
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHypoxia-activated NIR photosensitizer anchoring in the mitochondria for photodynamic therapy†
Feng
Xu
a,
Haidong
Li
a,
Qichao
Yao
a,
Haoying
Ge
a,
Jiangli
Fan
 a,
Wen
Sun
a,
Jingyun
Wang
b and
Xiaojun
Peng
*ac
a,
Wen
Sun
a,
Jingyun
Wang
b and
Xiaojun
Peng
*ac
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Road, Dalian 116024, P. R. China. E-mail: pengxj@dlut.edu.cn
bSchool of Life Science and Biotechnology, Dalian University of Technology, 2 Linggong Road, Dalian 116024, P. R. China
cShenzhen Research Institute, Dalian University of Technology, Nanshan District, Shenzhen 518057, P. R. China
First published on 2nd October 2019
Abstract
Photodynamic therapy is considered as a promising treatment for cancer, but still faces several challenges. The hypoxic environment in solid tumors, imprecise tumor recognition and the lack of selectivity between normal and cancer cells extremely hinder the applications of photodynamic therapy in clinics. Moreover, the “always on” property of photosensitizers also increases the toxicity to normal tissues when exposed to light irradiation. In this study, a hypoxia-activated NIR photosensitizer ICy-N was synthesized and successfully applied for in vivo cancer treatment. ICy-N is in the inactivated state with low fluorescence whereas its NIR emission (λem = 716 nm) was induced via reduction caused by nitroreductase at the tumor site. In addition, the reduced product ICy-OH was specially located in the mitochondria and demonstrated a high singlet oxygen production under 660 nm light irradiation, which efficiently induced cell apoptosis (IC50 = 0.63 μM). The in vivo studies carried out in Balb/c mice indicated that ICy-N was suitable for precise tumor hypoxia imaging and can work as an efficient photosensitizer for restraining tumor growth through the PDT process.
Introduction
In recent decades, cancer has gradually become the major cause of death in both developed and less developed countries due to the growth and aging of the population and the incidence rate is increasing every year.1,2 Therefore, multiple treatments including radiotherapy,3,4 chemotherapy,5,6 immunotherapy,7,8 photothermal therapy (PTT),9,10 photodynamic therapy (PDT)11–14 and so on have been developed for treating cancer. Among them, photodynamic therapy has attracted great interest due to its minimal invasiveness, negligible drug resistance, controllable treatment area selection and excellent therapeutic effects.15,16 A typical PDT process involves pretreatment with a photosensitizer and certain light irradiation on the tumour region which can activate the photosensitizer to produce reactive oxygen species (ROS) to damage cells.17,18 Thus, the capacity of photosensitizers to produce ROS and tumour recognition remain the most important factors for maximum therapeutic effects and minimum side effects. Up to now, numerous efficient photosensitizers have been developed based on various fluorophores.19–22 But little effort has been devoted to enhancing the specificity of photosensitizers for tumour cells. Because the photosensitizer is always “on”, ROS can be produced once it is exposed to light irradiation even in healthy cells, which consequently leads to undesired side effects.23 That's why patients should be kept out of light before being exposed to certain light irradiation. Hence, “stimuli-activatable” photosensitizers are urgently needed for precise photodynamic therapy and minimizing side effects.The hypoxic environment is an important feature of tumours due to the insufficient supply of oxygen. Several methods have been reported for hypoxic tumour therapy, such as those based on thermoresponsive nanoplatforms,24 decomposition of the endogenous H2O2 system,25,26 electron transfer mediated upconversion nanoparticles (UCNPs),27 light-controlled drug release complex conjugates28 and so on. Nitroreductase (NTR) is a well-known specific enzyme over-expressed in solid tumours due to the hypoxic microenvironment which can effectively reduce the nitroaromatic to the corresponding arylamine by using reduced nicotinamide adenine dinucleotide (NADH) as an electron donor.29,30 Based on this reduction reaction, many nitroaromatic decorated probes have been developed for hypoxia imaging.31–35 Li's group proved that nitroreductase was over-expressed when the O2 concentration was below 10% and the para-nitro benzoate group was an ideal substrate for NTR with a fast response, high sensitivity and excellent selectivity because of the strong hydrogen-bonding interaction and spatial match31 Herein, a hypoxia-activated photosensitizer ICy-N was designed, not only for hypoxic imaging, but also for PDT of cancer cells under hypoxic conditions, by decorating hemicyanine dye Cy-OH with 4-nitrobenzyl bromide as the recognition site and the introduction of iodine into the indole ring was used to improve the intersystem crossing (ISC) process so as to increase singlet oxygen production.36 As depicted in Scheme 1, in normal tissue, ICy-N was in a fluorescence-off state because the Intramolecular Charge Transfer (ICT) process was restrained and the energy of the excited state was mainly released through the vibration relaxation pathway due to the vibration of the nitro group37 and the ISC process was blocked. As a result, the ability to produce singlet oxygen was restricted and the toxicity was minimized in normal cells. But in the tumour region, ICy-N will be reduced by NTR and consequently ICy-OH will be released, serving as an efficient type II PS for photodynamic therapy. When irradiated with 660 nm light, ICy-OH can be excited and reach the singlet state (S1), which can be used for emitting fluorescence at 710 nm. Meanwhile, some of the excited molecules will reach the triplet state (T1) by the ISC process because of the “heavy atom effect” of the introduced iodine atom. Finally, the energy transfer from triplet states to the ground state of molecular oxygen results in the formation of singlet oxygen (1O2) which serves as a strong oxidant to induce cell apoptosis. Therefore, tumour recognition and precise PDT were simultaneously achieved.
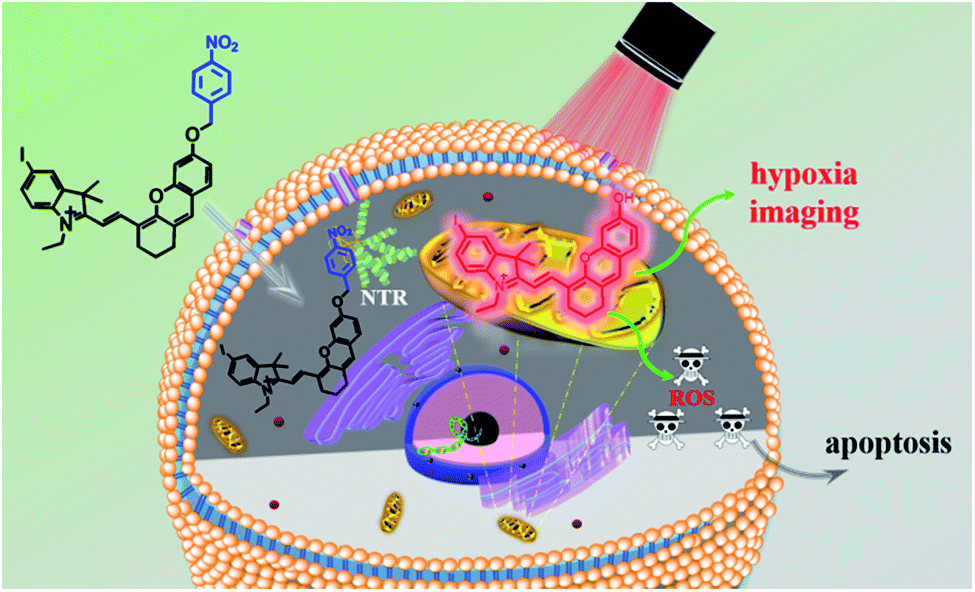 | ||
| Scheme 1 Proposed strategy of ICy-N for hypoxia imaging and cancer therapy. | ||
Results and discussion
Due to its unique properties including excellent photostability, controllable NIR emission and good water solubility, Cy-OH38 with iodine and 4-nitrobenzyl bromide substituents was selected for both precise tumour imaging and photodynamic therapy in this work. The water solubility and stability were characterized by transmission electron microscopy (TEM) and UV-vis spectroscopy. As shown in Fig. S1,†ICy-N showed slight aggregation in water and exhibited spherical morphology with a diameter of about 5 nm. Meanwhile, the absorbance measured during 6 h changed scarcely which indicated that ICy-N was stable and dispersed well in water solution. The recognition mechanism of the ICy-N response to nitroreductase is shown in Scheme 2. ICy-N was non-fluorescent in PBS buffer while its reduced product ICy-OH emitted strong fluorescence at 710 nm, which was extremely important for tumour hypoxia imaging (Fig. 1a). Along with the change of fluorescence spectra, the maximum absorption peak of ICy-N red shifted from 575 nm to 655 nm (Fig. 1b). This is a favorable result for PDT because longer wavelengths have deeper penetration in tissues when exposed to light irradiation during a therapeutic process. In addition, the quenching of the excited state by the nitro group of ICy-N may also lead to lower triplet state production which consequently resulted in a lower singlet oxygen quantum yield (ΦΔ) in contrast to ICy-OH. To confirm this hypothesis, the ability of ICy-N and ICy-OH to produce singlet oxygen (1O2) was tested and compared by using 1,3-diphenylisobenzofuran (DPBF) as the 1O2 trapping agent and methylene blue (MB) as a reference. As seen in Fig. 1d, the absorbance of DPBF at 415 nm decreased dramatically in the presence of ICy-OH under 660 nm light irradiation, while ICy-N only resulted in slight bleaching of DPBF under identical conditions (Fig. 1c). The singlet oxygen quantum yield (ΦΔ) was calculated to be 0.72% and 1.80% for ICy-N and ICy-OH respectively, suggesting that the reduced product ICy-OH displayed stronger ROS production capacity and thus may result in higher toxicity to cancer cells (Table S1†). What's more, the 1O2 production of ICy-OH was also proved by Singlet Oxygen Sensor Green (SOSG) and electron paramagnetic resonance (EPR) measurements (Fig. S5†), showing that ICy-OH can serve as an efficient photosensitizer under 660 nm light irradiation.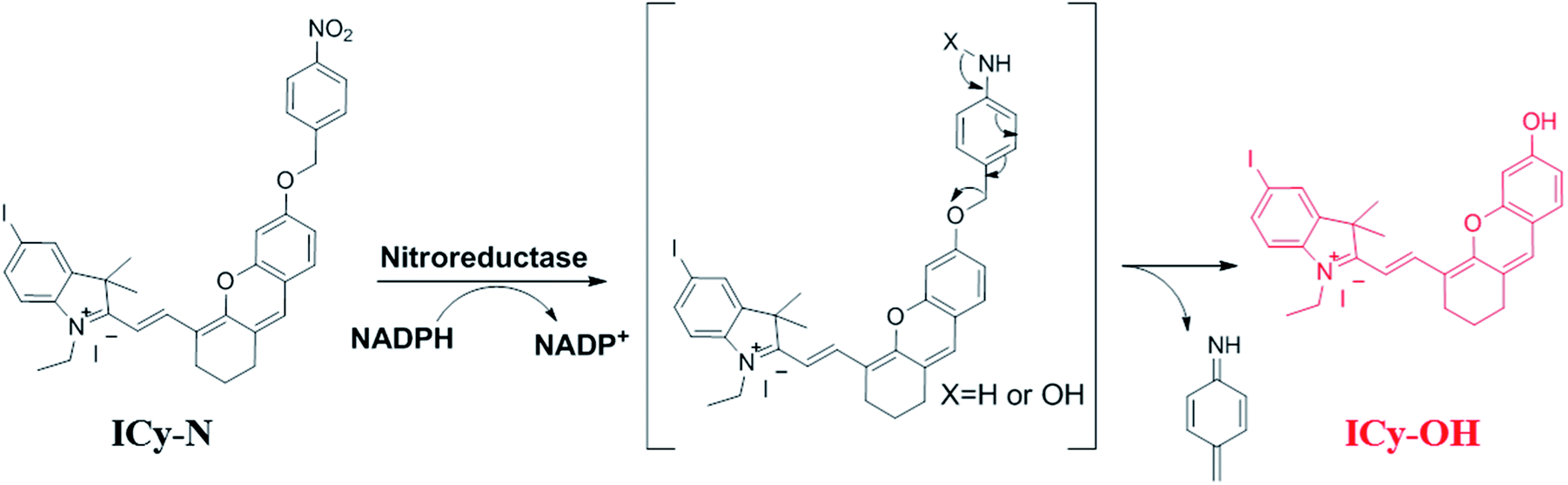 | ||
| Scheme 2 Recognition mechanism of ICy-N with NTR. | ||
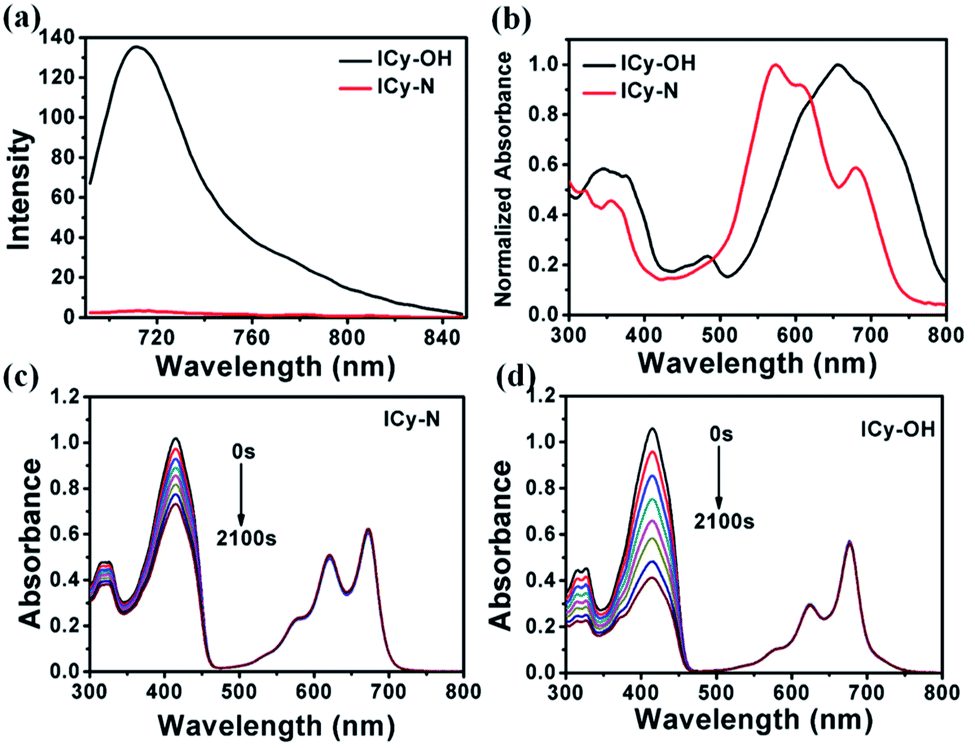 | ||
| Fig. 1 (a) Fluorescence and (b) UV-vis spectra of ICy-N and ICy-OH in PBS buffer (pH = 7.4, 10 mM). (c) Photodegradation curves of DPBF with ICy-N and (d) ICy-OH under 660 nm light irradiation for different times (0–2100 s). | ||
To confirm the feasibility of this protocol, 4T1 cells were preincubated under hypoxia (10%, 2% and <1% O2) conditions for 8 h respectively to induce the overexpression of nitroreductase. Cells that were incubated under normoxic conditions were also taken as the control group. Then the cells were stained with ICy-N and incubated for another 60 min before imaging under a laser scanning confocal microscope. As expected, no obvious fluorescence was observed under normoxic conditions (Fig. 2a) due to the low expression of nitroreductase, whereas the hypoxia (10%, 2% or <1% O2) induced overexpression of nitroreductase gave rise to a significant red fluorescence signal. The result demonstrated that ICy-N was reduced by nitroreductase, followed by releasing ICy-OH with red fluorescence when excited with a 635 nm laser (as illustrated in Scheme 2). To further investigate the subcellular organelle localization of the reduced product, various commercial trackers and ICy-N (2.5 μM) were applied to the cells that were pretreated under hypoxic conditions (2% O2) for 60 min respectively (Fig. 2b). Apparently, a poor colocalization region fluorescence signal was observed between the reduced product and LysoTracker Green (a lysosomal tracker) or Hoechst 33342 (a nuclear tracker). In contrast, the red fluorescence overlapped well with the MitoTracker Green (a mitochondrial tracker), indicating that ICy-N can be triggered by NTR efficiently and finally gathered in the mitochondria. When MCF-7 cells were treated with carbonyl cyanide 3-chlorophenylhydrazone (CCCP, a membrane potential uncoupler) to induce the loss of mitochondria membrane potential, the fluorescence signal of ICy-N decreased dramatically, whereas, the fluorescence signal of MitoTracker Green FM (a mitochondrial probe whose uptake is independent of membrane potential) remained unchanged (Fig. S6†). This result can be attributed to the negative membrane potential (−180 mV) of the mitochondria which facilitates the uptake of molecules with positive charge.39–41 It is quite important for improving the PDT efficiency of photosensitizers because mitochondrial damage and the following release of cytochrome C to trigger apoptosis was considered to be the main pathway during the PDT process.42
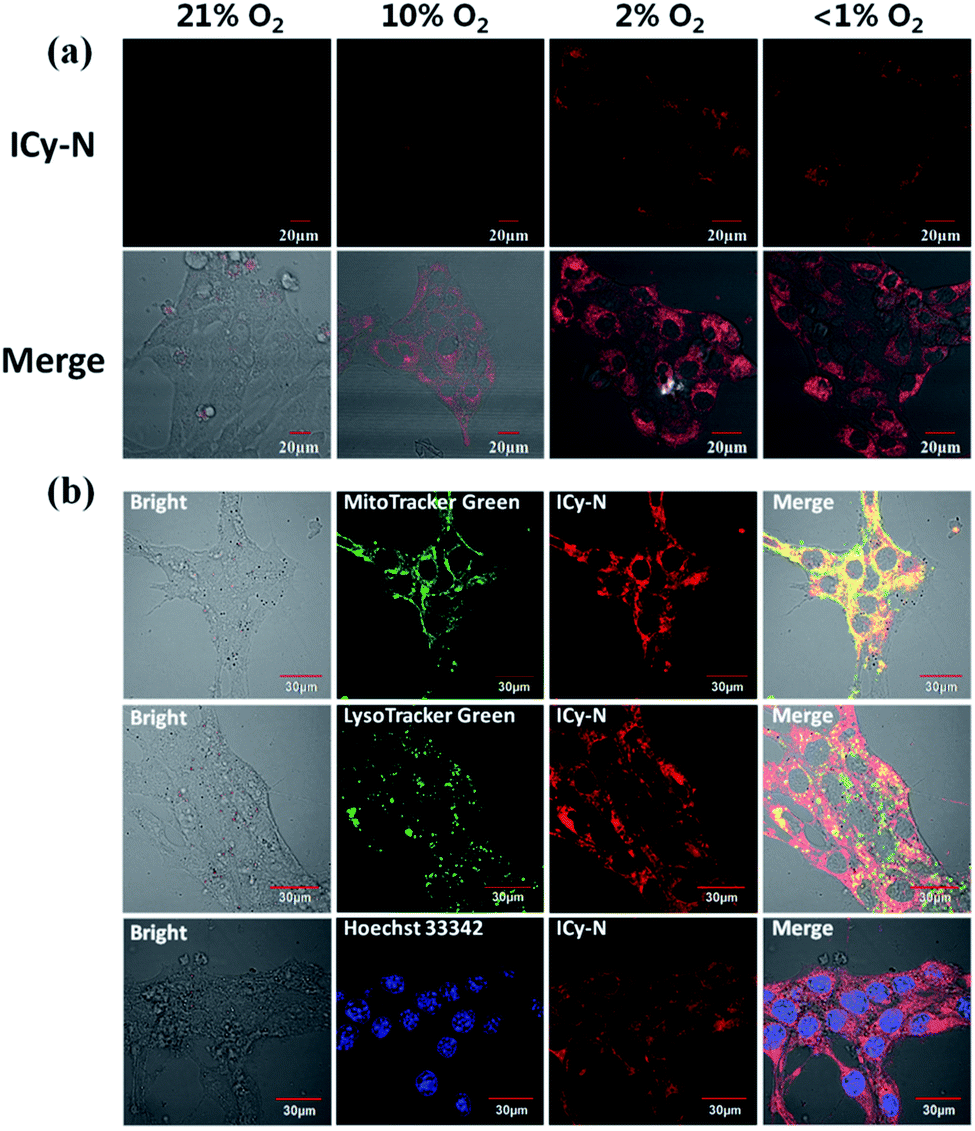 | ||
| Fig. 2 (a) Confocal microscopy imaging of 2.5 μM ICy-N in 4T1 cells under different O2 concentrations. (b) Subcellular colocalization assays of ICy-N in 4T1 cells. The green channel of MitoTracker Green and LysoTracker Green was excited at 488 nm and collected at 500–550 nm. The blue channel of Hoechst 33342 was excited at 405 nm and collected at 440–480 nm. | ||
Then, the PDT properties of the reduced product were studied with a 660 nm LED lamp. 4T1 cells were preincubated under hypoxic conditions (10% O2, 5% CO2) for 8 h and treated with 2.5 μM ICy-N for 60 min, followed by exposure to 660 nm light (12 mW cm−2) for 5 min. After that, annexin V-FITC and PI (Propidium Iodide) were utilized for assessing cell apoptosis induced after the treatment. When apoptosis occurs, phosphatidylserine inside the cytomembrane will turn over to the outside which can be recognized by annexin V with high affinity and specificity. With the development of apoptosis, the permeability of the cytomembrane changes so that PI can enter the cells and combine with DNA to emit red fluorescence, whereas it is inaccessible in living cells. As shown in Fig. 3a, the fluorescence signal of FITC was detected in the cytomembrane in the ICy-N + Light group, indicating that apoptosis was induced. Strong red fluorescence in the cell nucleus in the PI channel can also be observed, which indicated cell death after PDT. This phenomenon was in accordance with the one treated with ICy-OH under the same conditions (Fig. S8†). In contrast, no fluorescence signal was observed from FITC and PI channels in Light and ICy-N groups, because the cells were still surviving in these control groups. To get a better understanding of the cytotoxicity mechanism, the JC-1 assay was carried out in MCF-7 cells. As shown in Fig. S9,† when MCF-7 cells were treated with 2.5 μM ICy-N and irradiated with 660 nm light, the fluorescence signal of the red channel which belongs to the aggregation of JC-1 in the mitochondria decreased dramatically while the red fluorescence in the dark group was obvious, indicating mitochondrial damage. This result demonstrated that cell apoptosis was induced by mitochondrial dysfunction during the PDT process. All these results proved that the reduced product can work as an efficient photosensitizer and induce cell apoptosis under 660 nm light irradiation.
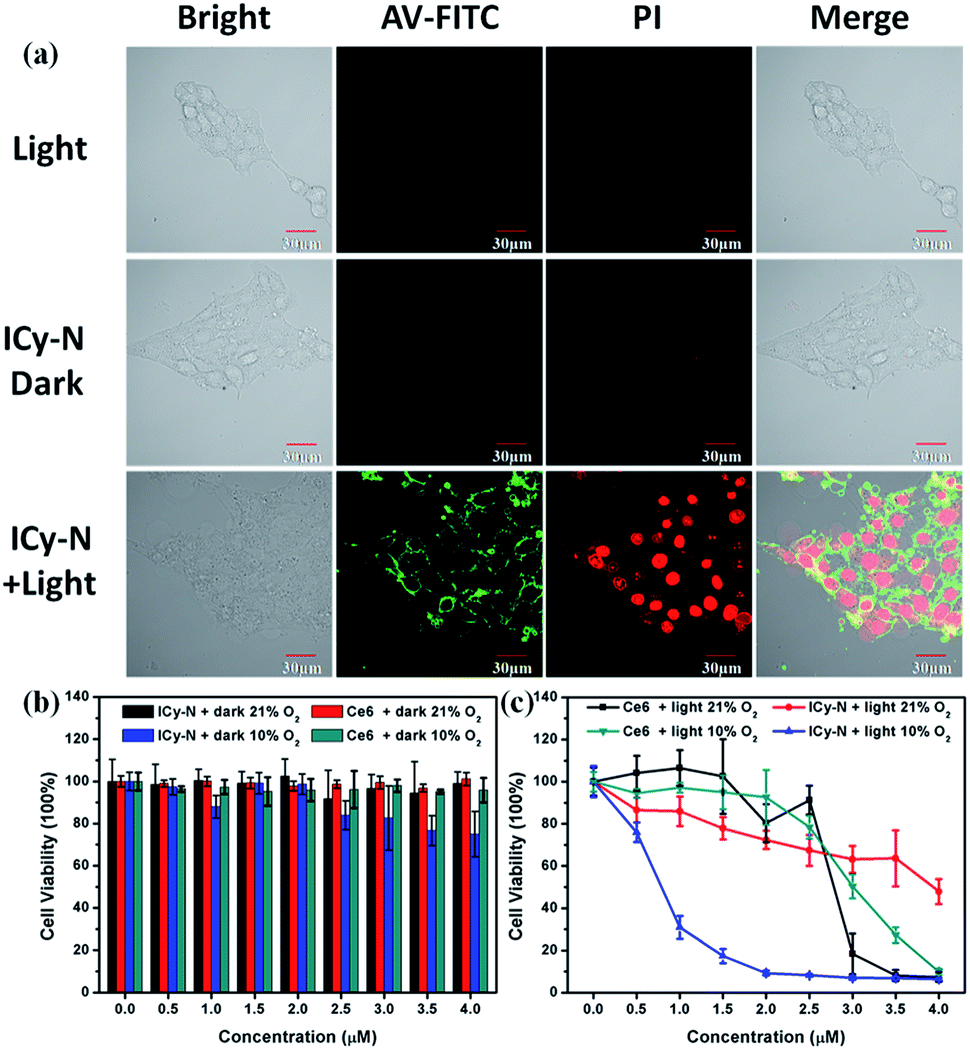 | ||
| Fig. 3 (a) Confocal microscopy imaging of annexin V – FITC and PI stained 4T1 cells after different treatments. The green channel of annexin V – FITC was excited at 488 nm and collected at 500–550 nm; the red channel of PI was excited at 561 nm and collected at 590–640 nm. Cytotoxicity of photosensitizers under dark conditions (b) and irradiation with 660 nm light (c) at various concentrations. | ||
To further investigate the cytotoxicity of ICy-N and the reduced product ICy-OH, the MTT assay was carried out (Fig. 3b and c). Negligible cytotoxicity was observed in the ICy-N treated group under 21% O2 and the viability of MCF-7 cells was still above 98% at a concentration of 4 μM under dark conditions. However, considerable damage was found after 12 h incubation in the 10% O2 group which was caused by the reduced product ICy-OH. The higher cytotoxicity of ICy-OH was beneficial for tumour therapy because only in tumour tissues can ICy-N be reduced to ICy-OH. Next, the PDT effect was studied under 660 nm light irradiation (12 mW cm−2) after incubation with ICy-N for 60 min under 21% and 10% O2 conditions, respectively. As shown in Fig. 3c, the photo-induced cytotoxicity of ICy-N was improved with increasing ICy-N concentration. But the PDT was still inefficient in the 21% O2 group in contrast to that treated with 10% O2. The cell viability decreased rapidly when the concentration of ICy-N increased under 10% O2 conditions and only 20% cells survived at a concentration of 1.5 μM, whereas, 80% cells were still alive in the 21% O2 treated group at the same concentration. This result was consistent with the hypothesis we proposed that a higher singlet oxygen quantum yield of ICy-OH can lead to stronger cytotoxicity so that the side effects in normal tissues can be minimized during the PDT process.
To have a better understanding of the PDT efficiency of ICy-N, a clinical photosensitizer Ce6 was used as a contrast. Similar cell viability was observed between ICy-N and Ce6 under dark conditions which further verified the good biocompatibility of ICy-N. However, quite different PDT efficiency was found in the light treated groups. Ce6 seems nontoxic at low concentrations (0–1.5 μM) while strong cytotoxicity was found in the group treated with ICy-N under 10% O2 conditions. When the concentration increased to 3 μM, comparative cell viability was observed and the IC50 values of ICy-N under 10% O2 conditions and Ce6 were determined to be 0.63 μM and 2.79 μM respectively, indicating that ICy-N demonstrates a better PDT efficiency than Ce6 under identical conditions.
To get more proof of the photo-induced cytotoxicity of the reduced product ICy-OH, DCFH-DA was used for tracking the ROS production during the PDT process. Striking contrast was found that strong fluorescence of DCF was observed after irradiation with 660 nm light, whereas the signal was low in the dark indicating that ICy-OH can work as an efficient photosensitizer and produce ROS under irradiation (Fig. S7†). In addition, the ROS induced apoptosis was visible even in the bright image (Fig. S10†). After being irradiated with a 635 nm laser for 1 min, obvious vesicles were observed with the extension of incubation time indicating the development of apoptosis. In contrast, when the cells without treatment with ICy-OH were exposed to the same laser irradiation, no morphological change was found. Then, the cell death was further proved by flow cytometry statistically. As seen in Fig. S11,† the PI signal in the ICy-OH treated group was negative. After irradiation with 660 nm light, a positive signal was observed, showing the death of MCF-7 cells.
All these results indicated that ICy-N was in a fluorescence-off state and had negligible cytotoxicity under dark conditions and low PDT efficiency without reduction by NTR (in normal tissues). But, it can be turned on under hypoxic conditions by NTR (in tumour tissues) and the reduced product ICy-OH showed higher singlet oxygen production. Thus, ICy-OH is an efficient photosensitizer and is promising for PDT under 660 nm light irradiation.
Inspired by the excellent performance of ICy-N during the PDT process in vitro, an in vivo study was carried out in 4T1 tumour bearing Balb/c mice. Firstly, Balb/c mice were implanted with a 4T1 tumour subcutaneously at the left armpit and then divided into four groups (Control, Light, ICy-N, and ICy-N + Light) when the tumour volume reached 200 mm3. The feasibility of ICy-N as an NIR fluorescent probe for hypoxia imaging was tested by intratumoral injection of ICy-N in PBS buffer in 4T1 tumour bearing Balb/c mice. As depicted in Fig. 4, an obvious fluorescence signal (680–720 nm) was observed in the tumour region (b1–b6) due to the overexpression of NTR under hypoxic conditions of tumours. As time went by, the intensity gradually increased and remained unchanged after 180 min, suggesting that the reduction of ICy-N by NTR was finished after 3 h, which was selected as the preincubation time before exposure to 660 nm light during the PDT process. As a comparison, Balb/c mice without tumour were injected with PBS subcutaneously and imaged at the same time period. No fluorescence signal was captured in spite of the extension of incubation time indicating that ICy-N was in a fluorescence-off state in normal tissues and can only be turned on by NTR specifically and the NIR fluorescence emission was suitable for bioimaging in vivo. Further, the PDT effect was assessed through the tumour volume growth and body weight of mice which were collected every two days (Fig. 4c and d). Distinctly, the tumour volume increased quickly in the PBS treated group whether the mice were irradiated with 660 nm light or not. Besides, the group treated with ICy-N under dark conditions showed a similar tumour growth rate to the control group indicating the low cytotoxicity of ICy-N under dark conditions. In contrast, the PDT group that was irradiated with a 660 nm lamp (100 mW cm−2) for 20 min after pretreatment with ICy-N for 3 h demonstrated evident suppression of tumour growth. The tumour volume remained almost unchanged for 14 days while other groups exhibited above 5-fold increase. The anticancer capability in vivo was as good as Ce6 (Fig. S12†). More importantly, scars were observed at the tumour surface in the PDT group, demonstrating that severe damage was caused in tumours through the PDT treatment (Fig. S13†).
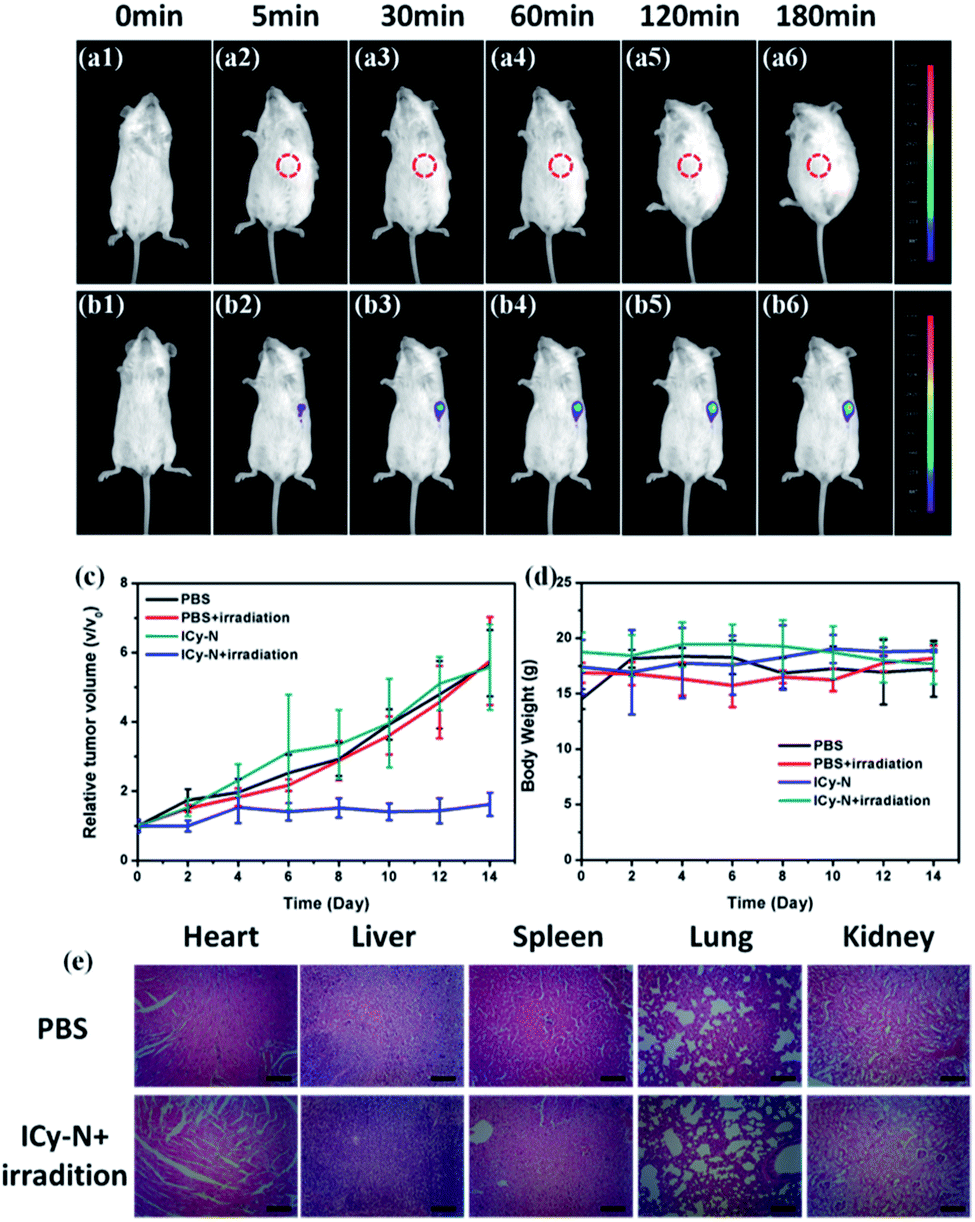 | ||
| Fig. 4 In vivo fluorescence imaging of nitroreductase in Balb/c mice. (a1–a6) PBS (100 μL) was injected subcutaneously and (b1–b6) ICy-N (100 μL, 50 μM in PBS) was injected intratumorally and observed with excitation at 660 nm and emission at 680–720 nm. The red circle in (a2–a6) represents the injected region. (c) Relative tumor volume and (d) body weight of mice after different treatments. (e) H&E staining of organs from the PBS group and PDT group after 14 days of treatment. Scale bar: 50 μm. | ||
The body weight of mice changed scarcely both in PDT and control groups indicating that no obvious injury was induced in mice during the treatment. To further prove this, the mice were euthanized after 14 days and dissected to get the organs of the four groups, which were made into paraffin slices and stained with hematoxylin and eosin (H&E). As seen in Fig. 4e and S9,† the organs in the PDT group didn't show any difference compared with the PBS treated one. Additionally, no physiological morphology abnormalities or histopathological lesions were observed, indicating low toxicity of ICy-N to normal tissues. All these results confirmed that ICy-N was suitable for precise tumour imaging and served as an efficient photosensitizer to restrain tumour growth under light irradiation.
Conclusions
In summary, an iodine substituted NIR hemicyanine dye ICy-N was synthesized and successfully applied for tumour hypoxia imaging and photodynamic therapy in this study. ICy-N is in a fluorescence-off state and has poor singlet oxygen quantum yield due to the vibration of the nitro group in normal tissues, whereas it can be reduced by the over-expressed nitroreductase in the tumour region. The reduced product ICy-OH displayed a strong fluorescence emission at 710 nm which is suitable for in vivo bioimaging. More importantly, the improved singlet oxygen production of ICy-OH makes it an efficient photosensitizer to induce cell apoptosis under 660 nm light irradiation. The in vivo antitumor experiment further confirms that ICy-N can serve as a promising NIR photosensitizer both for precise tumour hypoxia imaging and photodynamic therapy after the activation of nitroreductase. Overall, this work provides a novel strategy for establishment of an “activatable” photosensitizer for precise imaging and therapy.Experimental section
Materials and instruments
All reagents and solvents used were of reagent grade and were obtained from commercial suppliers. Flash column chromatography was performed with silica gel (200–300 mesh) and dichloromethane/methanol or petroleum ether/ethyl acetate was used as eluent. 1H NMR and 13C NMR spectra were obtained using a BrukerAvance II 400 MHz spectrometer. Chemical shift (δ) was reported as ppm in CDCl3 or DMSO-d6 with TMS as the internal standard. The mass spectrum was recorded on LTQ Orbitrap XL MS instruments. ICy-OH and ICy-N were dissolved in DMSO at a concentration of 1 mM respectively as the stock solution and stored at −20 °C for further use. UV-vis spectra and fluorescent spectra were achieved from an Agilent Technologies CARY 60 UV-vis spectrophotometer (serial no. MY1523004) and Varian CARY Eclipse fluorescence spectrophotometer (serial no. MY15210003) respectively. The excitation and emission slit widths were adjusted to obtain the fluorescence intensity in the appropriate range. Cell imaging was performed on Olympus FV1000 and FV3000 confocal microscopes (Olympus, Japan). Mitotracker Green, Lysotracker Green and Hoechst 33342 were purchased from Life Technologies Co. (USA). Reactive oxygen species detection kit DCFH-DA (2,7-dichlorodihydrofluorescein diacetate) and annexin V-FITC and PI apoptosis kits were purchased from Nanjing KeyGen Biotech. Co. Ltd. (China).Determination of singlet oxygen quantum yield (ΦΔ)
The singlet oxygen quantum yield was determined by the reported method.43 Briefly, Methylene Blue (MB) was used as the reference and 1,3-diphenylisobenzofuran (DPBF) was used as the singlet oxygen trapper. Firstly, the concentration of DPBF in 3 mL DCM was modified to adjust the absorbance at 415 nm close to 1.0. Then, appropriate concentrations of MB and samples were added respectively to make sure that the absorbance of the mixture at 660 nm was between 0.2 and 0.4. After that, the mixture solutions were exposed to 660 nm light irradiation and the absorption spectrum was recorded with a UV-vis spectrophotometer. At last, the singlet oxygen quantum yield was calculated with the following equation:| ΦΔ = ΦMB × (k(ps) × F(MB))/(k(MB) × F(PS)) |
| F = 1 − 10−OD |
Cell incubation
Breast cancer cells (MCF-7 cells and 4T1 cells) were purchased from the Institute of Basic Medical Sciences (IBMS) of the Chinese Academy of Medical Sciences and cultured with Dulbecco's modified Eagle's medium (DMEM, Invitrogen) containing 10% fetal bovine serum (Invitrogen) and 1% antibiotics (penicillin/streptomycin, 100 U mL−1) in an atmosphere of CO2/air = 5%/95% at 37 °C.Endogenous nitroreductase imaging in 4T1 cells
For hypoxia incubation, 4T1 cells were seeded in confocal culture dishes at an approximate concentration of 2 × 104 cells per mL and incubated in a cell incubator under normal conditions for 12 h and then the dishes were moved to a sealed box filled with 2% (or 10%) O2 and 5% CO2 for 8 h to induce the overexpression of NTR. As for lower O2 concentration, AnaeroPouch was placed with cell dishes to remove residual O2. Then, the 4T1 cells were incubated with 2.5 μM ICy-N at 37 °C for 60 min and washed with PBS 3 times before scanning under the confocal microscope. Finally, photosensitizers were excited with a 635 nm laser and the signal of the fluorescence channel at 700–750 nm was gathered with a 60× objective lens (N = 3).Apoptosis assessment with annexin V-FITC & PI under a confocal microscope
4T1 cells were pre-incubated under hypoxic conditions as mentioned before and incubated with 2.5 μM ICy-N for 60 min. Then, the cells were washed with DMEM (containing 10% FBS) 3 times and irradiated with 660 nm light for 10 min and incubated for another 30 min at 37 °C to induce apoptosis. After that, DMEM culture medium was replaced with 1× binding buffer and 5 μL annexin V and 5 μL PI were added and mixed slightly. After 30 min incubation at 37 °C, the cells were imaged under an Olympus FV-1000. The green channel (annexin V) was excited at 488 nm and the fluorescence signal at 500–550 nm was collected. The red channel (PI) was excited at 561 nm and the fluorescence signal at 590–640 nm was collected (N = 3).Detection of cellular ROS generation within DCFH-DA
MCF-7 cells were seeded at confocal culture dishes at a density of 5 × 104 and pre-incubated under hypoxic conditions. Then, various concentrations of ICy-N and 10 μM DCFH-DA were added and incubated for 60 min. Then, the cells were washed with PBS 3 times and irradiated with a 635 nm laser for 2 min and scanned under a confocal microscope at 488 nm excitation, whereas, the fluorescence channel at 500–550 nm was collected (N = 3).Cytotoxicity assay
The cytotoxicity in the presence or absence of light was evaluated by reducing MTT (3-(4,5)-dimethylthiazol-2-yl)-3,5-diphenyltetrazolium bromide) to formazan crystals with mitochondrial dehydrogenases. MCF-7 cells were seeded in a 96-well plate with a concentration of 1 × 105 cells per mL in 100 μL DMEM medium (containing 10% FBS). After 24 h attachment in a cell incubator, the cells were then treated with various concentrations of ICy-N, ICy-OH and Ce6 in 100 μL medium for 60 min (N = 6). Then the plate was irradiated with 660 nm light at an average optical density of 12 mW cm−2 for 10 min and then incubated in the cell incubator. As for the dark group, the cells were kept out of light with the other treatments unchanged. Cells cultured with free medium were used as control and six replicates were set for each group to eliminate contingency. After 12 h incubation, the medium was replaced with 100 μL MTT (5 mg mL−1 in DMEM) solution and cultured for another 4 h at 37 °C in the cell incubator. Finally, the MTT solution was removed carefully and the formed crystals were dissolved in 100 μL dimethyl sulfoxide (DMSO). The plate was then carefully shaken for 10 min and the absorbance was determined on a microplate reader (Thermo Fisher Scientific) at 570 nm and 630 nm. The viability was expressed as a percent of the controlled one using the following equation:| Cell viability (%) = (ODdye570 − ODdye630)/(ODcontrol570 − ODcontrol630) × 100 |
Visualization of nitroreductase and photodynamic therapy in the 4T1 tumor bearing mice model
All procedures were carried out in compliance with the Guide for the Care and Use of Laboratory Animal Resources and the National Research Council, and were approved by the Institutional Animal Care and Use Committee of the NIH. 4–5 week old Balb/c mice were chosen for transplanting 4T1 cells at a density of 5 × 106 under the armpit. When the tumour volume reached 200 mm3, the xenograft tumour mice were divided into 4 groups (Control, Light, ICy-N, and ICy-N + Light, N = 5). The tumour volume was measured using a vernier caliper and calculated using the following equation:| V = 1/2 × a × b2 |
As for nitroreductase imaging, the mice injected with 50 μM ICy-N were narcotized and imaged with a NightOWL II LB983 small animal in vivo imaging system with 660 nm excitation and 700 ± 20 nm emission filters. The light power for mice PDT was 100 mW cm−2.
Synthesis of ICy-N
The synthetic route to ICy-N is displayed in Scheme S1.†Synthesis of compound 1((4-iodophenyl) hydrazine)
10 g 4-iodoaniline (45.7 mmol) and 2 mL concentrated hydrochloric acid were dissolved in 40 mL water in a round-bottom flask and cooled to 0 °C. Then, 3.5 g (50.7 mmol) NaNO2 in 10 mL water was added to the turbid solution dropwise and stirred for 1 hour at that temperature. The reaction mixture was then heated to room temperature and 17.3 g SnCl2 (91.4 mmol) dissolved in 16 mL water and 4 mL concentrated hydrochloric acid was slowly added to the reaction mixture under continuous stirring. A light brown precipitate was obtained after 2 hour stirring and filtered under reduce pressure. After washing with water 3 times, the precipitate was dissolved in 200 mL saturated NaOH solution and extracted with ethyl acetate (50 mL × 3). The organic layer was then washed with Na2S2O3 and saline and dried with anhydrous Na2SO4. Finally, the organic layer was evaporated to dryness to give compound 1 as a brown solid (8.4 g, 78.5%). 1H NMR (400 MHz, DMSO-d6) δ 7.35 (d, J = 8.8 Hz, 2H), 6.89 (s, 1H), 6.62 (d, J = 8.8 Hz, 2H), 4.05 (s, 2H). ESI-MS: m/z calcd for C6H8IN2+ [M + H]+: 234.97, found: 234.97.Synthesis of compound 2 (5-iodo-2,3,3-trimethyl-3H-indole)
5g (4-iodophenyl) hydrazine (21.3 mmol) and 3.7 g 3-methyl-2-butanone (42.6 mmol) were dissolved in 50 mL glacial acetic acid and heated to reflux for 4 h under continuous stirring. Then, the reaction mixture was poured into 200 mL water and extracted with ethyl acetate (50 mL × 3). The organic layer was washed with saline and dried with anhydrous Na2SO4. Finally, the organic layer was evaporated to dryness and the crude product was purified by silica gel column chromatography to obtain 2 as a deep red gummy liquid (4.4 g, 72.4%). 1H NMR (400 MHz, CDCl3) δ 7.62 (dd, J = 8.0, 1.7 Hz, 1H), 7.59 (d, J = 1.3 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 2.25 (s, 3H), 1.28 (s, 6H). ESI-MS: m/z calcd for C11H13IN+ [M + H]+: 286.01, found: 286.05.Synthesis of compound 3 (1-ethyl-5-iodo-2,3,3-trimethyl-3H-indol-1-ium iodide)
4.4 g 2 (15.4 mmol) was dissolved in 20 mL toluene and heated to reflux. Then 7.2 g iodoethane (46.2 mmol) in 5 mL toluene was added through a syringe in batches under continuous stirring. A pink precipitate was obtained after stirring for 6 hours and filtered under reduced pressure. The precipitate was washed with ethyl acetate 3 times and dried over a vacuum oven to afford 3 (4.7 g, 69.1%). 1H NMR (400 MHz, DMSO-d6) δ 8.31 (s, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.78 (d, J = 8.2 Hz, 1H), 4.46 (q, J = 6.9 Hz, 2H), 2.80 (s, 3H), 2.50 (s, 6H), 1.41 (t, J = 7.1 Hz, 3H). ESI-MS: m/z calcd for C13H17IN+ [M + H]+: 314.04, found: 314.07.Synthesis of compound 4 (2-bromocyclohex-1-enecarbaldehyde)
12.4 mL PBr3 was dissolved in 20 mL CHCl3 and slowly added to a mixture of 11.2 mL anhydrous DMF and 30 mL CHCl3 during a period of 1 hour at 0 °C. Then 5 mL cyclohexanone (48.4 mmol) was added dropwise at that temperature. After stirring for 1 hour, the mixture was heated to room temperature and stirred for another 12 hours. The mixture was poured into a 200 mL ice-water mixture and neutralized with a NaHCO3 solid until the pH is approximately 7. The resulting solution was extracted with CH2Cl2 (50 mL × 3) and dried over anhydrous Na2SO4. Finally, the organic layer was concentrated in vacuo to give an orange oil (6.8 g) which was used without further purification.Synthesis of compound 5
200 mg 2-hydroxy-4-methoxybenzaldehyde (1.3 mmol) in 6 mL anhydrous DMF was added to 295 mg 4 (1.56 mmol) and 847 mg Cs2CO3 (2.6 mmol). The mixture was stirred at 25 °C for 12 hours in a N2 atmosphere and extracted with water (60 mL) and ethyl acetate (20 mL × 3). After being dried over anhydrous Na2SO4, the organic layer was evaporated and the crude product was purified by silica gel column chromatography to obtain 5 as a yellow solid (125 mg, 39.7%). 1H NMR (400 MHz, CDCl3) δ 10.31 (s, 1H), 7.08 (d, J = 9.1 Hz, 1H), 6.69–6.63 (m, 3H), 3.84 (s, 3H), 2.57 (t, J = 6.0 Hz, 2H), 2.44 (t, J = 6.0 Hz, 2H), 1.74–1.68 (m, 3H).Synthesis of compound 6
242 mg 5 (1 mmol), 441 mg 3 (1 mmol) and 0.2 mL piperidine were dissolved in 10 mL acetic anhydride and heated to 60 °C for 2 hours. The resulting blue mixture was poured into 100 mL water and extracted with CH2Cl2 (20 mL × 3). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was further purified by silica gel column chromatography to obtain 6 as a blue solid (432 mg, 64.9%). 1H NMR (400 MHz, DMSO-d6) δ 8.58 (d, J = 14.8 Hz, 1H), 8.15 (d, J = 1.3 Hz, 1H), 7.88 (dd, J = 8.3, 1.4 Hz, 1H), 7.62–7.56 (m, 2H), 7.48 (d, J = 8.4 Hz, 1H), 7.12 (d, J = 2.2 Hz, 1H), 7.04 (dd, J = 8.7, 2.3 Hz, 1H), 6.50 (d, J = 14.9 Hz, 1H), 4.38 (q, J = 7.1 Hz, 2H), 3.93 (s, 3H), 2.74 (t, J = 5.8 Hz 2H), 2.69 (t, J = 5.8 Hz, 2H), 1.87–1.79 (m, 2H), 1.75 (s, 6H), 1.34 (t, J = 7.1 Hz, 3H). ESI-MS: m/z calcd for C28H29INO2+ [M]+: 538.12, found: 538.28.Synthesis of compound ICy-OH
1g BBr3 (4.0 mmol) in 10 mL anhydrous CH2Cl2 was added dropwise to 20 mL anhydrous CH2Cl2 solution containing 200 mg 6 (0.3 mmol) at 0 °C with continuous stirring. After 1 hour stirring at that temperature, the solution was heated to room temperature and stirred for another 12 hours. Then the solution was poured into 100 mL water and extracted with CH2Cl2 (20 mL × 3). The organic layer was dried over anhydrous Na2SO4 and evaporated to give a blue solid. The crude product was then purified by silica gel column chromatography to obtain ICy-OH as a blue solid (180 mg, 92.1%). 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 14.4 Hz, 1H), 8.08 (d, J = 1.3 Hz, 1H), 7.81 (dd, J = 8.3, 1.4 Hz, 1H), 7.62 (s, 1H), 7.48 (d, J = 9.2 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 6.84–6.78 (m, 2H), 6.31 (d, J = 14.5 Hz, 1H), 4.27 (q, J = 7.0 Hz, 2H), 2.71 (t, J = 5.8 Hz, 2H), 2.67 (t, J = 5.8 Hz, 2H), 1.85–1.78 (m, 2H), 1.71 (s, 6H), 1.30 (t, J = 7.1 Hz, 3H). ESI-MS: m/z calcd for C27H27INO2+ [M]+: 524.11, found: 524.23.Synthesis of ICy-N
150 mg ICy-OH (0.23 mmol), 60 mg 1-(bromomethyl)-4-nitrobenzene and 95 mg K2CO3 (0.69 mmol) were dissolved in 10 mL DMF. The mixture was heated to 60 °C under a N2 atmosphere and monitored by TLC. When the reaction was completed, the mixture was washed with 60 mL water and extracted with CH2Cl2 (20 mL × 3). The organic layer was dried over dried over anhydrous Na2SO4 and evaporated in vacuo. The crude product was then purified by silica gel column chromatography to obtain ICy-N as a blue solid (86 mg, 47.6%). 1H NMR (400 MHz, DMSO-d6) δ 8.55 (d, J = 14.8 Hz, 1H), 8.32 (d, J = 8.5 Hz, 2H), 8.18 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.60 (d, J = 8.6 Hz, 1H), 7.56 (s, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.21 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.52 (d, J = 15.0 Hz, 1H), 5.49 (s, 2H), 4.40 (q, J = 7.2 Hz, 2H), 2.75–2.65 (m, 4H), 1.87–1.77 (m, 2H), 1.74 (s, 6H), 1.33 (t, J = 7.0 Hz, 3H).13C NMR (100 MHz, DMSO-d6) δ 176.68, 161.50, 161.30, 154.22, 147.65, 145.52, 144.87, 144.65, 141.39, 137.98, 134.09, 131.97, 129.58, 129.05, 127.79, 124.22, 116.39, 115.54, 114.97, 114.14, 104.54, 102.62, 92.90, 69.37, 50.85, 45.71, 28.89, 27.72, 24.15, 20.36, 13.03. ESI-MS: m/z calcd for C34H32IN2O4+ [M]+: 659.1407, found: 659.1405.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Science Foundation of China (project 21421005, 21576037 and U1608222).Notes and references
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87 CrossRef PubMed.
- W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66, 115 CrossRef PubMed.
- J. Du, Z. Gu, L. Yan, Y. Yong, X. Yi, X. Zhang, J. Liu, R. Wu, C. Ge, C. Chen and Y. Zhao, Adv. Mater., 2017, 29, 1701268 CrossRef PubMed.
- N. Ma, F.-G. Wu, X. Zhang, Y.-W. Jiang, H.-R. Jia, H.-Y. Wang, Y.-H. Li, P. Liu, N. Gu and Z. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 13037 CrossRef CAS PubMed.
- S. R. Chowdhury, S. Mukherjee, S. Das, C. R. Patra and P. K. Iyer, Chem. Sci., 2017, 8, 7566 RSC.
- M. Xiao, W. Sun, J. Fan, J. Cao, Y. Li, K. Shao, M. Li, X. Li, Y. Kang, W. Zhang, S. Long, J. Du and X. Peng, Adv. Funct. Mater., 2018, 28, 1805128 CrossRef.
- G. Lan, K. Ni, Z. Xu, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2018, 140, 5670 CrossRef CAS PubMed.
- S. L. Topalian, F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott, J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel, S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. Chen, W. H. Sharfman, R. A. Anders, J. M. Taube, T. L. McMiller, H. Xu, A. J. Korman, M. Jure-Kunkel, S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton and M. Sznol, N. Engl. J. Med., 2012, 366, 2443 CrossRef CAS PubMed.
- Z. Zhou, Y. Yan, L. Wang, Q. Zhang and Y. Cheng, Biomaterials, 2019, 203, 63 CrossRef CAS PubMed.
- J. Gao, M. Sanchez-Purra, H. Huang, S. Wang, Y. Chen, X. Yu, Q. Luo, K. Hamad-Schifferli and S. Liu, Sci. China: Chem., 2017, 60, 1219 CrossRef CAS.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun, K. Shao, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2019, 141, 2695 CrossRef CAS PubMed.
- L. Huang, Z. Li, Y. Zhao, Y. Zhang, S. Wu, J. Zhao and G. Han, J. Am. Chem. Soc., 2016, 138, 14586 CrossRef CAS PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795 CrossRef CAS PubMed.
- W. Chen, Y. Wang, M. Qin, X. Zhang, Z. Zhang, X. Sun and Z. Gu, ACS Nano, 2018, 12, 5995 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS PubMed.
- Z. Li, J. Wang, J. Chen, W. Lei, X. Wang and B. Zhang, Sci. China: Chem., 2010, 53, 1994 CrossRef CAS.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 106, 1098 CrossRef CAS PubMed.
- M. Li, S. Long, Y. Kang, L. Guo, J. Wang, J. Fan, J. Du and X. Peng, J. Am. Chem. Soc., 2018, 140, 15820 CrossRef CAS PubMed.
- J. C. Kennedy and R. H. Pottier, J. Photochem. Photobiol., B, 1992, 14, 275 CrossRef CAS.
- K. Y. Choi, G. Liu, S. Lee and X. Chen, Nanoscale, 2012, 4, 330 RSC.
- J. Zhang, C. Jiang, J. P. Figueiró Longo, R. B. Azevedo, H. Zhang and L. A. Muehlmann, Acta Pharm. Sin. B, 2018, 8, 137 CrossRef PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816 CrossRef CAS PubMed.
- S.-B. W. Kai Han, L. Qi, J.-Y. Zhu and X.-Z. Zhang, ACS Nano, 2015, 9, 10268 CrossRef PubMed.
- L. Feng, S. Gai, Y. Dai, F. He, C. Sun, P. Yang, R. Lv, N. Niu, G. An and J. Lin, Chem. Mater., 2018, 30, 526 CrossRef CAS.
- J. Xu, W. Han, P. Yang, T. Jia, S. Dong, H. Bi, A. Gulzar, D. Yang, S. Gai, F. He, J. Lin and C. Li, Adv. Funct. Mater., 2018, 28, 1803804 CrossRef.
- L. Feng, R. Xie, C. Wang, S. Gai, F. He, D. Yang, P. Yang and J. Lin, ACS Nano, 2018, 12, 11000 CrossRef CAS PubMed.
- Z. Hou, Y. Zhang, K. Deng, Y. Chen, X. Li, X. Deng, Z. Cheng, H. Lian, C. Li and J. Lin, ACS Nano, 2015, 9, 2584 CrossRef CAS PubMed.
- W. Sun, Y. Wen, R. Thiramanas, M. Chen, J. Han, N. Gong, M. Wagner, S. Jiang, M. S. Meijer, S. Bonnet, H. J. Butt, V. Mailänder, X. Liang and S. Wu, Adv. Funct. Mater., 2018, 28, 1804227 CrossRef.
- M. K. Wolpert, J. R. Althaus and D. G. Johns, J. Pharmacol. Exp. Ther., 1973, 185, 202 CAS.
- R. B. Elmes, Chem. Commun., 2016, 52, 8935 RSC.
- Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407 CrossRef CAS PubMed.
- R. H. Wong, T. Kwong, K. H. Yau and H. Y. Au-Yeung, Chem. Commun., 2015, 51, 4440 RSC.
- Z. Li, X. He, Z. Wang, R. Yang, W. Shi and H. Ma, Biosens. Bioelectron., 2015, 63, 112 CrossRef CAS PubMed.
- A. Chevalier, Y. Zhang, O. M. Khdour, J. B. Kaye and S. M. Hecht, J. Am. Chem. Soc., 2016, 138, 12009 CrossRef CAS PubMed.
- F. Song, R. Liang, J. Deng, Z. Liu and X. Peng, Chin. Chem. Lett., 2017, 28, 1997 CrossRef CAS.
- J. Atchison, S. Kamila, H. Nesbitt, K. A. Logan, D. M. Nicholas, C. Fowley, J. Davis, B. Callan, A. P. McHale and J. F. Callan, Chem. Commun., 2017, 53, 2009 RSC.
- X. Li, N. Zhao, L. Yu and Y.-A. Son, Mol. Cryst. Liq. Cryst., 2015, 608, 273 CrossRef CAS.
- L. Yuan, W. Lin, S. Zhao, W. Gao, B. Chen, L. He and S. Zhu, J. Am. Chem. Soc., 2012, 134, 13510 CrossRef CAS PubMed.
- Z. Xu and L. Xu, Chem. Commun., 2016, 52, 1094 RSC.
- N. K. Roopa, V. Bhalla and M. Kumar, Chem. Commun., 2015, 51, 15614 RSC.
- L. Yousif, K. Stewart and S. Kelley, ChemBioChem, 2009, 10, 1939 CrossRef CAS PubMed.
- D. Kessel and Y. Luo, Cell Death Differ., 1999, 6, 28 CrossRef CAS PubMed.
- H. Huang, S. Long, M. Li, F. Gao, J. Du, J. Fan and X. Peng, Dyes Pigm., 2018, 149, 633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03355f |
| This journal is © The Royal Society of Chemistry 2019 |