
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxygenative trifluoromethylthiolation of carboxylic acids†
Runze
Mao
 ,
Srikrishna
Bera
,
Alexis
Cheseaux
and
Xile
Hu
*
,
Srikrishna
Bera
,
Alexis
Cheseaux
and
Xile
Hu
*
Laboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), ISIC-LSCI, Lausanne 1015, Switzerland. E-mail: xile.hu@epfl.ch
First published on 26th August 2019
Abstract
Here we describe a deoxygenative trifluoromethylthiolation method that yields trifluoromethyl thioesters from readily available carboxylic acids. The method is built upon an “umpolung” strategy where triphenylphosphine is used to first activate an electrophilic trifluoromethylthiolating reagent and then serves as an oxygen acceptor for the deoxygenation. The method is mild, efficient, broad-scope, and tolerant. It can be applied for the late-stage functionalization of numerous natural products and drug molecules containing a carboxylic acid group. The trifluoromethyl thioesters can be converted into trifluoromethyl thioethers by Pd-catalyzed decarbonylation.
Organofluorine compounds have widespread applications in medicinal and materials sciences.1–4 Among fluorine-containing moieties, the trifluoromethylthio group (–SCF3) is of considerable interest because of its high lipophilic and electron-withdrawing nature. Significant progress has been made in direct trifluoromethylthiolation of C–H and C–X moieties.4–19 However, there are still few efficient methods for the synthesis of trifluoromethyl thioesters. Trifluoromethyl thioesters could be prepared by reactions of acid chlorides with Hg(SCF3)2, NMe4SCF3 or (bpy)CuSCF3 (Fig. 1A).17,20,21 These reactions were limited by the use of reactive or toxic reagents, or the generation of a stoichiometric amount of metallic by-products. The groups of Glorius18 and Shen19 reported elegant methods of accessing trifluoromethyl thioesters from aldehydes via a hydrogen atom transfer (HAT) process (Fig. 1A). Nevertheless, among organic carbonyl compounds aldehydes are relatively instable and less available. Carboxylic acids, on the other hand, are abundant, stable, and non-toxic. The deoxygenative trifluoromethylthiolation of carboxylic acids would represent an efficient and highly desirable approach to the synthesis of trifluoromethyl thioesters.
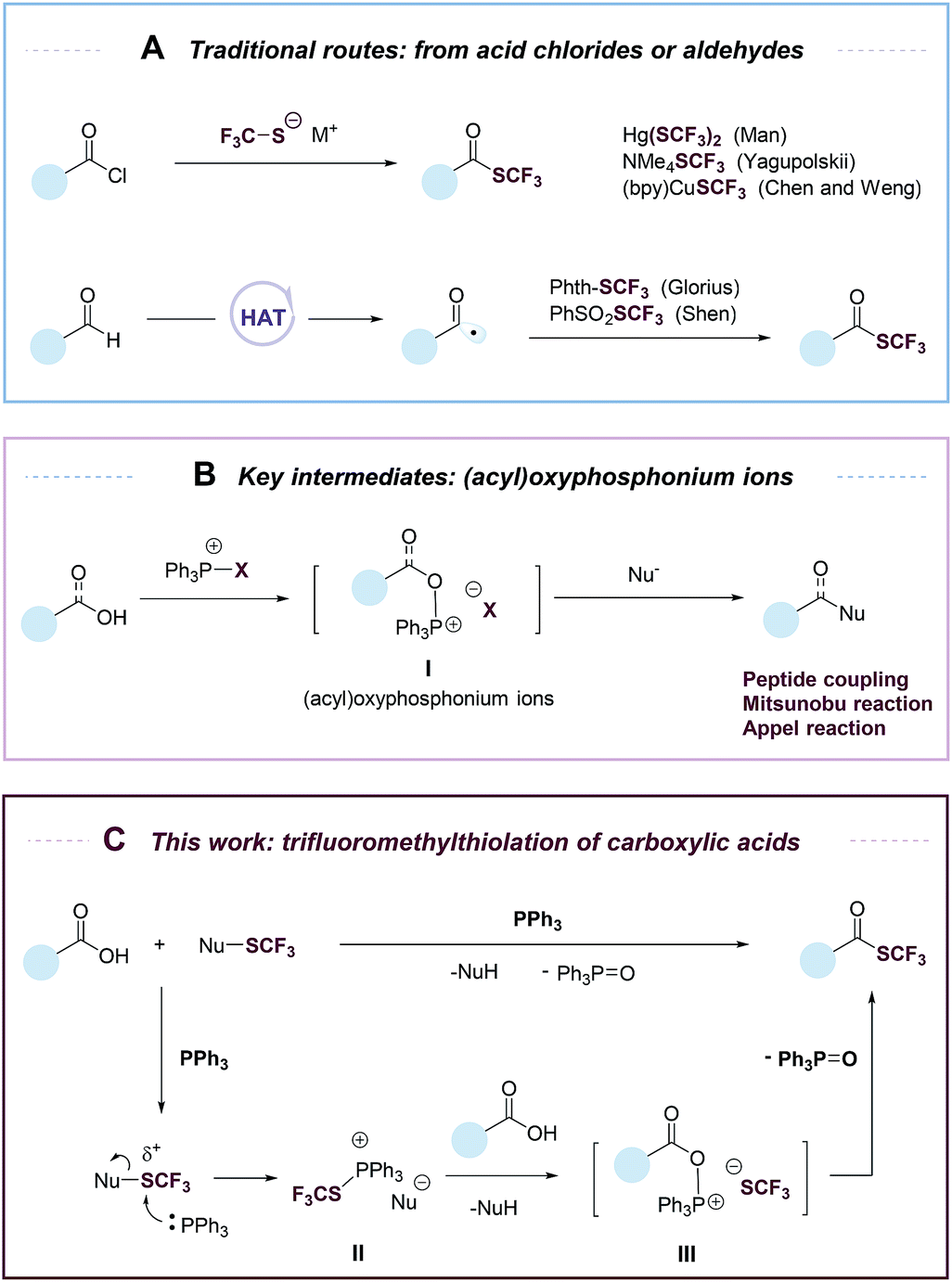 | ||
| Fig. 1 (A) Previous methods to synthesize trifluoromethyl thioesters from acid chlorides or aldehydes; (B) key intermidiates: (acyl)oxyphosphonium ions; (C) this work: deoxygenative trifluoromethylthiolation of carboxylic acids. | ||
Despite its conceptual simplicity, the deoxygenative trifluoromethylthiolation of carboxylic acids is challenging to achieve. It was reported that in the presence of a carboxylic acid, a CF3S− anion would lose an F− to form carbonothioic difluoride, which further reacted with carboxylic acids to give eventually an acyl fluoride.10,22 Inspired by the rich chemistry of phosphorus reagents and (acyl)oxyphosphonium ions I (Fig. 1B) in peptide coupling,23 Mitsunobu,24,25 and Appel26 reactions, we hypothesized that such intermediates could be possibly transformed into trifluoromethyl thioesters from carboxylic acids under suitable conditions. Here we describe an “umpolung” strategy that allows the use of electrophilic trifluoromethylthiolating reagents and avoids the decomposition of CF3S− anion by carboxylic acids (Fig. 1C). The “umpolung” is achieved with triphenylphosphine (PPh3), which first captures a CF3S+ cation to form a SCF3-phosphonium salt (II), followed by a metathesis reaction with a carboxylate to give an oxyphosphonium intermediate (III), which is probe to deoxygenative trifluoromethylthiolation to give a trifluoromethyl thioester while eliminating triphenylphosphine oxide (PPh3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (Fig. 1C). This strategy is applicable for the rapid synthesis of a diverse set of trifluoromethyl thioesters from readily available aromatic and aliphatic carboxylic acids, including many natural products and drugs.
O) (Fig. 1C). This strategy is applicable for the rapid synthesis of a diverse set of trifluoromethyl thioesters from readily available aromatic and aliphatic carboxylic acids, including many natural products and drugs.
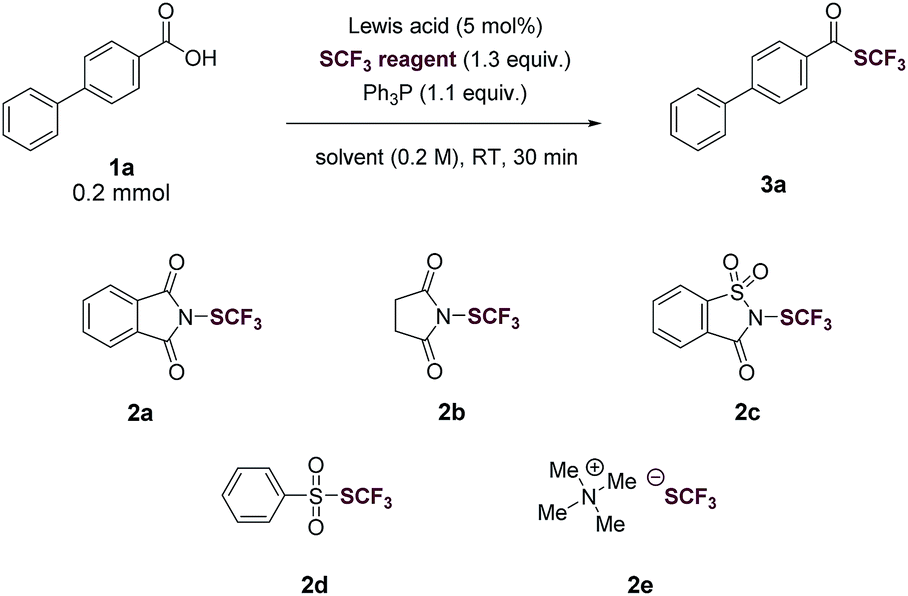
We began our investigation by optimizing the reaction of 4-phenylbenzoic acid (1a) with N-(trifluoromethylthio)phthalimide (2a) to give the corresponding trifluoromethyl thioester (3a). To our delight, the reaction proceeded in the presence of 1.1 equiv. of Ph3P in tetrahydrofuran (THF, 0.2 M) with a yield of 40% (Table 1, entry 1). The reaction was then optimized by varying reaction parameters (Table ESI, S1–S5†). A summary of key observations is shown in Table 1. THF was the best solvent (Table S2†). A small amount of Lewis acid could enhance the reactivity of 2a (Table 1, entries 2–6; Table S3†). The binding of a Lewis acid by the phthalimide group might polarize N-(trifluoromethylthio)phthalimide, facilitating the nucleophilic attack of PPh3 to N-(trifluoromethylthio)phthalimide and subsequent generation of the key intermediate SCF3-phosphonium salt (Fig. 1C, II). Anhydrous FeCl3 (5 mol%) was the best Lewis acid, giving a yield of 95% (Table 1, entry 2). Among various trifluoromethylthiolating agents,272a proved to be superior than other electrophilic trifluoromethylthiolating reagent 2b–2d (Table 1, entries 7–9). When the nucleophilic NMe4SCF3 (2e) was used, no product was formed (Table 1, entry 10). Addition of an external base slightly lowered the yields (Table 1, entries 11 and 12; Table S4†). Further optimization indicated Ph3P was the best mediator (Table 1, entries 2 and 13; Table S5†). It was worthy to note that the reaction completed within 30 minutes at room temperature.
|
|
|||
|---|---|---|---|
| Entry | SCF3 reagent | Lewis acid | Yield |
| a Yield determined by 19F NMR spectroscopy of the crude reaction mixture using α,α,α-trifluorotoluene as an internal standard. b BF3·OEt2 (10 mol%). c NaHCO3 (1.0 equiv.) as an external base. d 2,6-Lutidine (1.0 equiv.) as an external base. e Tricyclohexanephosphine (PCy3) in place of PPh3. | |||
| 1 | 2a | None | 40% |
| 2 | 2a | FeCl3 | 95% |
| 3 | 2a | FeCl3·6H2O | 71% |
| 4b | 2a | BF3·OEt2 | 64% |
| 5 | 2a | AlCl3 | 39% |
| 6 | 2a | Sc(OTf)3 | 71% |
| 7 | 2b | FeCl3 | 19% |
| 8 | 2c | FeCl3 | 51% |
| 9 | 2d | FeCl3 | 7% |
| 10 | 2e | FeCl3 | 0% |
| 11c | 2a | FeCl3 | 69% |
| 12d | 2a | FeCl3 | 68% |
| 13e | 2a | FeCl3 | 39% |
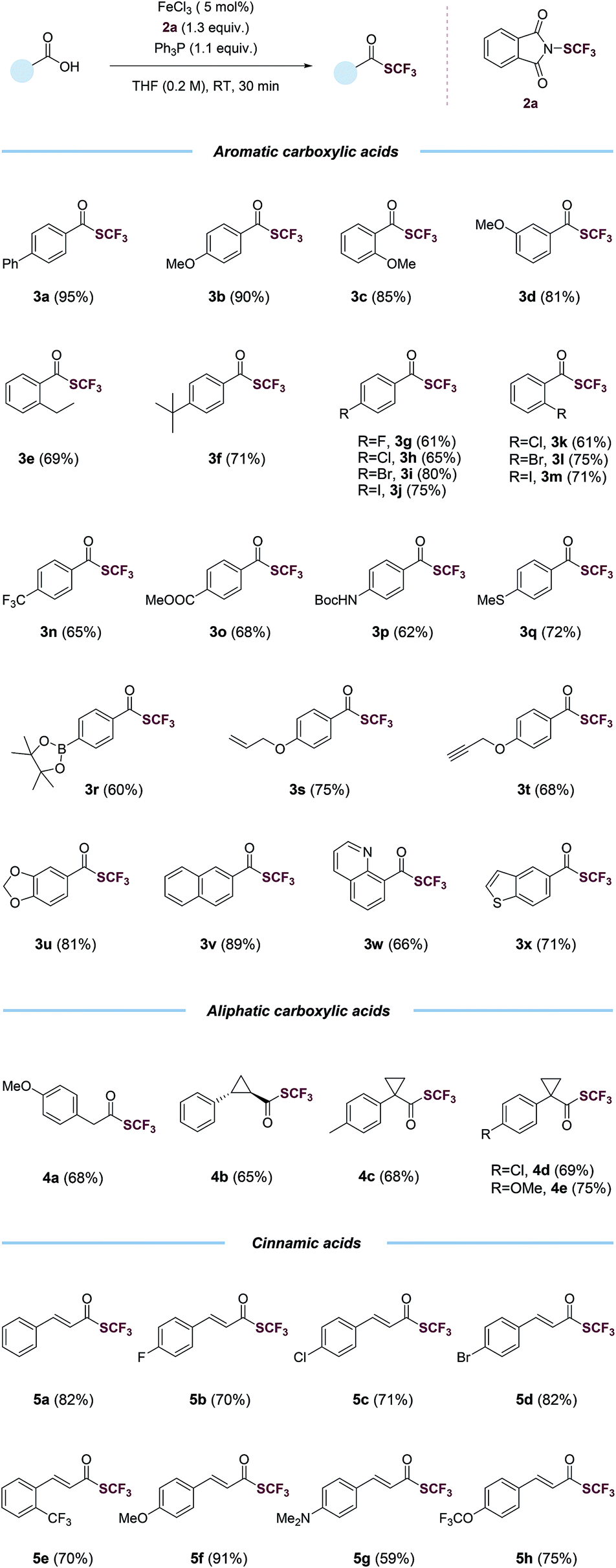
With the optimized conditions in hand (entry 2, Table 1), we probed the generality of this transformations (Table 2). A myriad of aryl carboxylic acids containing electron-donating (3b–3f) and electron-withdrawing (3g–3o) substituents were coupled to give the corresponding trifluoromethyl thioesters in moderate to excellent yields. Notable, aryl halides (3g–3m), including relatively reactive aryl iodides (3j, 3m), were tolerated in the reaction. Functional groups such as trifluoromethyl (3n), ester (3o), (tert-butoxycarbonyl)amino (3p), thiomethyl (3q), boronic ester (3r), alkene (3s), alkyne (3t), 1,3-benzodioxole (3u) and naphthalene (3v) were all compatible. The reactions also proceeded smoothly with various heteroaryl carboxylic acids, giving the desired products (3w–3x) in satisfying yields. Importantly, the reactions worked with aliphatic carboxylic acids as well. Primary, secondary, tertiary carboxylic acids were all suitable substrates, affording the corresponding trifluoromethyl thioesters (4a–4e) in good yields. The trifluoromethylthiolation was also successful for various cinnamic acids containing electron-neutral (5a), electron-withdrawing (5b–5e, 5h), and electron-donating (5f–5g) substituents. A variety of alcohols and substituted methyl benzoates were also tested as substrates (Table S7†). Reactions with methyl benzoates gave no products under the standard conditions, suggesting a crucial role of the O-nucleophilic site of carboxylic acids. Reactions of some alcohols, especially primary alcohols, gave the desired products in low yields, which were difficult to isolate amide various side products. Several amino acids were also used as substrates, however, yields were low (Table S8†), possibly due to the competition of N- and O-nucleophilic sites.
| a Carboxylic acid (1.0 equiv.), triphenylphosphine (Ph3P, 1.1 equiv.), N-(trifluoromethylthio)phthalimide (1.3 equiv.), FeCl3 (5 mol%) in THF (0.2 M), room temperature, 30 min, isolated yield. |
|---|
|
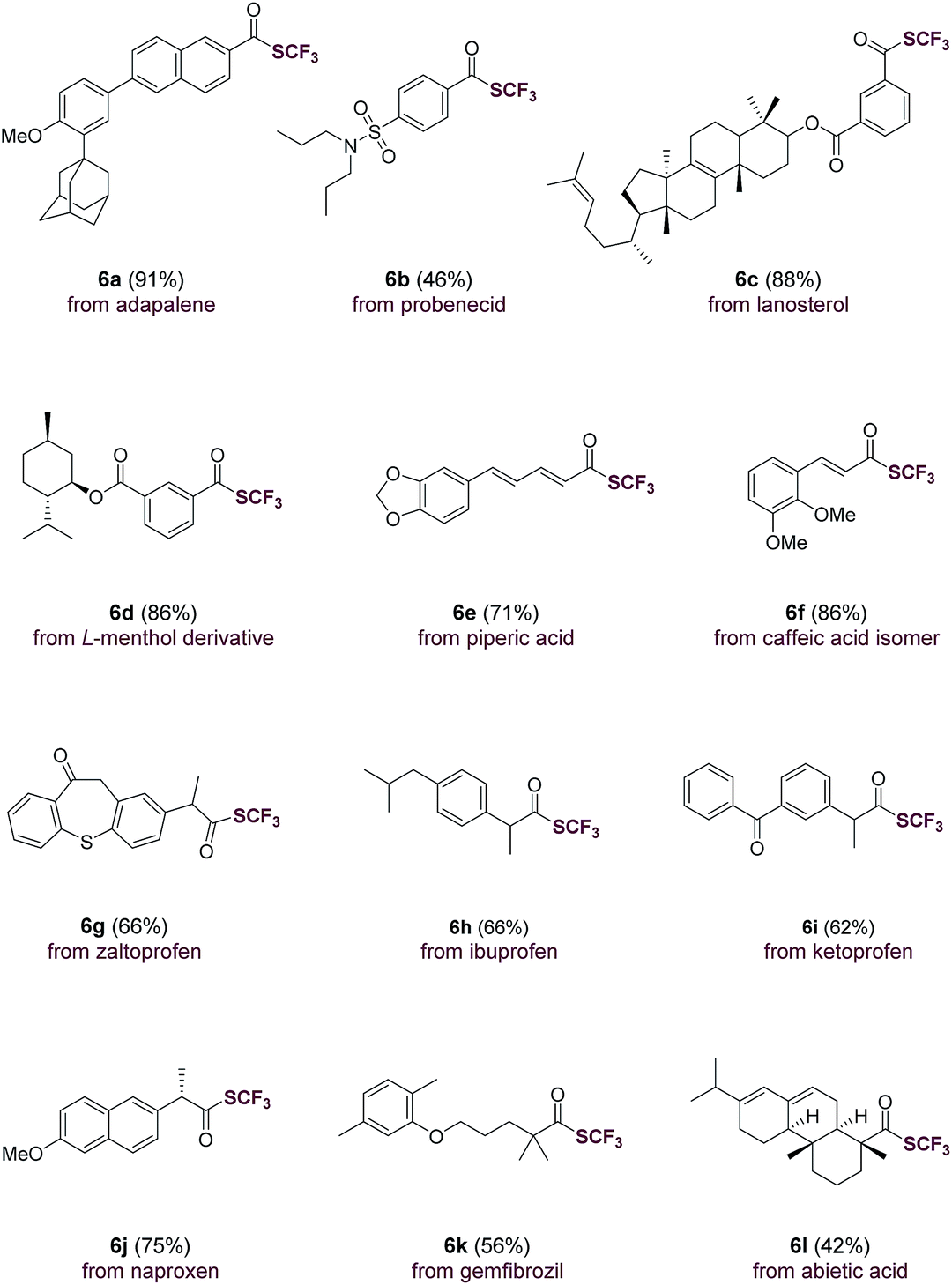
The direct use of carboxylic acids as substrates makes the current trifluoromethylthiolation method applicable for the rapid, late-stage modification of carboxylic acid-containing natural products and drug molecules (Table 3). Indeed, aromatic carboxylic acids such as adapalene (6a), probenecid (6b), lanosterol (6c), L-menthol derivative (6d) were trifluoromethylthiolated with ease. Moreover, natural-occurring cinnamic acids such as piperic acid (6e) and caffeic acid isomer (6f) underwent smooth transformations as well. Drug molecules and natural products containing an aliphatic carboxylic acid group such as zaltoprofen (6g), ibuprofen (6h), ketoprofen (6i), naproxen (6j), gemfibrozil (6k) and abietic acid (6l) were all easily converted to their corresponding trifluoromethyl thioesters. The successful late-stage functionalization of these natural products and drugs, many of which contain sensitive sulfonamide, alkene, carbonyl and heterocyclic groups, underscores the high chemoselectivity and functional group tolerance of the current method (Table 3).
| a Carboxylic acid (1.0 equiv.), triphenylphosphine (Ph3P, 1.1 equiv.), N-(trifluoromethylthio)phthalimide (1.3 equiv.), FeCl3 (5 mol%) in THF (0.2 M), room temperature, 30 min, isolated yield. |
|---|
|
To demonstrate the synthetic utility of trifluoromethyl thioesters, compound 3a was subjected to a Pd-catalyzed decarbonylation28,29 to give the corresponding trifluoromethyl thioether 7a in 91% yield (Fig. 2). Trifluoromethyl thioethers are ubiquitous in pharmaceutical and agrochemical compounds.4 Thus, our method enables the synthesis of trifluoromethyl thioethers from readily available carboxylic acids.
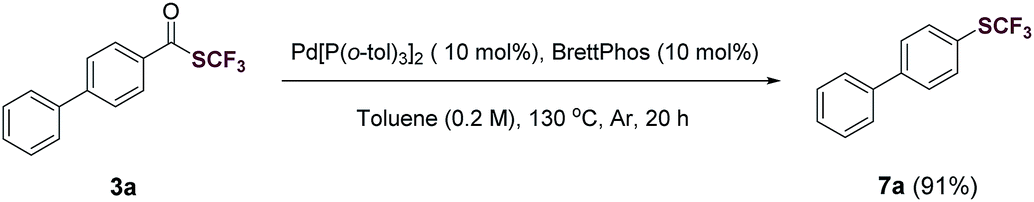 | ||
| Fig. 2 Conversion of a trifluoromethyl thioester (3a) to the corresponding trifluoromethyl thioether (7a) via a Pd-catalyzed decarbonylation. | ||
Based on results from the control experiments (Table 1 and S1–S5†),3031P NMR study (Fig. S51†), and previous reports,31,32 we propose a tentative mechanism for the deoxygenative trifluoromethylthiolation (Fig. 3). N-(Trifluoromethylthio)phthalimide (2a) coordinates to FeCl3via the phthalimide group. This coordination increases the electrophilicity of the SCF3 group, promoting the nucleophilic attack of PPh3. The latter generates a trifluoromethylthiophosphonium ion II, which reacts with a carboxylic acid to generate an acyloxyphosphonium CF3S− intermediate III. Intramolecular attack of the CF3S− anion on the acyl carbon of III then gives the thioester product as well as the Ph3PO byproduct.
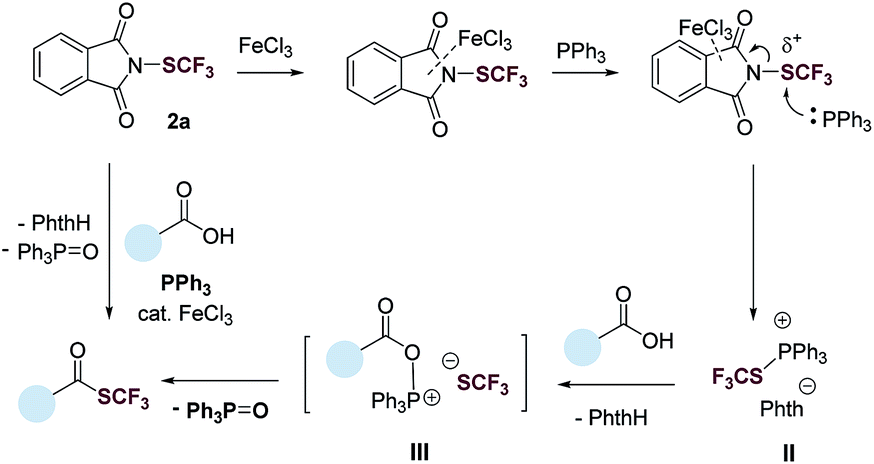 | ||
| Fig. 3 A tentative reaction pathway. | ||
Conclusions
In summary, by using PPh3 as a mediator to “umpolung” the electrophilic trifluoromethylthiolating agent 2a, we have achieved, for the first time, deoxygenative trifluoromethylthiolation of carboxylic acids. The reactions are rapid and occur at room temperature. They allow the access of a wide range of trifluoromethyl thioesters from readily available carboxylic acids. The method can be applied for the late-stage functionalization of many natural products and drug molecules. The trifluoromethyl thioesters can be converted into trifluoromethyl thioethers in one step by Pd-catalyzed decarbonylation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by the NoNoMeCat Marie Skłodowska-Curie training network funded by the European Union under the Horizon 2020 Program (675020-MSCA-ITN-2015-ETN) and the Swiss National Science Foundation.Notes and references
- I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons Ltd, 2009 Search PubMed.
- A. Tressaud and G. Haufe, Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals, Elsevier, London, 2008 Search PubMed.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- X. H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731–764 CrossRef CAS PubMed.
- F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415–2428 CrossRef CAS.
- V. N. Boiko, Beilstein J. Org. Chem., 2010, 6, 880–921 CrossRef PubMed.
- C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- F. Baert, J. Colomb and T. Billard, Angew. Chem., Int. Ed., 2012, 51, 10382–10385 CrossRef CAS PubMed.
- J. He, C. Chen, G. C. Fu and J. C. Peters, ACS Catal., 2018, 8, 11741–11748 CrossRef CAS PubMed.
- J. B. Liu, X. H. Xu, Z. H. Chen and F. L. Qing, Angew. Chem., Int. Ed., 2015, 54, 897–900 CrossRef CAS PubMed.
- S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200–16203 CrossRef CAS PubMed.
- X. Shao, C. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227–1236 CrossRef CAS PubMed.
- G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312–7314 CrossRef CAS PubMed.
- H. Wu, Z. Xiao, J. Wu, Y. Guo, J. C. Xiao, C. Liu and Q. Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 4070–4074 CrossRef CAS PubMed.
- Y. Zeng and J. Hu, Org. Lett., 2016, 18, 856–859 CrossRef CAS PubMed.
- C. P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183–185 CrossRef CAS.
- M. Zhang, J. Chen, Z. Chen and Z. Weng, Tetrahedron, 2016, 72, 3525–3530 CrossRef CAS.
- S. Mukherjee, T. Patra and F. Glorius, ACS Catal., 2018, 8, 5842–5846 CrossRef CAS.
- B. Xu, D. Li, L. Lu, D. Wang, Y. Hu and Q. Shen, Org. Chem. Front., 2018, 5, 2163–2166 RSC.
- E. H. Man, D. D. Coffman and E. L. Muetterties, J. Am. Chem. Soc., 1959, 81, 3575–3577 CrossRef CAS.
- M. M. Kremlev, W. Tyrra, D. Naumann and Y. L. Yagupolskii, Tetrahedron Lett., 2004, 45, 6101–6104 CrossRef CAS.
- T. Scattolin, K. Deckers and F. Schoenebeck, Org. Lett., 2017, 19, 5740–5743 CrossRef CAS PubMed.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- O. Mitsunobu, Synthesis, 1981, 1, 1–28 CrossRef.
- O. Mitsunobu and M. Yamada, Bull. Chem. Soc. Jpn., 1967, 40, 2380–2382 CrossRef CAS.
- R. Appel, Angew. Chem., Int. Ed., 1975, 14, 801–811 CrossRef.
- J. Zhang, J. D. Yang, H. Zheng, X. S. Xue, H. Mayr and J. P. Cheng, Angew. Chem., Int. Ed., 2018, 57, 12690–12695 CrossRef CAS PubMed.
- N. Ichiishi, C. A. Malapit, L. Wozniak and M. S. Sanford, Org. Lett., 2018, 20, 44–47 CrossRef CAS PubMed.
- C. A. Malapit, N. Ichiishi and M. S. Sanford, Org. Lett., 2017, 19, 4142–4145 CrossRef CAS PubMed.
- Lewis acids could enhance the yields of the reactions but they were not indispensable (Table 1, entries 1–6, Table S3†). However, PR3 and electrophilic SCF3 reagent were absolutely needed (Tables S1 and S5†).
- S. B. Munoz, H. Dang, X. Ispizua-Rodriguez, T. Mathew and G. K. S. Prakash, Org. Lett., 2019, 21, 1659–1663 CrossRef CAS PubMed.
- P. H. Huy and C. Mbouhom, Chem. Sci., 2019, 10, 7399–7406 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03396c |
| This journal is © The Royal Society of Chemistry 2019 |