
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic anti-counterfeiting security features using multicolor dianthryl sulfoxides†
Jennifer
Yuan
 ,
Peter R.
Christensen
and
Michael O.
Wolf
*
,
Peter R.
Christensen
and
Michael O.
Wolf
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: mwolf@chem.ubc.ca
First published on 18th September 2019
Abstract
A new concept for difficult-to-replicate security inks for use in advanced anti-counterfeiting applications is presented. Inks fabricated from a mixture of photoactive dyes result in a unique fluorescent color upon irradiation that differs from the starting fluorescence. The dyes are substituted 9,9′-dianthryl sulfoxides that undergo photochemical extrusion of a sulfoxide moiety (SO) to produce emissive red, blue, and green emitters. The resulting emissive feature has specific Commission international de l'éclairage (CIE) coordinates that are used for authentication. Additionally, the temporal evolution of the fluorescence can be monitored, introducing a dynamic nature to these security features. The three compounds show different rates of photoconversion dependent on the irradiation wavelength, allowing selective wavelengths for activation to be used for additional security. CIE coordinates can be extracted from patches containing the three compounds using an inexpensive, commercially available smartphone application (app) and compared against a known value to confirm the validity of the method.
Introduction
Counterfeit drugs can impose serious health complications to patients that are administered medicines with incorrect active ingredients or fake formulations.1–5 Genuine drug packaging is often labelled with basic security features such as watermarks, holograms and metal threads, where visual inspection of the marker can be used to verify products. Drugs can also be tracked using more sophisticated technology such as radio frequency identification (RFID).6,7 Improvements in computers, scanners and cameras have allowed counterfeiters to duplicate packaging details with great accuracy. Pharmaceutical counterfeiting is more common for products sold through an online marketplace, where it is estimated that 50% of drugs sold are counterfeit.8–10 Counterfeit drugs can enter the legitimate supply and distribution chain at any stage, therefore points of authentication should be implemented at multiple locations. Currently, products are readily validated at both the manufacturer and retailer stage.11 Items are marked with serial numbers at dispatch and checked systematically for inventory. Consumer, or end-user authentication is less common.12 Among the large selection of anti-counterfeiting technology currently available, molecular tags such as DNA and peptides present an interesting option.13–16 Their defined sequence acts as a molecular barcode and can be used in trace amounts to label and protect pharmaceuticals. However, these forensic methods are often susceptible to chemical and thermal degradation and must be identified through sequencing and mass spectrometry, which require specialized equipment and are typically not possible for the average consumer. Physical unclonable functions (PUF) or “digital fingerprints” are patterns of physical tags generated in a stochastic process and can be manufactured with chemical systems.11,17–19 As a result, PUF keys are intrinsically unique and difficult to predict, offering high security and are commonly used in cryptography. PUF keys are safe, but tedious, requiring a high level of expertise to decode, limiting practicality. There is a continuing need for new anti-counterfeiting materials and technologies that are difficult for even experienced counterfeiters to reproduce but are simple enough to be decoded by the general public at the point of sale.Luminescent materials offer a low-cost and high-throughput solution for advanced anti-counterfeiting.11,20 Fluorescent security inks can be used to conceal information under ambient lighting, where exposure to UV light results in photoluminescence (PL), which can be detected optically. Additionally, fluorescence can be read and decoded using the CCD camera of a smartphone which reduces the need for expensive and complex instruments for validation. A variety of inorganic complexes and nanomaterials have been studied for this application. For example, lanthanide complexes are recognized for their strong, narrow emission bands, pure colors and are currently used for banknote security.21–25 Quantum and carbon dots and other nanomaterials such as perovskite nanocrystals and metal–organic frameworks (MOFs) have also been utilized due to their unique emission characteristics such as upconversion, temperature-dependent and triple-mode emission.26–35
Typical covert designs employ small molecules that emit a single color, and in rare cases show dual-color emission.36–43 A significant disadvantage, however, to implementing fluorescent response as an anti-counterfeiting tool is their predictable output, which can be imitated by fluorescent molecules having similar emission characteristics. Stimuli-responsive conjugated molecular and polymeric systems have been put forward as options for the next-generation of security printing.44–52 Functional materials that respond to external stimuli such as light, heat or pressure are ideal candidates for protecting information and products because they offer an added level of complexity in security features as this behavior is more difficult to reproduce.53,54
We propose a new conceptual approach to luminescent security inks that are difficult to replicate, using a mixture of organic-based light responsive dyes which upon activation reveal a unique fluorescent color with specific Commission international de l'éclairage (CIE) coordinates. Compared to traditional authentication technology where information is stored in a marker such as a barcode, in this case information concerning the authenticity of an item is found in the resulting color, quantified by its CIE coordinates. The spectral response or emission color, upon activation, is dependent on the ratio of the compounds used initially and can vary depending on several external factors such as the irradiation wavelength of the activation source and duration of exposure. The compounds incorporate a second security feature in which the temporal evolution of the emission color during the activation process may be monitored visually. This can impart dual functionality, where the resulting fluorescence color and specific trajectory of color evolution is novel to this system. This dynamic feature utilizing photochemistry for security inks could be difficult to replicate, making this an attractive system for protecting high-value products.
This concept of temporal fluorescence evolution is based on photochemical behavior found in sulfur-bridged dianthracenes that is dependent on the oxidation state of the sulfur bridge.55,56 The weakly emissive 9,9′-dianthryl sulfoxide (1a) eliminates the SO bridge upon photoirradiation to form the strongly blue emissive molecule 9,9′-bianthracene (1b) (Fig. 1) either in solution, or in polymer host matrices with glass transition temperatures (Tg) below room temperature, such as poly(butylmethacrylate).57 The photochemical conversion of 1a to 1b can be modulated through UV exposure. As the overall emission intensity (i.e. a mixture containing 1a and 1b) can be controlled by length of irradiation time, inks containing 1a can be used to create photopatterned fluorescent images.57 We previously used this photochemical phenomenon to generate variable intensity security features. By doing so, we incorporated a third dimension into 2D barcodes, where the positional information can vary in intensity increasing the complexity of this straightforward technology. Lastly, we showed that fluorescence intensity can be extracted using a CCD detector found on a smartphone. Given the simplicity of this system, we have now expanded the scope of dyes to achieve full-color fluorescence. Red-green-blue (RGB) emission is required for reproduction of secondary colors within the gamut of the three primary colors.58
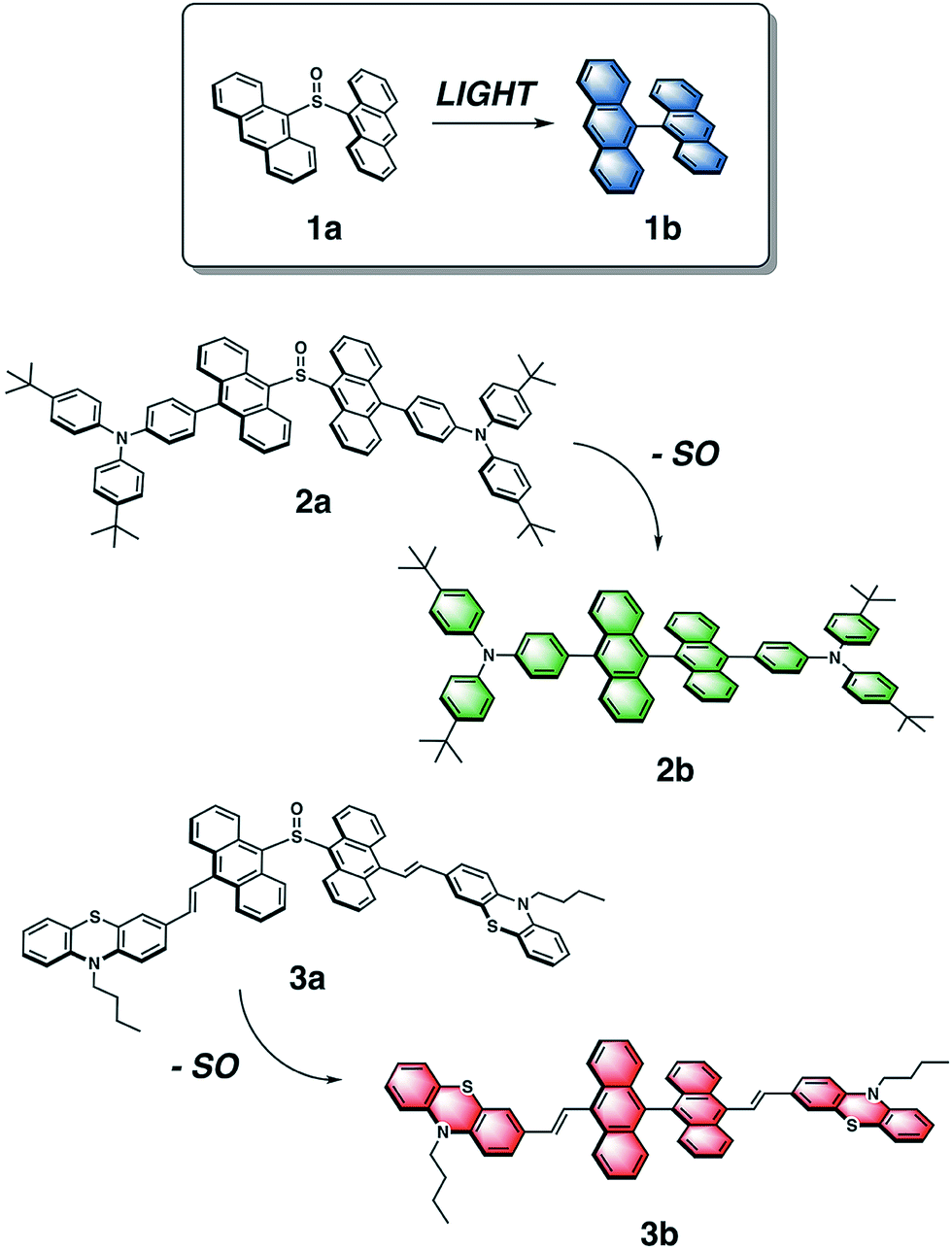 | ||
| Fig. 1 Chemical structures of dianthryl sulfoxides 1a, 2a and 3a, which can be converted to the respective bianthracenes 1b, 2b, 3b through light irradiation. | ||
To vary the emission color of bianthracene derivatives resulting from the photochemical extrusion of SO, we prepared two new chromophores, 2a and 3a, based on the sulfoxide-bridged dianthracene core used in 1a (Fig. 1). Based on reported bianthracenes known to have green and red emission, substitution at the 10,10′-positions of the dianthracene core with triarylamine and phenothiazine groups afford the desired green and red-emissive photoproducts (2b and 3b) upon elimination of the SO bridge when irradiated with light. The photochemical conversion kinetics for each dimer are dependent on the irradiation wavelength used for activation. Furthermore, we demonstrate the ability to modulate the chromaticity coordinates (x, y) of thin films containing varying compositions with different mixing ratios of dianthryl sulfoxides, based on the duration of light exposure demonstrating the temporal evolution of the emission color of this mixture. Lastly, we show that CIE coordinates can be extracted using an inexpensive, commercially available smartphone application (app) and compared against a known value to confirm the validity of the method. Utilizing the photochemistry of dianthryl sulfoxides is a simple yet effective strategy for the fabrication of security features which can be used for advanced anti-counterfeiting.
Experimental
General experimental considerations and synthetic details are described in the ESI.†Results and discussion
The emission color of the photoproduct is tunable based on functionalization of its bianthracene core. Triarylamine and phenothiazine are two common building blocks frequently used in the synthesis of organic luminescent materials. They were chosen to shift the emission spectrum of bianthracene into the green and red region based on reports by Schmittel59 and Lu,60 respectively. The general reaction scheme for the synthesis of substituted dianthryl sulfoxides is presented in the ESI (Scheme S1†). 9,10′-Dibromoanthracene was asymmetrically coupled to triarylamine 6via Suzuki cross-coupling to afford 7 in 75% yield. Likewise, vinyl-substituted phenothiazine 10 was installed via Heck cross-coupling to give a 54% yield of 11. Dimerization of the anthracene moieties 7 and 11 was achieved using two equivalents of the respective 9-anthryllithium and one equivalent of dimethyl sulfite as the electrophilic sulfoxide source to generate the desired sulfoxide-bridged species, 2a and 3a, in fair to good yields. The final products were purified by column chromatography in the dark with minimal light exposure and characterized using 1H, 13C{1H} NMR and IR spectroscopies, and high-resolution mass spectrometry (HR-MS). This methodology can be extended to other derivatives where donors can be easily incorporated synthetically by coupling reactions, followed by subsequent dimerization with dimethyl sulfite affords a direct one-step route to new substituted dianthryl sulfoxides.The photochemistry of 2a and 3a was probed by irradiating solutions of each dimer with 365 nm and 400 nm light in CDCl3 and C6D6, respectively, for approximately 30 min. The reactivity of 2a and 3a was monitored using 1H NMR spectroscopy (Fig. S13†). The 1H NMR spectra show the formation of new photoproducts, 2b and 3b. The photoproducts were confirmed as the corresponding bianthracene compounds 2b and 3b based on comparison with the spectra of closely related molecules and HR-MS data.59,60
The elimination of the SO bridge from 2a and 3a was also monitored by UV-vis and PL spectroscopies. The changes in UV-vis absorption for 2a and 3a in dilute CH2Cl2 solutions (4 × 10−6 M) is shown in Fig. 2. Initially, compounds 2a and 3a both exhibit broad, long wavelength absorption bands from 350–500 nm in CH2Cl2. Neither compound exhibits the characteristic long-wavelength structured bands that are attributed to π → π* transitions of the anthracene core.61 Introduction of the extended conjugation from triarylamine and phenothiazine donors results in a red-shift in absorption maximum in the visible region relative to the unsubstituted compound. After irradiation with 365 nm light, an increase in vibrational structure is observed in the absorption spectrum of 2a with an isosbestic point at 416 nm, consistent with the results observed in the formation of 1b from 1a.55 Compound 3a shows a blue-shift and small increase in intensity after irradiation, and 3b possesses a broad band centered at 419 nm in agreement with the spectra of reported derivatives.60
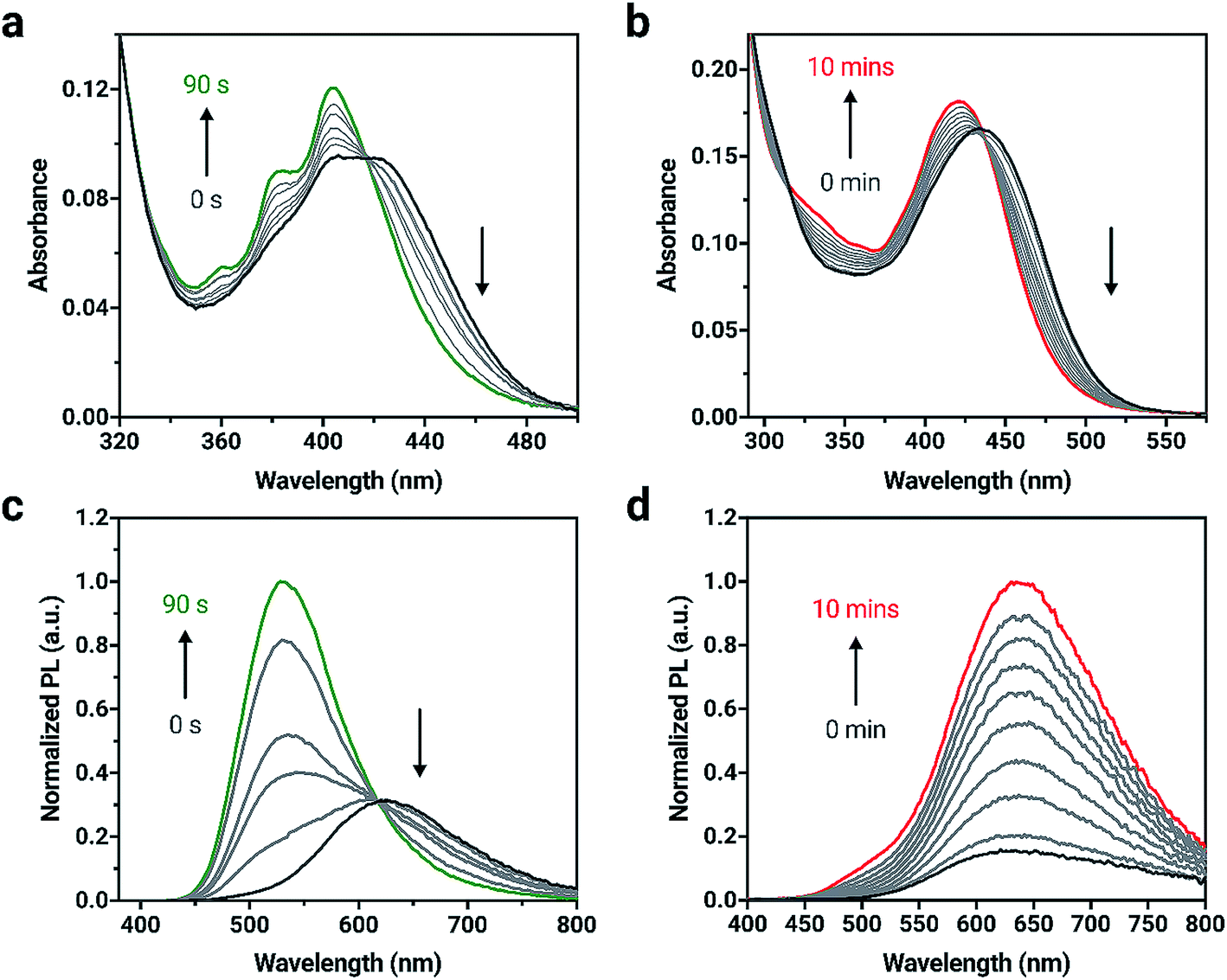 | ||
| Fig. 2 Changes in UV-vis absorption spectra for (a) 2a and (b) 3a when irradiated with 365 nm light in CH2Cl2. Changes in PL spectra for (c) 2a and (d) 3a when irradiated in CH2Cl2 (excitation wavelength = 365 nm). Irradiation was carried out in 10 to 30 s intervals for 2a and 1 min intervals for 3a until no further changes in the absorption spectrum were observed. Concentration = 4 × 10−6 M. | ||
Upon excitation with 365 nm light, both 2a and 3a initially show orange emission centered at 619 and 612 nm with PL quantum yields (ΦPL) of 0.54 and 0.12, respectively (Fig. 2). Other triarylamine chromophores also show high PL quantum yields, similar to 2a; chromophores that incorporate triarylamine-based motifs are commonly used in light-emitting applications for this reason.62 A change in PL intensity is observed when compounds 2a and 3a are irradiated with 365 nm light and the corresponding bianthracene photoproduct forms. A blue-shift of 91 nm in the emission maximum for 2a can be observed with a significant increase in emission intensity as 2b is formed. A red-shift of 21 nm is observed when 3a is converted to 3b. The photoproducts, 2b and 3b show green and red emission (528 and 631 nm, respectively) and are more emissive (ΦPL = 0.99 and ΦPL = 0.58, respectively) than 2a and 3a. The photophysical data are summarized in Table S1.†
Due to the unique absorption characteristics of the individual dianthryl sulfoxides, we investigated the capability of our system to undergo photoconversion by wavelength-specific irradiation. Light emitting diodes (LEDs) with peak wavelength outputs of 405, 470 and 500 nm were used to irradiate dilute solution samples of 1a, 2a, and 3a (∼3 to 6 × 10−6 M) in CH2Cl2, while changes in absorption at specific wavelengths were measured (Fig. S15†). The percent conversion of 1a, 2a, and 3a to bianthracene photoproduct after 60 s of irradiation was calculated (Table S3†). Although the absorption of all chromophores overlap in the UV region, 1a does not absorb at wavelengths longer than 450 nm and the lower energy absorption characteristics for 2a and 3a provide an opportunity to use irradiation wavelengths greater than 450 nm for selective conversion of two out of the three chromophores. Irradiation of compounds 1a and 2a with 405 nm light results in complete conversion to their respective bianthracene photoproducts in less than 30 s (Fig. 3). Conversion kinetics for 2a are faster than 1a as observed by the steeper initial rate. When a 470 nm LED is used for irradiation experiments of the same duration, 2a reaches full conversion whereas irradiation of 1a results in minimal conversion (<5%). In the same period of time, 3a did not reach full conversion when irradiated with the same wavelengths (405 and 470 nm) indicating a comparatively slower photoreaction rate than for 1a and 2a. Using lower energy light (500 nm) to initiate photoconversion results in no conversion in the case of 1a and some conversion for 2a (42%) and 3a (18%) after 60 s. This data demonstrates the possibility to preferentially control the photoconversion of different sulfoxide species to the bianthracene photoproduct and tune its conversion kinetics using specific irradiation wavelengths. This approach presents a new method for full-color tuning as the final emission color of a specific mixture of the three emitters depends on the duration of UV exposure and the irradiation wavelength.
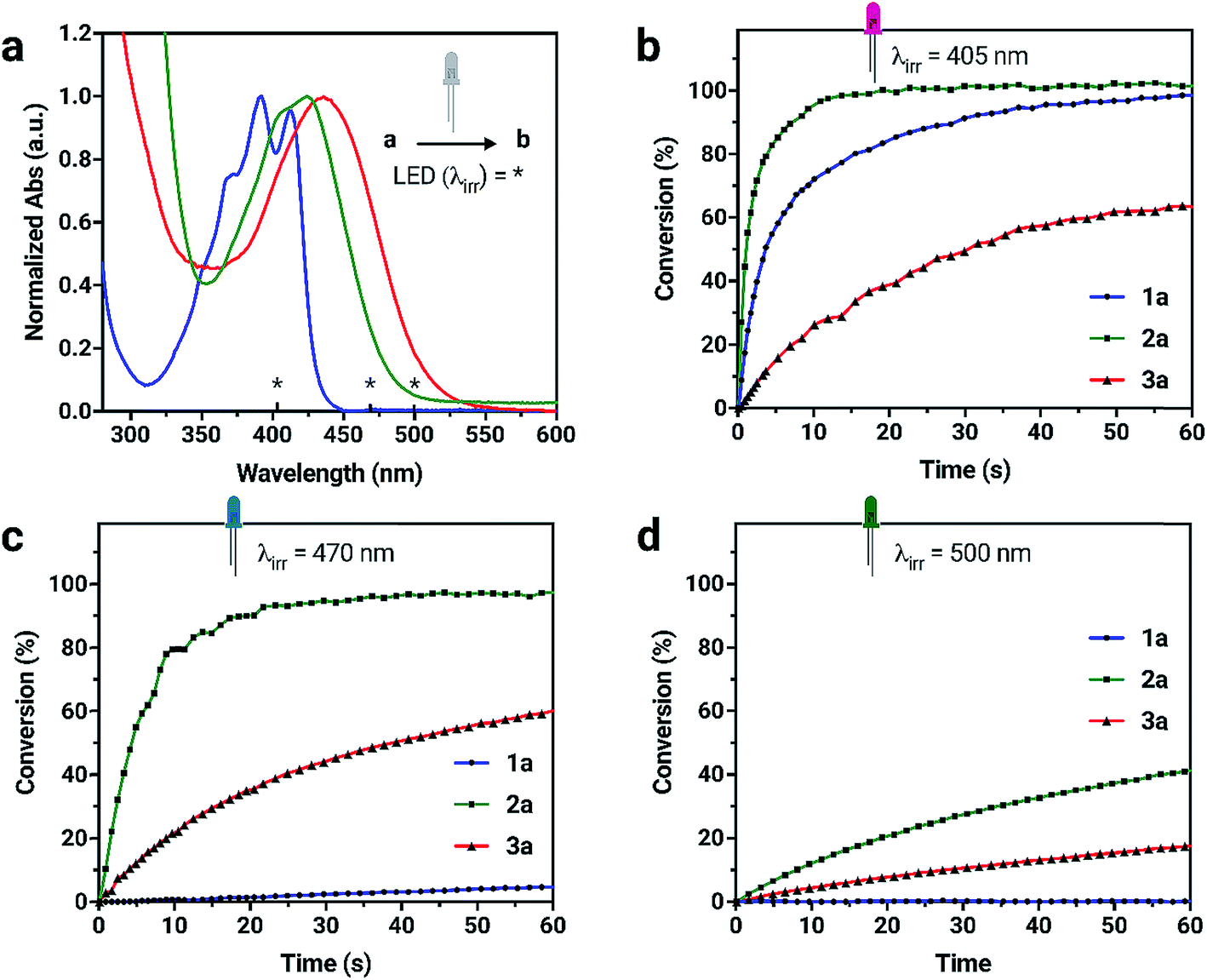 | ||
| Fig. 3 (a) UV-vis absorption spectra of 1a, 2a, 3a in CH2Cl2 with the irradiation wavelengths (*) highlighted. (b–d) Kinetic data for the formation of bianthracene photoproducts from individual dianthryl sulfoxides (a → b) by irradiation with LEDs as determined by UV-vis absorption spectroscopy. Different rates of conversion can be observed using selective irradiation wavelengths. | ||
The photostabilities of 1a, 2a and 3a were tested under ambient lighting conditions. The photoconversion was monitored using UV-vis spectroscopy. The initial spectrum of solutions at t = 0 min was recorded with minimal exposure to light. The samples were exposed to ambient light and spectra were recorded until no further change in absorbance was observed. Compound 2a converts to its respective bianthracene photoproduct quickly (20 min) compared to 1a and 3a which remains relatively photostable, taking over 300 min to fully convert into the photoproduct. The stability of 1a under ambient lighting conditions is the result of its absorption of higher-energy wavelength light. In contrast, 3a retains a longer photoconversion time than 2a under ambient lighting conditions. Although the compounds are sensitive to light, the conversion process is slow enough to make handling them in low light conditions possible.
Compounds 1b, 2b and 3b can also be characterized by their chromaticity coordinates, which graphically plots color from spectral measurements onto the CIE xyY color space (Fig. 4).58 This is widely used for describing chromaticity as numerical values. Since the production of bianthracene photoproduct can be controlled by measured exposure of light, the fluorescence intensity of a given mixture can be manipulated by light. For potential applications as an authentication tool, photochemistry is used as the method of verification. We propose that a “patch” consisting of a combination of the three dyes could be irradiated for a specific length of time to reveal particular CIE coordinates. In this approach, the CIE coordinates would be provided separately to the recipient to verify authenticity via a match against a known color. Additional security can be achieved if the trajectory of the color is also monitored during irradiation and compared against a time-lapse card illustrating the expected color change. We expect that this protocol will significantly increase the difficulty of counterfeiting these dynamic photopatterns, making them an attractive option as an authentication tool.
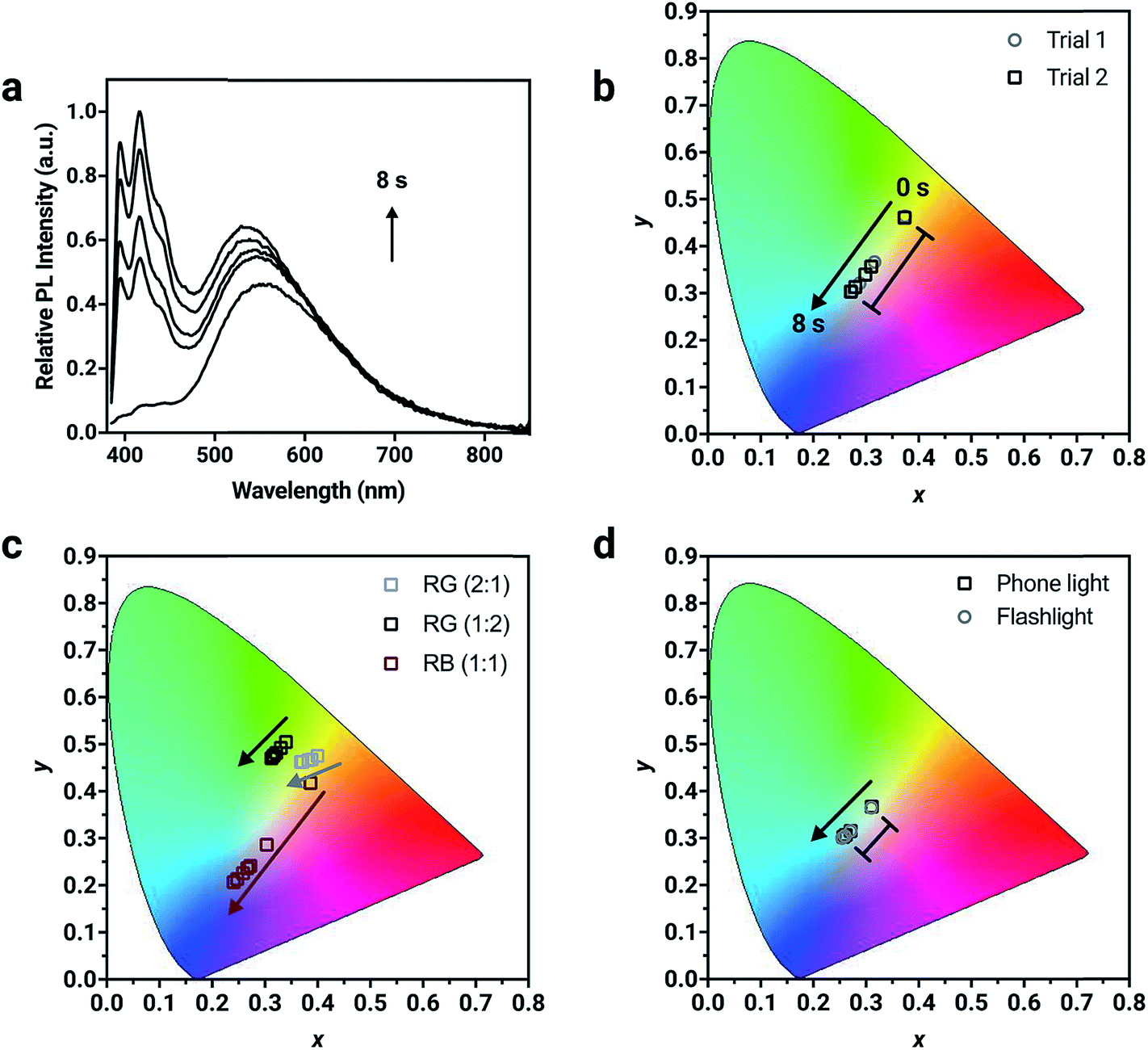 | ||
| Fig. 4 (a) Normalized solid-state PL spectrum of a thin film prepared with dianthryl sulfoxides doped into PMMA (total concentration = 1 wt%) showing the change in PL intensity with irradiated with 400 nm light after 8 s. (b–d) CIE 1931 chromaticity diagrams showing the change in coordinates of different thin films composed of varying formulations and using different light sources for activation. The magnitude of change is in good agreement between separate trials as shown in (b). | ||
Chromaticity coordinates can be calculated from emission spectra.58 Fluorescent “inks” were prepared by doping the chromophores into a poly(methylmethacrylate) (PMMA) host to achieve a total concentration of 1% (w/w) and spin coated onto glass substrates to make thin-films. The change in absorbance and fluorescence behavior of the dyes in the polymer host is similar to that in solution (Fig. S16†), suggesting that the photoconversion kinetics of the dyes in the solid-state mirrors the solution-state behavior. In a formulation containing the three compounds present in equal amounts (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 by weight), the resulting solid-state emission spectrum shows distinctive emission bands that correspond to the pure compounds demonstrating the potential to prepare full color emissive films (Fig. 4a). The change in chromaticity was monitored as a function of irradiation time. The thin-film was exposed to 400 nm light in 2 s intervals where the photoconversion of all chromophores present occurs and the resulting chromaticity was measured. The blue component at ∼420 nm from 1a in the corresponding solid-state emission spectrum increases to the greatest extent after 8 s of 400 nm UV exposure. Consequently, the chromaticity coordinates are initially in the orange region (0.37, 0.46) and move towards the blue region (0.27, 0.30) after 8 s of UV exposure (Table S4†). Using one specific mixture of the chromophores, five different chromaticity coordinates were accessed from one mixing ratio, depending on the length of UV irradiation. This temporal evolution of fluorescence color is demonstrated in Fig. S20 and Video S1.† The ink was drop cast using a pipette as a thin layer on a piece of filter paper. Under 365 nm light from a hand lamp, the fluorescence is initially pale orange, but with longer irradiation times turns first off-white and then sky-blue after 3 minutes. This trajectory monitored visually follows the same path as the data in the CIE color space.
:1:1 by weight), the resulting solid-state emission spectrum shows distinctive emission bands that correspond to the pure compounds demonstrating the potential to prepare full color emissive films (Fig. 4a). The change in chromaticity was monitored as a function of irradiation time. The thin-film was exposed to 400 nm light in 2 s intervals where the photoconversion of all chromophores present occurs and the resulting chromaticity was measured. The blue component at ∼420 nm from 1a in the corresponding solid-state emission spectrum increases to the greatest extent after 8 s of 400 nm UV exposure. Consequently, the chromaticity coordinates are initially in the orange region (0.37, 0.46) and move towards the blue region (0.27, 0.30) after 8 s of UV exposure (Table S4†). Using one specific mixture of the chromophores, five different chromaticity coordinates were accessed from one mixing ratio, depending on the length of UV irradiation. This temporal evolution of fluorescence color is demonstrated in Fig. S20 and Video S1.† The ink was drop cast using a pipette as a thin layer on a piece of filter paper. Under 365 nm light from a hand lamp, the fluorescence is initially pale orange, but with longer irradiation times turns first off-white and then sky-blue after 3 minutes. This trajectory monitored visually follows the same path as the data in the CIE color space.
For potential applications in anti-counterfeiting, it is important the change in chromaticity be reproducible. Variability in the photoirradiation environment will influence the rate at which the trajectory will change, consequently changing the exact position of the coordinates at specific time intervals. Factors such as distance from and intensity of the irradiation source used for activation, location of the detector and the surrounding ambient lighting conditions should be considered. However, if the final chromaticity coordinate of the trajectory from a specific mixture is consistent, then this methodology will be useful for this application. To demonstrate the reproducibility of the trajectory, irradiation of two thin-films prepared from the same ink mixture with 400 nm light resulted in a reproducible trajectory, where the initial (t = 0 s) and final (t = 8 s) chromaticity coordinates between different trials were in excellent agreement (Fig. S19†). The initial chromaticity coordinate is determined by the composition of the mixture. It can be challenging to precisely reproduce a mixing ratio by hand, however this problem can be resolved using automation. It is noteworthy that the change in chromaticity, defined as the magnitude of the vector from the initial to the final chromaticity coordinate, is consistent, regardless of the initial position of the chromaticity coordinate (Table S4†). For mixture 1 (Fig. 4b), the change in chromaticity between initial and final coordinates is 0.13 for both trial 1 and trial 2. This is the result of complete conversion of the mixture, regardless of the starting chromaticity position. Thin films were also irradiated with commercial light sources such as a smartphone light (iPhone 7) and UV flashlight with a peak wavelength output of 395 nm (Vansky). Using a mixture containing the compounds in a 1:1:1 ratio (by weight), the change in chromaticity was monitored. The thin-films were irradiated for 4 minutes using the smartphone light and for 2.5 minutes using the UV flashlight, until no further change in PL intensity was measured. The changes in chromaticity were calculated to both be 0.05 using the two common light sources, which were capable of converting all three chromophores simultaneously.
The direction of the trajectory will be influenced by the relative contribution of each compound in the mixture, which is controlled by the initial mixing ratio. Different blends of varying ratios of 1a, 2a and 3a were formulated and doped into a PMMA host matrix to verify the versatility of the method (Fig. 4d). Two different formulations containing 2a and 3a (red and green) were mixed in 2:1 and 1:2 ratios. The initial chromaticity coordinates differ due to their composition (0.40, 0.48 for the 2:1 mixing ratio versus 0.33, 0.49 for the 1:2 mixing ratio). Upon irradiation, the trajectory moves towards the green region of the color gamut and the magnitude of change (0.03 versus 0.02, respectively) differs despite the same chromophores being present. A composition containing 1a and 3a (red and blue) was also mixed in a 1:1 ratio and the change in chromaticity was monitored, where the trajectory was shown to move towards the blue region. Depending on the composition of the mixture, the shift in chromaticity moves towards the color of the compound that is converted to the photoproduct the fastest.
To demonstrate the performance of 1a, 2a and 3a for potential applications in anti-counterfeiting, fluorescent inks for paper substrates were mixed by doping the chromophores in equal contributions (1:1:1 by weight) into PMMA at a total concentration of 10% (w/w). An inexpensive, publicly available smartphone app called “Colorimeter”63 was used to measure color without a fluorimeter (Fig. 5). The app uses one-touch operation and is capable of taking measurements instantaneously and repetitively, provides common color parameters such as CIELAB, chroma, hue, RGB and assigns color names. In this case, we chose to report color numerically as CIELAB coordinates, as this model gives values that are device-independent. This color space is typically used for applications that include color specification or difference evaluation.64 Both CIELAB and CIE xyY models are derived from the same master CIE 1931 XYZ color space. Using this app, a match card was prepared as an end-user guide to illustrate the expected time-lapse of the color evolution. The app was used to capture the change in fluorescence color at different time intervals as the ink was activated by UV irradiation until no further change in color was captured. We report the final color of this ink mixture after approximately 1 minute of irradiation as the “true color” containing the CIELAB coordinates: L* = 86.1, a* = −34.8, b* = 10.2, measured by the “Colorimeter” app. Two patches were prepared where patch 1 was created using an incorrect formulation (RGB ratio = 2:1:3) and patch 2 was created using the correct formulation (RGB ratio = 1:1:1). The app measured the initial fluorescence color, an orange-brown, which appears qualitatively similar in both ink formulations but upon activation reveals two different colors, “powder blue” versus “aqua marine” as the result of different ink compositions. The app can readily distinguish between the two hues of blue/green. Color difference can be defined as the numerical comparison of the patches color to the standard or “true color”. The difference in absolute color coordinates is referred to as 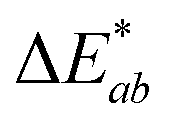 and is given by eqn (1):64
and is given by eqn (1):64
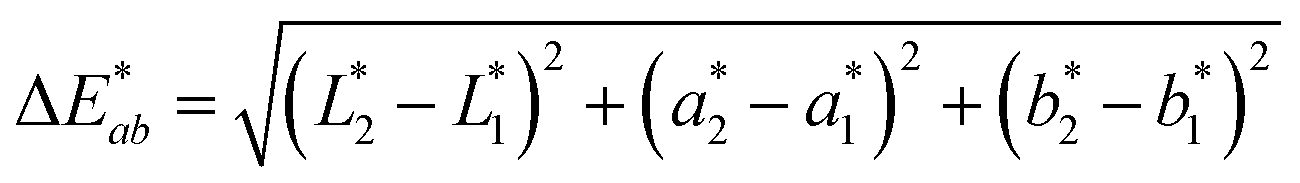 | (1) |
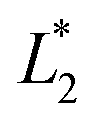 ,
, 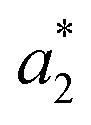 ,
, 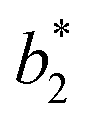 are the measured values from the patches and
are the measured values from the patches and 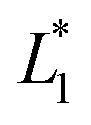 ,
, 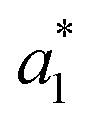 ,
, 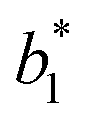 are the measured values of the true color. Using eqn (1) the total color difference calculated between the true color and patch 1 is 24.2 while for patch 2 the total color difference is 2.9. This difference can be evaluated against a predetermined color tolerance to determine the authenticity of a product. Color differences
are the measured values of the true color. Using eqn (1) the total color difference calculated between the true color and patch 1 is 24.2 while for patch 2 the total color difference is 2.9. This difference can be evaluated against a predetermined color tolerance to determine the authenticity of a product. Color differences 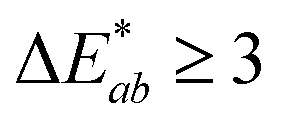 are considered perceptible, thus values that are greater than this tolerance level can be considered to be incorrect. This calculation provides a simple pass/fail formula, an efficient way for end-users to quickly verify authenticity of items at the point-of-sale.
are considered perceptible, thus values that are greater than this tolerance level can be considered to be incorrect. This calculation provides a simple pass/fail formula, an efficient way for end-users to quickly verify authenticity of items at the point-of-sale.
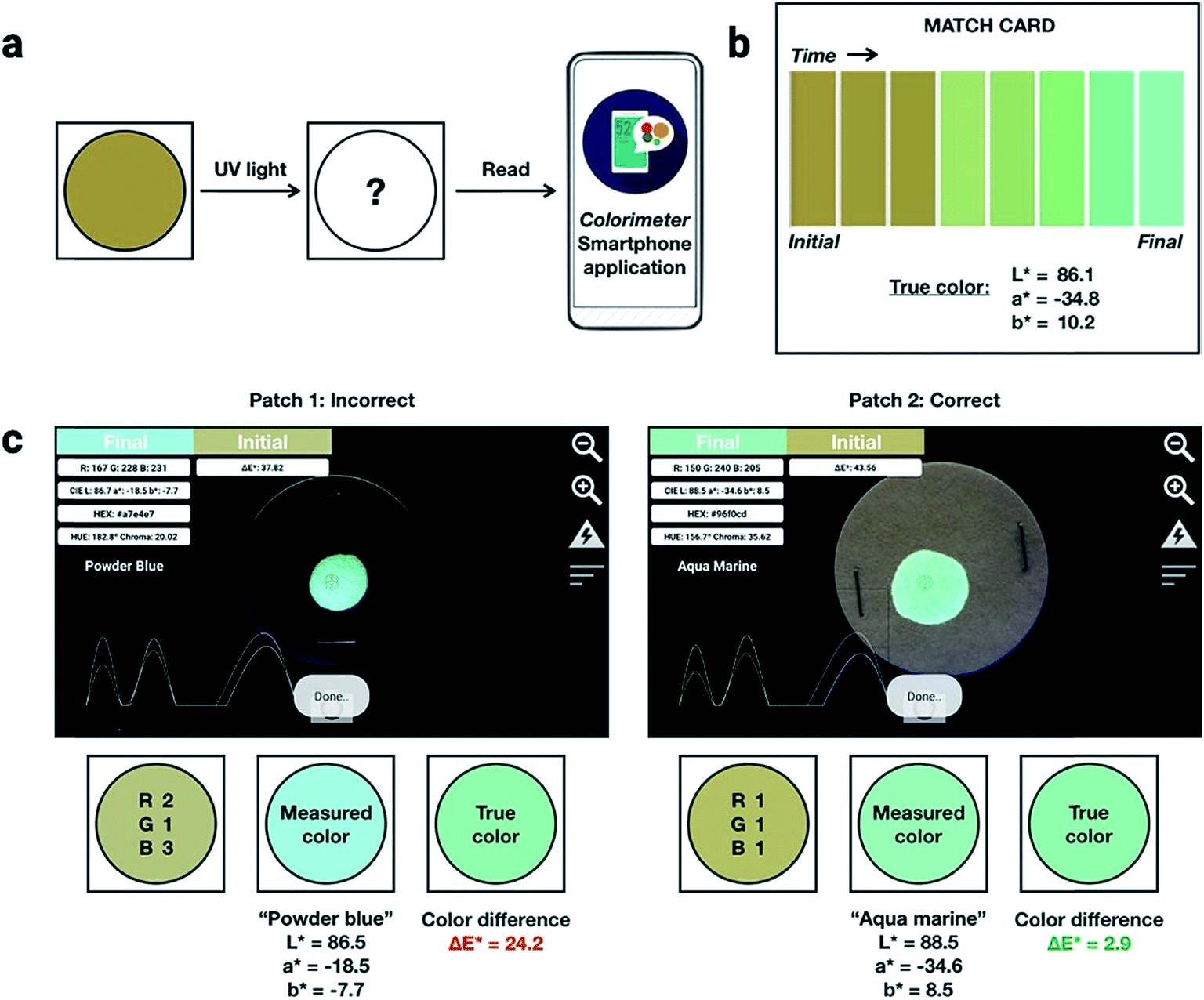 | ||
| Fig. 5 (a) Schematic illustration of how a patch fabricated with dianthryl sulfoxides can be used to authenticate a product. An unconverted patch on a product can be irradiated or activated using UV light. Using a smartphone equipped with a commercial application, the color can be read and compared to a known, true value found on a match card (b) provided to an end-user. (c) Photographs of the true (right) and false (left) patch as viewed through the software interface of the smartphone application. CIELAB coordinates can be measured using this application and the color difference can be calculated and compared to the true color found on the match card to determine the authenticity. | ||
Conclusions
In summary, we report a new approach using photochemistry based on multicolored dianthryl sulfoxides to develop applications in luminescent security printing. The emission color of the photoproduct is tuned by incorporating substituents on the dianthracene core, allowing formation of red, green, and blue fluorophores upon irradiation. The photoconversion of dianthryl sulfoxide to the corresponding bianthracene photoproduct can be modulated by both substitution at the 10,10′-positions, and by using different wavelengths of light for activation. Security features consisting of a mixture of the three compounds allows verification through the final CIE coordinates obtained on irradiation. In addition, a novel feature of this system is the ability to monitor the trajectory of the CIE coordinates as a function of time. Control of the excitation wavelength and/or length of light exposure allow different colors and trajectories to be obtained, adding a dynamic level of security that may be challenging to replicate. The photochemistry of the dianthryl sulfoxides offers increased complexity for potential applications in anti-counterfeiting due to the different number of parameters that can be manipulated, yet can be easily applied for end-user authentication as the activation can be performed with common light sources and the fluorescence color can be detected using a smartphone.Conflicts of interest
The authors have filed a provisional patent on this work.Acknowledgements
This research was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery and CREATE grants). The authors would like to thank Ms Candice C. Chua for assistance demonstrating the authentication method.References
- P. Aldhous, Counterfeit pharmaceuticals: murder by medicine, Nature, 2005, 434, 132–136 CrossRef CAS PubMed.
- C. Sheridan, Bad medicine, Nat. Biotechnol., 2007, 25, 707–709 CrossRef CAS PubMed.
- A. Mullard, The anticounterfeiter's technological tool kit, Nat. Med., 2010, 16, 361 CrossRef CAS.
- D. Bansal, S. Malla, K. Gudala and P. Tiwari, Anti-counterfeit technologies: a pharmaceutical industry perspective, Sci. Pharm., 2013, 81, 1–13 CrossRef CAS PubMed.
- Q. Bassat, M. Tanner, P. J. Guerin, K. Stricker and K. Hamed, Combating poor-quality anti-malarial medicines: a call to action, Malar. J., 2016, 15, 1–12 CrossRef PubMed.
- N. Zadbuke, S. Shahi, B. Gulecha, A. Padalkar and M. Thube, Recent trends and future of pharmaceutical packaging technology, J. Pharm. Bioallied Sci., 2013, 5, 98–110 CrossRef PubMed.
- L. Li, Technology designed to combat fakes in the global supply chain, Bus. Horizons, 2013, 56, 167–177 CrossRef.
- A. K. Deisingh, Pharmaceutical counterfeiting, Analyst, 2005, 130, 271–279 RSC.
- B. A. Liang, T. Kubic, T. K. Mackey and P. York, Counterfeit Drug Penetration into Global Legitimate Medicine Supply Chains: A Global Assessment, Am. J. Trop. Med. Hyg., 2015, 92, 59–67 CrossRef PubMed.
- T. K. Mackey and B. A. Liang, The global counterfeit drug trade: patient safety and public health risks, J. Pharm. Sci., 2011, 100, 4571–4579 CrossRef CAS PubMed.
- R. Arppe and T. J. Sørensen, Physical unclonable functions generated through chemical methods for anti-counterfeiting, Nat. Rev. Chem., 2017, 1, 1–13 CrossRef.
- R. Arppe-Tabbara, M. Tabbara and T. J. Sørensen, Versatile and Validated Optical Authentication System Based on Physical Unclonable Functions, ACS Appl. Mater. Interfaces, 2019, 11, 6475–6482 CrossRef CAS PubMed.
- D. Karamessini, T. Simon-Yarza, S. Poyer, E. Konishcheva, L. Charles, D. Letourneur and J. F. Lutz, Abiotic Sequence-Coded Oligomers as Efficient In Vivo Taggants for the Identification of Implanted Materials, Angew. Chem., Int. Ed., 2018, 57, 10574–10578 CrossRef CAS PubMed.
- C. Wolfrum and A. Josten, Oligonucleotides as coding molecules in an anti-counterfeiting system, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 1069–1074 CrossRef CAS.
- D. Paunescu, R. Fuhrer and R. N. Grass, Protection and deprotection of DNA - high-temperature stability of nucleic acid barcodes for polymer labeling, Angew. Chem., Int. Ed., 2013, 52, 4269–4272 CrossRef CAS PubMed.
- J. Gooch, B. Daniel, V. Abbate and N. Frascione, Taggant materials in forensic science: a review, Trends Anal. Chem., 2016, 83, 49–54 CrossRef CAS.
- B.-H. Wu, C. Zhang, N. Zheng, L.-W. Wu, Z.-K. Xu and L.-S. Wan, Grain Boundaries of Self-Assembled Porous Polymer Films for Unclonable Anti-Counterfeiting, ACS Appl. Polym. Mater., 2019, 1, 47–53 CrossRef CAS.
- C. Herder, M. D. Yu, F. Koushanfar and S. Devadas, Physical unclonable functions and applications: a tutorial, Proc. IEEE, 2014, 102, 1126–1141 Search PubMed.
- H. J. Bae, S. Bae, C. Park, S. Han, J. Kim, L. N. Kim, K. Kim, S. H. Song, W. Park and S. Kwon, Biomimetic microfingerprints for anti-counterfeiting strategies, Adv. Mater., 2015, 27, 2083–2089 CrossRef CAS PubMed.
- J. Andréasson and U. Pischel, Molecules for security measures: from keypad locks to advanced communication protocols, Chem. Soc. Rev., 2018, 47, 2266–2279 RSC.
- J. Andres, R. D. Hersch, J. E. Moser and A. S. Chauvin, A new anti-counterfeiting feature relying on invisible luminescent full color images printed with lanthanide-based inks, Adv. Funct. Mater., 2014, 24, 5029–5036 CrossRef CAS.
- M. R. Carro-Temboury, R. Arppe, T. Vosch and T. J. Sørensen, An optical authentication system based on imaging of excitation-selected lanthanide luminescence, Sci. Adv., 2018, 4, 1–8 Search PubMed.
- Y. Zhang, L. Zhang, R. Deng, J. Tian, Y. Zong, D. Jin and X. Liu, Multicolor barcoding in a single upconversion crystal, J. Am. Chem. Soc., 2014, 136, 4893–4896 CrossRef CAS PubMed.
- J. C. G. Bünzli and S. V. Eliseeva, Intriguing aspects of lanthanide luminescence, Chem. Sci., 2013, 4, 1939–1949 RSC.
- X. Liu, Y. Wang, X. Li, Z. Yi, R. Deng, L. Liang, X. Xie, D. T. B. Loong, S. Song, Di. Fan, A. H. All, H. Zhang, L. Huang and X. Liu, Binary temporal upconversion codes of Mn2+-activated nanoparticles for multilevel anti-counterfeiting, Nat. Commun., 2017, 8, 1–7 Search PubMed.
- C. Zhang, B. Wang, W. Li, S. Huang, L. Kong, Z. Li and L. Li, Conversion of invisible metal-organic frameworks to luminescent perovskite nanocrystals for confidential information encryption and decryption, Nat. Commun., 2017, 8, 1–9 Search PubMed.
- S. Kalytchuk, Y. Wang, K. Poláková and R. Zbořil, Carbon Dot Fluorescence-Lifetime-Encoded Anti-Counterfeiting, ACS Appl. Mater. Interfaces, 2018, 10, 29902–29908 CrossRef CAS PubMed.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Triple-Mode Emission of Carbon Dots: Applications for Advanced Anti-Counterfeiting, Angew. Chem., Int. Ed., 2016, 55, 7231–7235 CrossRef CAS PubMed.
- L. Xu, J. Chen, J. Song, J. Li, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, Double-Protected All-Inorganic Perovskite Nanocrystals by Crystalline Matrix and Silica for Triple-Modal Anti-Counterfeiting Codes, ACS Appl. Mater. Interfaces, 2017, 9, 26556–26564 CrossRef CAS PubMed.
- C. Sun, S. Su, Z. Gao, H. Liu, H. Wu, X. Shen and W. Bi, Stimuli-Responsive Inks Based on Perovskite Quantum Dots for Advanced Full-Color Information Encryption and Decryption, ACS Appl. Mater. Interfaces, 2019, 11, 8210–8216 CrossRef CAS PubMed.
- J. M. Meruga, W. M. Cross, P. Stanley May, Q. Luu, G. A. Crawford and J. J. Kellar, Security printing of covert quick response codes using upconverting nanoparticle inks, Nanotechnology, 2012, 23, 1–9 CrossRef PubMed.
- Z. Song, T. Lin, L. Lin, S. Lin, F. Fu, X. Wang and L. Guo, Invisible Security Ink Based on Water-Soluble Graphitic Carbon Nitride Quantum Dots, Angew. Chem., Int. Ed., 2016, 55, 2773–2777 CrossRef CAS PubMed.
- A. M. Kaczmarek, Y.-Y. Liu, C. Wang, B. Laforce, L. Vincze, P. Van Der Voort, K. Van Hecke and R. Van Deun, Lanthanide “Chameleon” Multistage Anti-Counterfeit Materials, Adv. Funct. Mater., 2017, 27, 1700258 CrossRef.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Red, green, and blue luminescence by carbon dots: full-color emission tuning and multicolor cellular imaging, Angew. Chem., Int. Ed., 2015, 127, 5450–5453 CrossRef.
- M. You, M. Lin, S. Wang, X. Wang, G. Zhang, Y. Hong, Y. Dong, G. Jin and F. Xu, Three-dimensional quick response code based on inkjet printing of upconversion fluorescent nanoparticles for drug anti-counterfeiting, Nanoscale, 2016, 8, 10096–10104 RSC.
- M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, A digital fluorescent molecular photoswitch, Nature, 2002, 420, 759–760 CrossRef CAS PubMed.
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Tunable solid-state fluorescent materials for supramolecular encryption, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- Z. Xu, D. Gonzalez-Abradelo, J. Li, C. A. Strassert, B. J. Jan and D. Guo, Supramolecular color-tunable photoluminescent materials based on a chromophore cascade as security inks with dual encryption, Mater. Chem. Front., 2017, 1, 1847–1852 RSC.
- B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y. Wu, Y. Zhang, C. Jin, P. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, White-light emission from a single heavy atom-free molecule with room temperature phosphorescence, mechanochromism and thermochromism, Chem. Sci., 2017, 8, 1909–1914 RSC.
- T. J. Wigglesworth and N. R. Branda, A family of multiaddressable, multicolored photoresponsive copolymers prepared by ring-opening metathesis polymerization, Chem. Mater., 2005, 17, 5473–5480 CrossRef CAS.
- J. E. Kwon, S. Park and S. Y. Park, Realizing molecular pixel system for full-color fluorescence reproduction: RGB-emitting molecular mixture free from energy transfer crosstalk, J. Am. Chem. Soc., 2013, 135, 11239–11246 CrossRef CAS PubMed.
- T. Panda, D. K. Maiti and M. K. Panda, Inkless Writing and Self-Erasing Security Feature of (Z)-1,2-Diarylacrylonitrile-Based Materials: A Confidential Data Communication, ACS Appl. Mater. Interfaces, 2018, 10, 29100–29106 CrossRef CAS PubMed.
- J. Chen, T. Yu, E. Ubba, Z. Xie, Z. Yang, Y. Zhang, S. Liu, J. Xu, M. P. Aldred and Z. Chi, Achieving Dual-Emissive and Time-Dependent Evolutive Organic Afterglow by Bridging Molecules with Weak Intermolecular Hydrogen Bonding, Adv. Opt. Mater., 2019, 7, 1801593 CrossRef.
- X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Reversible switching of the emission of diphenyldibenzofulvenes by thermal and mechanical stimuli, Adv. Mater., 2011, 23, 3261–3265 CrossRef CAS PubMed.
- H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Smart responsive phosphorescent materials for data recording and security protection, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- X. Zhu, R. Liu, Y. Li, H. Huang, Q. Wang, D. Wang, X. Zhu, S. Liu and H. Zhu, An AIE-active boron-difluoride complex: multi-stimuli-responsive fluorescence and application in data security protection, Chem. Commun., 2014, 50, 12951–12954 RSC.
- T. Han, X. Feng, D. Chen and Y. Dong, A diethylaminophenol functionalized Schiff base: crystallization-induced emission-enhancement, switchable fluorescence and application for security printing and data storage, J. Mater. Chem. C, 2015, 3, 7446–7454 RSC.
- Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, Solid-State Photoinduced Luminescence Switch for Advanced Anticounterfeiting and Super-Resolution Imaging Applications, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed.
- E. R. Thapaliya, Y. Zhang and F. M. Raymo, Fluorescence patterning with mild illumination in polymer films of photocleavable oxazines, J. Mater. Chem. C, 2017, 5, 1179–1183 RSC.
- J. Wang, B. Jin, N. Wang, T. Peng, X. Li, Y. Luo and S. Wang, Organoboron-Based Photochromic Copolymers for Erasable Writing and Patterning, Macromolecules, 2017, 50, 4629–4638 CrossRef CAS.
- Z. Gao, Y. Han and F. Wang, Cooperative supramolecular polymers with anthracene–endoperoxide photo-switching for fluorescent anti-counterfeiting, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- S. Basak, N. Nandi, S. Paul and A. Banerjee, Luminescent Naphthalene Diimide-Based Peptide in Aqueous Medium and in Solid State: Rewritable Fluorescent Color Code, ACS Omega, 2018, 3, 2174–2182 CrossRef CAS PubMed.
- B. Yoon, J. Lee, I. S. Park, S. Jeon, J. Lee and J. Kim, Recent functional material based approaches to prevent and detect counterfeiting, J. Mater. Chem. C, 2013, 1, 2388–2403 RSC.
- P. Kumar, S. Singh and B. K. Gupta, Future prospects of luminescent nanomaterial based security inks: from synthesis to anti-counterfeiting applications, Nanoscale, 2016, 8, 14297–14340 RSC.
- P. R. Christensen, B. O. Patrick, É. Caron and M. O. Wolf, Oxidation-state-dependent photochemistry of sulfur-bridged anthracenes, Angew. Chem., Int. Ed., 2013, 52, 12946–12950 CrossRef CAS PubMed.
- C. D. Cruz, J. Yuan, C. Climent, N. T. Tierce, P. R. Christensen, E. L. Chronister, D. Casanova, M. O. Wolf and C. J. Bardeen, Using Sulfur Bridge Oxidation to Control Electronic Coupling and Photochemistry in Covalent Anthracene Dimers, Chem. Sci., 2019, 10, 7561–7573 RSC.
- P. R. Christensen and M. O. Wolf, Photopatterned Multidimensional Fluorescent Images, Adv. Funct. Mater., 2016, 26, 8471–8477 CrossRef CAS.
- N. Ohta and A. R. Robertson, Colorimetry: Fundamentals and Applications, John Wiley & Sons Ltd., Chichester, West Sussex, England, 2005 Search PubMed.
- P. Natarajan and M. Schmittel, Photoluminescence, redox properties, and electrogenerated chemiluminescence of twisted 9,9′-bianthryls, J. Org. Chem., 2013, 78, 10383–10394 CrossRef CAS PubMed.
- P. Xue, B. Yao, X. Liu, J. Sun, P. Gong, Z. Zhang, C. Qian, Y. Zhang and R. Lu, Reversible mechanochromic luminescence of phenothiazine-based 10,10′-bianthracene derivatives with different lengths of alkyl chains, J. Mater. Chem. C, 2015, 3, 1018–1025 RSC.
- M. Chen, L. Yan, Y. Zhao, I. Murtaza, H. Meng and W. Huang, Anthracene-based semiconductors for organic field-effect transistors, J. Mater. Chem. C, 2018, 6, 7416–7444 RSC.
- H. Yen and G. Liou, Novel Thermally Stable Triarylamine-Containing Aromatic Polyamides Bearing Anthrylamine Chromophores for Highly Efficient, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7354–7368 CrossRef CAS.
- R. Ravindranath, A. P. Periasamy, P. Roy, Y. W. Chen and H. T. Chang, Smart app-based on-field colorimetric quantification of mercury via analyte-induced enhancement of the photocatalytic activity of TiO2–Au nanospheres, Anal. Bioanal. Chem., 2018, 410, 4555–4564 CrossRef CAS PubMed.
- Encyclopedia of Color Science and Technology, Springer, New York, NY, 1st edn, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03948a |
| This journal is © The Royal Society of Chemistry 2019 |