
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of γ-substituted carbonyl compounds from DMSO-mediated oxidation of enynamides: mechanistic insights and carbon- and hetero-functionalizations†
Quynh H.
Nguyen
 ,
Nguyen H.
Nguyen
,
Hanbyul
Kim
and
Seunghoon
Shin
*
,
Nguyen H.
Nguyen
,
Hanbyul
Kim
and
Seunghoon
Shin
*
Department of Chemistry, Research Institute for Natural Sciences and Center for New Directions in Organic Synthesis (CNOS), Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Korea. E-mail: sshin@hanayng.ac.kr
First published on 12th August 2019
Abstract
Oxidative coupling of 1,3-enynamides using DMSO as a terminal oxidant has been developed. Carbon as well as unmodified heteroatom nucleophiles, including aliphatic alcohols, thiols, and hydrazides, could be efficiently alkylated at the γ-position in a highly regioselective fashion. The kinetic analysis suggested a nucleophile-dependent mechanism ranging from a concerted SN2′′ to a carbocationic mechanism. Thus, the remote site-selectivity was ascribed to the partial positive charge developing at the terminal carbocationic center.
Introduction
Inverting natural polarity (Umpolung) allows for an outstanding opportunity to streamline redox efficient processes and enables novel disconnections with complementary selectivity to the traditional methods.1 For example, under conditions of normal polarity, α-hetero-functionalization of carbonyl compounds often requires strongly basic conditions and/or elaborate, pre-oxidized heteroatom-electrophiles.2 In contrast, Umpolung protocols allow for the use of unmodified heteroatom donors, which greatly facilitates the synthesis.Towards umpoled enolate (a2) synthons, oxidation of alkynes through transition metal catalysis (Au, Rh, Ir, Ru, and Zn)3 and the hypervalent iodine chemistry of carbonyl compounds4 have led the way (Scheme 1a). More recently, we5 and others6,7 have reported a non-metal protocol involving Brønsted acid that can catalyze the oxidation of ynamides 1 and induce subsequent addition of carbon nucleophiles, furnishing α-heteroaryl imides 3 (Scheme 1b). In this process, it was unambiguously proved that the key intermediate, adduct 2, underwent a concerted SN2′ alkylation by the observation of chirality transfer from optically active N-oxides.5a
 | ||
| Scheme 1 Vinylogous enolonium chemistry: γ-(or ε-) functionalizations. | ||
Compared to enolonium equivalents, direct access to umpoled a4 synthons is rare. Although synthesis of γ-substituted-α,β-carbonyl compounds has been demonstrated using PhNO, DEAD, or oxygen gas,8 generally applicable methods for the introduction of O-, S-, N- as well as carbon nucleophiles either at the γ- or ε-position are highly desirable as they are found in the scaffolds of numerous biologically active agents.9 To this end, we envisioned that oxidation of 1,3-enynamides 4 would generate dienolonium, such as 6 that corresponds to an umpoled version of silyl dienol ethers in Mukaiyama reactions. It would be interesting to explore the unknown regioselectivity in the alkylation,10,11 and to elucidate the underlying mechanism. Herein, we have developed an oxidative alkylation of enynamides 4 and dienynamides 5 that occurs with remarkable selectivity for the remote substitution furnishing γ-substituted 8 or ε-substituted 9, respectively, and disclosed that the reaction proceeded through a mechanism ranging from concerted SN2′′ or through a dienolonium carbocation intermediate 7, depending on the types of nucleophiles.
Meanwhile, one inherent disadvantage of the alkyne oxidation protocols is the necessity to use pyridine-N-oxide which liberates pyridine, complicating the acid catalysis. In this context, dimethyl sulfoxide (DMSO) is advantageous in that the byproduct (Me2S) is easily removable (bp = 37 °C) and not so basic (pKa = −5.4 in water).12–14 Initially, we were concerned that the nucleophiles may attack the O-dienoxysulfenium intermediate 6 (Scheme 1c) at the S-atom (SN2) rather than at C4 (SN2′′) as in interrupted Pummerer reactions.15 Notwithstanding, a selective oxygenative coupling at the remote position was obtained and the usefulness of this oxidation is further demonstrated by the introduction of exceptionally diverse nucleophiles, ranging from carbon (indoles, furans, and silyl enol ethers) to heteroatom donors (aliphatic alcohols, thiols, and hydrazides) with good to excellent remote regioselectivity.
Results and discussion
Survey of reaction conditions and Umpolung alkylation with carbon nucleophiles
For the examination of site selectivity in the enynamide oxidation, we initially chose 1,3-enynamide 4a bearing sterically encumbering o-MeO-C6H4 at C4 as a substrate that presents a regioselectivity challenge (Table 1). For initial screening, 3 equiv. of N-Me-indole along with 4 equiv. of N-oxides (Ox1-4) was employed. 2-Cl-Pyridine-N-oxide, Ox1 mediated the formation of 8a with a remarkable efficiency in only 5 min at rt, although the regioselectivity remained mediocre (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :γ = 1:4.5, entry 1).16 Interestingly, diphenyl sulfoxide Ox5 was also found to be a suitable oxidant for enynamides, while previous studies on the ynamide oxidation documented that [3,3]-sigmatropic rearrangement occurred dominantly.14b To our delight, DMSO (Ox7) displayed an excellent reactivity with α:γ = 1:5.6 (entry 7).17 Change of solvents (entries 8–10) to 1,4-dioxane slightly improved the ratio to α:γ = 1:8.7, albeit at the expense of the reactivity (entry 10).
:γ = 1:4.5, entry 1).16 Interestingly, diphenyl sulfoxide Ox5 was also found to be a suitable oxidant for enynamides, while previous studies on the ynamide oxidation documented that [3,3]-sigmatropic rearrangement occurred dominantly.14b To our delight, DMSO (Ox7) displayed an excellent reactivity with α:γ = 1:5.6 (entry 7).17 Change of solvents (entries 8–10) to 1,4-dioxane slightly improved the ratio to α:γ = 1:8.7, albeit at the expense of the reactivity (entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Oxidant | Conditions | 2 (%) | α:γc |
| a 4 (0.1 mmol), oxidant 3 (4 equiv.), N-Me-indole (3 equiv.) and HNTf2 (10 mol%) in solvents (0.1 M). b Isolated yields of 8 after chromatographic separation. c Determined from the crude 1H NMR spectra. d Hydration byproduct was observed. e Hydration byproduct (74% yield). f N-Me-indole (1.2 equiv.) and DMSO (1.2 equiv.) were used in the presence of 2.5 mol% of HNTf2. g With 3.77 mmol of 4b. | |||||
| 1 | 4a | Ox1 | DCE, 5 min | 8aa, 90 | 1:4.5 |
| 2 | 4a | Ox2 | DCE, 15 min | 8aa, 71 | 1:6.7 |
| 3 | 4a | Ox3 | DCE, 6 h | 8aa, 35 | 1:5.2 |
| 4 | 4a | Ox4 | DCE, 10 h, 60 °C | 8aa, 41 | 1:4.5 |
| 5 | 4a | Ox5 | DCE, 5 min | 8aa, 70 | 1:5.8 |
| 6 | 4a | Ox6 | DCE, 5 min | 8aa, 77 | 1:5.1 |
| 7 | 4a | Ox7 | DCE, 5 min | 8aa, 95 | 1:5.6 |
| 8 | 4a | Ox7 | CH3CN, 15 min | 8aa, 62 | 1:6.8 |
| 9 | 4a | Ox7 | MTBE, 0.5 h | 8aa, 42d | 1:7.8 |
| 10 | 4a | Ox7 | 1,4-Dioxane, 0.5 h | 8aa, 66 | 1:8.7 |
| 11 | 4b | Ox7 | DCE, 5 min | 8ba, 96 | 1:10 |
| 12 | 4c | Ox7 | DCE, 10 min | 8ca, 83 | 1:2.7 |
| 13 | 4d | Ox7 | DCE, 3 h | 8da, 62 | 1:1 |
| 14 | 4e | Ox7 | DCE, 7 h | 8ea, 63 | 1:1 |
| 15 | 4f | Ox7 | DCE, 3 h | —e | — |
| 16f | 4b | Ox7 | DCM, 3 h | 8ba, 97 | 1:10 |
| 17f,g | 4b | Ox7 | DCM, 3 h | 8ba, 93 | 1:10 |
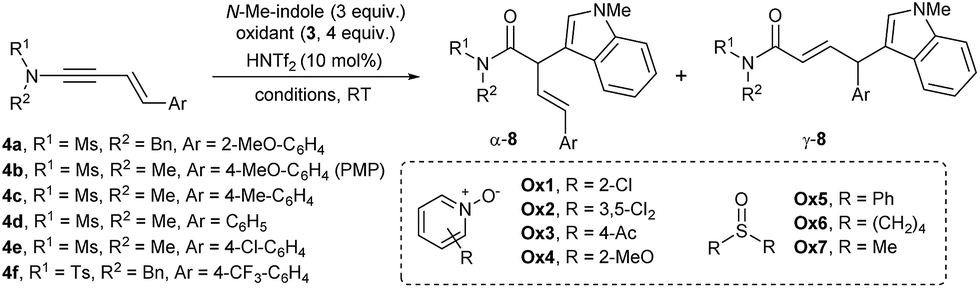
At this point, we turned to other derivatives of enynamides 4b–f having different aryl groups at C4 (entries 11–15). It was revealed that both the α:γ selectivity and the reactivity in the γ-arylation with indoles critically depended on the electron-density of the Ar group at C4. For example, the reaction of 4-MeO-derivatives 4b delivered 8ba with a good ratio (α:γ = 1:10) in an excellent yield (entry 11). However, p-tolyl (4c), Ph (4d), 4-Cl-C6H4 (4e) and 4-CF3-C6H4 (4f) derivatives showed decreasing reactivity and regioselectivity in the order, suggesting that a carbocation stabilizing group is needed at the C4 position. Remarkably, under the optimized conditions (entry 16), the oxidative coupling of 4b occurred with only 1.2 equiv. of N-Me-indole and 1.2 equiv. of DMSO at 2.5 mol% of HNTf2, without adversely affecting the yield (entry 16). Under these conditions, a gram-scale reaction (3.8 mmol of 4b) proceeded smoothly as well (entry 17).
With this initial reaction profile and the optimized conditions, we then inspected the reaction scope with various carbon nucleophiles (Table 2). The variation of N-alkyl and N-sulfonyl groups as well as electron-rich Ar groups (8ga–8ka) was all well accommodated with good reactivity and regioselectivity. Various indoles could also be incorporated without any event as in 8bb–8bf. Notably, heterocyclic motifs such as 2-furyl (8bg), N-acyl indole (8la), or 2-thiophene (8ma) could be easily incorporated into the products. Notably, the formation of a quaternary center as in 8na and 8oa was very efficient with a useful level of γ-selectivity: even phenyl derivative 8oa was obtained with higher selectivity compared to 8da (entry 13, Table 1).18 Subsequently, we went on extending the conjugation by preparing dienynamide 5a (n = 2; R1 = Ts, R2 = Me; R3 = H; Ar = 4-MeO-C6H4). To our delight, its reaction went smoothly to furnish the corresponding α,β,γ,δ-unsaturated imide 9aa with high ε-selectivity.
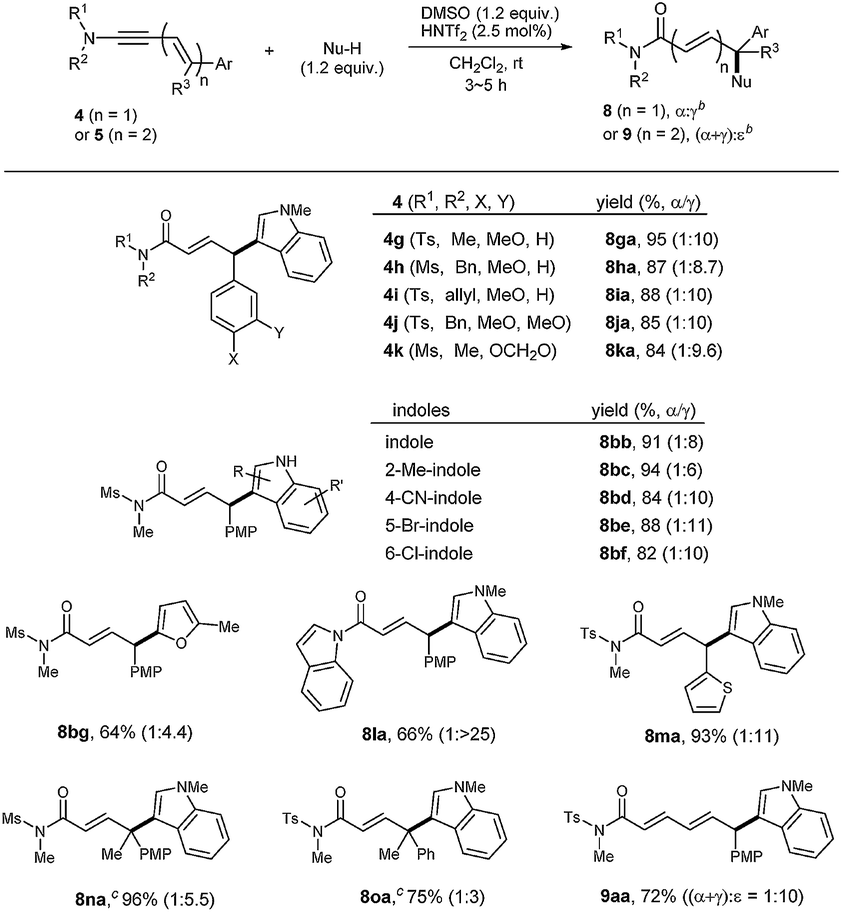
On the other hand, the reaction of silyl enol ethers was much slower (Table 3). For example, the reaction of 4b with di-substituted silyl enol ether 10a in the presence of 20 mol% of HNTf2 at 60 °C was incomplete even after 36 h, from which 11ba was obtained in 53% yield. Tri-substituted silyl enol ethers 10b, 10c were more reactive, giving higher yields of 11bb and 11bc, respectively, in the presence of 5 mol% of HNTf2. However, tetra-substituted silyl enol ether 10d, reacted sluggishly. These indicated that a balance between the electron-density and the steric hindrance is necessary for an efficient coupling. Remarkably, all the reactions of silyl enol ethers were completely regioselective (α:γ = 1:>25).
Umpolung alkylation with heteroatom nucleophiles
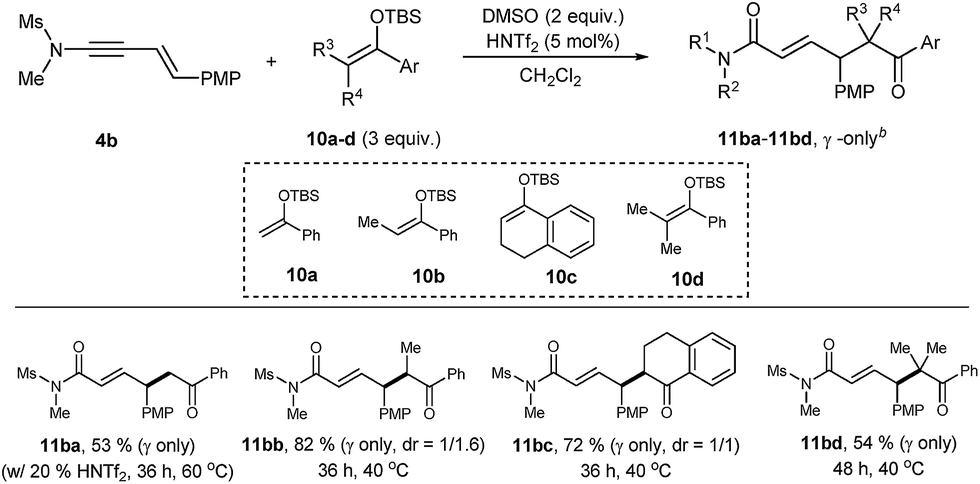
Heteroatom-functionalization at the a4 or a6 site may perhaps be the most important application of this Umpolung chemistry.2,4b Intermolecular Umpolung reaction leading to α-hetero-functionalization has been studied with various nucleophiles, including (thio)phenols,19a oxyacids,19b,c halogen,19d thiocyanate,19d azides,19d,e amines,4f,19f,g and TEMPO.19h–j Although unmodified phenols/thiols19a and amines19f,g could be used as nucleophiles, use of oxidizable aliphatic (primary and secondary) alcohols/thiols is surprisingly rare.19When benzyl alcohol (3 equiv.) was tested as a nucleophile in the presence of DMSO (4 equiv.) and HNTf2 (2.5 mol%), we were delighted to observe the formation of 67% of 12ba with a complete γ-selectivity in less than 5 min at room temperature (Table 4). Previous hypervalent iodine chemistry allowed for the use of MeOH, EtOH and iPrOH, but required these alcohols as solvents.20 In contrast, the current protocol can operate with only 3 equivalents of nucleophiles and is thus suitable for the more elaborate alcohol donors. Gratifyingly, the transformation was general not only for simple primary alcohols (12ba–12bd), but also for furfuryl, allyl, propargyl, and cinnamyl alcohols, furnishing 12be, 12bf, 12bg, and 12bh, respectively. Products with secondary alcohols 12bi, 12bj were also obtained in reasonable yields, but tert-butanol gave only a trace amount of the adduct (not shown). Notably, all the reactions of 4b to form 12ba–12bj were complete in less than 1 h and an exclusive γ-regioselectivity was obtained, in contrast to the carbon nucleophiles in Table 2.
The faster rates with alcohol nucleophiles allowed us to use a less electron-rich aryl group at C4. For example, the reaction with 4c (Ar = p-tol) with benzyl alcohol proceeded uneventfully. However, the reaction with 4d (Ar = Ph) was very slow and required 5 mol% of HNTf2 and a longer time (6 h) for completion, giving reasonable yields of 12dc and 12dj with an exclusive γ-selectivity. Alkoxylation products with a tetra-substituted center, 12nb and 12oh were also obtained in reasonable yields, employing 5 mol% HNTf2. Finally, the reaction could be extended to dienynamides 5a, 5b, affording completely ε-selective alkoxylation products 13ab and 13ba in good yields.
Encouraged by the success of alcohol nucleophiles, the reaction scope was extended to S- and N-nucleophiles as well (Table 5). Gratifyingly, with these nucleophiles, only a slight excess of donors (1.2 equiv.) and DMSO (1.2 equiv.) in the presence of HNTf2 (2.5 mol%) were sufficient. Aliphatic thiols were suitable nucleophiles, furnishing 14ba, 14bb and 14bc with exclusive γ-selectivity, and the dienimide 15ab with exclusive ε-selectivity. However, in contrast to Zn(OTf)2-catalyzed oxidative coupling,19a,d aromatic thiol derivatives (PhSH) failed to deliver the corresponding product 14 (not shown).
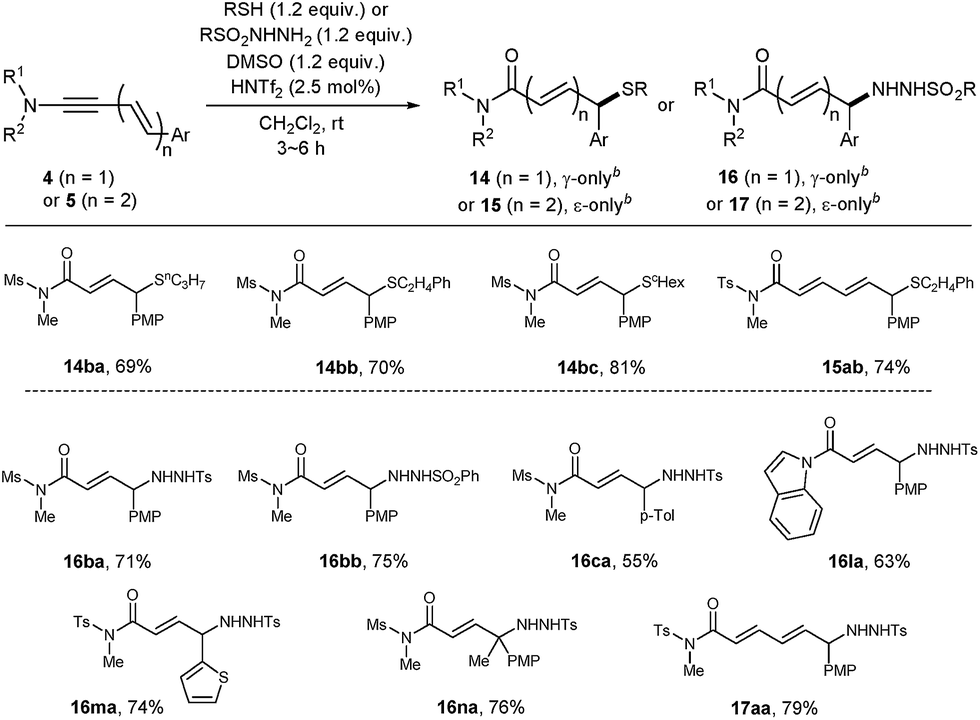
Towards N-functionalization, various N-nucleophile donors were tested, including amines, amides, and hydroxylamines. Among them, sulfonyl hydrazides were found to be effective nucleophiles. With p-toluenesulfonyl- or benzenesulfonyl hydrazide, the corresponding products 16 or 17 were obtained in a completely regioselective manner. The connectivity of a hydrazide adduct 16ma was unambiguously confirmed by single crystal X-ray crystallography.21
Kinetic study and mechanistic insights
To gain a mechanistic insight into the remote Umpolung functionalizations and to probe the nature of the intermediates involved, kinetic analysis was conducted. To the best of our knowledge, kinetic analysis on the Brønsted acid-catalyzed oxidative coupling of ynamides that can be used to check the validity of the DFT theoretical studies13e,22 is unavailable. We set out to perform a Hammett study with a set of enynamides differing in para-substituents, 4b (4-OMe), 4c (4-Me), 4d (4-H), 4e (4-Cl), 4p (4-Ph), and 4q (4-Br) to probe the electronic effect of aryl groups at C4 according to eqn (1) (Scheme 2). In the substitution with N-Me-indole, the log(kR/kH) displayed a linear correlation with σ+ parameters, with a negative ρ value (−1.3) (Scheme 2a). Upon changing the nucleophile to an alcohol, Ph(CH2)2OH, the rate became significantly more sensitive to σ+ with ρ = −3.7 (Scheme 2b), while the reaction with 2-Me-furan nucleophile showed a decreased sensitivity (ρ = −0.8) (Scheme 2c). Interestingly, the observed regioselectivity in the reaction of enynamides 4b (R = 4-MeO) with nucleophiles was in good accordance with the reaction constant ρ. For example, increasing α/γ ratio was observed in the order of 2-Me-indole (1/4.4 for 8bg), N-Me-indole (1/10 for 8ba), and Ph(CH2)2OH (0/100 for 12bd), where the corresponding ρ values were −0.8, −1.3, and −3.7, in the order. The observed reaction constant ρ reflects the build-up of a positive charge at C4 in the turnover-limiting step (TLS) and based on this, we formulated a tentative mechanism as in Scheme 3. At one extreme, Ph(CH2)2OH (ρ = −3.7) would favor a route where S–O cleavage occurs first, forming an enolonium carbocation 20, which is then trapped to yield 21. At the other end of the continuum, 2-Me-furan (ρ = −0.8) will follow a route in which a more synchronous cleavage of the S–O bond and the formation of a new C–C bond (TS19–22) occur and as a result, less charge builds up at C4.23 Here, the γ-selectivity in the alkoxylation may be due to the formation of the more stable conjugated product and this tendency weakens with the SN2′′ pathway.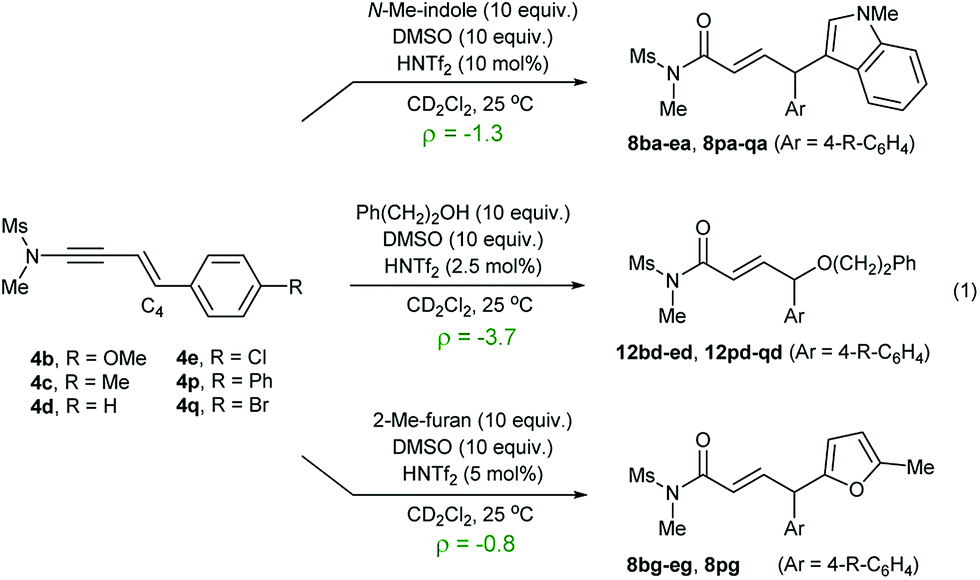
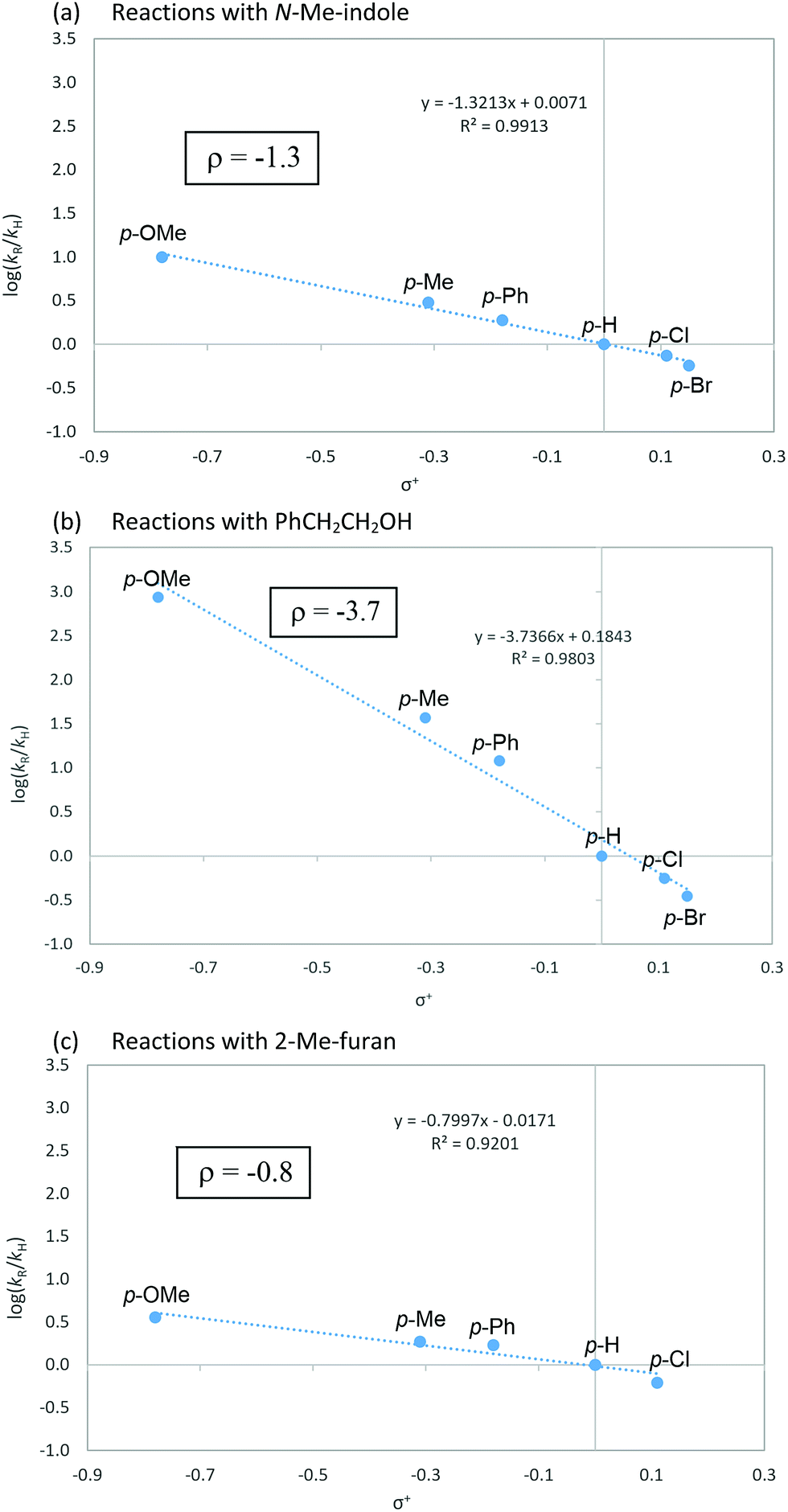 | ||
| Scheme 2 Hammett correlation with N-Me-indole, Ph(CH2)2OH and 2-Me-furan as nucleophiles according to eqn (1). | ||
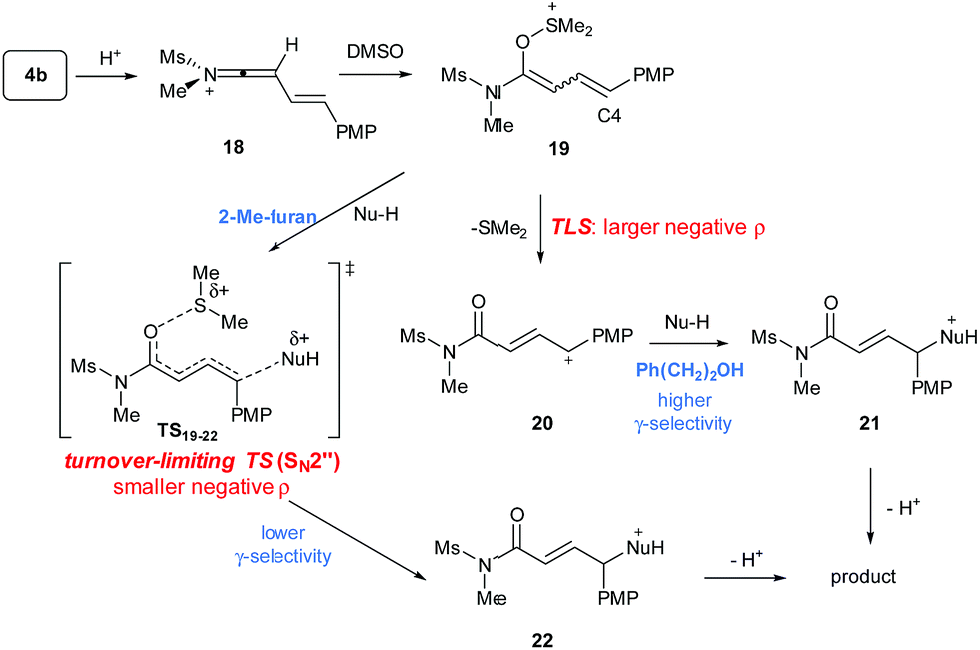 | ||
| Scheme 3 A continuum mechanism between SN2′′ and via an enolonium cation. | ||
Next, the temperature dependence of the reaction rate (kobs) for the reaction of 4b with N-Me-indole was examined in the range of 298–328 K (Fig. 1).15 From the Eyring equation, the activation parameters were determined to be ΔH‡ = 83.5 kJ mol−1 and ΔS‡ = −37.6 J K−1 mol−1. The large negative entropy of activation is consistent with an associative transition state, such as TS19–22.24 Other associative steps, such as protonation of the enynamides (4b to 18) or addition of DMSO (18 to 19) were excluded, because if these earlier steps were turnover-limiting, different sensitivity (ρ) depending on the nucleophiles (Scheme 2) should not have been observed.
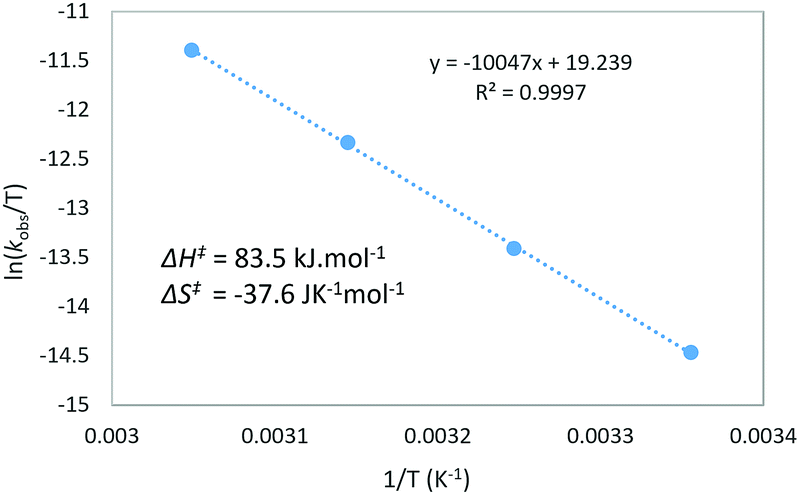 | ||
| Fig. 1 Eyring plot at 298–328 K in the reaction of 4b with N-Me-indole to form 8ba. | ||
Finally, the reaction order was examined with respect to various reactants. The rate (kobs) of the formation of 8ba was found to be inversely proportional to the concentration of N-Me-indole (Fig. S5†), which, at first, seemed contradictory to the turnover-limiting trapping of 19 to form 22. However, this should be due to the depletion of HNTf2 by the basicity of indoles and the indole moiety in the product 8ba, as kobs is linearly related to the concentration of HNTf2 (Fig. S7†). In fact, for PhCH2CH2OH, kobs was apparently independent of the concentration of the alcohol (Fig. S6†), likely because the increased concentration of the nucleophile counter-balanced the buffering of the superacid (HNTf2). Meanwhile, the reaction rate (kobs) was zeroth order in DMSO, which suggested that once the keteniminium 18 is formed, trapping with DMSO to yield 19 is very rapid and much faster than the dissociation to 20 or attack by N-Me-indole. This accounts for the chemoselectivity of this Umpolung alkylation process that occurs efficiently in a situation of multiple possible nucleophiles.5b,d
Synthetic applications
The current Umpolung alkylation provides unique structures that are difficult to obtain otherwise. Therefore, we examined their synthetic utility which is summarized in Scheme 4. Initially, 13ba (from 1.91 mmol of 4b, 69%) and 16ba (1.13 mmol of 4b, 75%) were prepared on a larger scale without any event. Esterification of these occurred chemoselectively to furnish the corresponding ε-alkoxydienoates 23 or γ-hydrazidoenoates 24, respectively, in good yields (eqn (2)). The imide product 8ba could be converted into a Weinreb amide 25 under basic conditions (eqn (3)).25 The ε-alkoxy-α,β,γ,δ-unsaturated imide 13ba underwent Diels–Alder cycloaddition with N-methyl maleimide to give 26 with an exclusive endo-selectivity and a good level of substrate-controlled diastereoselectivity (dr = 8:1, eqn (4)). Conjugate addition with thiolate gave 27 with dr = 2:1. Subsequently, attempts to cleave the N–N bond in this mixture with SmI2 failed and instead 28 was obtained as a side product, presumably because of the Lewis acidity of Sm(III). Interestingly, 28 was obtained as a single diastereomer,26 from the major diastereomer of 27. The N–N bond cleavage could then be achieved through a non-reductive method comprising alkylation of 28 and the in situ E1cb-elimination to form the lactam 29 (86%) (eqn (5)).27 Interestingly, the compound 16ba was found to be quite sensitive to bases and, upon treatment with Et3N, cyclic hydrazone 30 was obtained in an excellent yield. Presumably, this occurs through a series of events, comprising conjugate addition of Et3N, proton transfer, ring closure, elimination of Et3N and isomerization (eqn (6)).
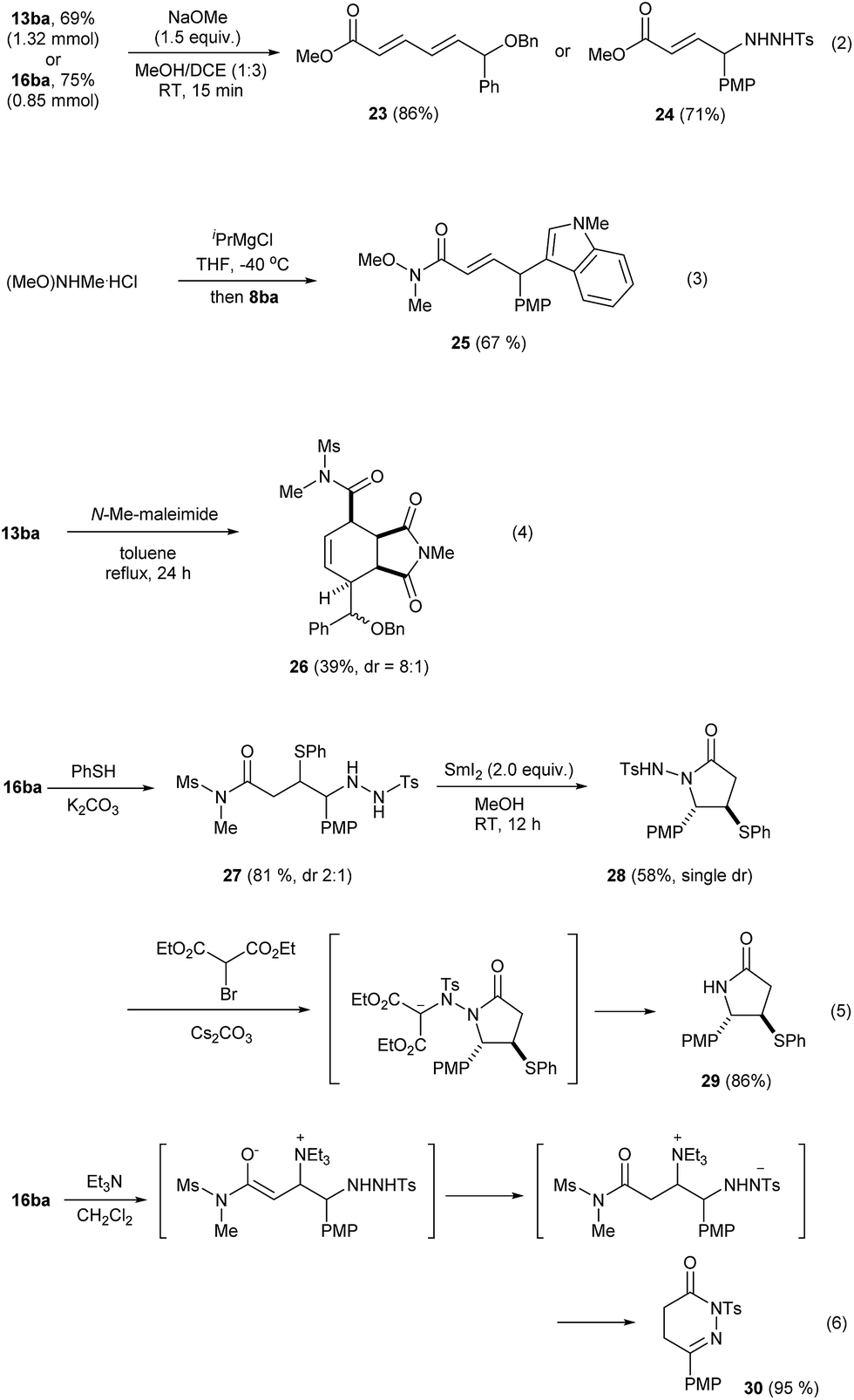 | ||
| Scheme 4 Synthetic applications of the products. | ||
Conclusions
Herein, we reported a generally applicable synthesis of γ-substituted-α,β-unsaturated carbonyl compounds. This DMSO-based oxidation protocol can accommodate diverse carbon- and heteronucleophiles, including oxidizable heteroatom donors, in a highly chemo- and regio-selective manner. The origin of the remote-selectivity was interrogated by kinetic analysis and this suggested that the reaction proceeds through an SN2′′ or via a dienolonium carbocation, depending on the nucleophiles. This may provide mechanistic insights which may be of use in devising strategies for the future enantioselective processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Research Foundation of Korea (grant numbers NRF-2012M3A7B4049653, NRF-2014-011165 and NRF-2017R1A2B4010888) for generous financial support.Notes and references
- (a) D. Seebach, Angew. Chem., Int. Ed., 1979, 16, 239–336 CrossRef; (b) O. Miyata, T. Miyoshi and M. Ueda, ARKIVOC, 2013, 60–81 Search PubMed.
- A. de la Torre, V. Tona and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 12416–12423 CrossRef CAS PubMed.
- [Au]: (a) H.-S. Yeom and S. Shin, Acc. Chem. Res., 2014, 47, 966–977 CrossRef CAS PubMed; (b) L. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS PubMed; (c) D. B. Huple, S. Ghorpade and R.-S. Liu, Adv. Synth. Catal., 2016, 358, 1348–1367 CrossRef CAS; (d) A. Mukherjee, R. B. Dateer, R. Chaudhuri, S. Bhunia, S. N. Karad and R.-S. Liu, J. Am. Chem. Soc., 2011, 133, 15372–15375 CrossRef CAS PubMed; (e) D. Vasu, H.-H. Hung, S. Bhunia, S. A. Gawade, A. Das and R.-S. Liu, Angew. Chem., Int. Ed., 2011, 50, 6911–6914 CrossRef CAS PubMed; (f) L. Li, C. Shu, B. Zhou, Y.-F. Yu, X.-Y. Xiao and L.-W. Ye, Chem. Sci., 2014, 5, 4057–4064 RSC; (g) C. Shu, Y.-H. Wang, B. Zhou, X.-L. Li, Y.-F. Ping, X. Lu and L.-W. Ye, J. Am. Chem. Soc., 2015, 137, 9567–9570 CrossRef CAS PubMed; [Rh]: (h) R. Liu, G. N. Winston-McPherson, Z.-Y. Yang, X. Zhou, W. Song, I. A. Guzei, X. Xu and W. Tang, J. Am. Chem. Soc., 2013, 135, 8201–8204 CrossRef CAS PubMed; [Ir]: (i) G. Song, D. Chen, Y. Su, K. Han, C.-L. Pan, A. Jia and X. Li, Angew. Chem., Int. Ed., 2011, 50, 7791–7796 CrossRef CAS PubMed; [Ru]: (j) K. Patil and R.-S. Liu, Chem. Commun., 2009, 5233–5235 RSC; [Zn]: (k) L. Li, B. Zhou, Y.-H. Wang, C. Shu, Y.-F. Pan, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2015, 54, 8245–8249 CrossRef CAS PubMed.
- For reviews: (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (b) E. A. Merritt and B. Olofsson, Synthesis, 2011, 517–538 CAS; for selected examples: (c) S. Arava, J. N. Kumar, S. Maksymenko, M. A. Iron, K. N. Parida, P. Fristrup and A. M. Szpilman, Angew. Chem., Int. Ed., 2017, 56, 2599–2603 CrossRef CAS PubMed; (d) O. S. Shneider, E. Pisarevsky, P. Fristrup and A. M. Szpilman, Org. Lett., 2015, 17, 282–285 CrossRef CAS PubMed; (e) S. Maksymenko, K. N. Parida, G. K. Pathe, A. A. More, Y. B. Lipisa and A. M. Szpilman, Org. Lett., 2017, 19, 6312–6315 CrossRef CAS PubMed; (f) P. Mizar and T. Wirth, Angew. Chem., Int. Ed., 2014, 53, 5993–5997 CrossRef CAS PubMed.
- (a) D. V. Patil, S. W. Kim, Q. H. Nguyen, H. Kim, S. Wang, T. Hoang and S. Shin, Angew. Chem., Int. Ed., 2017, 56, 3670–3674 CrossRef CAS PubMed; (b) S. W. Kim, T.-W. Um and S. Shin, Chem. Commun., 2017, 53, 2733–2736 RSC; (c) S. W. Kim, T.-W. Um and S. Shin, J. Org. Chem., 2018, 83, 4703–4711 CrossRef CAS PubMed; (d) D. V. Patil and S. Shin, Asian J. Org. Chem., 2019, 8, 63–73 CrossRef CAS.
- (a) D. Kaldre, B. Maryasin, D. Kaiser, O. Gajsek, L. González and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 2212–2215 CrossRef CAS PubMed; (b) Y. Zhao, Y. Hu, C. Wang, X. Li and B. Wan, J. Org. Chem., 2017, 82, 3935–3942 CrossRef CAS PubMed; (c) Y. Zhao, Y. Hu, X. Li and B. Wan, Org. Biomol. Chem., 2017, 15, 3413–3417 RSC.
- For a related enolonium chemistry from dehydration of amides: (a) D. Kaiser, A. de la Torre, S. Shaaban and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 5921–5925 CrossRef CAS PubMed; (b) S. Shaaban, V. Tona, B. Peng and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 10938–10941 CrossRef CAS PubMed; (c) N. Takeda, E. Futaki, Y. Kobori, M. Ueda and O. Miyata, Angew. Chem., Int. Ed., 2017, 56, 16342–16346 CrossRef CAS PubMed; (d) Z. Zhang, Y. Luo, H. Du, J. Xu and P. Li, Chem. Sci., 2019, 10, 5156–5161 RSC.
- (a) S. Bertelsen, M. Marigo, S. Brandes, P. Dinér and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973–12980 CrossRef CAS PubMed; (b) A. J. Fraboni and S. E. Brenner-Moyer, Org. Lett., 2016, 18, 2146–2149 CrossRef CAS PubMed; (c) H. J. Zhang, A. W. Schuppe, S.-T. Pan, J.-X. Chen, B. R. Wang, T. R. Newhouse and L. Yin, J. Am. Chem. Soc., 2018, 140, 5300–5310 CrossRef CAS PubMed; (d) X. Chen, X. Liu and J. T. Mohr, Org. Lett., 2016, 18, 716–719 CrossRef CAS PubMed.
- (a) S. Kölker, J. Inherited Metab. Dis., 2018, 41, 1055–1063 CrossRef PubMed; (b) M. Chebib and G. A. R. Johnston, Clin. Exp. Pharmacol. Physiol., 1999, 26, 937–940 CrossRef CAS PubMed; (c) E. Szabadi, J. Psychopharmacol., 2015, 29, 744–749 CrossRef CAS PubMed; (d) J. D. Griffin and L. Ellman, Semin. Thromb. Hemostasis, 1978, 5, 27–40 CrossRef CAS.
- Lithium dienolates are known to react preferentially at the α position, while alkylation of silyl dienol ethers occurs at the γ carbon: (a) S. E. Denmark, J. R. Heemstra Jr and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682–4698 CrossRef CAS PubMed; (b) M. Frías, W. Cieślik, A. Fraile, A. Rosado-Abón, A. F. Garrido-Castro, F. Yuste and J. Alemán, Chem.–Eur. J., 2018, 24, 10906–10933 CrossRef PubMed.
- For γ-functionalization of enynamides: (a) A. M. Jadhav, S. A. Gawade, D. Vasu, R. B. Dateer and R.-S. Liu, Chem.–Eur. J., 2014, 20, 1813–1817 CrossRef CAS PubMed; (b) A. M. Jadhav, D. B. Huple, R. R. Singh and R.-S. Liu, Adv. Synth. Catal., 2016, 358, 1017–1022 CrossRef CAS; (c) R. R. Singh and R.-S. Liu, Adv. Synth. Catal., 2016, 358, 1421–1427 CrossRef CAS; (d) S. S. Giri, L.-H. Lin, P. D. Jadhav and R.-S. Liu, Adv. Synth. Catal., 2017, 359, 590–596 CrossRef CAS.
- For an excellent review on the redox chemistry of sulfoxides: A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef CAS PubMed.
- For oxygen atom transfer from sulfoxides: (a) C.-W. Li, K. Pati, G.-Y. Lin, S. M. A. Sohel, H.-H. Hung and R.-S. Liu, Angew. Chem., Int. Ed., 2010, 49, 9891–9894 CrossRef CAS PubMed; (b) S. Mori, M. Takubo, T. Yanase, T. Maegawa, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2010, 352, 1630–1634 CrossRef CAS; (c) C.-F. Xu, M. Xu, Y.-X. Jia and C.-Y. Li, Org. Lett., 2011, 13, 1556–1559 CrossRef CAS PubMed; (d) H.-S. Yeom and S. Shin, Org. Biomol. Chem., 2013, 11, 1089–1092 RSC; (e) T. Stopka, M. Niggemann and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 13270–13274 CrossRef CAS PubMed.
- Use of Ph2S
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O often resulted in [3,3]-sigmatropic rearrangement rather than O-atom transfer:
(a) A. B. Cuenca, S. Montserrat, K. M. Hossain, G. Mancha, A. Lledós, M. Medio-Simón, G. Ujaque and G. Asensio, Org. Lett., 2009, 11, 4906–4909 CrossRef CAS PubMed;
(b) B. Peng, X. Huang, L.-G. Xie and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 8718–8721 CrossRef CAS PubMed.
O often resulted in [3,3]-sigmatropic rearrangement rather than O-atom transfer:
(a) A. B. Cuenca, S. Montserrat, K. M. Hossain, G. Mancha, A. Lledós, M. Medio-Simón, G. Ujaque and G. Asensio, Org. Lett., 2009, 11, 4906–4909 CrossRef CAS PubMed;
(b) B. Peng, X. Huang, L.-G. Xie and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 8718–8721 CrossRef CAS PubMed. - These reactions would produce a by-product apparently identical to the one via hydration of 4. For interrupted Pummerer reactions, see: Z. He, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2019, 58, 7813–7817 CrossRef CAS PubMed and references therein.
- Assignment of the α- and γ-isomers was based on the analysis of 1H NMR, COSY, and the nOe experiments (8ga, 8ha, 8na, 12bd, and 16ma). See the ESI.†.
- In contrast, the corresponding oxygenative alkylation of ynamides (according to ref. 5a) did not proceed when the oxidant changed from Ox1·(2-Cl-pyridine-N-oxide) to Ox7 (DMSO).
- However, the absence of an aryl group at C4 (for example, n = 1; R1 = Ms, R2 = Me, R3/Ar = (CH2)4) did not provide the corresponding product, indicating that an aryl group at C4 is necessary.
- (a) P.-P. Ruan, C.-H. Shen, L. Li, C.-Y. Liu and L.-W. Ye, Org. Chem. Front., 2016, 3, 989–993 RSC; (b) G. F. Koser, A. G. Relenyi, A. N. Kalos, L. Rebrovic and R. H. Wettach, J. Org. Chem., 1982, 47, 2487–2489 CrossRef CAS; (c) K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508–6512 CrossRef CAS PubMed; (d) F. Pan, X.-L. Li, X.-M. Chen, C. Shu, P.-P. Ruan, C.-H. Shen, X. Lu and L.-W. Ye, ACS Catal., 2016, 6, 6055–6062 CrossRef CAS; (e) V. Tona, A. de la Torre, M. Padmanaban, S. Ruider, L. Gonzalez and N. Maulide, J. Am. Chem. Soc., 2016, 138, 8348–8351 CrossRef CAS PubMed; (f) D. H. Miles, J. Guasch and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7632–7635 CrossRef CAS PubMed; (g) X. Xia, B. Chen, X. Zeng and B. Xu, Org. Biomol. Chem., 2018, 16, 6918–6922 RSC; (h) A. de la Torre, D. Kaiser and N. Maulide, J. Am. Chem. Soc., 2017, 139, 6578–6581 CrossRef CAS PubMed; (i) S. Taninokuchi, R. Yazaki and T. Ohshima, Org. Lett., 2017, 19, 3187–3190 CrossRef CAS PubMed; (j) X. Li, F. Lin, K. Huang, J. Wei, X. Li, X. Wang, X. Geng and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 12307–12311 CrossRef CAS PubMed.
- J. Yu, J. Tian and C. Zhang, Adv. Synth. Catal., 2010, 352, 531–546 CrossRef CAS.
- CCDC 1895889 (16ma).†.
- V. Tona, S. A. Ruider, M. Berger, S. Shaaban, M. Padmanaban, L.-X. Xie, L. González and N. Maulide, Chem. Sci., 2016, 7, 6032–6040 RSC.
- However, both the reactions of N-Me-indole and BnOH had some degree of ‘concertedness’, as suggested by the asymmetric induction test (14% ee and 29% ee, respectively), employing chiral N,N′-dioxide (ref. 5a; eqn (S4)†).
- A reviewer suggested a possibility that the reaction with indoles may follow a concerted SN2′′ substitution and such a reaction may display a large negative entropy of activation. For a similarly large negative entropy of activation in the SN2 reaction: K. Okamoto, S. Fukui, I. Nitta and H. Shingu, Bull. Chem. Soc. Jpn., 1967, 40, 2350–2353 CrossRef CAS.
- F. Sirindi, S. Miaskiewicz, N. Kern, A. Lalaut, A.-S. Felten, J. –M. Weibel, P. Pale and A. Blanc, Tetrahedron, 2017, 73, 5096–5106 CrossRef.
- An nOe experiment supported the relative stereochemistry in 27.
- J. Ferreira, S. C. M. Rees-Jones, V. Ramaotsoa, A. Msutu and R. Hunter, Org. Biomol. Chem., 2016, 14, 1545–1549 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1895889. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03663f |
| This journal is © The Royal Society of Chemistry 2019 |