig. S38†). Due to the strong binding of Cu2+ to the N3S ligand (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K ≈ 16),85 the Cu2+ complex will remain intact and impose no significant cytotoxicity similar to many other copper-based bioimaging probes.86–89 Our fluorescence studies also showed that no detectable amount of free ROS would be generated (see above), further demonstrating the biocompatibility of CAP488.
K ≈ 16),85 the Cu2+ complex will remain intact and impose no significant cytotoxicity similar to many other copper-based bioimaging probes.86–89 Our fluorescence studies also showed that no detectable amount of free ROS would be generated (see above), further demonstrating the biocompatibility of CAP488.
Imaging of dopamine depletion in a cellular Parkinson's disease model using CAP488
CAP488 was also examined in a cellular model of Parkinson's disease (PD), which is a major neurological disorder characterized by death of dopaminergic neurons in the midbrain.90 Previously, the decrease in dopamine levels in PD has been characterized by techniques involving tracers that mimic the uptake and retention of dopamine.91–93 To directly study changes of intracellular dopamine in a PD model, CAP488 was first differentiated with NGF and then challenged with 1-methyl-4-phenylpyridinium (MPP+), which is a common neurotoxin for generating cellular and animal models of PD.94,95 The neurotoxicity of MPP+ is multifold and includes dopamine redistribution and efflux.96 As expected, a change in the cell morphology and integrity of the neurites was observed, consistent with the expected neurotoxicity of MPP+.96 Fluorescence imaging with CAP488 also showed a significant decrease in the intracellular fluorescence in MPP+-treated cells, consistent with the expected depletion of the neurotransmitter as a neurotoxic effect of MPP+ (Fig. 4d). Moreover, MPP+-treatment on undifferentiated cells under the same conditions resulted in no statistically different fluorescence as compared with that of control cells, consistent with the selective vulnerability of dopaminergic neurons to MPP+ (Fig. 4b).96 Again, these experiments have established CAP488 as a convenient and effective imaging tool for the direct evaluation of dopamine content in a cellular PD model, and reinforced that the complementary use of the molecular and morphological imaging can be an integrative approach for studying neuronal processes.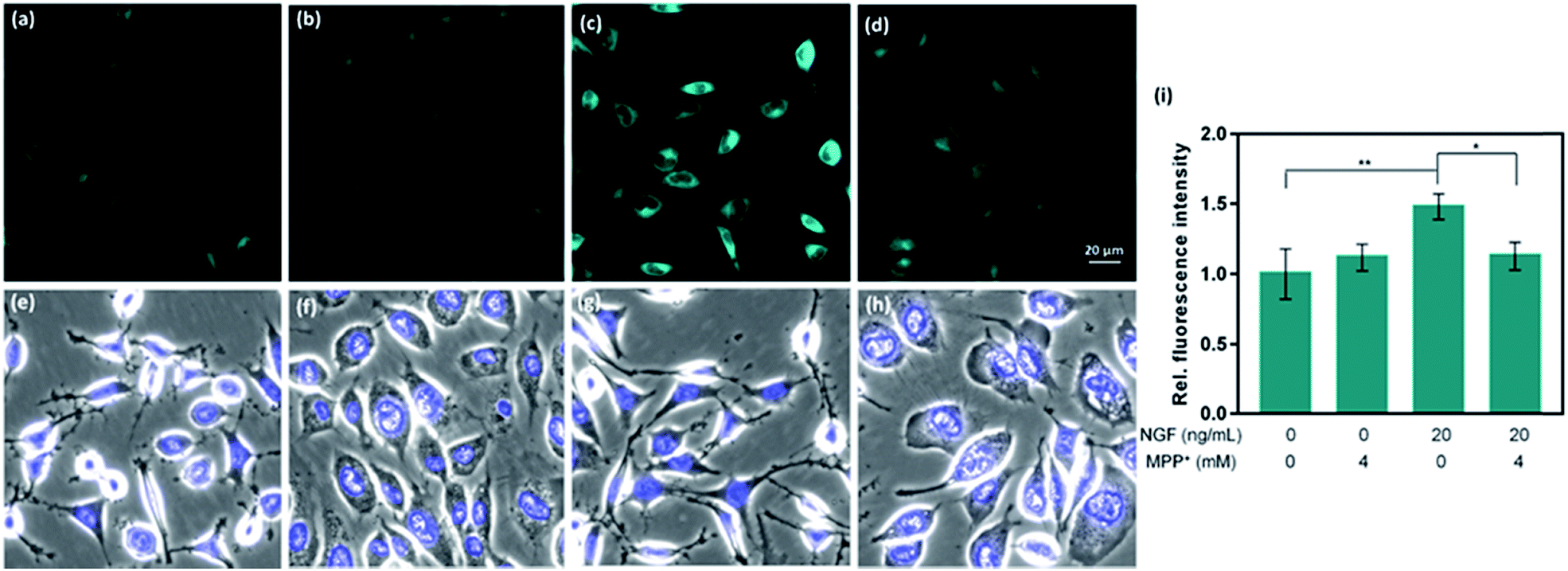 | ||
| Fig. 4 Representative fluorescence images of PC12 cells treated with NGF/MPP+ and stained with CAP488. (a) Control cells; and cells treated with (b) 4 mM MPP+; (c) 20 ng ml−1 NGF; (d) 20 ng ml−1 NGF and then 4 mM MPP+; (e–h) overlays of bright field and Hoechst 33342 in (a–d); (i) average fluorescence intensity of PC12 cells treated under conditions in (a)–(d) measured across the entire field from triplicate experiments. Scale bar = 20 μm. Error bars are ±SD (n = 3). Statistical analysis was performed with one-way ANOVA followed by the Tukey test. *: p < 0.05; **: p < 0.01. | ||
Conclusions
In summary, the bioinspired development of CAP488 as a first generation of activity-based catecholamine fluorescent probes is described. The probe features a catecholamine-triggered, copper-based oxidative cleavage to release a fluorescent reporter with high sensitivity and selectivity. The combination of small molecule reactivity and metal-based oxidation has significantly expanded the available chemical reactivity for designing effective activity-based recognition and detection systems for challenging targets. This cleavage-based approach is highly modular and the catecholamine-responsive N3S–Cu2+ complex can be installed on other reporters to give other catecholamine probes (e.g.CAP-R) of desired analytical, photophysical or biological properties. In addition to aqueous buffer and plasma, CAP488 can also be applied as an imaging agent in live cells for the direct visualization of catecholamine content in models of neuronal differentiation and Parkinson's disease, showing for the first time that molecular and morphology imaging can be performed in a single imaging experiment to complementarily correlate changes of intracellular catecholamine to different stages of neuronal development. Detailed mechanistic studies on the catecholamine-triggered, copper-based oxidation and further development of catecholamine probes with improved selectivity, sensitivity, reaction kinetics and tailored bioanalytical properties for specific applications are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Croucher Foundation and grants from the Research Grants Council of the Hong Kong Special Administrative Region, China to H. Y. Au-Yeung (HKU 106150183) and J. Rong (HKU 17120915 and HKU 17146216), and grants from the Health and Medical Research Fund (15161731) and National Natural Science Foundation of China (81703726) to J. Zhao. K. Y. Tong is a recipient of a Postgraduate Scholarship from The University of Hong Kong. We thank Dr Lei Yun for initial synthetic studies, Dr K. H. Low for X-ray crystallography, Dr C. N. Lok for LC/MS/MS studies, Dr Wei Cui (Ningbo University) for initial animal experiments and Profs. C. C. L. Wong and E. H. Y. Chan for helpful discussions. Special thanks are due to Prof. C. M. Che, C. J. Chang and J. Chan for their support and advices in the project. We acknowledge UGC funding administered by The University of Hong Kong for supporting the Electrospray Ionization Quadrupole Time-of-Flight Mass Spectrometry Facilities under the support for Interdisciplinary Research in Chemical Science. Hong Kong Red Cross Blood Transfusion Service is gratefully acknowledged for donating the human plasma samples.Notes and references
- R. A. Webster, Neurotransmitters, Drugs and Brain Function, John Wiley&Sons, New York, 2001 Search PubMed.
- C. Rangel-Barajas, I. Coronel and B. Florán, Aging Dis., 2015, 6, 349–368 CrossRef PubMed.
- K. L. Davis, R. S Kahn, G. Ko and M. Davidson, Am. J. Psychiatry, 1991, 148, 1474–1486 CrossRef CAS PubMed.
- M. D. Tricklebank, Trends Pharmacol. Sci., 1989, 10, 127–129 CrossRef CAS PubMed.
- J. Biederman and T. Spencer, Biol. Psychiatry, 1999, 46, 1234–1242 CrossRef CAS.
- Y. Kobayashi, H. Kako and H. Yokogoshi, Cell. Mol. Neurobiol., 2010, 30, 173–184 CrossRef CAS PubMed.
- S. J. Kish, K. Shannak and O. Hornykiewicz, N. Engl. J. Med., 1988, 318, 876–880 CrossRef CAS PubMed.
- G. L. Ming and H. Song, Annu. Rev. Neurosci., 2005, 28, 223–250 CrossRef CAS PubMed.
- S. Kolusheva, O. Molt, M. Herm, T. Schrader and R. Jelinek, J. Am. Chem. Soc., 2005, 127, 10000–10001 CrossRef CAS PubMed.
- S. Kolusheva, T. Shahal and R. Jelinek, J. Am. Chem. Soc., 2000, 122, 776–780 CrossRef CAS.
- F. M. Raymo and M. A. Cejas, Org. Lett., 2002, 4, 3183–3185 CrossRef CAS.
- D. R. Radu, C. Y. Lai, J. W. Wiench, M. Pruski and V. S. Y. Lin, J. Am. Chem. Soc., 2004, 126, 1640–1641 CrossRef CAS PubMed.
- H. P. Wu, T. L. Cheng and W. L. Tseng, Langmuir, 2007, 23, 7880–7885 CrossRef CAS PubMed.
- R. Freeman, L. Bahshi, T. Finder, R. Gill and I. Willner, Chem. Commun., 2009, 764–766 RSC.
- T. Patriarchi, J. R. Cho, K. Merten, M. W. Howe, A. Marley, W. H. Xiong, R. W. Folk, G. J. Broussard, R. Liang, M. J. Jang and H. Zhong, Science, 2018, 360, eaat4422 CrossRef PubMed.
- F. Sun, J. Zeng, M. Jing, J. Zhou, J. Feng, S. F. Owen, Y. Luo, F. Li, H. Wang, T. Yamaguchi and Z. Yong, Cell, 2018, 174, 481–496 CrossRef CAS PubMed.
- H. Wang, M. Jing and Y. Li, Curr. Opin. Neurobiol., 2018, 50, 171–178 CrossRef CAS PubMed.
- S. Kruss, D. P. Salem, L. Vuković, B. Lima, E. Vander Ende, E. S. Boyden and M. S. Strano, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1789–1794 CrossRef CAS PubMed.
- A. G. Beyene, K. Delevich, J. T. D. B. ODonnell, D. J. Piekarski, W. C. Lin, A. W. Thomas, S. J. Yang, P. Kosillo, D. Yang, L. Wilbrecht and M. P. Landry, bioRxiv, 2018, 356543.
- J. T. D. Bonis-O'Donnell, R. H. Page, A. G. Beyene, E. G. Tindall, I. R. McFarlane and M. P. Landry, Adv. Funct. Mater., 2017, 27, 1702112 CrossRef.
- P. Feng, Y. Chen, L. Zhang, C. G. Qian, X. Xiao, X. Han and Q. D. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 4359–4368 CrossRef CAS.
- L. Zhang, X. A. Liu, K. D. Gillis and T. E. Glass, Angew. Chem., Int. Ed., 2019, 58, 7611–7614 CrossRef CAS PubMed.
- K. S. Hettie, X. Liu, K. D. Gillis and T. E. Glass, ACS Chem. Neurosci., 2013, 4, 918–923 CrossRef CAS PubMed.
- K. E. Secor and T. E. Glass, Org. Lett., 2004, 6, 3727–3730 CrossRef CAS PubMed.
- J. Bergquist, A. Ściubisz, A. Kaczor and J. Silberring, J. Neurosci. Methods, 2002, 113, 1–13 CrossRef CAS PubMed.
- R. T. Peaston and C. Weinkove, Ann. Clin. Biochem., 2004, 41, 17–38 CrossRef CAS PubMed.
- J. Bicker, A. Fortuna, G. Alves and A. Falcão, Anal. Chim. Acta, 2013, 768, 12–34 CrossRef CAS PubMed.
- K. Jackowska and P. Krysinski, Anal. Bioanal. Chem., 2013, 405, 3753–3771 CrossRef CAS PubMed.
- E. S. Bucher and R. M. Wightman, Annu. Rev. Anal. Chem., 2015, 8, 239–261 CrossRef CAS PubMed.
- N. G. Gubernator, H. Zhang, R. G. Staal, E. V. Mosharov, D. B. Pereira, M. Yue, V. Balsanek, P. A. Vadola, B. Mukherjee, R. H. Edwards and D. Sulzer, Science, 2009, 324, 1441–1444 CrossRef CAS PubMed.
- P. C. Rodriguez, D. B. Pereira, A. Borgkvist, M. Y. Wong, C. Barnard, M. S. Sonders, H. Zhang, D. Sames and D. Sulzer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 870–875 CrossRef CAS PubMed.
- D. B. Pereira, Y. Schmitz, J. Mészáros, P. Merchant, G. Hu, S. Li, A. Henke, J. E. Lizardi-Ortiz, R. J. Karpowicz Jr, T. J. Morgenstern and M. S. Sonders, Nat. Neurosci., 2016, 19, 578–586 CrossRef CAS PubMed.
- M. Lee, N. G. Gubernator, D. Sulzer and D. Sames, J. Am. Chem. Soc., 2010, 132, 8828–8830 CrossRef CAS PubMed.
- A. Henke, Y. Kovalyova, M. Dunn, D. Dreier, N. G. Gubernator, I. Dincheva, C. Hwu, P. Šebej, M. S. Ansorge, D. Sulzer and D. Sames, ACS Chem. Neurosci., 2018, 9, 925–934 CrossRef CAS PubMed.
- J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J. M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783–4804 RSC.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- Z. H. Yu, C. Y.-S. Chung, F. K. Tang, T. F. Brewer and H. Y. Au-Yeung, Chem. Commun., 2017, 53, 10042–10045 RSC.
- M. Taki, S. Iyoshi, A. Ojida, I. Hamachi and Y. Yamamoto, J. Am. Chem. Soc., 2010, 132, 5938–5939 CrossRef CAS PubMed.
- H. Y. Au-Yeung, E. J. New and C. J. Chang, Chem. Commun., 2012, 48, 5268–5270 RSC.
- H. Y. Au-Yeung, J. Chan, T. Chantarojsiri and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 15165–15173 CrossRef CAS PubMed.
- Y. Hitomi, T. Takeyasu, T. Funabiki and M. Kodera, Anal. Chem., 2011, 83, 9213–9216 CrossRef CAS PubMed.
- L. C. Stewart and J. P. Klinman, Annu. Rev. Biochem., 1988, 57, 551–590 CrossRef CAS PubMed.
- B. K. Santra, P. A. N. Reddy, M. Nethaji and A. R. Chakravarty, Inorg. Chem., 2002, 41, 1328–1332 CrossRef CAS PubMed.
- S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304, 864–867 CrossRef CAS PubMed.
- W. Lin, L. Long and W. Tan, Chem. Commun., 2010, 46, 1503–1505 RSC.
- A. Decker and E. I. Solomon, Curr. Opin. Chem. Biol., 2005, 9, 152–163 CrossRef CAS PubMed.
- J. U. Rohde, J. H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Science, 2003, 299, 1037–1039 CrossRef CAS PubMed.
- A. Decker, J. U. Rohde, L. Que and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378–5379 CrossRef CAS PubMed.
- N. Lehnert, R. Y. Ho, L. Que and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 8271–8290 CrossRef CAS PubMed.
- (a) D.-H. Lee, L. Q. Hatcher, M. A. Vance, R. Sarangi, A. E. Milligan, A. A. Narducci Sarjeant, C. D. Incarvito, A. L. Rheingold, K. O. Hodgson, B. Hedman, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2007, 46, 6056–6068 CrossRef CAS PubMed; (b) S. Kim, J. Y. Lee, R. E. Cowley, J. W. Ginsbach, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2015, 137, 2796–2799 CrossRef CAS PubMed; (c) M. Bhadra, J. Y. C. Lee, R. E. Cowley, S. Kim, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2018, 140, 9042 CrossRef CAS PubMed.
- Model complexes for N2S-cyan and N3qS-cyan revealed respectively that the Cu2+ is coordinated by an N2S and N3 donor set, with the thioether in the latter not coordinating to the metal in its first coordination sphere (Fig. S84 and S85†). For X-ray structures of copper(II) supported by tris(2-pyridylmethyl)amine, see: W. T. Eckenhoff and T. Pintauer, Inorg. Chem., 2010, 49, 10617–10626 CrossRef CAS PubMed.
- ESI-MS analysis of CAP488 prepared from CuCl2 showed [CuII(1)Cl]+ (m/z = 691.8) as the major ion whereas [CuII(1)(HCOO)]+ (m/z = 702.2, formate ion came from HPLC buffer) was observed when other Cu2+ salts were used, suggesting that the chloride is not involved in the reaction.
- L. E. McQuade, J. Ma, G. Lowe, A. Ghatpande, A. Gelperin and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8525–8530 CrossRef CAS PubMed.
- N. Wilson, L. H. Mak, A. Cilibrizzi, A. D. Gee, N. J. Long, R. Woscholski and R. Vilar, Dalton Trans., 2016, 45, 18177–18182 RSC.
- K. K. Y. Yuen and K. A. Jolliffe, Chem. Commun., 2013, 49, 4824 RSC.
- A. Ojida, Y. Mito-oka, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 2454–4826 CrossRef CAS PubMed.
- H. Y. Au-Yeung, C. Y. Chan, K. Y. Tong and Z. H. Yu, J. Inorg. Biochem., 2017, 177, 300–312 CrossRef CAS PubMed.
- F. Champloy, N. Benali-Cherif, P. Bruno, I. Blain, M. Pierrot, M. Reglier and A. Michalowicz, Inorg. Chem., 1998, 37, 3910–3918 CrossRef CAS PubMed.
- L. Q. Hatcher, D.-H. Lee, M. A. Vance, A. E. Milligan, R. Sarangi, K. O. Hodgson, B. Hedman, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2006, 45, 10055–10057 CrossRef CAS PubMed.
- M. C. Heffern, H. M. Park, H. Y. Au-Yeung, G. C. Van de Bittner, C. M. Ackerman, A. Stahl and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14219–14224 CrossRef CAS PubMed.
- (a) A. T. Aron, M. C. Heffern, Z. R. Lonergan, M. N. Vander Wal, B. R. Blank, B. Spangler, Y. Zhang, H. M. Park, A. Stahl, A. R. Renslo, E. P. Skaar and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12669–12674 CrossRef CAS PubMed; (b) A. T. Aron, M. O. Loehr, J. Bogena and C. J. Chang, J. Am. Chem. Soc., 2016, 138(43), 14338–14346 CrossRef CAS PubMed.
- M. K. Leach, Y. I. Naim, Z. Q. Feng, C. C. Gertz and J. M. Corey, J. Neurosci. Methods, 2011, 199, 192–198 CrossRef PubMed.
- A. Abeliovich and R. Hammond, Dev. Biol., 2007, 304, 447–454 CrossRef CAS PubMed.
- E. W. Miller, O. Tulyathan, E. Y. Isacoff and C. J. Chang, Nat. Chem. Biol., 2007, 3, 263–267 CrossRef CAS PubMed.
- A. Rukavishnikov, K. R. Gee, I. Johnson and S. Corry, Anal. Biochem., 2011, 419, 9–16 CrossRef CAS PubMed.
- R. Álvarez-Diduk and A. Galano, J. Phys. Chem. B, 2015, 119, 3479–3491 CrossRef PubMed.
- Analogous reactions of CAP488 with other catecholamines similarly produced new peaks at m/z = 911.1426 (adrenaline) and m/z = 897.1229 (noradrenaline) assignable to CAP488-catecholamine adducts.
- A. S. Al-Ayed, H. A. Al-Lohedan, M. Rafiquee, M. S. Ali and Z. A. Issa, Transition Met. Chem., 2013, 38, 173–181 CrossRef CAS.
- R. Serna-Guerrero and A. Sayari, Chem. Eng. J., 2010, 161, 182–190 CrossRef CAS.
- A. S. Tischler, Ann. N. Y. Acad. Sci., 2002, 971, 366–370 CrossRef PubMed.
- R. H. S. Westerink and A. G. Ewing, Acta Physiol., 2008, 192, 273–285 CrossRef CAS PubMed.
- L. A. Greene and G. Rein, Brain Res., 1977, 129, 247–263 CrossRef CAS PubMed.
- J. Cremins, J. A. Wagner and S. Halegoua, J. Cell Biol., 1986, 103, 887–893 CrossRef CAS PubMed.
- H. Hatanaka, Brain Res., 1981, 222, 225–233 CrossRef CAS PubMed.
- Y. Obara, A. Yamauchi, S. Takehara, W. Nemoto, M. Takahashi, P. J. Stork and N. Nakahata, J. Biol. Chem., 2009, 284, 23564–23573 CrossRef CAS PubMed.
- R. R. Lim, M. R. Bloomfield, A. M. Johnson and J. M. Allen, Biochem. Soc. Trans., 1995, 23, 46S CrossRef CAS PubMed.
- P. R. Dunkley, L. Bobrovskaya, M. E. Graham, E. I. Von Nagy-Felsobuki and P. W. Dickson, J. Neurochem., 2004, 91, 1025–1043 CrossRef CAS PubMed.
- S. J. Chinta and J. K. Andersen, Int. J. Biochem. Cell Biol., 2005, 37, 942–946 CrossRef CAS PubMed.
- E. T. Kavanagh, J. P. Loughlin, K. R. Herbert, P. Dockery, A. Samali, K. M. Doyle and A. M. Gorman, Biochem. Biophys. Res. Commun., 2006, 351, 890–895 CrossRef CAS PubMed.
- C. M. Huang and L. S. Kao, J. Neurochem., 1996, 66, 124–130 CrossRef CAS PubMed.
- P. Tapley, F. Lamballe and M. Barbacid, Oncogene, 1992, 7, 371–381 CAS.
- W. J. Friedman and L. A. Greene, Exp. Cell Res., 1999, 253, 131–142 CrossRef CAS PubMed.
- E. A. Ambundo, M. V. Deydier, A. J. Grall, N. Aguera-Vega, L. T. Dressel, T. H. Cooper, M. J. Heeg, L. A. Ochrymowycz and D. B. Rorabacher, Inorg. Chem., 1999, 38, 4233–4242 CrossRef CAS.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536–5537 CrossRef CAS PubMed.
- Z. J. Tonzetich, L. E. McQuade and S. J. Lippard, Inorg. Chem., 2010, 49, 6338–6348 CrossRef CAS PubMed.
- U. P. Apfel, D. Buccella, J. J. Wilson and S. J. Lippard, Inorg. Chem., 2013, 52, 3285–3294 CrossRef CAS PubMed.
- Y. Zhou, K. Liu, J. Y. Li, Y. Fang, T. C. Zhao and C. Yao, Org. Lett., 2011, 13, 1290–1293 CrossRef CAS PubMed.
- W. Poewe, K. Seppi, C. M. Tanner, G. M. Halliday, P. Brundin, J. Volkmann, A. E. Schrag and A. E. Lang, Nat. Rev. Dis. Primers, 2017, 3, 17013 CrossRef PubMed.
- K. L. Leenders, E. P. Salmon, P. Tyrrell, D. Perani, D. J. Brooks, H. Sager, T. Jones, C. D. Marsden and R. S. Frackowiak, Arch. Neurol., 1990, 47, 1290–1298 CrossRef CAS PubMed.
- G. Chinaglia, F. J. Alvarez, A. Probst and J. M. Palacios, Neuroscience, 1992, 49, 317–327 CrossRef CAS PubMed.
- Parkinson Study Group, J. Am. Med. Assoc., 2002, 287, 1653–1661 CrossRef PubMed.
- J. W. Langston and I. Irwin, Clin. Neuropharmacol., 1986, 9, 485–507 CrossRef CAS PubMed.
- J. Bove and C. Perier, Neuroscience, 2012, 211, 51–76 CrossRef CAS PubMed.
- S. J. Choi, A. Panhelainen, Y. Schmitz, K. E. Larsen, E. Kanter, M. Wu, D. Sulzer and E. V. Mosharov, J. Biol. Chem., 2015, 290, 6799–6809 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, fluorescence, LCMS, HRMS, NMR, cell and X-ray data. CCDC 1853672, 1896522 and 1896525. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03338f |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |