
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiroptical inversion of a planar chiral redox-switchable rotaxane†
Marius
Gaedke
a,
Felix
Witte
a,
Jana
Anhäuser
b,
Henrik
Hupatz
a,
Hendrik V.
Schröder
 a,
Arto
Valkonen
c,
Kari
Rissanen
c,
Arne
Lützen
b,
Beate
Paulus
a and
Christoph A.
Schalley
*a
a,
Arto
Valkonen
c,
Kari
Rissanen
c,
Arne
Lützen
b,
Beate
Paulus
a and
Christoph A.
Schalley
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: c.schalley@schalley-lab.de
bKekulé-Institut für Organische Chemie und Biochemie, Universität Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany
cUniversity of Jyvaskyla, Department of Chemistry, P.O. Box 35, 40014 Jyväskylä, Finland
First published on 4th September 2019
Abstract
A tetrathiafulvalene (TTF)-containing crown ether macrocycle with Cs symmetry was designed to implement planar chirality into a redox-active [2]rotaxane. The directionality of the macrocycle atom sequence together with the non-symmetric axle renders the corresponding [2]rotaxane mechanically planar chiral. Enantiomeric separation of the [2]rotaxane was achieved by chiral HPLC. The electrochemical properties – caused by the reversible oxidation of the TTF – are similar to a non-chiral control. Reversible inversion of the main band in the ECD spectra for the individual enantiomers was observed after oxidation. Experimental evidence, conformational analysis and DFT calculations of the neutral and doubly oxidised species indicate that mainly electronic effects of the oxidation are responsible for the chiroptical switching. This is the first electrochemically switchable rotaxane with a reversible inversion of the main ECD band.
Introduction
Evidenced by the homochirality in our biosphere,1–3 chirality is a fundamental principle, which governs the molecular recognition and activity of virtually all biomolecules. Therefore, gaining control over the preferred isomer of a molecule or an assembly by carefully designing a molecular system is a worthwhile endeavour.The term “chiroptical switch” has been used by Canary to refer to molecules, which are capable of “changes in their interaction with polarized light”.4 Potential applications are information processing, data storage and sensing. In this context, the ground breaking work of Feringa and co-workers5–7 on overcrowded alkenes, which act as light triggered chiroptical switches was awarded with the Nobel Prize in chemistry 2016 “for the design and synthesis of molecular machines”8 and underlines the general interest in this topic.
Mechanically interlocked molecules (MIMs)9–12 consist of parts that can move relative to each other guided by intramolecular forces. Therefore, we envisioned them to be ideal candidates for chiroptical switches in which co-conformational or even configurational changes in the MIM occur.
An achiral wheel with directionality in its atom sequence forms a chiral [2]rotaxane, when threaded onto a directional axle (Scheme 1). In 1997, Vögtle et al. reported on the first resolution of a racemate of such mechanically planar chiral rotaxanes.13 Chiral rotaxanes may be chiral from inclusion of classical stereogenic elements or by virtue of being mechanically planar chiral. Since then, several examples followed,14–25 in which the mechanically interlocked structure was used to induce directionality in polymers,26–28 for sensing,29–31 and to act as an enantioselective catalyst.32 Today, sophisticated synthetic protocols allow an efficient enantioselective synthesis. For example, Goldup and co-workers33,34 described elegant protocols to synthesise planar chiral enantiopure [2]rotaxanes using readily available chiral auxiliaries. However, switchable planar chiral rotaxanes remain rare. So far, the modulation of chirality relies on heat,21 the choice of solvent, anion exchange,35 or pH.36
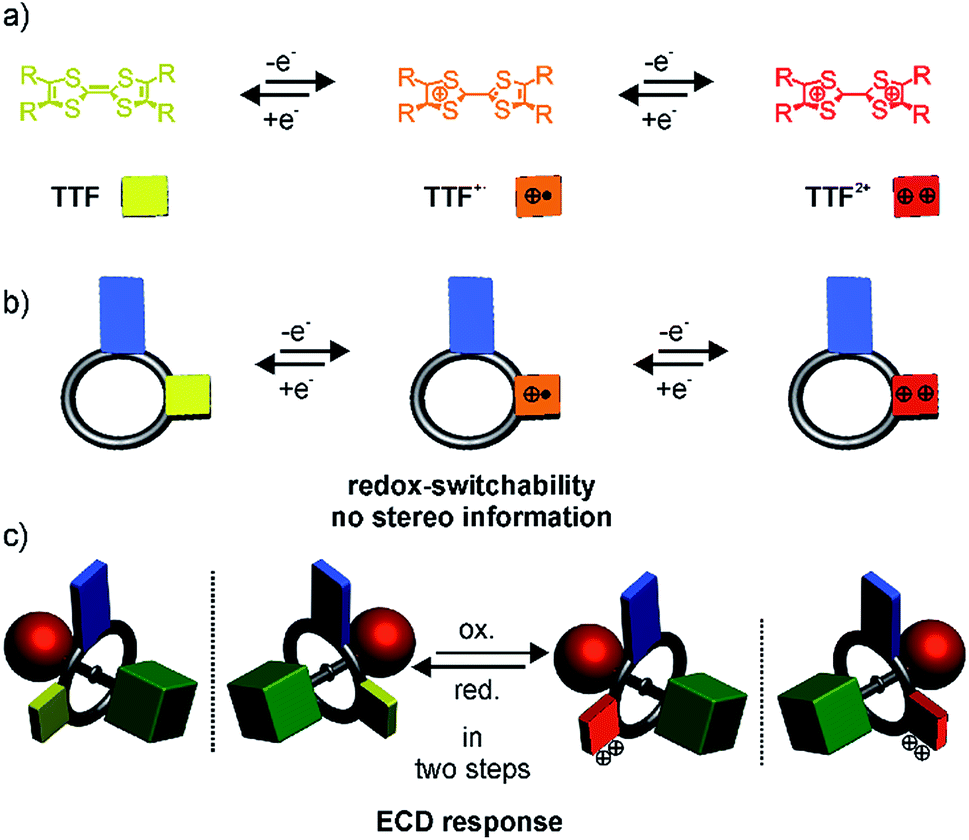 | ||
| Scheme 1 (a) Reversible one-electron oxidations of the TTF moiety, (b) reversible oxidation of a directional crown ether wheel bearing a TTF unit, (c) chiroptical switching of the planar chiral [2]rotaxane enantiomers. | ||
Recently, we described redox-switchable rotaxanes, in which the wheels are decorated with tetrathiafulvalenes (TTF).37–41 TTF can be reversibly oxidised to the TTF˙+ and TTF2+ states (Scheme 1). Large-amplitude motion and co-conformational changes in (oligo)rotaxanes were triggered by redox chemistry.38,42–50 Apart from rotaxanes, TTF derivatives with covalently bound chiral substituents exhibited a chiroptical response to a change of their redox-state.51–57 Hence, our switchable rotaxanes display ideal optoelectronic properties since they are air stable in their neutral and oxidised form and show a clear-cut optical output,37 which is even visible by the naked eye.
In this paper, we report the synthesis, characterisation and optical resolution of a new mechanically planar chiral tristable [2]rotaxane based on the 24-crown-8/secondary ammonium binding motif.58 The rotaxane consists of the directional wheel dTTFC8 (Scheme 2), which is derived from a C2v-symmetric TTF-decorated crown ether TTFC8 (Scheme 2) published by our group recently.38 ECD measurements show reversible chiroptical switching, which can be explained mainly by electronic changes. The measurements are supported by quantum chemical calculations, which were also used to determine the absolute configuration. To the best of our knowledge, this is the first example of a chiroptical switch with a complete sign reversal of the main band in the ECD spectra based on electronic changes in a mechanically bound assembly.
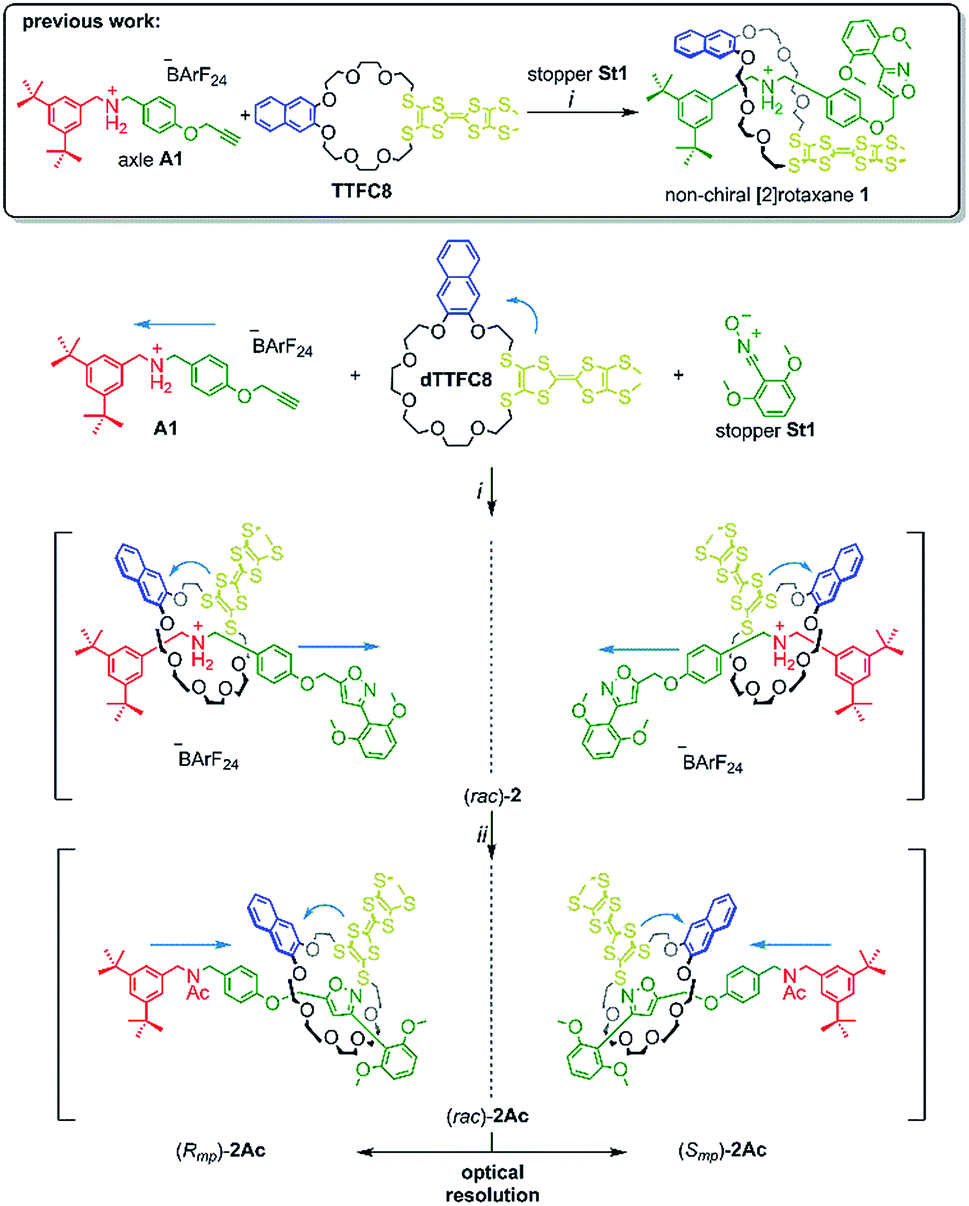 | ||
| Scheme 2 Synthesis of rotaxanes (rac)-2 and (rac)-2Ac. Conditions and reagents: (i) DCM, 35 °C, 12 h (73%); (ii) Ac2O, NEt3, ACN, 12 h, r.t. (95%). | ||
Results and discussion
Synthesis and characterisation
The prerequisite for rotaxane formation is a sufficiently high binding constant between the crown ether and the ammonium axle. ITC experiments revealed an association constant of Ka = (3.6 ± 0.3) 105 M−1 and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry for pseudorotaxane formation from dTTFC8 and axle A1 (Scheme 2). The binding constant is very similar to that of our previous non-directional TTF-decorated wheel TTFC8 (Ka = (4.4 ± 0.4) 105 M−1, for thermodynamic parameters see ESI,† Section 4),38 which indicates the positional change of the TTF unit not to significantly affect the binding properties of the wheel.
:1 stoichiometry for pseudorotaxane formation from dTTFC8 and axle A1 (Scheme 2). The binding constant is very similar to that of our previous non-directional TTF-decorated wheel TTFC8 (Ka = (4.4 ± 0.4) 105 M−1, for thermodynamic parameters see ESI,† Section 4),38 which indicates the positional change of the TTF unit not to significantly affect the binding properties of the wheel.
As for the non-chiral [2]rotaxane 1, rotaxane formation was achieved with nitrile-oxide stopper St1 using a catalyst-free end-capping protocol established by Takata and co-workers59 yielding a racemic mixture of rotaxane (rac)-2 (73%). The non-ionic version (rac)-2Ac (95%) was obtained through N-acylation with Ac2O60 (Scheme 2). The 1H NMR spectra of (rac)-2 and (rac)-2Ac (Fig. 1) reveal a diastereotopic splitting of the macrocycle's methylene protons as well as of the axle methylene protons Hh.37,38 The splitting of both macrocycle and axle protons is characteristic for the formation of a chiral, yet racemic [2]rotaxane. Isoxazole formation during stopper attachment leads to a strong downfield shift of 3.88 ppm for proton Hi.
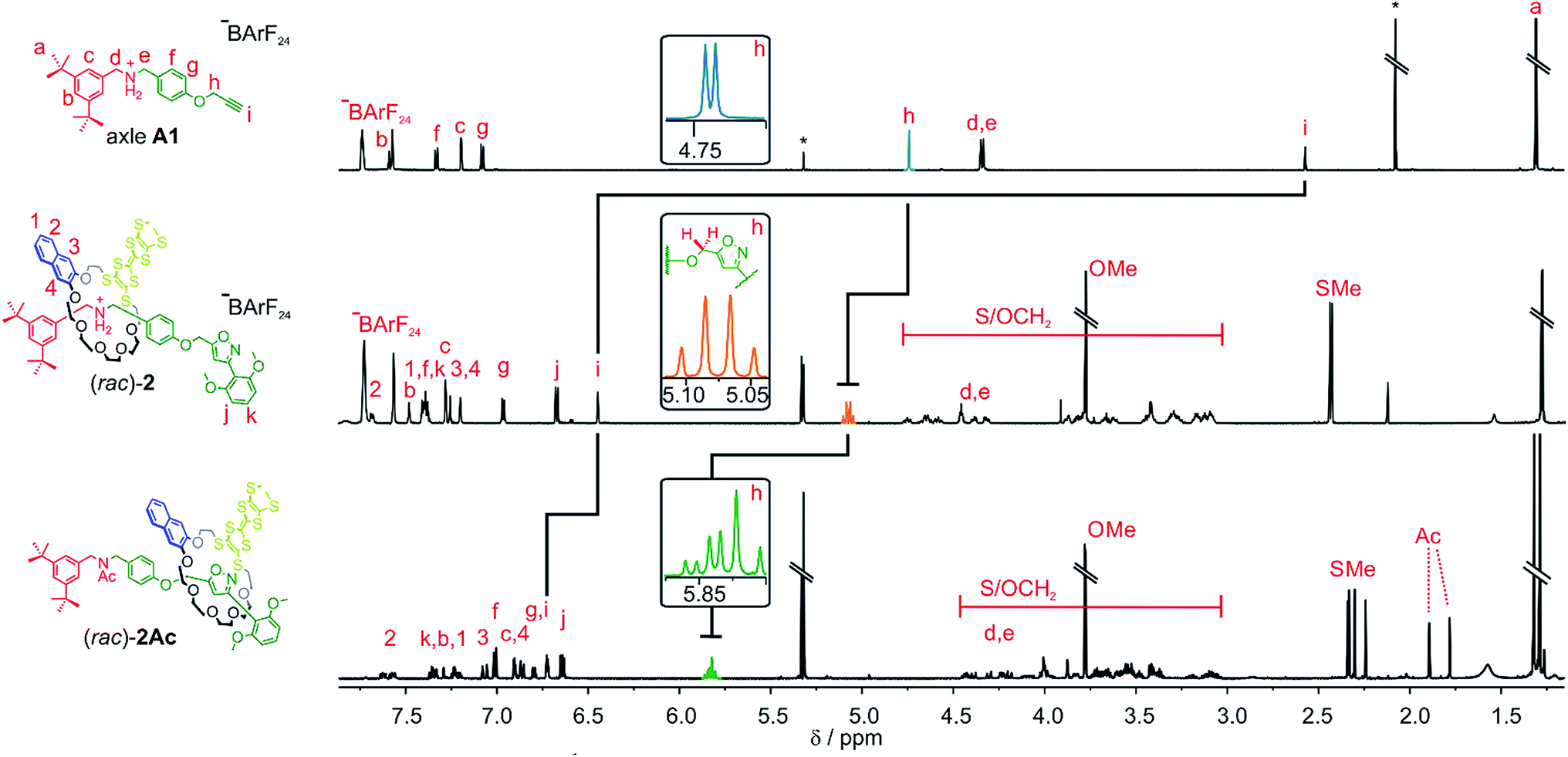 | ||
| Fig. 1 Comparison of the shifts and splitting in the partial 1H NMR spectra of the methylene groups on the axle A1 (top), rotaxane (rac)-2 (middle) and acetylated rotaxane (rac)-2Ac (bottom) (700 MHz, 298 K, CD2Cl2). | ||
In (rac)-2, the S-methyl protons on dTTFC8 split into two singlets of the same intensity. Comparable rotaxanes also showed this behaviour on the same position.27,28 HR-ESI mass and tandem MS experiments support the interlocked architecture (Fig. S1†).
For non-ionic (rac)-2Ac, the shift of Hi (Δδ = +0.28 ppm) and Hh (Δδ = +0.76 ppm) relative to (rac)-2 suggests that the wheel translates towards the isoxazole moiety in the absence of attractive interactions with the ammonium ion. Two sets of signals are observed due to the cis–trans isomerism of the amide bond in (rac)-2Ac. Variable temperature NMR experiments (Fig. S3†) in DMSO-d6 reveal the same barrier (ΔG‡ = 74 ± 2 kJ mol−1) for amide cis–trans isomerisation as observed for a similar acetylated rotaxane.41
Optoelectronic properties
Photometric titrations of (rac)-2 and (rac)-2Ac with Fe(ClO4)3 (Fig. 2a and b) show similar bands for the three redox states (TTF, TTF+˙ and TTF2+)61–63 of both rotaxanes with distinct isosbestic points. These findings are consistent with structurally related rotaxanes featuring a non-directional TTF-decorated wheel.38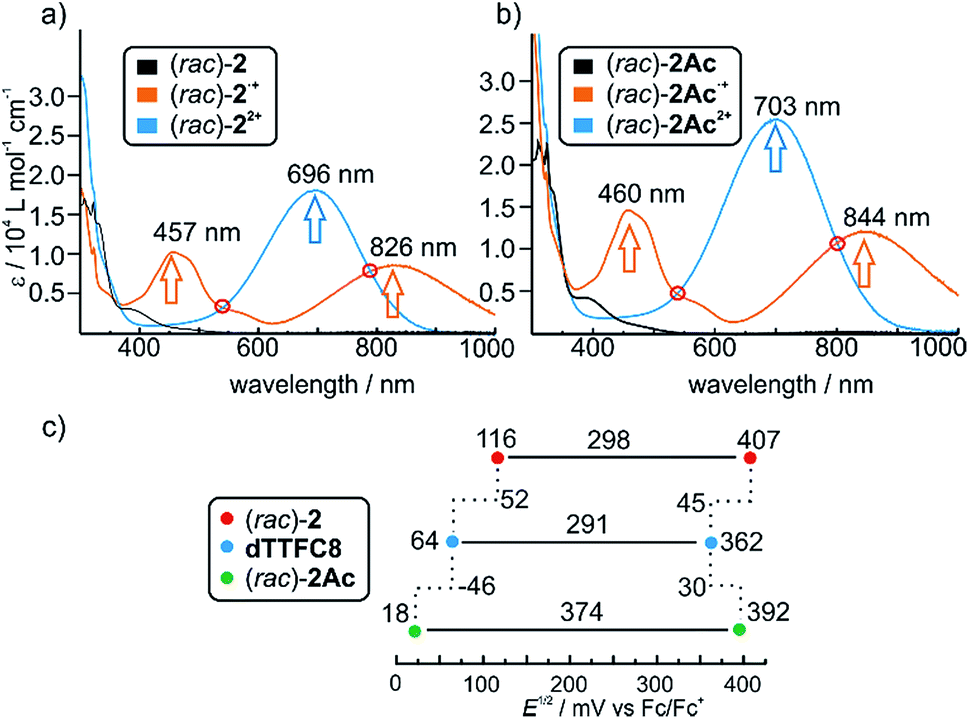 | ||
| Fig. 2 UV/Vis spectra of (a) ionic (rac)-2 and (b) non-ionic (rac)-2Ac in different oxidation states. Isosbestic points indicating a clean transition from TTF˙+ to TTF2+ are highlighted with red circles. Spectra were obtained with 25 μM solutions in CH2Cl2 using bulk Fe(ClO4)3 as the oxidant; (c) correlation diagram of half-wave potentials obtained by cyclic voltammetry for the first and second redox process of (rac)-2, dTTFC8 and (rac)-2Ac (each 1 mM) in CH2Cl2 referenced against Fe(Cp)20/+ with NBu4PF6 (0.1 M) as the electrolyte. | ||
Cyclovoltammetric (CV) experiments were conducted with dTTFC8, (rac)-2 and (rac)-2Ac in dichloromethane (Fig. 2c). The potentials for (rac)-2 (116 mV and 407 mV) are considerably higher for both oxidation steps as compared to dTTFC8 (64 mV and 362 mV). Both oxidations are thus energetically disfavoured because of the charge repulsion between the TTF cation radical as well as the TTF dication and the ammonium station. In case of (rac)-2Ac (18 mV and 392 mV) the first oxidation is more easily accomplished in comparison to the free macrocycle and the second oxidation is disfavoured. We attribute this behaviour to a stabilising interaction with the isoxazole moiety on the axle for the first oxidation.
For the second oxidation, the limited accessibility of the TTF2+ by counterions caused by the steric demand of the axle needs to be taken into account. Again these trends were already observed for the non-directional macrocycle and rotaxanes thereof.38 The reversibility of the redox-waves of (rac)-2 and (rac)-2Ac strongly indicated that the interlocked structures remain intact during the redox switching, however it is reasonable to assume conformational changes to occur due to charge repulsion and charge stabilisation. The data does not show any significant change in the electrochemical properties by introducing directionality into the TTF decorated wheel.
Enantiomer separation on chiral HPLC and CD spectroscopy
The two enantiomers of (rac)-2Ac could be separated using HPLC with a CHIRALPAK® IA stationary phase. The optical purity was determined (>99% ee; Fig. 3a) and mirror-image CD spectra were obtained for the neutral enantiomers with bands at 242 nm and 325 nm (Fig. 3b). We assigned the absolute configuration based on the computational results (see below). The oxidised species 2Ac˙+ and 2Ac2+ show bands at the same wavelengths. While no sign inversion occurs at 325 nm, the band at 242 nm exhibits a sign inversion during the first and a significant intensity increase during the second oxidation step. To exclude decomposition to be responsible for the switching, 2Ac2+ was reduced back to the neutral state using Zn dust and then showed the initial CD spectrum again (Fig. 3d and e dashed lines). Surprisingly, no other CD signals are observed at a higher wavelength, although the change in UV/Vis absorption is most pronounced at 460 nm and 844 nm for the radical cation and at 703 nm for the dication (Fig. 2b). The reason for the sign change remains ambiguous. In fact, conformational changes were observed to induce transitions in CD spectra of non-interlocked TTF derivatives with centrochiral elements earlier.53,64 Other examples show varying intensities54 or shifts of the maxima56 upon oxidation of the TTF attached.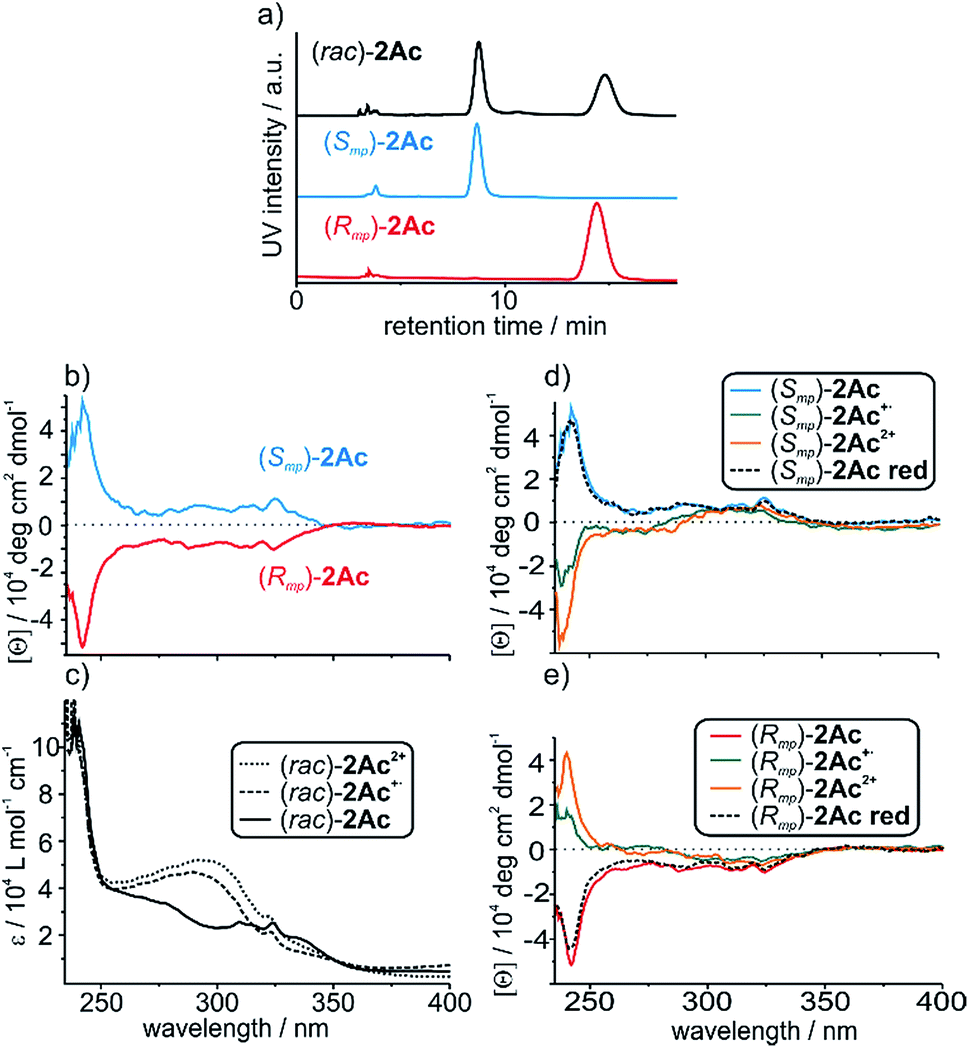 | ||
| Fig. 3 (a) Traces of analytical chiral HPLC. The chromatographic resolution of (rac)-2Ac was realised by chiral phase HPLC on a CHIRALPAK® IA column using methyl tert-butylether/CH2Cl2 80:20 (v/v) as the eluent. (b) CD spectra of the individual neutral enantiomers and (c) partial UV/Vis spectra of the neutral, singly and doubly oxidized (rac)-2Ac. (d and e) CD spectra of the individual enantiomers in their three oxidation states and after reduction to the neutral state. Spectra were taken from 160 μM solutions in CH2Cl2 using bulk Fe(ClO4)3 as the oxidant and Zn dust as the reductant. | ||
Nevertheless, no TTF derivative is reported that shows a sign reversal in the maximum of an ECD spectrum without a shift in the wavelength. Apart from TTF derivatives, chiroptical switching via a redox process can be achieved with catechol,65 viologen,66 and tetraarylethylene67 building blocks. Intense switching with a sign reversal was also observed for a viologen-type dicationic helquat.68 Chiral inversion can also be achieved with metal ion complexation69,70 acid–base-71 and photoswitching.6,68,72
Computational results
To investigate whether the redox-induced sign inversion at 242 nm in the ECD spectra of (Rmp)-2Ac is due to a change in its electronic properties or to a (co-)conformational change, density functional theory (DFT) calculations were performed at the TPSS-D3(BJ)73–75 and ωB97X-D3 (ref. 76) levels. Conformational analyses reveal the structure depicted in Fig. 4 (left) to be the most stable one for (Rmp)-2Ac. It is at least 18 kJ mol−1 more favourable than any other possible conformation found by theory (see Table S2†). For (Rmp)-2Ac2+, there are two conformations relatively close in electronic energy: Conformer A (Fig. 4 middle) and B (Fig. 4 right) with a flipped naphthalene unit, ca. 9 kJ mol−1 more stable than A. This conformational change is explained by the oxidation of (Rmp)-2Ac occuring fairly localised at the TTF unit.77 The emerging charge of the oxidised TTF moiety is then stabilised by the naphthalene that moves into close proximity of the TTF2+. Additionally, an atoms-in-molecules (AIM) analysis suggests that the electrostatic attraction between the naphthalene and TTF moieties outweighs all other non-covalent interactions for (Rmp)-2Ac2+, while the maximisation of non-covalent interactions (C–H⋯π and π–π-stacking) is the most important factor in the neutral state (see ESI† for details).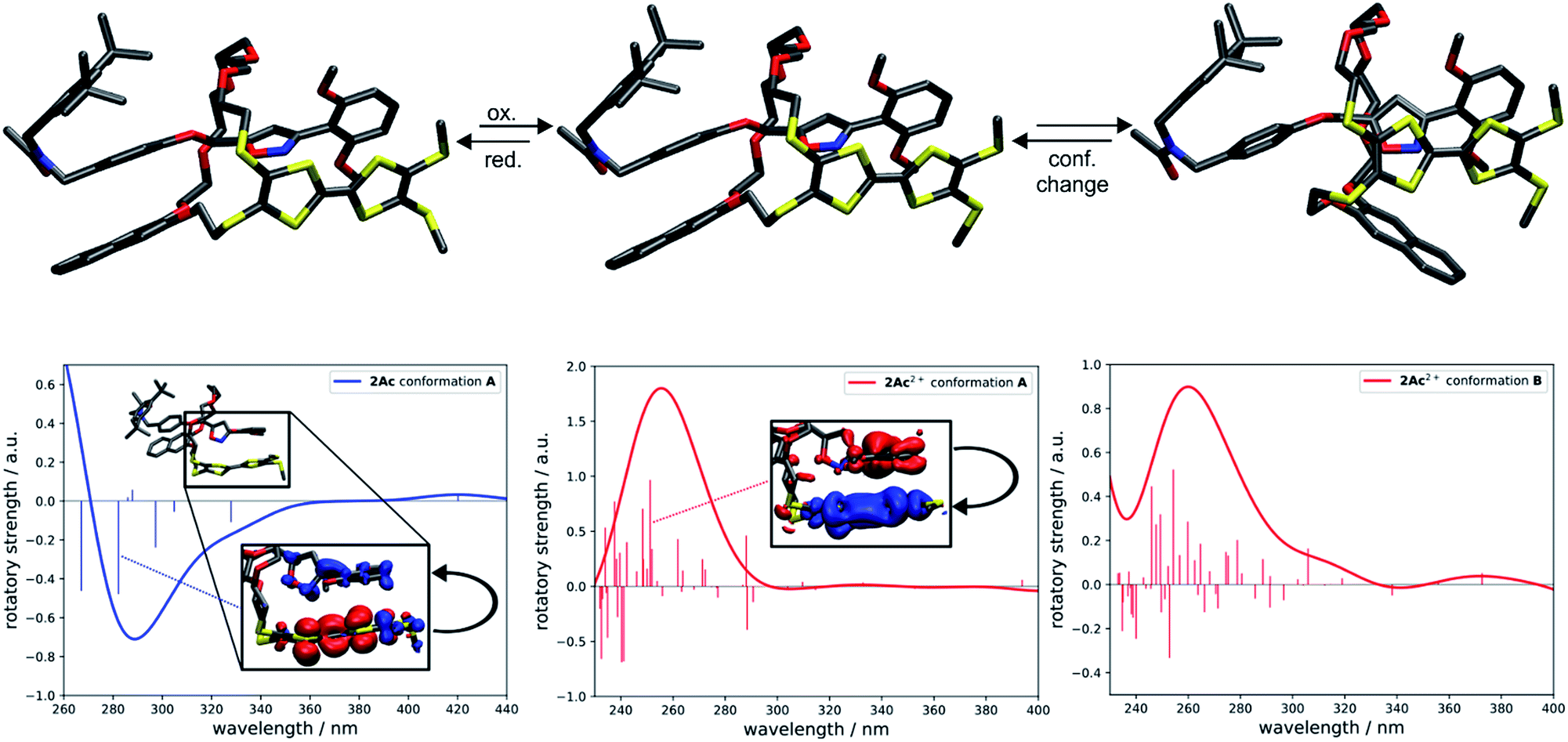 | ||
| Fig. 4 Structural and spectral comparison of the most stable conformation of (Rmp)-2Ac and the two most stable conformations, A and B, of (Rmp)-2Ac2+. Oxidation induces a flip of the naphthalene moiety in A towards the TTF unit yielding conformation B. The difference in electronic energy between A and B is around 9 kJ mol−1. All structures were obtained at the TPSS-D3(BJ) level. Corresponding simulated CD spectra with excited state difference densities of selected transitions (insets) visualising the change in electronic structure upon photoexcitation of (Rmp)-2Ac (left) and (Rmp)-2Ac2+ (middle). The difference in the CD spectra due to the conformational change of (Rmp)-2Ac2+ is negligible in the region of interest. The spectra were obtained at the ωB97X-D3 level using sTD-DFT. Gaussian line broadening with σ = 20 nm was applied. Insets: blue and red zones correspond to areas of electron enhancement and electron depletion, respectively. Isovalue = 0.001 a0−3. | ||
The simulated CD spectra in Fig. 4 were obtained using simplified time-dependent DFT78 at the ωB97X-D3 level. The spectrum of (Rmp)-2Ac shows a deviation of around 40–50 nm, while that of (Rmp)-2Ac2+ is off by less than 20 nm compared to experiment. The experimentally detected sign inversion at 242 nm is reproduced well by the calculations. The conformational change of (Rmp)-2Ac upon oxidation, however, hardly influences the shape of the CD spectra as both conformations yield very similar CD spectra in the region between 230 and 400 nm. Therefore, we exclude the conformational change as the prime origin of the sign inversion.
To rationalise the optical behaviour of (Rmp)-2Ac and (Rmp)-2Ac2+, we examined its valence electronic structure, which is, as expected, dominated by orbitals localised at the TTF moiety (see Fig. S20†). Analysing the electronic transitions in the spectral region between 230 and 400 nm reveals that practically every excitation involves the TTF unit to some extent. While many transitions are of local nature, i.e., between orbitals in close proximity, quite a few display a charge-transfer-like behaviour (insets Fig. 4 and S21†). For neutral (Rmp)-2Ac, the vast majority of these transitions can be described by advancing an electron from an orbital centred at the TTF core, usually the HOMO, into an orbital located in another part of the rotaxane (e.g. the dimethoxy-phenyl moiety). For (Rmp)-2Ac2+, the corresponding transitions progress from some orbital in the molecule into an orbital localised at the TTF moiety, usually the LUMO or LUMO+1. This induces differently oriented magnetic dipole transition moments leading to different signs in the CD spectrum. Hence, we conclude that the sign inversion in the CD spectra upon oxidation can be exclusively attributed to the change of the electronic structure.
Conclusions
In conclusion, electrochemically switchable crown ether/ammonium [2]rotaxanes bearing a directional wheel are reported. The wheel features a redox-switchable TTF unit. The directionality had no observable impact on the electrochemical and optical properties of the racemic mixtures determined by UV/Vis spectroscopy and CV measurements. Instead, the pure enantiomers of the acetylated non-ionic derivatives display a redox-induced reversible inversion of the sign in the ECD spectrum without a change of absolute configuration. The mechanism and the absolute configuration of this chiroptical switch has been examined by computational methods. While co-conformational changes have hardly any impact on the ECD spectra, the changes in electronic structure induced by oxidation play a pivotal role. These results underline the impact of the mechanical bond, which allows the construction of intriguing switchable chemical assemblies with unexpected properties. This is the first in class example of a redox-controlled chiroptical switch with a complete sign reversal based on a mechanically planar chiral rotaxane. In the future, these properties could be employed in materials science to construct novel optoelectronic building blocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (CRC 765) and Academy of Finland (KR proj. no. 309399, AV proj. no. 314343) for funding. We are grateful to the Alexander von Humboldt-Foundation for support of KR (AvH research award). JA thanks the Studienstiftung des deutschen Volkes for a doctoral scholarship. Furthermore, we thank Dr Lucia Volbach for help with the chiral separation and Dr Rakesh Puttreddy for help with the crystallisation.Notes and references
- R. Breslow and Z. L. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9144–9146 CrossRef CAS PubMed.
- F. Jafarpour, T. Biancalani and N. Goldenfeld, Phys. Rev. E, 2017, 95, 032407 CrossRef PubMed.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2010, 2, a002147 Search PubMed.
- J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC.
- N. Koumura, R. W. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- B. L. Feringa, R. A. van Delden and M. K. J. ter Wiel, Pure Appl. Chem., 2003, 75, 563–575 CAS.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- J. E. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2016, 53, 298–312 RSC.
- C. Yamamoto, Y. Okamoto, T. Schmidt, R. Jäger and F. Vögtle, J. Am. Chem. Soc., 1997, 119, 10547–10548 CrossRef CAS.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- N. H. Evans, Chem.–Eur. J., 2018, 24, 3101–3112 CrossRef CAS PubMed.
- P. E. Glen, J. A. T. O'Neill and A.-L. Lee, Tetrahedron, 2013, 69, 57–68 CrossRef CAS.
- N. Kameta, K. Hiratani and Y. Nagawa, Chem. Commun., 2004, 466–467 RSC.
- J. Niemeyer and N. Pairault, Synlett, 2018, 29, 689–698 CrossRef.
- Y. Makita, N. Kihara, N. Nakakoji, T. Takata, S. Inagaki, C. Yamamoto and Y. Okamoto, Chem. Lett., 2007, 36, 162–163 CrossRef CAS.
- Y. Tachibana, N. Kihara, Y. Ohga and T. Takata, Chem. Lett., 2000, 29, 806–807 CrossRef.
- Y. Mochizuki, K. Ikeyatsu, Y. Mutoh, S. Hosoya and S. Saito, Org. Lett., 2017, 19, 4347–4350 CrossRef CAS PubMed.
- M. Asakawa, G. Brancato, M. Fanti, D. A. Leigh, T. Shimizu, A. M. Z. Slawin, J. K. Y. Wong, F. Zerbetto and S. Zhang, J. Am. Chem. Soc., 2002, 124, 2939–2950 CrossRef CAS PubMed.
- T. Ogoshi, D. Yamafuji, T. Aoki, K. Kitajima, T. A. Yamagishi, Y. Hayashi and S. Kawauchi, Chem.–Eur. J., 2012, 18, 7493–7500 CrossRef CAS PubMed.
- G. Bottari, D. A. Leigh and E. M. Pérez, J. Am. Chem. Soc., 2003, 125, 13360–13361 CrossRef CAS PubMed.
- P. R. Ashton, J. A. Bravo, F. M. Raymo, J. F. Stoddart, A. J. P. White and D. J. Williams, Eur. J. Org. Chem., 1999, 899–908 CrossRef CAS.
- Y.-G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2010, 43, 4070–4080 CrossRef CAS.
- S. Suzuki, F. Ishiwari, K. Nakazono and T. Takata, Chem. Commun., 2012, 48, 6478–6480 RSC.
- F. Ishiwari, K. Nakazono, Y. Koyama and T. Takata, Angew. Chem., Int. Ed., 2017, 56, 14858–14862 CrossRef CAS PubMed.
- N. Kameta, Y. Nagawa, M. Karikomi and K. Hiratani, Chem. Commun., 2006, 3714–3716 RSC.
- J. Y. C. Lim, I. Marques, V. Felix and P. D. Beer, Angew. Chem., Int. Ed., 2018, 57, 584–588 CrossRef CAS PubMed.
- K. Hirose, M. Ukimi, S. Ueda, C. Onoda, R. Kano, K. Tsuda, Y. Hinohara and Y. Tobe, Symmetry, 2018, 10, 20 CrossRef.
- Y. Tachibana, N. Kihara and T. Takata, J. Am. Chem. Soc., 2004, 126, 3438–3439 CrossRef CAS PubMed.
- R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed.
- M. A. Jinks, A. de Juan, M. Denis, C. J. Fletcher, M. Galli, E. M. G. Jamieson, F. Modicom, Z. Zhang and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 14806–14810 CrossRef CAS PubMed.
- S. Corra, C. de Vet, J. Groppi, M. La Rosa, S. Silvi, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2019, 141, 9129–9133 CrossRef CAS PubMed.
- C. E. Gell, T. A. McArdle-Ismaguilov and N. H. Evans, Chem. Commun., 2019, 55, 1576–1579 RSC.
- H. V. Schröder, H. Hupatz, A. J. Achazi, S. Sobottka, B. Sarkar, B. Paulus and C. A. Schalley, Chem.–Eur. J., 2017, 23, 2960–2967 CrossRef PubMed.
- H. V. Schröder, S. Sobottka, M. Nößler, H. Hupatz, M. Gaedke, B. Sarkar and C. A. Schalley, Chem. Sci., 2017, 8, 6300–6306 RSC.
- H. V. Schröder, J. M. Wollschläger and C. A. Schalley, Chem. Commun., 2017, 53, 9218–9221 RSC.
- H. V. Schröder, A. Mekic, H. Hupatz, S. Sobottka, F. Witte, L. H. Urner, M. Gaedke, K. Pagel, B. Sarkar, B. Paulus and C. A. Schalley, Nanoscale, 2018, 10, 21425–21433 RSC.
- H. V. Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 3496–3500 CrossRef PubMed.
- H. V. Schröder and C. A. Schalley, Beilstein J. Org. Chem., 2018, 14, 2163–2185 CrossRef PubMed.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- A. Jana, M. Ishida, J. S. Park, S. Bahring, J. O. Jeppesen and J. L. Sessler, Chem. Rev., 2017, 117, 2641–2710 CrossRef CAS PubMed.
- M. Fumanal, M. Capdevila-Cortada, J. S. Miller and J. J. Novoa, J. Am. Chem. Soc., 2013, 135, 13814–13826 CrossRef CAS PubMed.
- M. Yoshizawa, K. Kumazawa and M. Fujita, J. Am. Chem. Soc., 2005, 127, 13456–13457 CrossRef CAS PubMed.
- M. R. Bryce, J. Mater. Chem., 2000, 10, 589–598 RSC.
- D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC.
- J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS PubMed.
- A. Saad, F. Barriere, E. Levillain, N. Vanthuyne, O. Jeannin and M. Fourmigue, Chem.–Eur. J., 2010, 16, 8020–8028 CrossRef CAS PubMed.
- M. Hasegawa, J. Endo, S. Iwata, T. Shimasaki and Y. Mazaki, Beilstein J. Org. Chem., 2015, 11, 972–979 CrossRef CAS PubMed.
- F. Pop, S. Laroussi, T. Cauchy, C. J. Gomez-Garcia, J. D. Wallis and N. Avarvari, Chirality, 2013, 25, 466–474 CrossRef CAS PubMed.
- E. Gomar-Nadal, J. Veciana, C. Rovira and D. B. Amabilino, Adv. Mater., 2005, 17, 2095–2098 CrossRef CAS.
- Y. Zhou, D. Zhang, L. Zhu, Z. Shuai and D. Zhu, J. Org. Chem., 2006, 71, 2123–2130 CrossRef CAS PubMed.
- T. Biet, A. Fihey, T. Cauchy, N. Vanthuyne, C. Roussel, J. Crassous and N. Avarvari, Chem.–Eur. J., 2013, 19, 13160–13167 CrossRef CAS PubMed.
- F. Riobe and N. Avarvari, Chem. Commun., 2009, 3753–3755 RSC.
- P. R. Ashton, P. J. Campbell, P. T. Glink, D. Philp, N. Spencer, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Angew. Chem., Int. Ed., 1995, 34, 1865–1869 CrossRef CAS.
- T. Matsumura, F. Ishiwari, Y. Koyama and T. Takata, Org. Lett., 2010, 12, 3828–3831 CrossRef CAS PubMed.
- Y. Tachibana, H. Kawasaki, N. Kihara and T. Takata, J. Org. Chem., 2006, 71, 5093–5104 CrossRef CAS PubMed.
- S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2007, 129, 828–838 CrossRef CAS PubMed.
- M. B. Kirketerp, L. A. Leal, D. Varsano, A. Rubio, T. J. Jorgensen, K. Kilsa, M. B. Nielsen and S. B. Nielsen, Chem. Commun., 2011, 47, 6900–6902 RSC.
- V. Khodorkovsky, L. Shapiro, P. Krief, A. Shames, G. Mabon, A. Gorgues and M. Giffard, Chem. Commun., 2001, 2736–2737 RSC.
- T. Cauchy, F. Pop, J. Cuny and N. Avarvari, Chimia, 2018, 72, 389–393 CrossRef CAS PubMed.
- M. Fukui, T. Mori, Y. Inoue and R. Rathore, Org. Lett., 2007, 9, 3977–3980 CrossRef CAS PubMed.
- J. Deng, N. Song, Q. Zhou and Z. Su, Org. Lett., 2007, 9, 5393–5396 CrossRef CAS PubMed.
- T. Mori and Y. Inoue, J. Phys. Chem. A, 2005, 109, 2728–2740 CrossRef CAS PubMed.
- L. Pospisil, L. Bednarova, P. Stepanek, P. Slavicek, J. Vavra, M. Hromadova, H. Dlouha, J. Tarabek and F. Teply, J. Am. Chem. Soc., 2014, 136, 10826–10829 CrossRef CAS PubMed.
- E. Lee, H. Ju, I. H. Park, J. H. Jung, M. Ikeda, S. Kuwahara, Y. Habata and S. S. Lee, J. Am. Chem. Soc., 2018, 140, 9669–9677 CrossRef CAS PubMed.
- A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Gorecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052 RSC.
- T.-Y. Tai, Y.-H. Liu, C.-C. Lai, S.-M. Peng and S.-H. Chiu, Org. Lett., 2019, 21, 5708–5712 CrossRef CAS PubMed.
- C. Petermayer and H. Dube, J. Am. Chem. Soc., 2018, 140, 13558–13561 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures including full characterisation of new compounds, electrochemical data, crystallographic data and mass spectrometry data. CCDC 1910670. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03694f |
| This journal is © The Royal Society of Chemistry 2019 |