
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective hydrogenation of amides catalysed by a molybdenum pincer complex: scope and mechanism†
Thomas
Leischner
a,
Lluis
Artús Suarez
 b,
Anke
Spannenberg
a,
Kathrin
Junge
a,
Ainara
Nova
*b and
Matthias
Beller
*a
b,
Anke
Spannenberg
a,
Kathrin
Junge
a,
Ainara
Nova
*b and
Matthias
Beller
*a
aLeibniz Institut für Katalyse e. V., Albert-Einstein-Straße 29a, Rostock, 18059, Germany. E-mail: Matthias.Beller@catalysis.de
bHylleraas Centre for Quantum Molecular Sciences, Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N-0315, Oslo, Norway. E-mail: Ainara.nova@kjemi.uio.no
First published on 8th October 2019
Abstract
A series of molybdenum pincer complexes has been shown for the first time to be active in the catalytic hydrogenation of amides. Among the tested catalysts, Mo-1a proved to be particularly well suited for the selective C–N hydrogenolysis of N-methylated formanilides. Notably, high chemoselectivity was observed in the presence of certain reducible groups including even other amides. The general catalytic performance as well as selectivity issues could be rationalized taking an anionic Mo(0) as the active species. The interplay between the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O reduction and the catalyst poisoning by primary amides accounts for the selective hydrogenation of N-methylated formanilides. The catalyst resting state was found to be a Mo–alkoxo complex formed by reaction with the alcohol product. This species plays two opposed roles – it facilitates the protolytic cleavage of the C–N bond but it encumbers the activation of hydrogen.
O reduction and the catalyst poisoning by primary amides accounts for the selective hydrogenation of N-methylated formanilides. The catalyst resting state was found to be a Mo–alkoxo complex formed by reaction with the alcohol product. This species plays two opposed roles – it facilitates the protolytic cleavage of the C–N bond but it encumbers the activation of hydrogen.
Introduction
The reduction of carboxylic acid derivatives via catalytic homogeneous hydrogenation represents an attractive atom-economic and environmentally benign methodology.1,2 To date, the vast majority of homogeneous catalysts for these transformations rely on noble metals.3 The limited availability of these elements along with their toxicity and pollutive nature initiated efforts for their replacement. Significant progress in this direction has been achieved in the past decade, in particular with respect to iron,4 manganese5 and cobalt6 based systems. Thus, several examples of base metal catalysed hydrogenations of aldehydes, ketones, carboxylic acids, esters and nitriles have been reported in recent years, some of them with remarkable activities and selectivities.2a,7 On the contrary, hydrogenation of amides is known to a much less extent.8 The latter can be attributed to the extremely low electrophilicity of the carbonyl group, which renders their hydrogenation particularly challenging.In general, catalytic hydrogenation of amides can proceed via either C–N (hydrogenolysis) or C–O (hydrogenation) bond cleavage of the intermediate hemiaminal (Scheme 1). While the C–O bond scission results in the formation of the alkylated/benzylated amine with H2O as the only by-product, the C–N bond cleavage leads to the free amine and the corresponding alcohol. Recently, an additional amide hydrogenation pathway was demonstrated, where the alkylated/benzylated amine is produced by a hydrogen borrowing/autotransfer mechanism from the initially formed alcohol and amine under specific acidic reaction conditions.9 Until today, the development of catalytic systems that enable these chemoselective transformations continues to be challenging and therefore are subject of ongoing research.
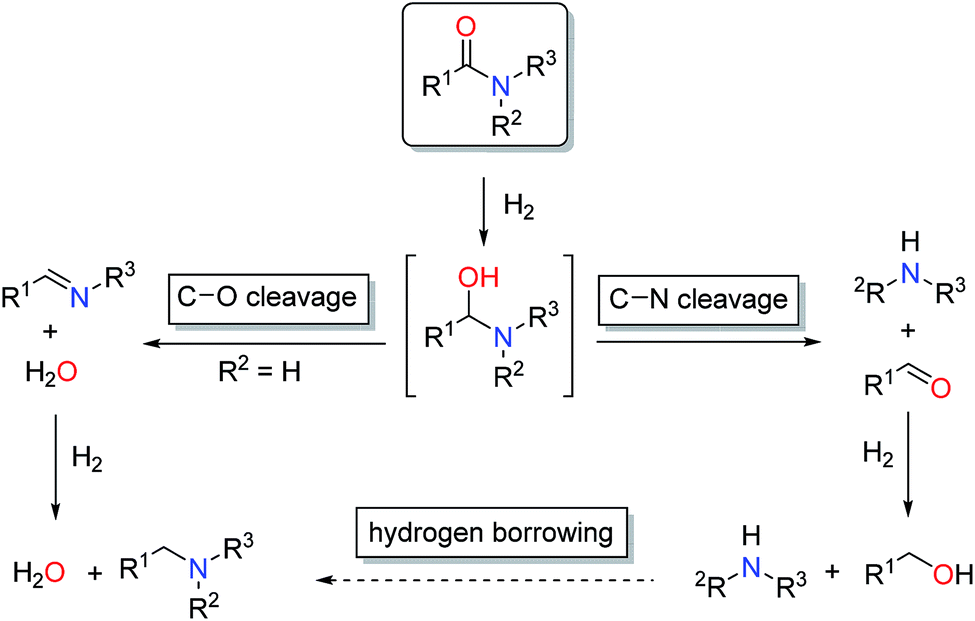 | ||
| Scheme 1 Pathways for amide reduction. | ||
Initial efforts in this direction mainly focused on homogeneous ruthenium catalysts.10 Since the inspiring report by Cole-Hamilton and co-workers in 2012, various Ru-based systems for the highly selective scission of either the C–N or the C–O bond have been described.10
In sharp contrast, reports on homogeneous base metal catalysts for this important reaction are particularly scarce. Pioneering work in this area was published by the groups of Milstein, Langer and Sanford only as late as 2016.11–13 For the first time, they could demonstrate the ability of certain iron PNP pincer complexes (Fe-1 as well as Fe-2a/b, Scheme 2) to promote the C–N bond cleavage in a number of different amides.
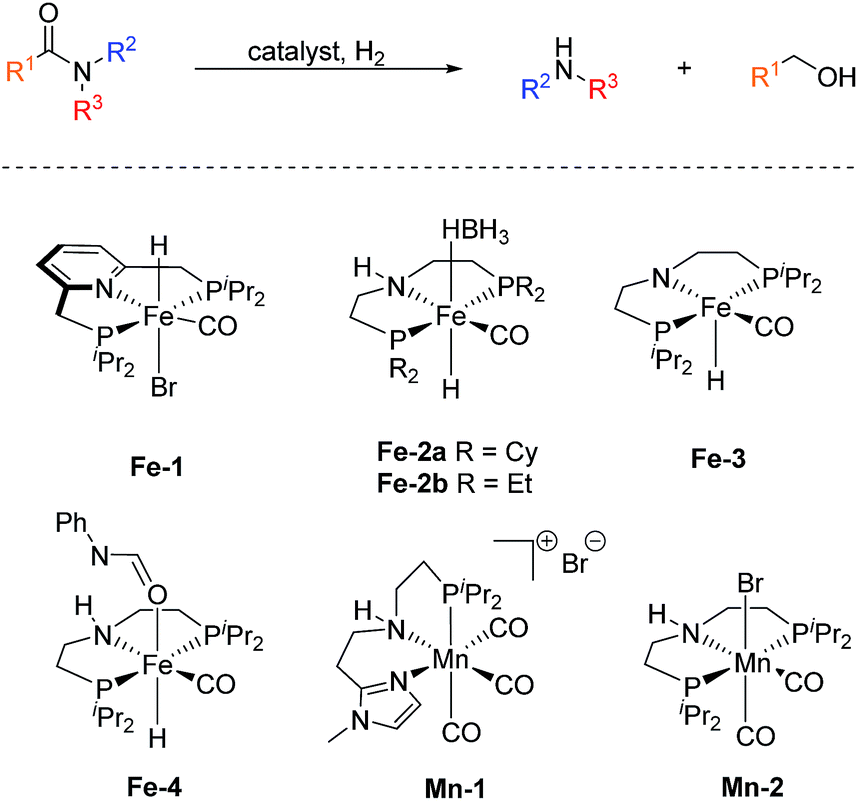 | ||
| Scheme 2 Base metal catalysts reported for the hydrogenolysis (C–N bond cleavage) of amides. | ||
More specifically, Milstein and co-workers reported, that Fe-1, after activation with KHMDS, induced the hydrogenolysis of activated aliphatic and aromatic 2,2,2-trifluoroacetamides. However, no reaction was observed, with more common substrates such as N-phenylacetamide and N-phenylbenzamide.11 The protocols described by Sanford (Fe-2a) and Langer (Fe-2b) showed more general substrate scopes and obtained notable conversions and yields also for unactivated amides.12,13
Additionally, Bernskoetter and co-workers showed that the pentavalent iron PNP-pincer complex Fe-3 is particularly active for the hydrogenolysis of a number of secondary formanilides and N-formylmorpholine (Scheme 2). The system stands out due to its extremely low catalyst loading (0.018–0.07 mol%) and notably operates under base-free conditions. Interestingly, the group of Bernskoetter demonstrated that an addition of 20 equivalents of formanilide resulted in a significantly improved activity of the system towards otherwise almost unreactive N-methylformanilide. Based on NMR experiments, the authors concluded that the catalyst adopts a different resting state in the presence of the additive (Fe-4, Scheme 2) and thus is less prone towards deactivating side reactions.14 The computational study of this reaction also suggested that the formanilide additive is involved in the C–N bond cleavage of the hemiaminal intermediate, which is the rate limiting step.15
Recently, our group reported the very first example of a manganese catalysed deaminative hydrogenation of amides under relatively mild conditions.16 After activation with exogenous base, the PNN pincer complex Mn-1 (Scheme 2) exhibits remarkable activity for the hydrogenation of a broad scope of secondary and tertiary amides to the corresponding alcohols and amines. Notably, also more challenging primary amides were successfully cleaved in modest yields, tough more forcing conditions were shown to be necessary. The generality of the system was finally highlighted by the cleavage of the amide bond in the herbicide diflufenican. To date, Mn-1 represents one of the most active and broadly applicable non-noble metal catalysts for amide hydrogenation. In a related study, Prakash and co-workers demonstrated that the manganese PNP pincer complex Mn-2 is a suitable catalyst for the hydrogenation of formamides. The reaction proceeds via cleavage of the C–N bond to produce methanol and the corresponding amine.17
In 2018, we published the synthesis of a number of structurally related molybdenum PNP pincer complexes. Among the described complexes, Mo-1a (Table 1) was shown to be active in the catalytic hydrogenation of different acetophenones and styrenes.18 Similar Mo-systems have also been used for the hydrogenation of CO2, imines and nitriles.19 Based on these reports and our previous work, we became interested in the behaviour of such base-metal catalysts for the reductive cleavage of amides. Herein, we demonstrate its suitability for the hydrogenolysis of N-methylated formanilides under relatively mild conditions. To the best of our knowledge, PNP pincer supported molybdenum complexes have not been described for such transformations. Interestingly, the optimal catalyst exhibits a high selectivity for formamides. This preference has been rationalized by means of DFT calculations, which suggest that the produced MeOH reacts with the catalyst and changes the mechanism and rate limiting step of the reaction. This result, which is not observed in related Fe-catalysts, indicates that the catalyst design strategy should be adapted to the nature of the metal centre.
|
|
||||
|---|---|---|---|---|
| Entrya,b | [Mo] | T [°C] | Convc. [%] | 2a [%] |
| a Standard reaction conditions: N-methylformanilide 1a (67.6 mg, 0.5 mmol), NaBHEt3 (50 μL, 0.05 mmol, 10 mol%), 2 mL toluene, 50 bar H2, 24 h. b Yield of 3 was not determined. c Conversion of 1a and yield of 2a were determined by GC using hexadecane as internal standard. d No catalyst was used. e Reaction was performed with 2.5 mol% of Mo catalyst. | ||||
| 1 | Mo-1a | 130 | >99 | 99 |
| 2 | Mo-1b | 130 | >99 | 99 |
| 3 | Mo-1c | 130 | >99 | 99 |
| 4 | Mo-2 | 130 | 10 | 9 |
| 5d | — | 130 | 10 | 8 |
| 6 | Mo-1a | 100 | >99 | 98 |
| 7 | Mo-1b | 100 | >99 | 99 |
| 8 | Mo-1c | 100 | 76 | 73 |
| 9 | Mo-1a | 80 | 89% | 86% |
| 10 | Mo-1b | 80 | 87% | 84% |
| 11e | Mo-1a | 80 | 49 | 47 |
| 12e | Mo-1b | 80 | 46 | 46 |
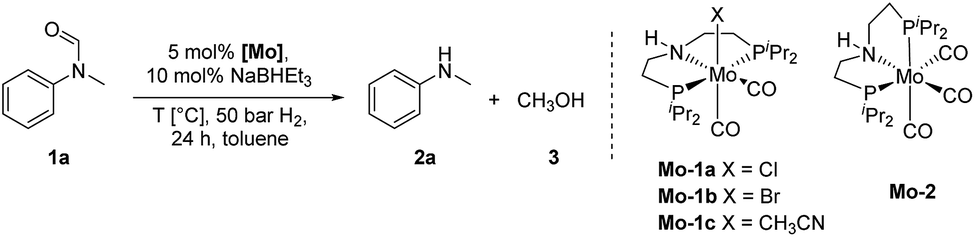
Results and discussion
Catalytic hydrogenation of amides using molybdenum pincer complexes
At the outset of our study, we explored molybdenum-based PNP pincer complexes Mo-1a–c and Mo-2 (Table 1), recently synthesised by our group, as potential catalysts for the hydrogenation of amides. Using N-methylformanilide 1a as benchmark substrate, preliminary experiments were conducted using 5 mol% of Mo catalyst in toluene at 50 bar H2 and 130 °C, in the presence of 10 mol% of NaBHEt3. The reaction proceeded smoothly for complexes Mo-1a–c to afford N-methylaniline 2a in quantitative yield along with methanol as the only by-product (Table 1, entries 1–3). However, complex Mo-2 failed to display any catalytic activity (Table 1, entry 4). Next, the activity of the complexes was tested at reduced temperatures (Table 1, entries 6–10). It was found, that complexes Mo-1a as well as Mo-1b were equally efficient, when the reaction was conducted at 100 °C. Catalyst Mo-1c, however, gave a somewhat lower conversion and yield. Further reduction of the reaction temperature to 80 °C resulted once again in similar conversions and yields for Mo-1a and Mo-1b, respectively. Based on these observations, the catalyst loading was reduced to 2.5 mol% under otherwise identical reaction conditions (Table 1, entries 11 and 12). It turned out, that changing this parameter also led to almost identical outcomes for both catalytic systems. Therefore we concluded that, under reaction conditions, Mo-1a and Mo-1b very likely form the same active species. On the basis of the obtained results and due to the more challenging synthesis of Mo-1b, we decided to focus on catalyst Mo-1a in the due course of the study.Selecting 80 °C reaction temperature and 5 mol% of Mo-1a (Table 1, entry 8) as the optimal setting for further optimization, we tested several different solvents. In contrast to previous work on manganese catalysed hydrogenolysis of amides, toluene was found to give the best results. Cyclohexane yielded slightly lower activities, while n-heptane as well as polar solvents, were shown to be significantly less suitable for the attempted transformation (Fig. 1).
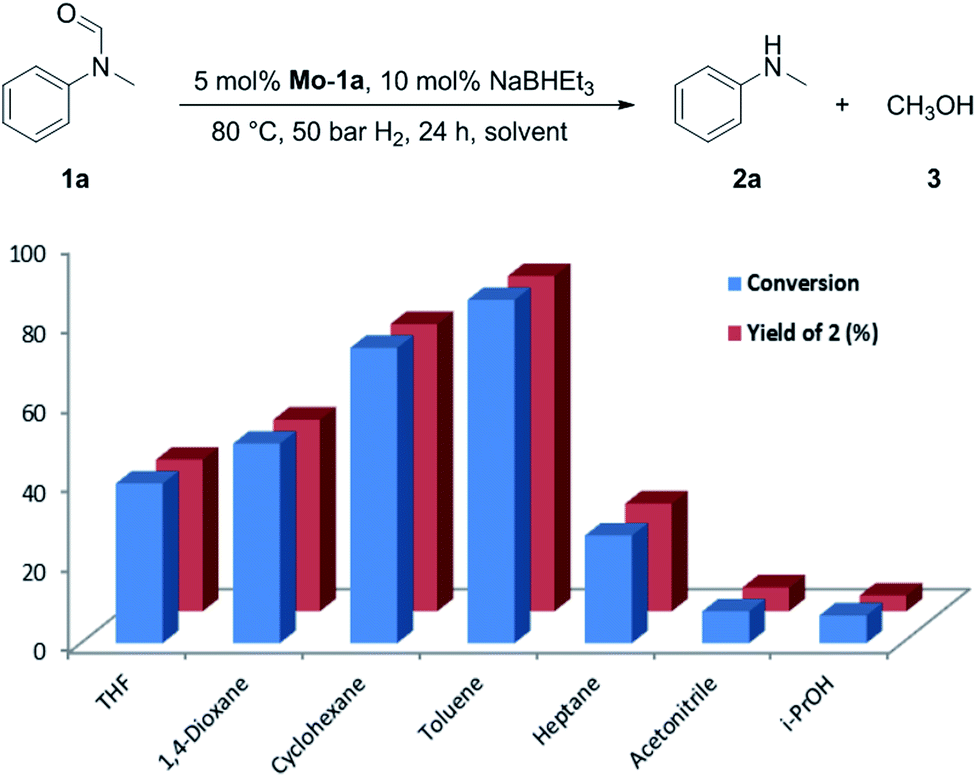 | ||
| Fig. 1 Study of the solvent effect in the hydrogenation of N-methylformanilide 1a to N-methylaniline 2a and methanol 3 catalysed by Mo-1a. | ||
Subsequently, we studied the influence of dihydrogen pressure, catalyst loading as well as the amount of additive used on the reaction outcome (Table 1, see ESI†). Lowering the pressure to 30 bar H2 resulted in a sharp drop in activity. However, no loss of reactivity was observed when the amount of NaBHEt3 was decreased to 5 mol%. A rise of the reaction temperature to 100 °C resulted in full conversion of the benchmark substrate to N-methylaniline in the presence of 5 mol% NaBHEt3 and Mo-1a, respectively. Further mitigation of the catalyst loading as well as the amount of NaBHEt3, however, had negative effects on the catalytic performance of the system.
Having optimised conditions in hand, we proceeded to the application of Mo-1a in the hydrogenation of a variety of different N-methylformanilides to the corresponding anilines and methanol (Table 2).
|
|
|||
|---|---|---|---|
| Entrya,b | Formamide | Convc. (%) | Yieldd of 2 (%) |
| a Standard reaction conditions: N-methylformanilide (0.5 mmol), Mo-1a (12.5 mg, 5 mol%), NaBHEt3 (50 μL, stock solution 0.5 M in THF, 5 mol%), 2 mL toluene, 50 bar H2, 24 h. b Yield of 3 was not determined. c Conversions of N-methylformanilides were determined by GC using hexadecane as internal standard. d Isolated yields. e Reaction was carried out at 130 °C. f Yields were determined by GC using hexadecane as internal standard. g Yield was determined based on the hydrochloride salt. | |||
| 1 |
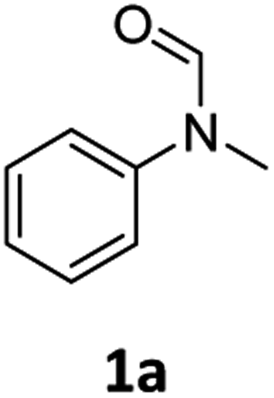
|
>99 | 94 |
| 2e |
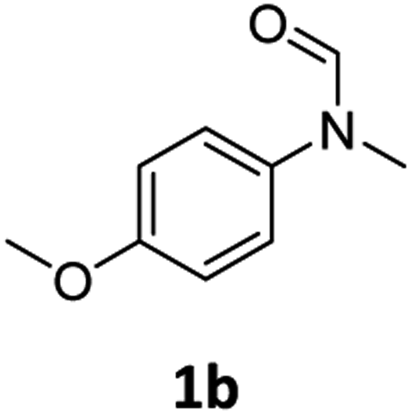
|
>99 | 96 |
| 3 |
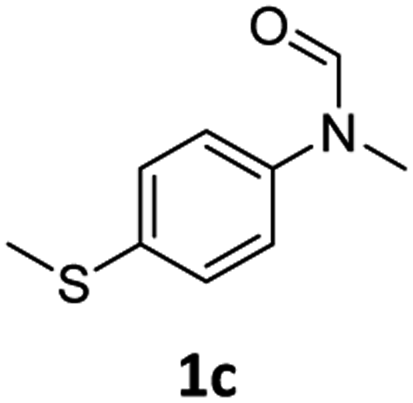
|
>99 | 95 |
| 4 |
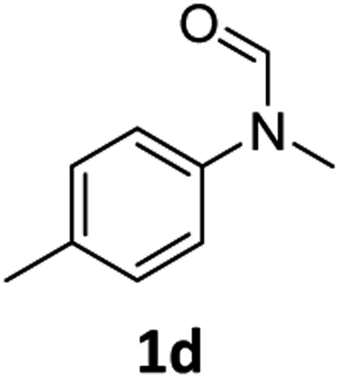
|
83 | 80 |
| 5 |
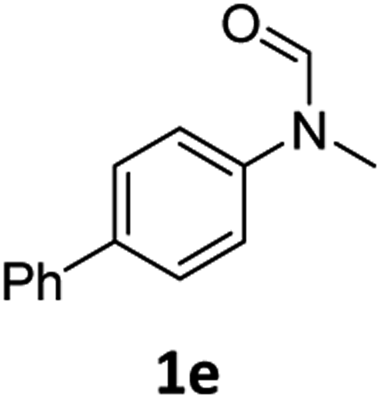
|
87 | 84 |
| 6e |
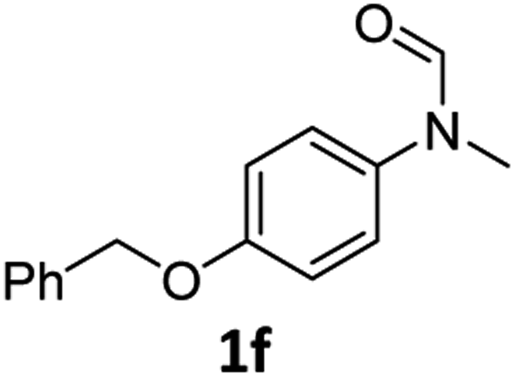
|
56 | 52 |
| 7eg |
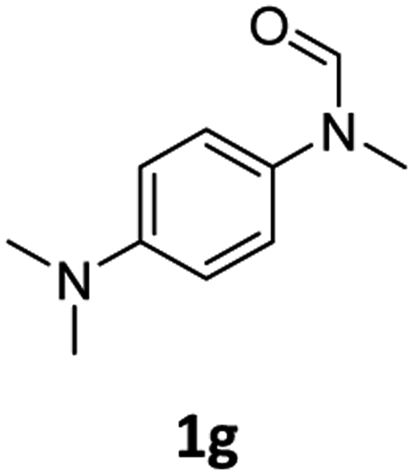
|
46 | 43 |
| 8 |
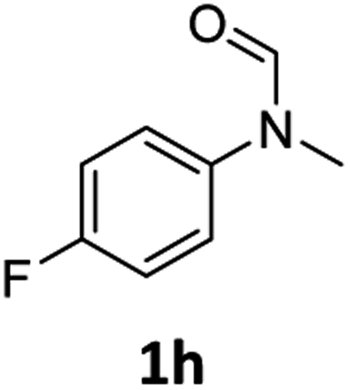
|
98 | 93 |
| 9 |
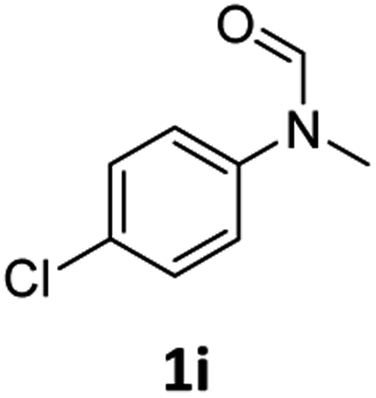
|
40 | 34f |
| 10 |
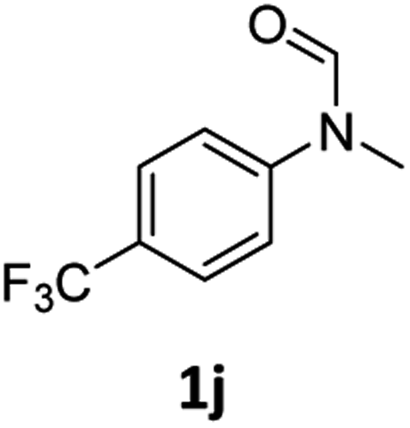
|
>99 | >99 |
| 11e |
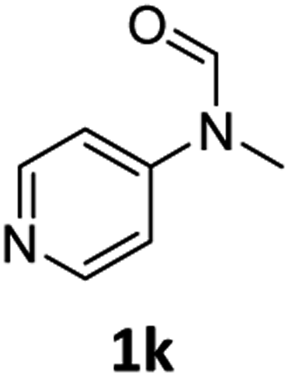
|
52 | 50 |
| 12 |
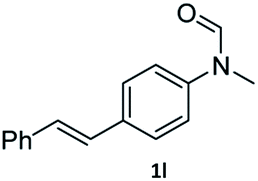
|
95 | 92 |
| 13 |
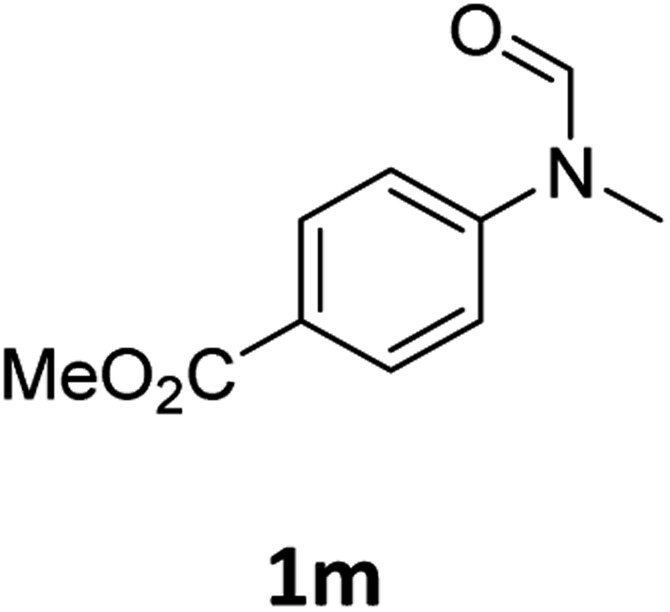
|
>99 | 97 |
| 14e |
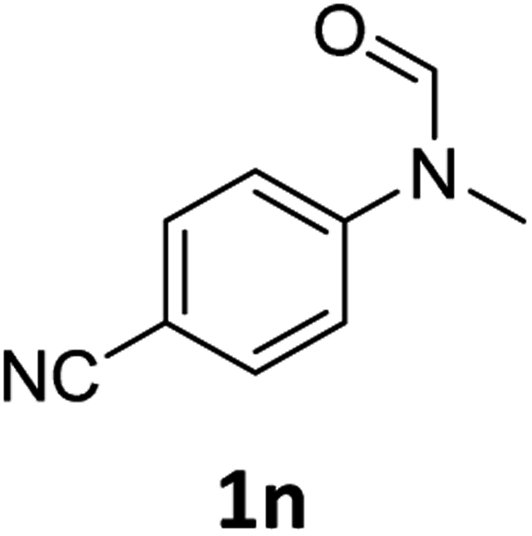
|
14 | 12f |
| 15e |
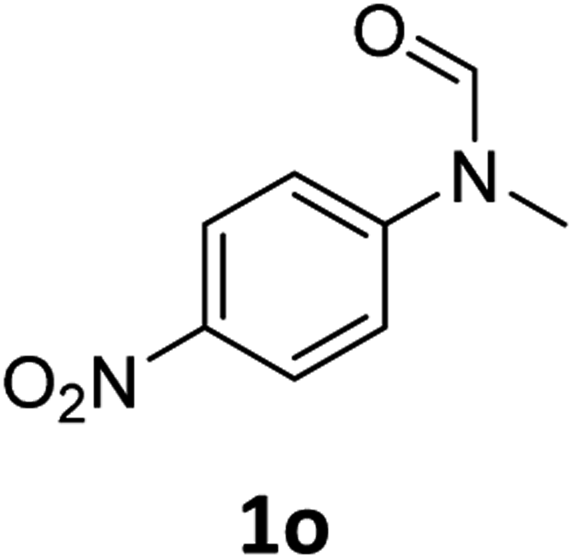
|
8 | 6f |
| 16 |
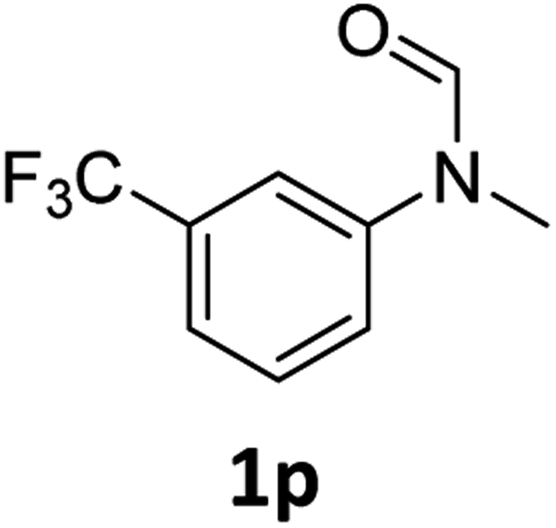
|
>99 | 97 |
| 17 |
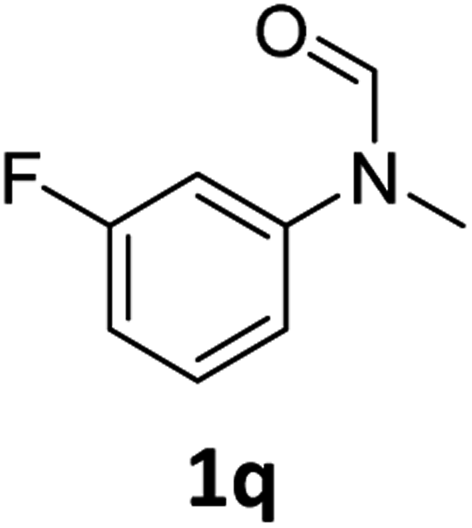
|
>99 | 98 |
| 18 |
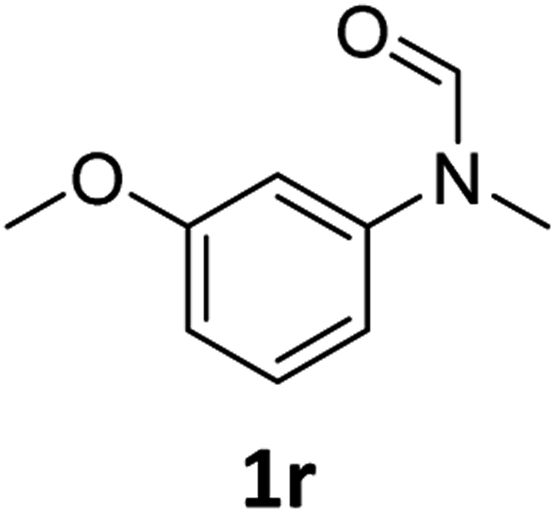
|
>99 | 93 |
| 19e |
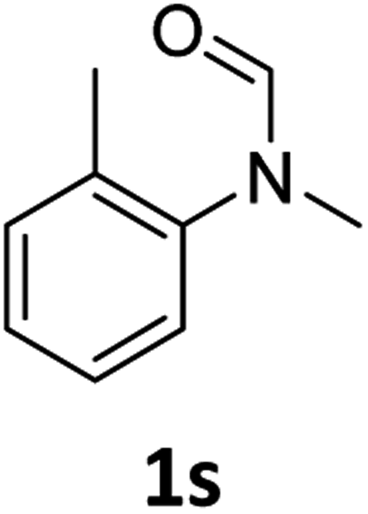
|
12 | 9f |
| 20e |
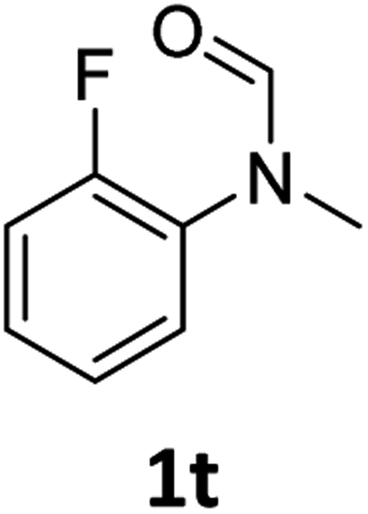
|
18 | 15f |
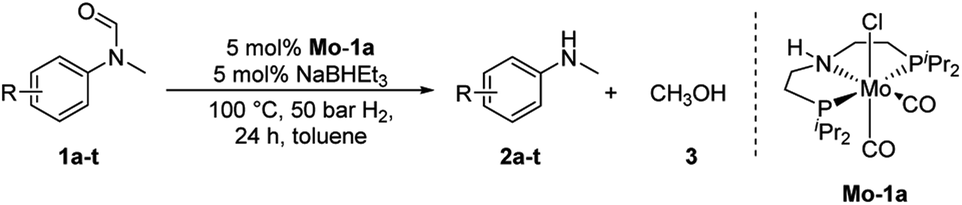
Most substrates were hydrogenated in good to excellent yields under optimised conditions at 100 °C and 50 bar H2 over 24 h, using toluene as solvent. In general, meta- and para-substitution were well tolerated, while substituents in ortho-position (Table 2, entries 19 and 20) appeared to be troublesome, probably due to steric hindrance. Amides containing electron donating groups were less reactive under standard conditions as compared to the benchmark substrate. In some cases higher reaction temperatures were required, in order to achieve good conversions (Table 2, entries 2, 6, 7). Notably, the thiomethyl substituted derivative (Table 2, entry 3) was fully hydrogenated and no catalyst poisoning effect was observed. Moreover, the system tolerated fluoro-substituents (Table 2, entries 8, 17, 20) and no dehalogenation products were detected. Interestingly, the system showed a good functional group tolerance towards substrates containing other reducible moieties such as benzyl ethers, CC double bonds and esters (Table 2, entries 6, 12, 13). Noteworthy, no double bond isomerisation occurred during the reduction of a stilbene derivative (Table 2, entry 12). Additionally, pyridines, nitriles and nitro arenes remained unaffected under our reaction conditions; however, only poor to modest conversions were observed when the reaction was carried out at 130 °C (Table 2, entries 11, 14, 15). Presumably, this effect originates from substrate coordination to the metal centre and subsequent catalyst deactivation. The system turned out to be sensitive towards halides other than fluorine. Hence, during one of the hydrogenations, small amounts of the dehalogenation product were detected (Table 2, entry 9).
Subsequently, we investigated the more general applicability of our PNP pincer complex Mo-1a in the hydrogenation of other amides. Initial experiments focussed on the role of the nitrogen substitution on the reaction outcome. For this purpose, a series of different secondary and tertiary formanilides were subjected to our protocol (Scheme 3). The presence of an NH moiety turned out to be detrimental, as was observed for the parental formanilide (4a). This is in sharp contrast with the results obtained with Fe pincer complexes, in which formanilide derivatives give the highest conversion.14 In order to further validate this, 2,2,2-trifluoroacetanilide (6a) and simple benzanilide (7a) were employed and results comparable to formanilide (4a) were obtained. Likewise, only low conversions and yields were obtained in the case of N-iPr- (4b) and N-allylformanilide (4c), respectively. Surprisingly, when N-allylformanilide was tested as substrate, the formation of N-allylaniline was only observed in traces. The main product was identified to be aniline, thus hinting at a deallylation pathway that additionally takes place to the envisaged hydrogenolysis. In contrast, N,N-diphenylformanilide (4d) was reduced smoothly and N,N-diphenylamine was isolated in excellent yield. Next, the hydrogenation of N-methylacetanilide (5) and the more activated 2,2,2-N-methyl-trifluoroacetanilide (6b), respectively, were attempted. In either case, only poor conversions were determined demonstrating the high preference of this complex for specific formanilides. This was further supported by the low reactivity of N-methylbenzanilide (7b) and some aliphatic formamides (see Table 2, ESI†).
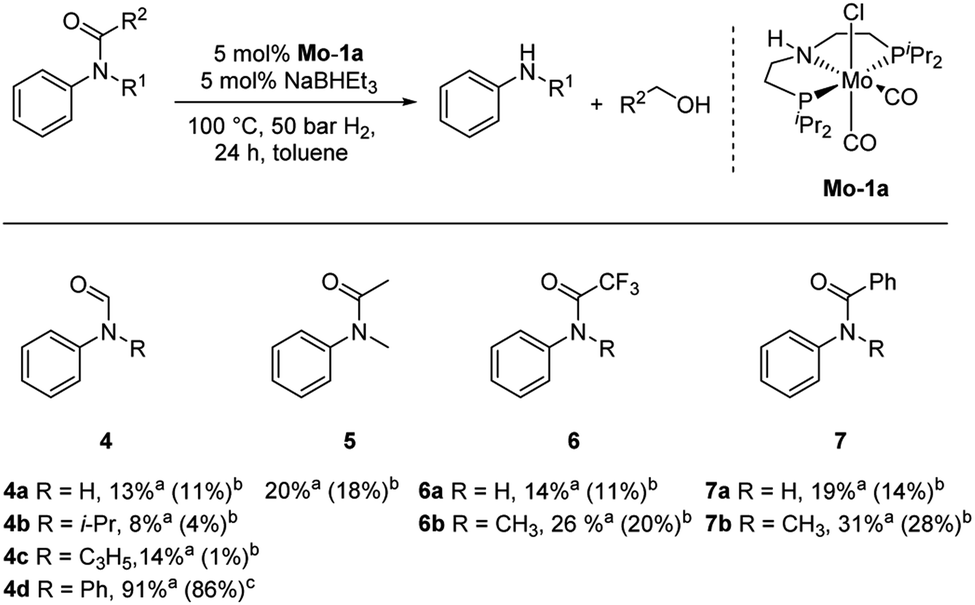 | ||
| Scheme 3 Hydrogenation of different amides (4–7) to the corresponding amines and alcohols catalysed by Mo-1a. aConversions of amides were determined by GC using hexadecane as internal standard. bYields were determined by GC using hexadecane as internal standard and refer to anilines, yields of alcohols were not determined. cIsolated yields of anilines. | ||
Based on these observations, we were curious to demonstrate selective formamide reduction in the presence of other amide moieties. In a proof of concept experiment, the hydrogenation of the benchmark amide in the presence of benzamide 7a was conducted (Scheme 4, eqn (a)). It could be shown that Mo-1a was capable to cleave N-methylformanilide (1a) with extremely high preference. Notably, the reaction still proceeded with 80% conversion with respect to N-methylformanilide (1a). To further highlight the scope of our system, we designed model substrate 9 combining two amide functionalities in one structure. After 24 h reaction, the intended hydrogenolysis of the formamide moiety in 9 had occurred smoothly and the target molecule 10 was isolated in a very high yield (92%). Notably, no cleavage of the benzamide was observed.
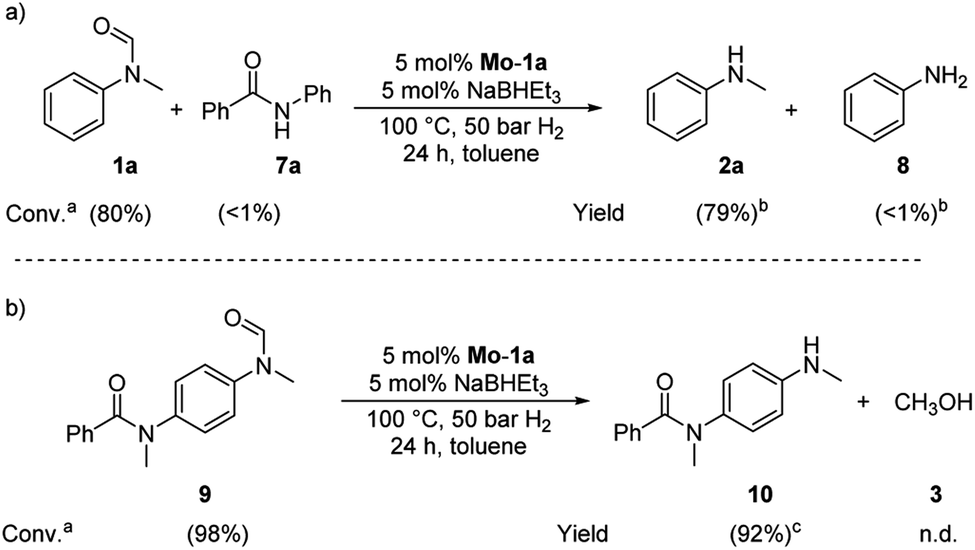 | ||
| Scheme 4 Selective hydrogenations of (a) N-methylformanilide 1a in the presence of benzamide 7a and (b) N-methyl-N-(4-(N-methylformamide)phenyl)benzamide 9. Standard conditions: substrate(s) 0.5 mmol (each), Mo-1a (12.5 mg, 0.025 mmol, 5 mol%), NaBHEt3 (50 μL, 0.5 M stock solution in THF, 0.025 mmol, 5 mol%), toluene (2 mL), 50 bar H2, 100 °C, 24 h. aConversions determined by GC using hexadecane as internal standard. bYields determined by GC using hexadecane as internal standard. cIsolated yield. | ||
We believe these results could pave the way towards new and selective deprotection strategies in organic synthesis mediated by this base metal PNP pincer complex.
Reaction mechanism
In order to understand the general reactivity of Mo-1a and its performance with different amides, DFT calculations and supporting experiments were conducted. Scheme 5 shows the experiments performed to determine the active catalyst species. Treatment of Mo-1a with NaBHEt3 resulted in rapid hydrogen evolution. The nature of the gas was determined in a scale up experiment (100 μmol of Mo-1a) using GC-analysis. This observation prompted us to assume that the obtained reaction product was likely to be a pincer amido species such as Mo-3, in which Mo(I) has been reduced to Mo(0). This conclusion was further supported by HR-ESI mass spectrometry of the corresponding reaction mixture. When the distinct reactivity of the catalyst towards formanilide was studied, we isolated Mo-4 in form of colorless needles from the reaction mixture (Fig. 2; for detailed experimental procedure see ESI†).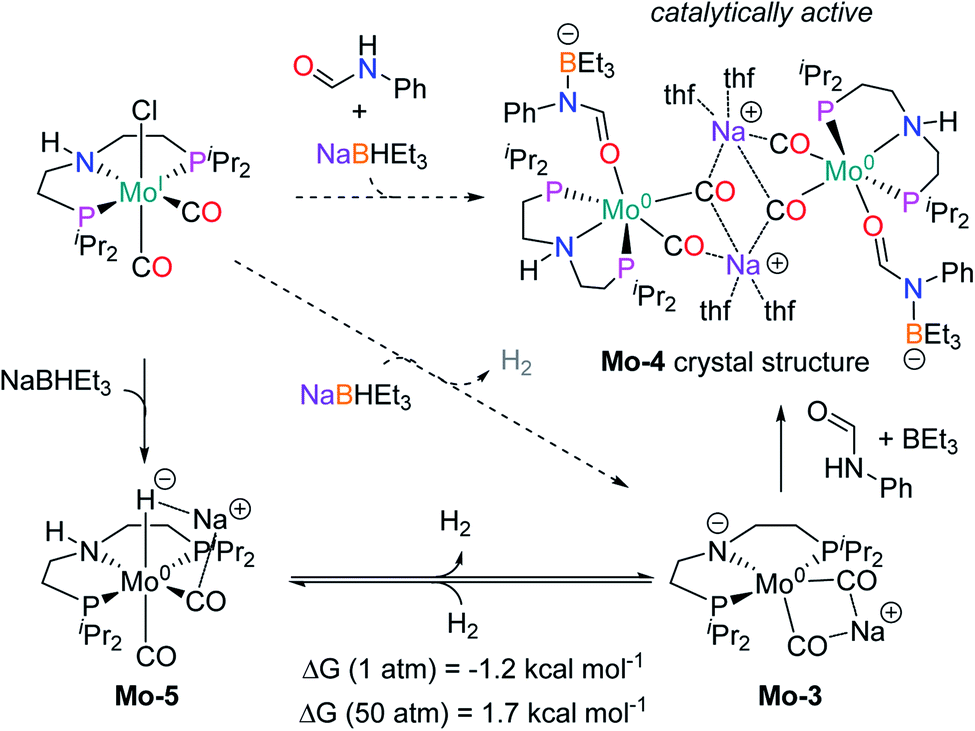 | ||
| Scheme 5 Reactions performed to get insight on the active catalytic species (in dashed arrows) with the experimental observed products (H2 and the crystal structure of Mo-4, in color) and the intermediates proposed (Mo-3 and Mo-5). Gibbs energies calculated for the de-hydrogenation of Mo-5 at different pressure. | ||
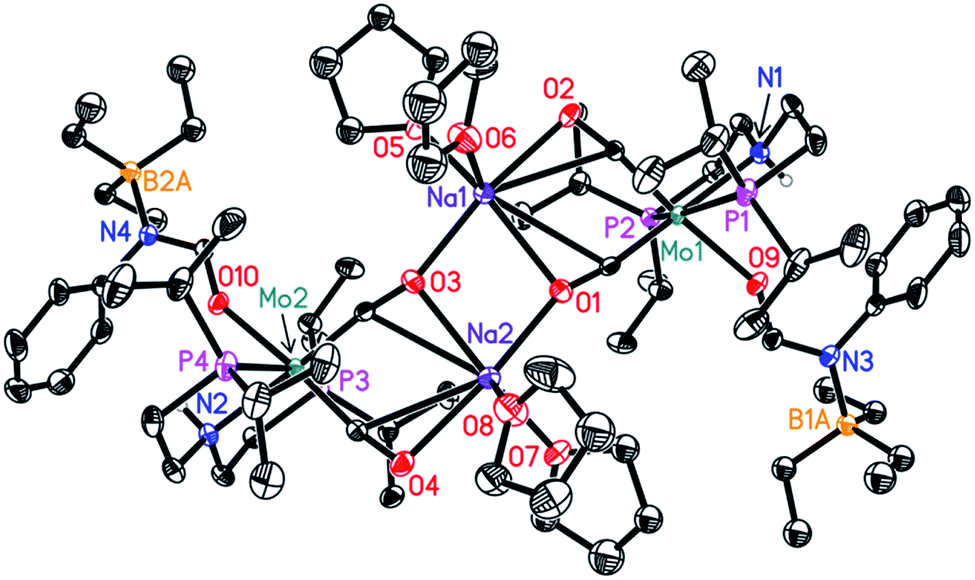 | ||
| Fig. 2 Molecular structure of Mo-4 in the crystal (see Scheme 5 for a graphical representation). Displacement ellipsoids correspond to 30% probability. Hydrogen atoms except the N-bound are omitted for clarity. | ||
Notably, the crystal structure of Mo-4 (Fig. 2 and Scheme 5) features two anionic Mo(0) complexes neutralized by two Na+ cations interacting with the CO ligands. In order to investigate, whether Mo-4 is involved in the catalytic cycle, the reduction of N-methylformanilide was carried out using 2.5 mol% of Mo-4 under conditions optimized for Mo-1a. In fact, we observed full conversion of the substrate and isolated N-methylaniline in 92% yield. Thus, we conclude, that the catalytically active species contains a Mo(0) center. This is also consistent with the EPR-silent nature of the product formed in the activation of Mo-1a by NaBHEt3.
The observed activity of Mo-4 suggests that the Mo(0)-complexes Mo-3 and Mo-5, shown in Scheme 5, are presumably the main catalytic intermediates. Similar species have been proposed for the isoelectronic Fe(II)-complexes Fe-2, Fe-3 and the Mn(I)-complex Mn-2 (Scheme 2).20
Based on these results, DFT calculations, with the M06 functional, including toluene solvation with the SMD model, were used to get further insights into the reaction mechanism (see computational details and ESI for details†). The hydrogenation of Mo-3 to yield Mo-5, was found to be almost isoenergetic, with a small preference for Mo-3 at 1 bar and Mo-5 at 50 bar (Scheme 5). These energies agree with the bubbling of H2 observed experimentally during the catalyst activation reaction.
As represented in Scheme 1, amide hydrogenolysis is proposed to consist in three steps: amide CO reduction, C–N bond protonolysis of the formed hemiaminal, and aldehyde CO reduction. These steps were computed for N-methylformanilide and the energy profiles for the preferred pathways are given in Fig. 3 and 5, and the ESI.†
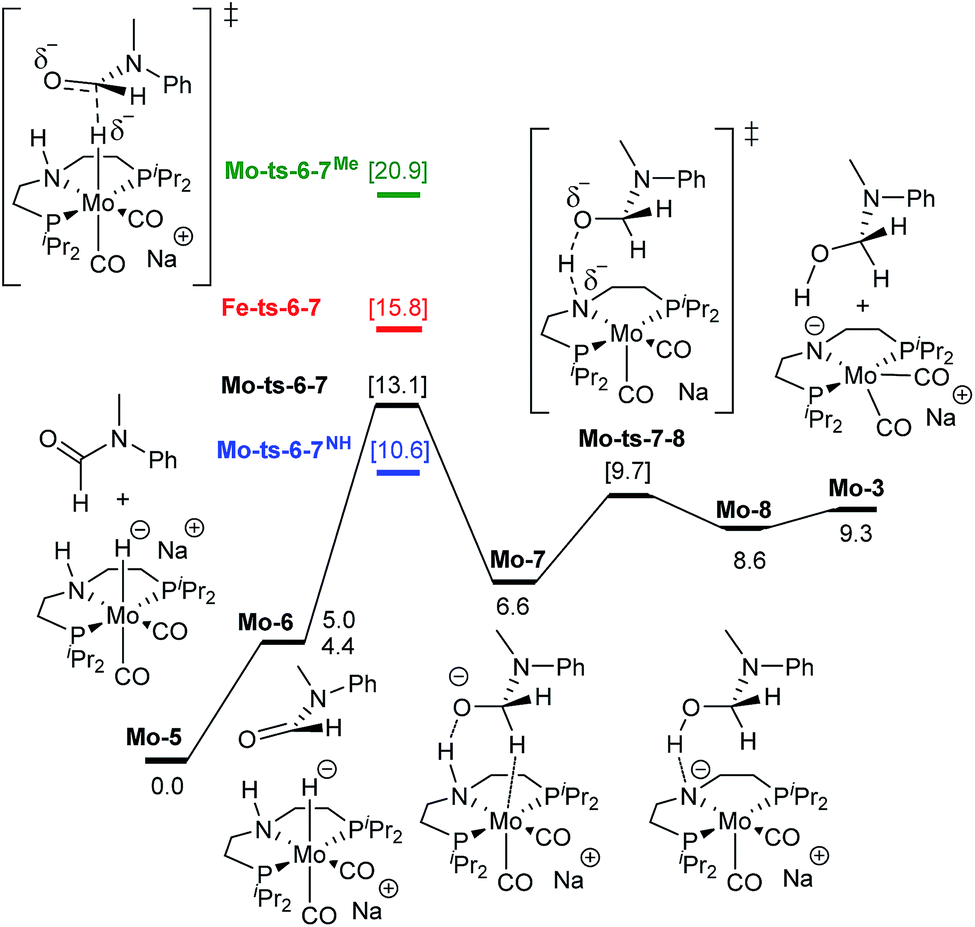 | ||
| Fig. 3 Reaction pathway for the hemiaminal formation from the N-methyl formanilide with Mo-5. Gibbs energies in toluene (SMD) at 50 atm and 373 K are given in kcal mol−1. In blue and green, energies for the hydride transfer using formanilide and N-methylacetanilide, respectively. In red, energy for the hydride transfer using the reported Fe-3 complex at 30 atm (Scheme 2).15 | ||
The mechanism for the amide CO hydrogenation by Mo-5 consists of the hydride transfer from Mo to the amide carbonyl group (Mo-ts-6-7), followed by proton transfer from the ligand nitrogen to the amide oxygen (Mo-ts-7-8). This pathway was computed for formanilide (Mo-ts-6-7NH in Fig. 3) and N-methylformanilide. With both substrates, the hydride transfer has the highest energy barrier (10.6 kcal mol−1 with formanilide and 13.1 kcal mol−1 with N-methylformanilide). Interestingly, these energies are lower than those reported by us for the analogous Fe catalyst with formanilide (15.8 kcal mol−1, Fe-ts-6-7 in Fig. 3).15
The mechanism for the C–N bond cleavage from the formed hemiaminal (Scheme 1) was also investigated. In the case of Fe-3, this step was reported to proceed via the transition state Fe-ts-CH–NH (Fig. 4).15 With Mo and N-methylformanilide, the same pathway involves a Gibbs energy barrier of 22.9 kcal mol−1 (Mo-ts-CH–NMe). An increase of less than 1 kcal mol−1 is observed by changing the substrate to N-methylacetanilide (Mo-ts-CMe–NMe).
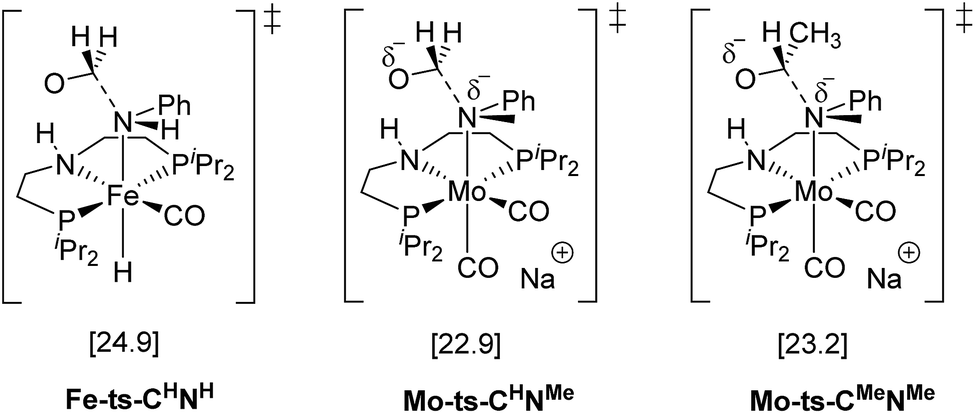 | ||
| Fig. 4 TSs for the C–N bond cleavage step via the mechanism previously reported for Fe-3.15 | ||
The similar energy barriers obtained with these substrates did not account for the large differences in yield observed experimentally (99% Conv. in N-methylformanilide vs. 20% Conv. in N-methylacetanilide). In addition, the lower energy barriers obtained with Mo compared to Fe are inconsistent with the higher H2 pressure and time required to accomplish amide hydrogenation with Mo-1a compared to Fe-3.14
These discrepancies were explained by considering the reaction of Mo-3 with methanol leading to the Mo-methoxy intermediate Mo-9a (Fig. 5). This reaction, which involves the deprotonation of MeOH by the amido ligand (Mo-ts-3-9a), has a low energy barrier (ΔG‡ = 2.8 kcal mol−1) and is highly exergonic (ΔG = −11.4 kcal mol−1). The formation of related M-methoxy species have been observed for similar Fe, Ru, Os and Mn PNP-pincer complexes.20c,21,22 This species can promote the protonolysis of the C–N bond by assisting the OH-deprotonation and N-protonation of the hemiaminal intermediate (Mo-ts-11-9a). The highest energy of this process is 10.8 kcal mol−1, which corresponds to the zwitterion hemiaminal intermediate interacting with the methoxide–Mo complex (Mo-11). This energy is lower than the energy barrier for the hydride transfer (13.1 kcal mol−1), indicating that the C–N bond cleavage is not the rate limiting step once MeOH is formed (note: for a comparison of this mechanism with Mo and Fe-systems see ESI†).
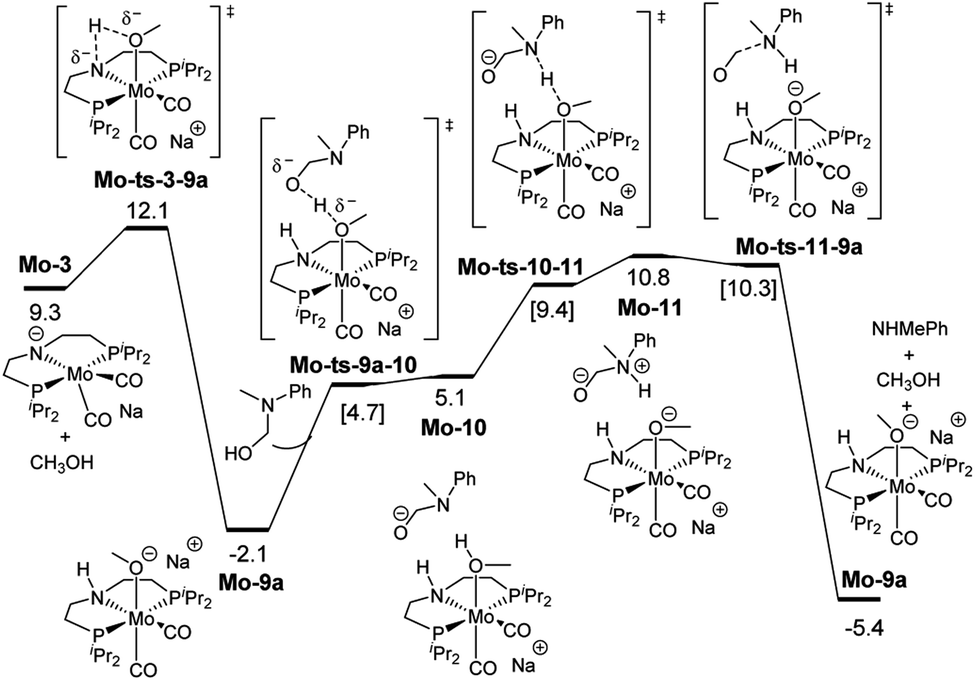 | ||
| Fig. 5 Reaction pathway of the MeOH assisted hemiaminal proton transfer and posterior C–N bond cleavage. Gibbs energies in toluene (SMD) at 50 atm and 373 K are given in kcal mol−1. | ||
The reaction of Mo-5 with MeOH yields hydrogen and is exergonic (ΔG = −9.7 kcal mol−1, Scheme 6). The methoxy intermediate Mo-9a is thus the resting state of the catalyst.
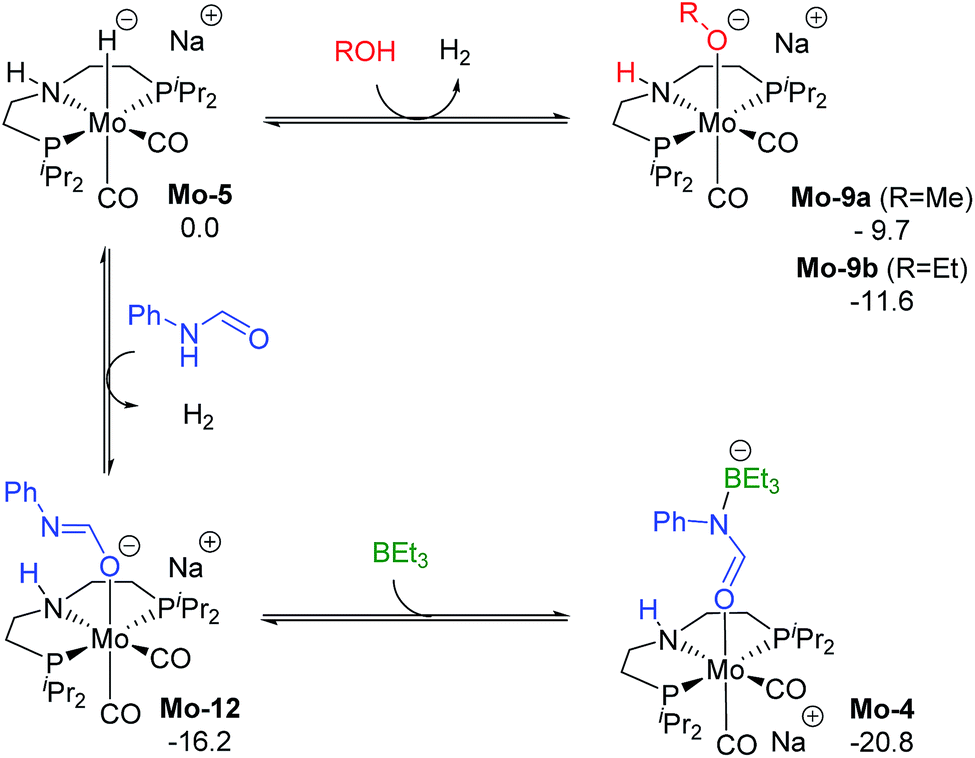 | ||
| Scheme 6 Calculated Gibbs energies (kcal mol−1) for the substitution of H2 in Mo-5 by methanol, ethanol, formanilide and BEt3 yielding Mo-9a, b, Mo-10 and Mo-4, respectively. | ||
Formanilide, and other secondary amides, can also displace H2 from the catalyst (Mo-12 in Scheme 6). This reaction is even more exergonic (ΔG = −16.2 kcal mol−1) than with MeOH increasing the global energy barrier for the hydride transfer from 10.6 to 26.8 kcal mol−1 with formanilide. This energy may increase to 31.4 kcal mol−1 by reaction with BEt3 (Mo-4). In contrast, with N-methylformanilide, the only penalty to pay is the addition of MeOH. Therefore, the energy barrier for the hydride transfer increases from 13.1 to 22.9 kcal mol−1, which is lower than the barrier for formanilide, consistent with the larger conversion obtained with N-methylformanilide. In the case of N-methylacetanilide, the addition of ethanol instead of methanol is expected. The higher stability of the ethoxide complex Mo-9b compared to Mo-9a by ca. 2 kcal mol−1 (Scheme 6), together with the higher energy barrier for the hydride transfer with this substrate (ΔG = 20.9 kcal mol−1, Fig. 3), is consistent with the low yields obtained experimentally with N-methylacetanilide.
The mechanism of catalyst recovery by addition of H2 to the methoxide complex Mo-9a is shown in Fig. S3.† In this pathway, methanol assists the activation of the Mo–H2 complex (Mo-14) by acting as a proton-shuttle with a global energy barrier of 23.0 kcal mol−1. Similar mechanisms have been proposed with Ru–N and Fe–N complexes (see ESI†).21b,23
The results from the computational study can be summarized in the catalytic cycle represented in Fig. 6. In the absence of alcohol, the Mo-catalyst is involved in the hemiaminal C–N bond cleavage after the amide CO reduction (blue cycle). This reaction yields amine and formaldehyde, which is reduced to alcohol by the catalyst Mo-5 in a subsequent reaction (in red). In the presence of alcohol, a Mo-alkoxo intermediate is formed, Mo-9a. This species, which becomes the catalyst resting state, is involved in the hemiaminal C–N bond cleavage. Finally, the catalyst recovery takes place by the displacement of alcohol by H2. The nature of the catalyst resting state may change with secondary amides, which reacts with the catalyst forming an adduct (Mo-4, in green) that hampers the reaction.
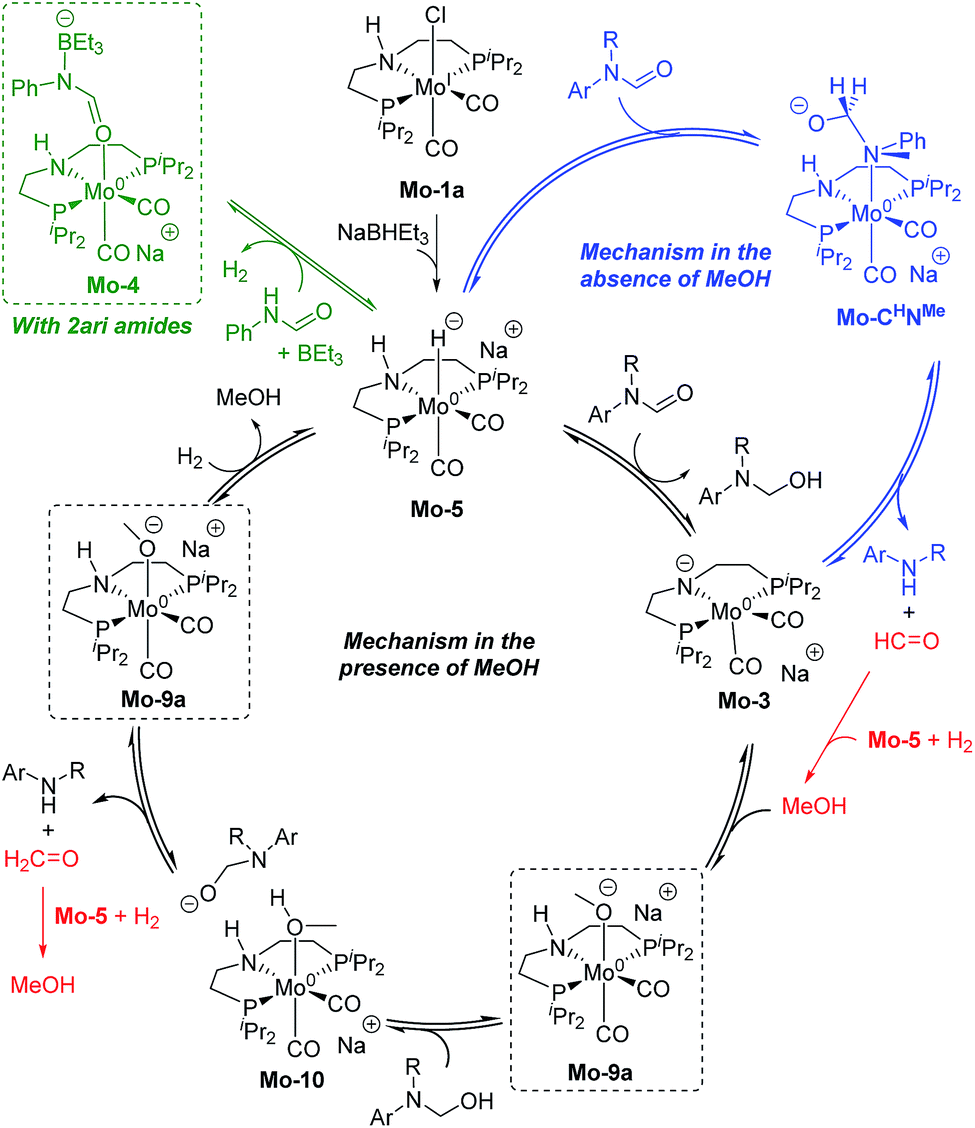 | ||
| Fig. 6 General mechanism for the amide hydrogenation in the absence (in blue) and presence (in black) of methanol with the formaldehyde hydrogenation in red. Dashed squares indicate the catalyst resting state in the presence of MeOH and 2ari amides (in green). | ||
In order to validate this mechanism and the nature of Mo(0) active species, the role of the counter-cation in this reaction was explored computational and experimentally by using LiHBEt3, NaHBEt3, and KHBEt3. Carrying out the benchmark reaction at 80 °C, 5 mol% of the alkali metal hydrides were added to activate Mo-1a. It could be shown, that for NaBHEt3 and KBHEt3 similar conversions of N-methylformanilide (1a) (76% and 77%, respectively) and yields of 2a (75% and 73%, respectively) were obtained. However, when LiBHEt3 was used, only 10% conversion of 1a and 9% yield of N-methylaniline 2a was obtained. These results were in agreement with the trends on the energy barriers obtained for the amide CO reduction step, which are 22.9, 23.0 and 28.8 kcal mol−1 with Na+, K+ and Li+, respectively, taking Mo-9a as energy reference. The stronger electrostatic interaction of Li+ with the methoxide intermediate (Mo-9aLi), accounts for the highest energy barrier predicted for this system (see ESI†).
Next, the role of the alcohol was explored by adding different amounts of ethanol to the benchmark system. In the presence of 50 mol% of EtOH, 96% conversion of N-methylformanilide (1a) and 93% product yield were obtained. However, the addition of 200 mol% resulted in a sharp decrease in conversion and yield (35% conversion, 32% yield). Thus, it was concluded that ethanol has a detrimental effect on the performance of the catalytic system. Notably, these trends were reproduced with a microkinetic model based on the general mechanism represented in Fig. 6 (in Fig. 7). This model predicted 100% conversion after 24 h of reaction for both 0% and 50% concentrations of ethanol. In contrast, and in line with the experiments, the same model predicted a significant decrease of conversion to 64% with an ethanol concentration of 200% (see ESI for further details†).
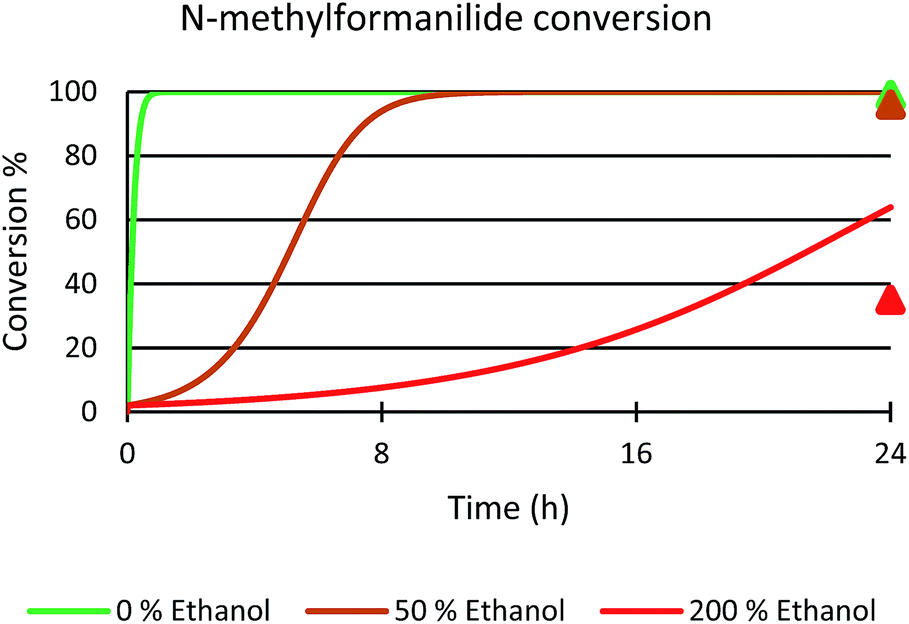 | ||
| Fig. 7 Microkinetic simulation of N-methylformanilide 1a conversion with 0% (green), 50% (brown) and 200% (red) ethanol in solution. The initial concentration of reactants were the same as those used in the experiments; i.e. 0.25 M N-methylformanilide 1a, 0.207 M of dihydrogen and 12.5 mM of Mo-5. Experimental values at 24 hours represented with triangles. | ||
Conclusions
Well-defined molybdenum–PNP pincer complexes have been used for the first time in the hydrogenation of a range of amides to the corresponding alcohols and amines. N-Alkylated and N-arylated formamides can be hydrogenated to the corresponding products in good to high yields. Applying complex Mo-1a high selectivity for the hydrogenation of formamides was observed in the presence of other reducible groups. These results pave the way for potential applications of this type of complexes in synthetic methodologies.The DFT study shows that the active Mo(0) species (Mo-5) reduces the CO group of the amide through low-energy barriers, compared to Fe-based systems. However, the alcohol product and secondary amides react with the catalyst forming stable adducts encumbering catalyst recovery and increasing the overall barrier for the reduction of the CO group. These results suggest that further catalyst design should focus on preventing the formation of these adducts, while keeping the high hydricity of the complex.
Experimental details
General experimental information
All hydrogenation reactions were set up under Ar in a 300 mL autoclave (PARR Instrument Company). In order to avoid unspecific reductions, all catalytic experiments were carried out in 4 mL glass vials, which were set up in an alloy plate and placed inside the autoclave.In a glove box, a 4 mL glass vial containing a stirring bar was charged with complex Mo-1a (12.5 mg; 5 mol%). Toluene (2 mL) was added and the corresponding brown suspension was treated with NaBHEt3 (0.5 M in THF; 50 μL; 10 mol%). The reaction mixture was stirred for 10 minutes and the corresponding substrate was subsequently added. Afterwards, the vial was capped and transferred into an autoclave. Once sealed, the autoclave was purged three times with 10 bar of hydrogen, then pressurized to the desired hydrogen pressure (50 bar), and placed into an aluminum block that was preheated to the desired temperature (100 °C). After 24 h, the autoclave was cooled in an ice bath and the remaining gas was released carefully. The solution was subsequently diluted with ethyl acetate and filtered through a small pad of Celite (1 cm in a Pasteur pipette). The Celite was washed with methanol (2 mL) and the combined filtrates were subsequently evaporated to dryness. The remaining residue was purified by column chromatography (SiO2, heptane/EtOAc, gradient 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 → 0:100). In the case of substrate 7, the purified product was dissolved in 5 mL of Et2O and subsequently treated with 1 mL of HCl (2 M in Et2O). The reddish precipitate was filtered off, washed three times with 5 mL of Et2O and finally dried in vacuo. For the characterization of the products of the catalysis, see ESI.†
:0 → 0:100). In the case of substrate 7, the purified product was dissolved in 5 mL of Et2O and subsequently treated with 1 mL of HCl (2 M in Et2O). The reddish precipitate was filtered off, washed three times with 5 mL of Et2O and finally dried in vacuo. For the characterization of the products of the catalysis, see ESI.†
Computational details
DFT calculations were carried out with Gaussian 0924 with the M0625 functional and the double-z LANL2DZ (on Mo, including relativistic effects)26 and 6-31+G** (on all other elements)27 basis sets. Calculations were done using the full system. The location of the Na+ cation was evaluated in some of the intermediates, and the preferred position is represented in figures and schemes of the manuscript (see ESI†). The geometry optimization and energies of the possible spin states of Mo-1a and Mo-4 were consistent with a doublet and singlet ground state, respectively (see ESI†). Vibrational frequencies were computed at the same level of theory to obtain the thermochemistry corrections (zero-point, thermal and entropy energies) at the experimental p = 50 atm and T = 373.15 K. The energy of the optimized geometries was refined by single point calculations with triple-z quality basis sets, including the LANL2TZ26 on Mo and the 6-311+G** on all other elements.28 The energies reported in the text were obtained by adding the thermochemistry corrections to the refined potential energies. The solvation effects of toluene were included in both the geometry optimizations and energy refinements using the continuum SMD model.29 The ultrafine (99590) grid was used in all calculations for higher numerical accuracy. A repository containing all input and output files is available on-line from ioChem BD at https://iochem-bd.bsc.es/browse/handle/100/193698.30 Microkinetic models were simulated with the COPASI software31 using the LSODA algorithm. See ESI for further details.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. Leischner, K. Junge and M. Beller thank the analytical department (S. Schareina, S. Buchholz, A. Lehmann) of the Leibniz-Institute for Catalysis, Rostock. Ll. A. and A. N. thank the support from the Research Council of Norway (FINATEK Grant No. 250044 and Center of Excellence Grant No 262695), the Norwegian Metacenter for Computational Science (NOTUR, nn4654k) and the ‘Nordic Consortium for CO2 Conversion’ (NordForsk project no 85378, http://site.uit.no/nordco2).Notes and references
- (a) P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Hönke, Ullmann's Encyclopedia of Industrial Chemistry, Germany, 2000 Search PubMed; (b) P. G. Jessop, The Handbook of Homogeneous Hydrogenation, Germany, 2006 Search PubMed.
- For selected examples of carboxylic acid derivatives hydrogenation, see: (a) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. J. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed; (b) T. vom Stein, M. Meureusch, D. Limper, M. Schmitz, M. Holscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed; (c) D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed; (d) X. J. Cui, Y. H. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed; (e) T. J. Korstanje, J. I. van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–301 CrossRef CAS PubMed; (f) M. Naruto and S. Saito, Nat. Commun., 2015, 6, 8140 CrossRef PubMed; (g) J. R. Cabrero-Antonino, I. Sorribes, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 387–391 CrossRef CAS PubMed.
- (a) B. Cornils and W. A. Herrmann, Applied Homogenous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Two Volumes, Germany, 1996 CrossRef; (b) D. L. Dodds and D. L. Cole-Hamilton, Sustainable Catalysis, United States of America, 2013 Search PubMed.
- D. Wie, C. Netkaew and C. Darcel, Eur. J. Inorg. Chem., 2019, 2471–2487 Search PubMed.
- M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 30, 4344–4362 CrossRef.
- G. A. Filonenko, R. van Putten, E. J. M. Jensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- For selected examples see: (a) R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120–2124 CrossRef CAS PubMed; (b) N. Gorgas, B. Stöger, L. F. Veiros, E. Pittenauer, G. Allmaier and K. Kirchner, Organometallics, 2014, 33, 6905–6914 CrossRef CAS PubMed; (c) T. Zell, Y. Ben-David and D. Milstein, Catal. Sci. Technol., 2015, 5, 822–826 RSC; (d) N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2016, 6, 2664–2672 CrossRef CAS PubMed; (e) S. Elangovan, B. Wendt, C. Topf, S. Bachmann, M. Scalone, A. Spannenberg, H. Jiao, W. Baumann, K. Junge and M. Beller, Adv. Synth. Catal., 2016, 358, 820–825 CrossRef CAS; (f) S. Lange, S. Elangovan, C. Cordes, A. Spannenberg, H. Jioa, H. Junge, S. Bachmann, M. Scalone, C. Topf, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 4768–4772 RSC; (g) S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- (a) P. M. Rylander, Hydrogenation Methods, Academic Press, London, 1985 Search PubMed; (b) J. Hartwig, Organotransition Metal Chemistry, University Science Books, 2010 Search PubMed.
- (a) D. Cantillo, Eur. J. Inorg. Chem., 2011, 19, 3008–3013 CrossRef; (b) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS; (c) A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- (a) A. A. Nunez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 30, 3154–3156 RSC; (b) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed; (c) J. M. John and S. H. Bergens, Angew. Chem., Int. Ed., 2011, 50, 10377–10380 CrossRef CAS PubMed; (d) J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinksi, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem.–Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed; (e) T. Miura, I. E. Held, S. Oishi, M. Naruto and S. Saito, Tetrahedron Lett., 2013, 54, 2674–2678 CrossRef CAS; (f) T. vom Stein, M. Meureusch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed; (g) J. R. Cabrero-Antonino, E. Alberico, H. J. Drexler, W. Baumann, K. Junge, H. Junge and M. Beller, ACS Catal., 2016, 6, 47–54 CrossRef CAS; (h) L. Shi, X. Tan, J. Long, X. Xiong, S. Yang, P. Xue, H. Lv and X. Zhang, Chem.–Eur. J., 2017, 23, 546–548 CrossRef CAS PubMed.
- J. A. Garg, S. Chakraborty, Y. Ben-David and D. Milstein, Chem. Commun., 2016, 52, 5285–5288 RSC.
- N. M. Rezayee, D. C. Samblanet and M. S. Sanford, ACS Catal., 2016, 6, 6377–6383 CrossRef CAS.
- F. Schneck, M. Assmann, M. Balmer, K. Harms and R. Langer, Organometallics, 2016, 35, 1931–1943 CrossRef CAS.
- U. Jayaranthe, Y. Zhang, N. Hazari and W. Bernskoetter, Organometallics, 2017, 36, 409–416 CrossRef.
- L. A. Suarez, Z. Culacova, D. Balcells, W. H. Bernskoetter, O. Eisenstein, K. I. Goldberg, N. Hazari, M. Tilset and A. Nova, ACS Catal., 2018, 8, 8751–8762 CrossRef.
- V. Papa, J. R. Cabrero-Antonino, E. Alberico, A. Spannenberg, K. Junge, H. Junge and M. Beller, Chem. Sci., 2017, 8, 3576–3585 RSC.
- S. Kar, A. Goeppert, J. Kothandaraman and J. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- T. Leischner, A. Spannenberg, K. Junge and M. Beller, Organometallics, 2018, 37, 4402–4408 CrossRef CAS.
- (a) S. Chakraborty, O. Blacque, T. Fox and H. Berke, Chem.–Asian J., 2014, 9, 328–337 CrossRef CAS PubMed; (b) S. Chakraborty, O. Blacque and H. Berke, Dalton Trans., 2015, 44, 6560–6570 RSC; (c) S. Chakraborty and H. Berke, ACS Catal., 2014, 4, 2191–2194 CrossRef CAS; (d) Y. Zhang, P. G. Williard and W. H. Bernskoetter, Organometallics, 2016, 35, 860–865 CrossRef CAS.
- (a) S. Chakraborty, P. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS; (b) E. A. Bielinski, P. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari, S. Schneider and P. O. Lagaditis, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed; (c) D. Nguyen, X. Trivelli, F. Capet, J. F. Paul, F. Dumeignil and R. M. Gauvin, ACS Catal., 2017, 7, 2022–2032 CrossRef CAS.
- (a) Z. Shao, Y. Wang, Y. Liu, Q. Wang, X. Fu and Q. Liu, Org. Chem. Front., 2018, 5, 1248–1256 RSC; (b) X. Chen, Y. Jing and X. Yang, Chem.–Eur. J., 2016, 22, 1950–1957 CrossRef CAS PubMed; (c) E. Bielinski, M. Föster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- D. Gusev, ACS Catal., 2017, 7, 6656–6662 CrossRef CAS.
- (a) G. Zhang and S. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC; (b) Z. Wie, A. De Aguirre, K. Junge, M. Beller and H. Jiao, Catal. Sci. Technol., 2018, 8, 3649–3665 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreve, J. A. Montgomery, J. E. Peralta, F. Ogliare, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghvachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Fakas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) P. Hay and W. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (b) W. Wadt and P. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (b) W. Kohn, A. D. Becke and R. G. Parr, J. Phys. Chem., 1996, 1, 12974–12980 CrossRef.
- A. McLean and G. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- A. Marenich, C. Cramer and D. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Álvarez-Moreno, C. De Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef PubMed.
- S. Hoops, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummez, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03453f |
| This journal is © The Royal Society of Chemistry 2019 |