
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-functionalised graphene nanoflakes as tumour-targeting theranostic drug-delivery vehicles†
Jennifer
Lamb
a,
Eliane
Fischer
a,
Martin
Rosillo-Lopez
b,
Christoph G.
Salzmann
 b and
Jason P.
Holland
*a
b and
Jason P.
Holland
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057, Zurich, Switzerland. E-mail: jason.holland@chem.uzh.ch; Web: http://www.hollandlab.org Tel: +41 44 63 53 990
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 13th August 2019
Abstract
Graphene nanoflakes (GNFs) consist of a graphene sheet approximately 30 nm in diameter with a pristine aromatic system and an edge terminated with carboxylic acid groups. Their high water solubility and relative ease of functionalisation using carboxylate chemistry means that GNFs are potential scaffolds for the synthesis of theranostic agents. In this work, GNFs were multi-functionalised with derivatives of (i) a peptide-based Glu-NH-C(O)-NH-Lys ligand that binds prostate-specific membrane antigen (PSMA), (ii) a potent anti-mitotic drug (R)-ispinesib, (iii) the chelate desferrioxamine B (DFO), and (iv) an albumin-binding tag reported to extend pharmacokinetic half-life in vivo. Subsequent 68Ga radiochemistry and experiments in vitro and in vivo were used to evaluate the performance of GNFs in theranostic drug design. Efficient 68Ga-radiolabelling was achieved and the particle-loading of (R)-ispinesib and Glu-NH-C(O)-NH-Lys was confirmed using cellular assays. Using dose–response curves and FACS analysis it was shown that GNFs loaded with (R)-ispinesib inhibited the kinesin spindle protein (KSP) and induced G2/M-phase cell cycle arrest. Cellular uptake and blocking experiments demonstrated that GNFs functionalised with the Glu-NH-C(O)-NH-Lys ligand showed specificity toward PSMA expressing cells (LNCaP). The distribution profile and excretion rates of 68Ga-radiolabelled GNFs in athymic nude mice was evaluated using time–activity curves derived from dynamic positron-emission tomography (PET). Image analysis indicated that GNFs have low accumulation and retention in background tissue, with rapid renal clearance. In summary, our study shows that GNFs are suitable candidates for use in theranostic drug design.
Introduction
The underlying concept of a ‘theranostic’ drug is to combine chemotherapeutic and diagnostic capabilities in a single compound. Such an agent would allow the distribution of the drug to be tracked in real time and the dose delivered to the lesion to be determined accurately.1–3 In recent years, graphene-based nanomaterials have been studied for potential use in various biological applications.4–9 For instance, modified graphene nanomaterials and carbon nanotubes show potential in anti-cancer treatments, as drug delivery vehicles, and in diagnostic imaging.10–14 If these different applications can be combined, then graphene nanomaterials could be used in the synthesis of ‘theranostics’.15The covalent modification of graphene sheets and carbon nanotubes is chemically challenging. Disrupting the delocalised π-bonding network of the graphene sheet is thermodynamically unfavourable due to steric and electronic factors.16 Most reactions that lead to graphene or nanotube functionalisation overcome these issues by exposing the material to harsh reagents and conditions.17 As a result, the activation chemistry often forms a heterogeneous surface consisting of different graphene oxide (GO) and reduced graphene oxide (rGO) groups.18–20 Modification of the oxygen-containing functional groups on GOs and rGOs is relatively simple and the increased polarity improves water solubility. For example, Shi et al. modified rGO with polyethylene glycol (PEG), a tumour targeting vector, and a 64Cu-radiolabelled complex, creating a targeted positron emission tomography (PET) radiotracer.12
While GO and rGO are promising scaffolds for drug design, the heterogenous chemical nature of the graphene materials is an issue. Specifically, standard GO and rGO materials contain varying amounts of oxide, hydroxide and epoxide groups scattered throughout their structures. This makes it challenging to control the chemo- and regioselectivity of functionalisation reactions. In addition, heterogeneity in the starting materials hinders batch-to-batch reproducibility by increasing variability in the product.
In an effort to circumvent some of these issues, we recently synthesised edge-carboxylated graphene nanoflakes (GNFs) via the oxidative breakdown of chemical vapour deposition (CVD) multi-walled carbon nanotubes.21,22 Unlike GO and rGO materials, GNFs consist of a single graphene sheet approximately 30 nm in diameter with a pristine aromatic system. The GNF edge is terminated with carboxylic acid groups that can form carboxylic anhydrides (Fig. 1A).22 An atomic force microscopy image of GNFs spin-coated from aqueous dispersion onto highly oriented pyrolytic graphite is shown in Fig. 1B. The high solubility in water, negative zeta potential,22 and relative ease of functionalisation using carboxylate chemistry means that GNFs are potential platforms for the design of theranostic agents, where reactions can be restricted to the carboxylated edge.
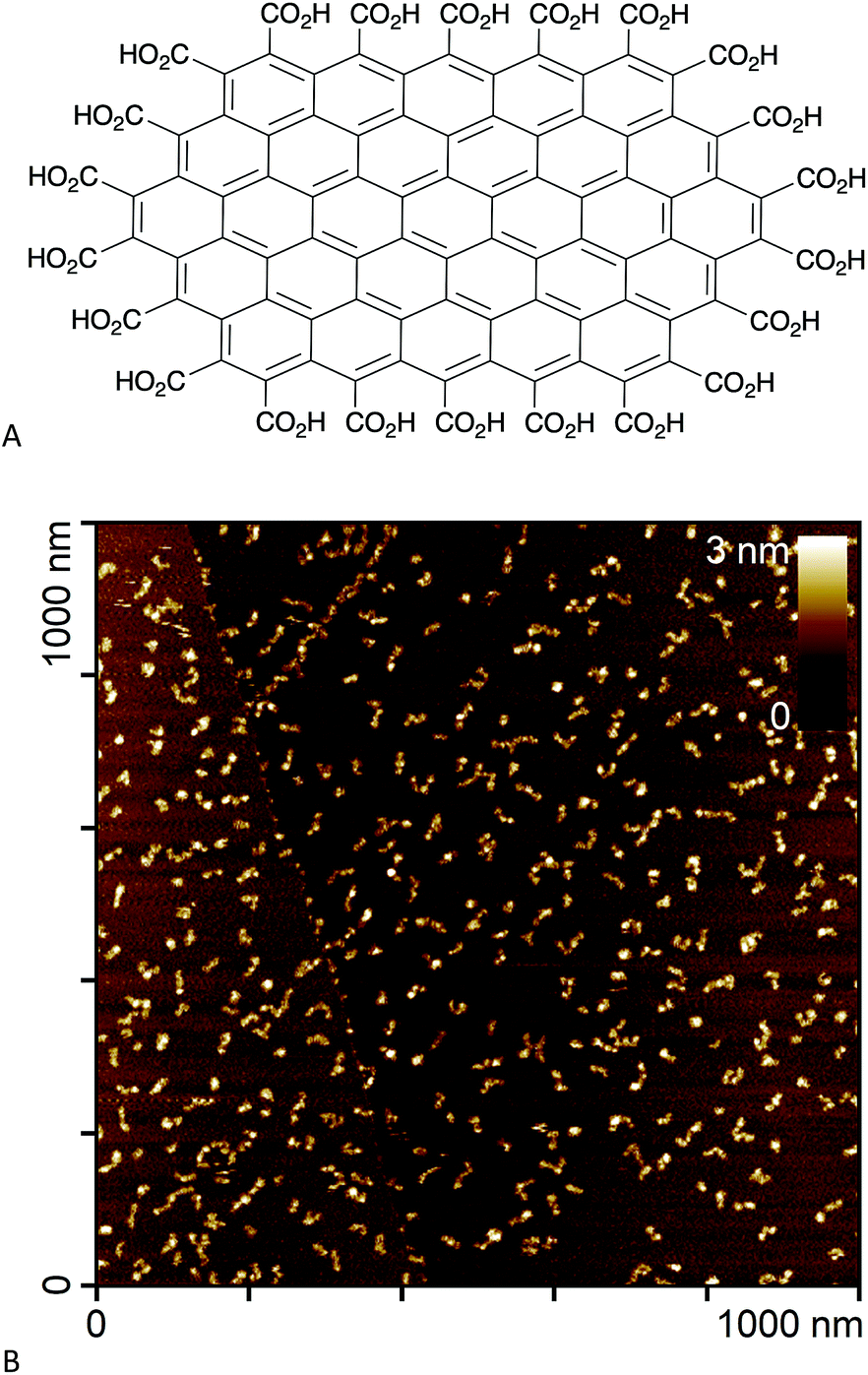 | ||
| Fig. 1 (A) Chemical structure of a small GNF (not to scale). (B) Atomic force microscopy image of GNFs spin-coated onto highly oriented pyrolytic graphite. | ||
In this work, we used pristine GNFs to synthesise a series of theranostic agents for targeted drug delivery and PET imaging. With this in mind, GNFs were decorated with several different components. Desferrioxamine B (DFO) is a well-known chelate for Fe3+, Zr4+ and Al3+ ions (among others) but also binds gallium with high affinity (formation constant, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β = 28.2).2368Ga is a positron emitting radionuclide suitable for PET imaging and is readily accessible via commercially available 68Ge/68Ga generators. (R)-Ispinesib is an extremely potent anti-mitotic, anti-cancer drug that targets the kinesin spindle protein (KSP); a motor protein that plays a critical role in mitosis by mediating centrosome separation and bipolar spindle formation.24 Inhibiting KSP causes cell cycle arrest at the mitotic (M) phase, eventually leading to apoptosis.25 Alongside its potency, (R)-ispinesib is a convenient choice of drug for GNF modification due to its free primary amine which lies outside of the KSP binding pocket and is available for chemical modification.24 An additional goal was to incorporate a biologically active vector that targets a characteristic protein on prostate cancer cells. For this purpose, we selected the urea-based Glu-NH-C(O)-NH-Lys group, which has a high affinity and specificity toward prostate-specific membrane antigen (PSMA). Finally, we also functionalised GNFs with a pharmacokinetic modifying group (an albumin binding tag) that alters the interaction with systemic proteins and can modulate distribution profiles in vivo. In summary, the synthesis, in vitro and in vivo evaluation of theranostic GNF vehicles with up to four different functionalities was explored.
β = 28.2).2368Ga is a positron emitting radionuclide suitable for PET imaging and is readily accessible via commercially available 68Ge/68Ga generators. (R)-Ispinesib is an extremely potent anti-mitotic, anti-cancer drug that targets the kinesin spindle protein (KSP); a motor protein that plays a critical role in mitosis by mediating centrosome separation and bipolar spindle formation.24 Inhibiting KSP causes cell cycle arrest at the mitotic (M) phase, eventually leading to apoptosis.25 Alongside its potency, (R)-ispinesib is a convenient choice of drug for GNF modification due to its free primary amine which lies outside of the KSP binding pocket and is available for chemical modification.24 An additional goal was to incorporate a biologically active vector that targets a characteristic protein on prostate cancer cells. For this purpose, we selected the urea-based Glu-NH-C(O)-NH-Lys group, which has a high affinity and specificity toward prostate-specific membrane antigen (PSMA). Finally, we also functionalised GNFs with a pharmacokinetic modifying group (an albumin binding tag) that alters the interaction with systemic proteins and can modulate distribution profiles in vivo. In summary, the synthesis, in vitro and in vivo evaluation of theranostic GNF vehicles with up to four different functionalities was explored.
Experimental
Full experimental methods and materials are reported in the ESI.†Results and discussion
Functionalisation of GNFs
Four different GNF constructs (GNF-1, -2, -3 and -4, Fig. 2) were synthesised to evaluate the use of GNFs as a platform for the construction of theranostic agents. GNF-1 was produced by pre-activating the carboxylated edge using HATU, and sequential conjugating to DFO via amide bond formation. The DFO chelate can be radiolabelled readily with 68Ga3+ ions using established methods, allowing the use of radiochemistry to determine the success of the chemical reaction.26 Radiolabelling (see below) confirmed that the DFO functionalisation reaction and the purification step were successful. With this information, we then used the same conjugation chemistry to functionalise GNFs with DFO, Glu-NH-C(O)-NH-Lys, and (R)-ispinesib to produce GNF-2. The free amine on all components allowed functionalisation via a one-pot, multi-component reaction. Purification was achieved on silica gel. Interestingly, when water is used as the mobile phase, the modified GNFs ran at the solvent front, allowing efficient separation from other compounds in the reaction mixture which were retained on silica. | ||
| Fig. 2 Schematic structures of [68Ga]GNF-1, -2, -3, and -4. In each case, the GNFs are functionalised with multiple copies of each component. (A) GNF with the chelate DFO. (B) GNF with DFO, (R)-ispinesib, and the PSMA ligand. (C) GNF with DFO, (R)-ispinesib, and the PEG4-PSMA ligand spaced with a PEG4 linker. (D) GNF with DFO, (R)-ispinesib, PEG4-PSMA, and an albumin binding tag. | ||
GNF-3 and GNF-4 were synthesised using the same chemistry. We hypothesised that by creating some distance between the GNF and Glu-NH-C(O)-NH-Lys, PSMA binding would be improved, and hence, increasing the specific uptake in target cells (LNCaP [PSMA +ve]). Therefore, GNF-3 included a PEG4 linker between the GNF and the PSMA ligand. The synthesis of Glu-NH-C(O)-NH-Lys-NH-C(O)-PEG4-NH2 (Scheme S1†) allowed the free amine to be used in the equivalent HATU-mediated amide bond formation as before.
Next, we speculated that the relatively small size, negative zeta (ζ) potential, and low lipophilicity of the GNFs would cause fast excretion of the compounds in vivo. The structure of GNF-4 sees the addition of N-(4-aminobutyl)-6-(4-iodophenyl)butylamide – a recognised affinity tag for albumin – in an attempt to modulate circulation times. Antibody fragments with similar derivatisation have been reported to bind albumin, and in turn, showed increased circulation half-life in vivo.27
Following the synthesis and isolation of the four GNF constructs, the ζ potentials were measured. GNF-1 and GNF-2 exhibited similar ζ potentials (−36.0 ± 4.87 and −39.5 ± 8.12 mV respectively) compared to non-modified GNFs (−37 ± 10.2 mV, note: this measured value is consistent with previous reports22). GNF-3 had a decreased ζ potential (−60.7 ± 10.3 mV) consistent with the additional PEG4 ether groups causing an increase in negative charge at the slipping plane. Despite also containing the PEG4 linker, GNF-4 displayed a less negative ζ potential (−42.3 ± 7.80 mV) which is consistent with addition of the neutral, lipophilic N-(4-aminobutyl)-6-(4-iodophenyl)butylamide group.
Radiochemistry
DFO modification allowed for efficient radiolabelling of GNF-1, -2, -3 and -4, using the same general procedure for each construct. An aliquot of stock [68Ga][Ga(H2O)6]Cl3(aq.) (∼40 MBq) was added to an aqueous solution of the functionalised GNFs (25 μg) buffered with NaOAc (0.2 M, pH 4.4), and the mixtures were incubated 10 min at 21 °C (n = 3 independent reactions per compound). Radiochemical reactions were monitored by using radioactive instant thin-layer chromatography (radio-iTLC) and size-exclusion chromatography (SEC) (Fig. 3). Radiochemical conversions (RCCs) for the synthesis of 68Ga-labelled GNF-1, -2, -3 and -4 were typically >97% (measured by radio-iTLC), with specific activities ranging from 8.7 ± 2.4 GBq mg−1 to 10.4 ± 3.0 GBq mg−1 (Table 1). Extending reaction times and heating the mixtures to 70 °C did not improve the percentage of radiochemical conversion. Chelate-free radiolabelling of rGO with 64Cu2+ has been reported.28 A chelate-free approach can be beneficial as the addition of chelates to a nanoparticle (or other biological vector) can often alter the properties of the graphene, hence changing results in vivo.29 The intrinsic radiolabelling of the GNFs with 68Ga was attempted, but no labelling was observed, which also provided further confirmation that the DFO functionalisation was successful.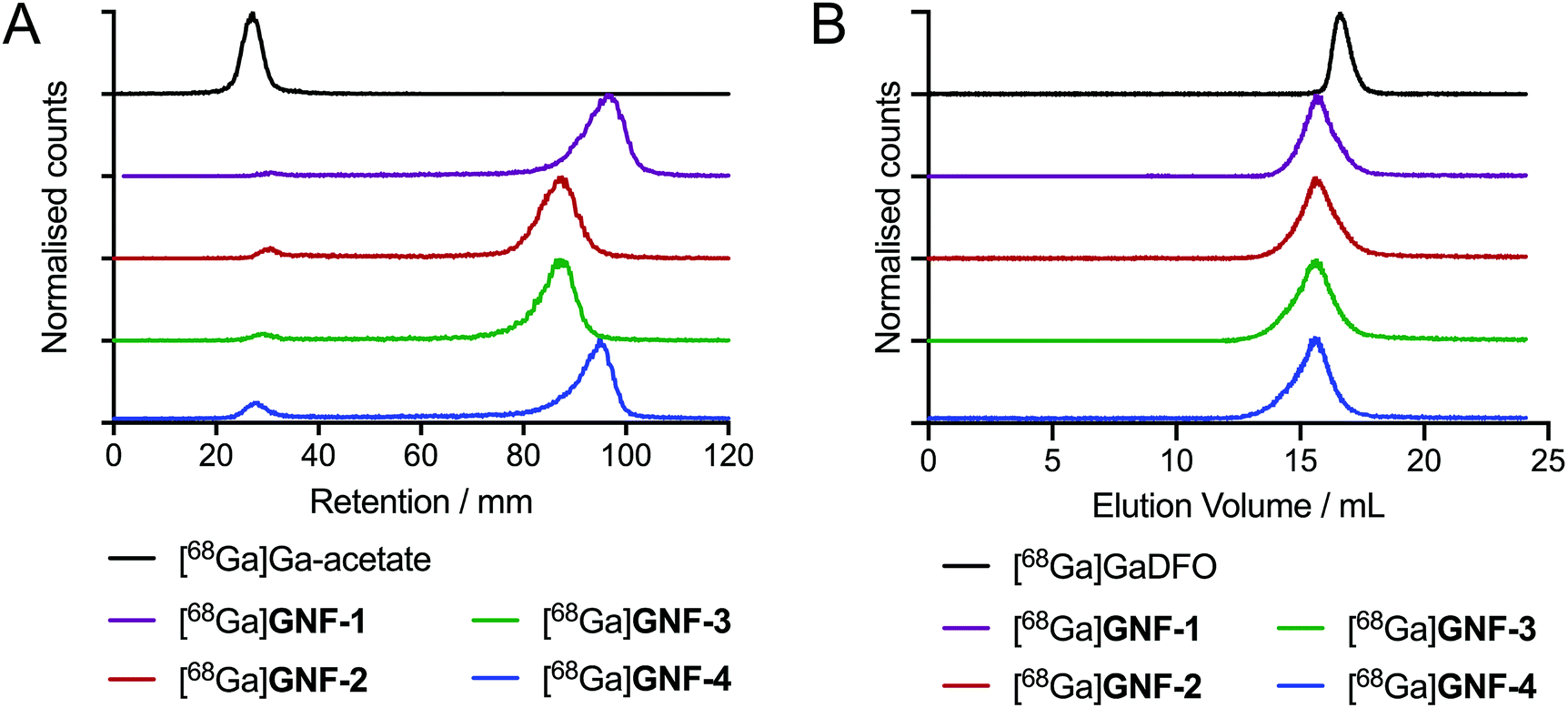 | ||
| Fig. 3 Radiochemical data on the 68Ga-radiolabelling of GNF-1 to -4. (A) Radio-iTLC with silica gel as stationary phase and H2O as mobile phase. (B) Radio-SEC-HPLC with 200 mM arginine in PBS as mobile phase. | ||
| [68Ga]GNF-1 | [68Ga]GNF-2 | [68Ga]GNF-3 | [68Ga]GNF-4 | |
|---|---|---|---|---|
| RCC/% | 99.1 ± 0.7 | 98.6 ± 0.2 | 97.4 ± 0.6 | 97.3 ± 0.4 |
| Specific activity/GBq mg−1 | 9.08 ± 2.59 | 8.79 ± 0.4 | 10.41 ± 2.98 | 8.7 ± 2.44 |
| Mole of DFO per mass of GNF/nmol mg−1 | 46.1 ± 13.1 | 44.6 ± 2.26 | 52.8 ± 15.1 | 44.4 ± 12.4 |
| logD value |
−3.34 ± 0.02 | −2.48 ± 0.01 | −3.14 ± 0.06 | −1.75 ± 0.07 |
| Zeta (ζ) potential/mV | −36.0 ± 4.87 | −39.5 ± 8.12 | −60.7 ± 10.3 | −42.3 ± 7.80 |
By using the experimentally measured molar activity of the [68Ga][Ga(H2O)6]Cl3(aq.) stock solutions, radioactive titrations allowed the quantification of the amount of DFO loaded per unit mass of GNF nanoparticle. Titration experiments showed that the functionalised compounds GNF-1, -2, -3, and -4 contained similar amount of DFO (ranging from ∼44–53 nmol DFO per mg GNF) indicating that the conjugation reactions proceeded with similar efficiency for each compound and that adding multiple components to the different GNF constructs did not compromise reaction efficiency (Table 1, Fig. S1†).
Estimates of the lipophilicity of the samples were obtained by measuring distribution coefficients (logD) using standard n-octanol/PBS (pH 7.4) partition experiments (Table 1).30 As expected, radiolabelled compounds [68Ga]GNF-1, -2, -3 and -4, were found to have negative logD values in the range −1.75 to −3.34. Functionalisation of the GNF nanoparticles with Glu-NH-C(O)-NH-Lys and (R)-ispinesib (GNF-2) reduced the hydrophilicity (logD value of −2.48 ± 0.01, n = 3), which suggests that the lipophilic quinazolinone drug was loaded onto the particles. For GNF-3, the presence of the PEG4-spacer between the nanoparticle and the Glu-NH-C(O)-NH-Lys group increased the hydrophilicity (logD = −3.14 ± 0.06, n = 3). Also, as anticipated, loading the nanoparticles with the N-(4-aminobutyl)-6-(4-iodophenyl)butylamide tag made construct GNF-4 less hydrophilic (logD = −1.75 ± 0.07, n = 3).
Stability studies in vitro confirmed that the 68Ga-radioactivity remained bound to the particles when incubated in solutions of saline (2 h), PBS (2 h) or human serum (2.5 h) at 37 °C (Tables S1 and S2, Fig. S2†).
Cellular studies
To determine the presence and activity of the (R)-ispinesib and Glu-NH-C(O)-NH-Lys on GNF-2, -3, and -4, a range of different cellular assays were performed. The PSMA expressing LNCaP cell line was used in all cell assays with PC-3 (PSMA negative) cells used in appropriate control experiments.To determine the success of the functionalisation of the GNF with (R)-ispinesib, we exposed cells to GNF-2 to observe the effect on cellular proliferation. GNF-2 exhibited a similar biphasic profile to the drug alone, indicating that the nanoflake was successfully modified and was also a potent inhibitor of cellular growth and proliferation (Fig. 4A). To determine if further functionalisation of a graphene nanoflake with PEG4 linkers and additional pharmacokinetic modifying groups (the albumin binder) had an impact on the anti-proliferative activity, equivalent MTT assays were conducted using GNF-3 and GNF-4 (Fig. 4B and C, respectively). Again, characteristic biphasic profiles were observed which suggested more extensive derivatisation of the nanoflakes did not alter the drug loading capacity or biological activity. Similar to (R)-ispinesib, two separate growth inhibition values could be extracted from the biphasic curves giving values centred in the micro- (GI50(1)/μg mL−1) and nano- (GI50(2)/ng mL−1) mass concentration ranges for GNF-2, -3 and -4 (Table 2). It is worth noting that anti-proliferation assays with (R)-ispinesib alone require the use of DMSO to solubilise the compound. The high aqueous solubility of GNFs meant that DMSO was unnecessary when using the GNF constructs. This fact provides additional support for development of nanoflakes as potential drug-delivery vehicles.
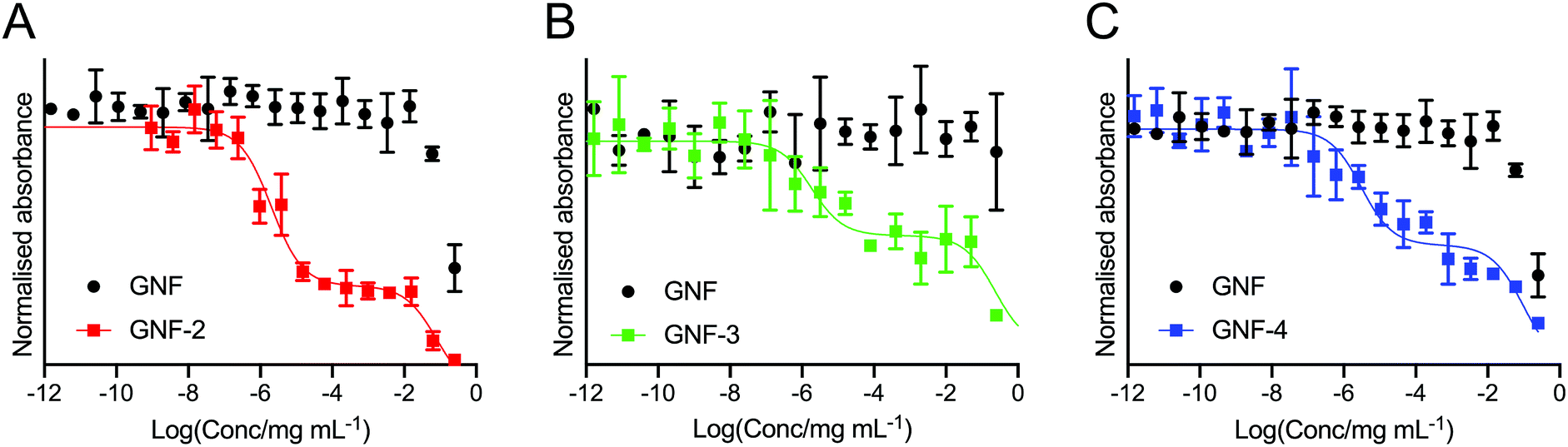 | ||
| Fig. 4 Cellular anti-proliferation (MTT) curves showing the response of LNCaP (PSMA +ve) cells to treatment with (A) GNF-2, (B) GNF-3, and (C) GNF-4. In each plot, the black data points correspond to treatment of the LNCaP cells with the native (non-functionalised) GNF in the same formulation as the active compounds. | ||
| GNF-2 | GNF-3 | GNF-4 | |
|---|---|---|---|
| log(GI50(1)/mg mL−1) | −1.11 ± 0.41 | −0.63 ± 0.50 | −1.02 ± 0.18 |
| log(GI50(2)/mg mL−1) | −5.70 ± 0.11 | −5.73 ± 0.73 | −5.52 ± 0.18 |
 | ||
| Fig. 5 Cell cycle analysis derived from FACS analysis with concentrations (A) 120 ng mL−1 and (B) 12 ng mL−1 of compounds. 2N refers to cells in the G0/G1 phase whilst 4N refers to cells in the G2/M phase. | ||
| Sample | Mass concentration/ng mL−1 | Cells associated with each cell cycle phase/% | ||
|---|---|---|---|---|
| G0/G1 | S | G2/M | ||
| Vehicle | N/A | 74.1 | 15.8 | 8.97 |
| GNF | 120 | 71.2 | 10.6 | 7.24 |
| 12 | 69.2 | 11.6 | 10.2 | |
| GNF-2 | 120 | 4.92 | 5.21 | 87.3 |
| 12 | 38.2 | 9.51 | 29.3 | |
| GNF-3 | 120 | 4.15 | 4.06 | 89.6 |
| 12 | 4.43 | 10.8 | 78.7 | |
| GNF-4 | 120 | 5.79 | 6.49 | 77.6 |
| 12 | 36.4 | 12.5 | 36.7 | |
 | ||
| Fig. 6 Confocal microscopy images showing the effects of treatment of LNCaP cells with GNF, GNF-2, GNF-3, GNF-4. Microtubules stained with Alexa Fluor 568 (red) and DNA stained with Hoechst 33342 (blue). (Inset) Scale bars correspond to 10 μm. | ||
 | ||
| Fig. 7 (A) Cellular binding assay of [68Ga]GNF-1–4 with the LNCaP (PSMA +ve) and PC-3 (PSMA −ve) cell lines. Data given as the percentage of activity bound normalised per 1 mg mL−1 of total protein. All cell media contained 0.1% azide and protein content was measured using a BCA assay. For the LNCaP cell line: [68Ga]GNF-1vs. [68Ga]GNF-2, p ≤ 0.01; [68Ga]GNF-1vs. [68Ga]GNF-3p ≤ 0.001; [68Ga]GNF-1vs. [68Ga]GNF-4, p ≤ 0.0001. Comparison of cell lines: PC-3 vs. LNCaP [68Ga]GNF-2, p ≤ 0.01; [68Ga]GNF-3, p ≤ 0.001; and [68Ga]GNF-4, p ≤ 0.0001. (B) Blocking assay with the LNCaP (PSMA +ve) cell line pre-treated with free Glu-NH-C(O)-NH-Lys ligand (5 μM) before addition of radiotracers. Note: Student's t-test analysis: ns = not significant, * = P-value < 0.05, ** = P-value < 0.01, *** = P-value < 0.001. | ||
We hypothesised that increasing the distance between the GNFs and the Glu-NH-C(O)-NH-Lys targeting vector using a PEG4 linker may increase the cellular uptake and specificity for LNCaP cells. However, no difference was observed between the cell-associated uptake of [68Ga]GNF-2 and that [68Ga]GNF-3 (Fig. 7A). It is possible that the short PEG4 chain is insufficient to shift the uptake mechanism away from non-specific accumulation and toward specific PSMA-mediated binding. Also of note, the slightly higher cell-associated activity observed for [68Ga]GNF-4 in LNCaP cells was simply attributed to an increased non-specific uptake which was also seen in the data using PC-3 cells.
In vivo studies
Next, to gauge the potential of using nanoflakes in vivo, we performed dynamic PET imaging to measure the pharmacokinetic (PK) distribution and excretion profiles of [68Ga]GNF-3 and [68Ga]GNF-4 in a limited number of athymic nude mice bearing subcutaneous LNCaP tumours (n ≥ 3 mice per compound). The main goal was to measure time–activity curves to obtain a baseline PK profile of GNFs, and to evaluate if functionalisation of the GNF constructs with an albumin binding tag could modulate blood pool concentrations, circulation times and excretion pathways.Pilot PET imaging experiments indicated that the small size and high hydrophilicity of the [68Ga]GNF-3 and [68Ga]GNF-4 facilitated rapid clearance from the blood pool by a renal excretion pathway and elimination through the bladder (Fig. 8). The observation that [68Ga]GNF-3 and [68Ga]GNF-4 are cleared through the kidneys is an interesting finding that may help minimise radiation burden to background tissues and facilitate further optimisation of radiotracers based on GNFs. Time–activity curves (TACs) showed [68Ga]GNF-3 was essentially first-pass extracted from the blood pool by the kidneys. Peak uptake in the kidney reached 38 ± 4% ID cm−3 within in the first two minutes post-administration. Concordantly, accumulation of 68Ga-radioactivity in the bladder increased immediately after injection confirming that the [68Ga]GNF-3 particles are not retained in the kidney (Fig. 8B and S4A†). In contrast, the peak in radioactivity in the kidneys for [68Ga]GNF-4 occurred between 4–6 minutes post-administration with a similar time delay observed before activity was seen to accumulate in the bladder (Fig. 8C and S4B†). For both [68Ga]GNF-3 and [68Ga]GNF-4, blood pool activity (as measured by drawing volumes of interest over the left ventricle of the heart) showed a peak in the first frame after bolus injection of the radiotracer, and a rapid decrease over the imaging time window. In comparison to [68Ga]GNF-3, the slight delay in extraction of the [68Ga]GNF-4 activity from the blood pool by the kidney, gave a higher blood pool peak activity of 48 ± 5% ID cm−3. Kinetic analysis of the TACs found that the effective half-life of [68Ga]GNF-3 in the blood pool (heart) was 3.85 ± 0.35 min whereas for [68Ga]GNF-4 the half-life increased to 5.22 ± 0.61 min (p < 0.05). These TAC data indicate that modification of the GNF particles with the albumin binding tag has a small but significant effect on prolonging the circulation time and reducing the elimination rate of [68Ga]GNF-4.
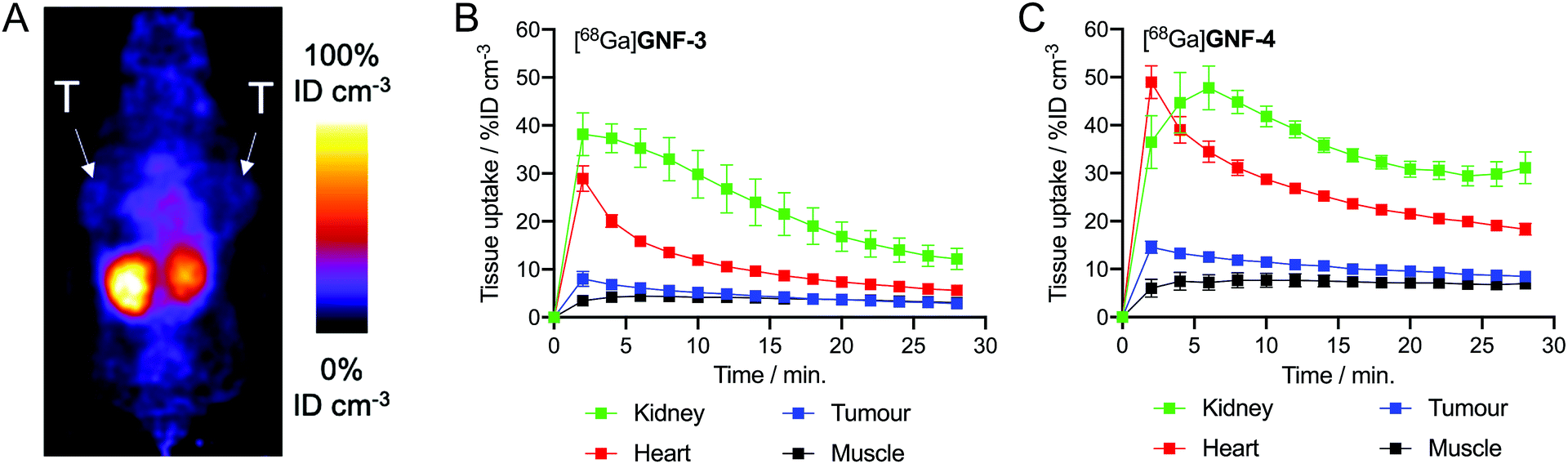 | ||
| Fig. 8 PET images recorded in mice bearing LNCaP tumours, T = tumour. (A) 2 h coronal PET image recorded following injection with [68Ga]GNF-4. TACs plotted with data extracted from 30 min dynamic PET analysis (15 × 2 min scans) with injection of (B) [68Ga]GNF-3 (n = 3) and (C) [68Ga]GNF-4 (n = 4) at t = 2 min VOI analysis was performed using VivoQuant™. | ||
For both [68Ga]GNF-3 and [68Ga]GNF-4, tumour-associated radioactivity was found to be low and non-specific. The TAC profiles of VOIs drawn over the tumours showed that the PK profile followed the same trend as the blood pool, indicating that the constructs do not show specific accumulation in the tumours on the time scale of the experiment. Factors leading to this lack of specific tumour uptake include the relatively high component of non-specific binding found for GNFs on LNCaP cells, the rapid excretion profile, and potentially, a reduced access or affinity of the Glu-NH-C(O)-NH-Lys binding ligand for the PSMA target when bound to the GNF particles. In spite of the absence of tumour uptake, the PET images provide valuable information which will aid further optimisation of the GNFs as potential drug delivery vehicles and theranostic agents. In particular, modification of the particles with higher affinity albumin tags or an increased number of tags per particle may increase circulation times, and more work on improving the cellular binding profile will likely allow increased cellular uptake and specificity.
Conclusions
GNFs were used as design scaffolds for the synthesis of targeted theranostic agents. Multi-functionalised GNFs can be prepared with a diverse array of compounds including metal-binding chelates, tumour-targeting ligands, cytotoxic drugs and pharmacokinetic modification groups. Derivatisation of GNFs with the potent drug molecule (R)-ispinesib gave constructs that remained pharmacologically active, and cellular assays confirmed that the mode of action was consistent with mitotic phase arrest induced by KSP motor protein inhibition. An advantage of using GNFs as a drug carrier was that it helped to solubilise (R)-ispinesib in water. In addition, GNF particles functionalised with the Glu-NH-C(O)-NH-Lys group exhibited enhanced cellular uptake. Although a significant fraction of cellular association was assigned to non-specific interactions, it was found that targeted GNFs showed specificity toward PSMA expressing cells. Dynamic PET imaging found that functionalised GNFs have rapid blood pool clearance and renal excretion, but experiments also demonstrated that functionalising with an albumin-binding tag resulted in a significant modulation of the pharmacokinetic profile. Further optimisation is required to enhance the uptake kinetics and distribution in vivo but our study supports the conclusion that GNFs are a highly versatile platform for theranostic drug development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JPH thanks the Swiss National Science Foundation (SNSF Professorship PP00P2_163683), the Swiss Cancer League (Krebsliga Schweiz; KLS-4257-08-2017), and the University of Zurich (UZH) for financial support. This project has received funding from the European Union's Horizon 2020 research and innovation programme/from the European Research Council under the Grant Agreement No. 676904, ERC-StG-2015, NanoSCAN. We thank all members of the Radiochemistry and Imaging Science group at UZH for helpful discussions. We thank Florian Gribi for synthesis of (R)-ispinesib. CGS thanks the Royal Society for a University Research Fellowship (UF100144) and the European Research Council for funding under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 725271).Notes and references
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- F. Chen, E. B. Ehlerding and W. Cai, J. Nucl. Med., 2014, 1919–1923 CrossRef CAS PubMed.
- S. Goel, C. G. England, F. Chen and W. Cai, Adv. Drug Delivery Rev., 2017, 113, 157–176 CrossRef CAS PubMed.
- S. Priyadarsini, S. Mohanty, S. Mukherjee, S. Basu and M. Mishra, J. Nanostruct. Chem., 2018, 8, 123–137 CrossRef CAS.
- X. Ding, H. Liu and Y. Fan, Adv. Healthcare Mater., 2015, 4, 1451–1468 CrossRef CAS PubMed.
- E. K. Wujcik and C. N. Monty, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 233–249 CAS.
- F. Menaa, A. Abdelghani and B. Menna, J. Tissue Eng. Regener. Med., 2012, 9, 1321–1338 CrossRef PubMed.
- S. Kumar and K. Chatterjee, ACS Appl. Mater. Interfaces, 2016, 8, 26431–26457 CrossRef CAS PubMed.
- Kenry, W. C. Lee, K. P. Loh and C. T. Lim, Biomaterials, 2018, 155, 236–250 CrossRef CAS PubMed.
- M. Wojtoniszak, K. Urbas, M. Peruzynska, M. Kurzawski, M. Drozdzik and E. Mijowska, Chem. Phys. Lett., 2013, 569, 151–156 CrossRef.
- J. Chen, S. Chen, X. Zhao, L. V. Kuznetsova, S. S. Wong and I. Ojima, J. Am. Chem. Soc., 2008, 130, 16778–16785 CrossRef CAS PubMed.
- S. Shi, K. Yang, H. Hong, H. F. Valdovinos, T. R. Nayak, Y. Zhang, C. P. Theuer, T. E. Barnhart, Z. Liu and W. Cai, Biomaterials, 2013, 34, 3002–3009 CrossRef CAS PubMed.
- A. Ramachandra, K. Sasikala, R. G. Thomas, A. R. Unnithan, B. Saravanakumar and Y. Y. Jeong, Nat. Publ. Gr., 2016, 6, 1–14 Search PubMed.
- S. Y. Hong, G. Tobias, K. T. Al-Jamal, B. Ballesteros, H. Ali-Boucetta, S. Lozano-Perez, P. D. Nellist, R. B. Sim, C. Finucane, S. J. Mather, M. L. H. Green, K. Kostarelos and B. G. Davis, Nat. Mater., 2010, 9, 485–490 CrossRef CAS PubMed.
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- J. Johns and M. Hersam, Acc. Chem. Res., 2014, 46, 77–86 CrossRef PubMed.
- B. C. G. Salzmann, S. A. Llewellyn, G. Tobias, M. A. H. Ward, Y. Huh and M. L. H. Green, Adv. Mater., 2007, 19, 883–887 CrossRef.
- J. Zhu, J. Hiltz, M. A. Mezour, V. Bernard-gauthier, R. B. Lennox and R. Schirrmacher, Chem. Mater., 2014, 26, 5058–5062 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4086–4814 CrossRef PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- C. G. Salzmann, V. Nicolosi and M. L. H. Green, J. Mater. Chem., 2010, 20, 314–319 RSC.
- M. Rosillo-lopez, J. Lee, M. Bella, M. Hart and C. G. Salzmann, RSC Adv., 2015, 5, 104198–104202 RSC.
- A. Evers, R. D. Hancock, A. E. Martell and R. J. Motekaitis, Inorg. Chem., 1989, 27, 2189–2195 CrossRef.
- J. P. Holland, M. W. Jones, S. Cohrs, R. Schibli and E. Fischer, Bioorg. Med. Chem., 2013, 21, 496–507 CrossRef CAS PubMed.
- V. Sarli and A. Giannis, Clin. Cancer Res., 2008, 14, 7583–7588 CrossRef CAS PubMed.
- E. Gourni, L. Del Pozzo, M. Bartholoma, Y. Kiefer, P. T. Meyer, H. R. Maecke and J. P. Holland, Mol. Imaging, 2017, 16, 1–11 CrossRef CAS PubMed.
- S. Trussel, C. Dumelin, K. Frey, A. Villa, F. Buller and D. Neri, Bioconjugate Chem., 2009, 20, 2286–2292 CrossRef CAS PubMed.
- S. Shi, C. Xu, K. Yang, S. Goel, H. F. Valdovinos, H. Luo, E. B. Ehlerding, C. G. England, L. Cheng, F. Chen, R. J. Nickles, Z. Liu and W. Cai, Angew. Chem., Int. Ed., 2017, 56, 2889–2892 CrossRef CAS PubMed.
- J. Lamb and J. P. Holland, J. Nucl. Med., 2018, 59, 382–390 CrossRef CAS PubMed.
- A. A. Wilson, L. Jin, A. Garcia, J. N. Dasilva and S. Houle, Appl. Radiat. Isot., 2001, 54, 203–208 CrossRef CAS PubMed.
- M. Venere, C. Horbinski, J. F. Crish, X. Jin, A. Vasanji, J. Major, A. C. Burrows, C. Chang, J. Prokop, Q. Wu, P. A. Sims, P. Canoll, M. K. Summers, S. S. Rosenfeld and J. N. Rich, Sci. Transl. Med., 2015, 7, 1–12 Search PubMed.
- L. Lad, L. Luo, J. D. Carson, K. W. Wood, J. J. Hartman, R. A. Copeland and R. Sakowicz, Biochemistry, 2008, 47, 3576–3585 CrossRef CAS PubMed.
- B. T. Ristau, D. S. O'Keefe and D. J. Bacich, Urol. Oncol., 2014, 32, 272–279 CrossRef PubMed.
- A. Afshar-Oromieh, A. Malcher, M. Eder, M. Eisenhut, H. G. Linhart, B. A. Hadaschik, T. Holland-Letz, F. L. Giesel, C. Kratochwil, S. Haufe, U. Haberkorn and C. M. Zechmann, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, 486–495 CrossRef CAS PubMed.
- A. Afshar-oromieh, J. W. Babich, C. Kratochwil, F. L. Giesel, M. Eisenhut, K. Kopka and U. Haberkorn, J. Nucl. Med., 2016, 57, 79–89 CrossRef PubMed.
- K. Kopka, M. Benesova, C. Ba, U. Haberkorn and J. W. Babich, J. Nucl. Med., 2019, 58, 17–26 CrossRef PubMed.
- M. Eder, M. Schäfer, U. Bauder-Wüst, W. Hull, C. Wängler, W. Mier, U. Haberkorn and M. Eisenhut, Bioconjugate Chem., 2012, 23, 688–697 CrossRef CAS PubMed.
- R. A. Faouri, R. Henry, A. S. Biris, R. Sleezer and G. J. Salamo, J. Appl. Toxicol., 2017, 37, 1346–1353 CrossRef PubMed.
- V. Castagnola, W. Zhao, L. Boselli, M. C. Lo Giudice, F. Meder, E. Polo, K. R. Paton, C. Backes, J. N. Coleman and K. A. Dawson, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed methods, equipment and characterisation data are provided. See DOI: 10.1039/c9sc03736e |
| This journal is © The Royal Society of Chemistry 2019 |