
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHAMA: a multiplexed LC-MS/MS assay for specificity profiling of adenylate-forming enzymes†
Aleksa
Stanišić
,
Annika
Hüsken
and
Hajo
Kries
 *
*
Independent Junior Research Group Biosynthetic Design of Natural Products, Leibniz Institute for Natural Product Research and Infection Biology e.V., Hans Knöll Institute (HKI Jena), Beutenbergstr. 11a, 07745 Jena, Germany. E-mail: hajo.kries@leibniz-hki.de
First published on 3rd October 2019
Abstract
Adenylation enzymes selecting substrates for ribosomal and nonribosomal protein and peptide biosynthesis have been popular targets of enzyme engineering. Previous standard assays for adenylation specificity have been cumbersome and failed to reflect the competition conditions inside a cell because they measure substrates one at a time. We have developed an adenylation assay based on hydroxamate quenching and LC-MS/MS detection of hydroxamate products testing dozens of competing amino acid substrates in parallel. Streamlined specificity profiling of adenylation enzymes will facilitate engineering and directed evolution of ribosomal and nonribosomal peptide synthesis.
Adenylate forming enzymes control the substrate selection process in ribosomal and nonribosomal peptide synthesis (Fig. 1A and B).1–3 Hence, aminoacyl-tRNA synthetases (aaRSs) from ribosomal protein synthesis and nonribosomal adenylation (A) domains have been extensively engineered4–6 in order to change their specificity for incorporation of alternative substrates. Non-natural building blocks have been of particular interest for enriching the functional spectrum of peptides and proteins, for instance with handles for bio-orthogonal cross-linking.7,8 While aaRSs function as standalone enzymes, A domains are embedded in a large biosynthetic scaffold together with thiolation (T), condensation (C), thioesterase (Te) and a number of editing domains. Outside these cellular peptide synthesis machineries, adenylating enzymes have recently found application as biocatalysts for amide bond formation, one of the most important reactions in pharmaceutical chemistry.9,10
 | ||
| Fig. 1 (A) Adenylation domains and (B) aaRSs activate amino acids (AAs) for peptide synthesis. (C) Aminoacyl hydroxamate (HA) release after quenching of aminoacyl adenylates provides an alternative assay reaction to PPi release and PPi exchange. (D) Hydroxamates are quantified by UPLC-MS/MS to obtain full specificity profiles of adenylation enzymes in a single chromatographic run. | ||
Although the importance of adenylating enzymes has long been recognized, labour intensive specificity profiling hinders engineering efforts.11 None of the available assays can detect adenylation activity under substrate competition which is critical for intracellular reactions. The widely used pyrophosphate (PPi) exchange assay sensitively detects incorporation of radioactive 32P-PPi in the reverse reaction but handling of radioactivity and laborious sample work-up are major drawbacks.11–13 Non-radioactive but still discontinuous is the mass-spectrometric detection of exchange between PPi and γ-18O4-ATP.14 Release assays detecting the liberation of PPi in the forward reaction allow continuous data collection and are more convenient in terms of instrumentation and handling but suffer from low sensitivity and strong product inhibition.15–18 In order to overcome inhibition by tightly binding aminoacyl-adenylates in release assays, hydroxylamine has been added as a quencher19,20 and resulting hydroxamates have been detected as iron complexes.21
A salient feature of many nonribosomal adenylation domains is a high degree of substrate promiscuity which can serve as a springboard for natural enzyme evolution22 and engineering in the laboratory.23–28 Given the availability of vast numbers of structurally related amino acid substrates, adenylation enzymes are highly suitable for studying enzyme promiscuity.29 However, specificity profiles of adenylating enzymes must be measured one substrate at a time24,27,30 which poorly reflects the situation in a cell where numerous amino acids and carboxylic acids compete for the active site. Consequently, the natural promiscuity of adenylate forming enzymes is imperfectly approximated unless specificity constants (kcat/KM) are determined for each substrate in saturation kinetics.31 It follows from an extension of Michaelis–Menten kinetics to competition conditions that product formation rates are proportional to the kcat/KM of the respective substrate times the substrate concentration (ESI eqn (1)–(3)†).32 It is hence possible to predict the performance of an adenylation enzyme inside the cell from kcat/KM values and intracellular substrate concentrations.
Here, we present an adenylation assay that allows deduction of kcat/KM ratios from hydroxamate product concentrations after reacting a mixture of substrates in the presence of hydroxylamine. In the multiplexed hydroxamate assay (HAMA), product mixtures are disentangled via highly specific and sensitive LC-MS/MS. Since the experimental effort is minimal for determining a full specificity profile including dozens of substrates, this assay has great potential for exploring and evolving the promiscuity of adenylate forming enzymes.
Results and discussion
In order to test whether formation of hydroxamates in an adenylation reaction would yield meaningful specificity constants (kcat/KM), we used Phe specific TycA, the first module from tyrocidine synthetase, as a reference. In a thorough kinetic analysis based on PPi exchange, full Michaelis–Menten kinetics have previously been determined for a range of substrates.31 First, we confirmed that TycA would be stable in the presence of the hydroxylamine concentration (150 mM) required for efficient quenching.33 TycA maintained full activity after treatment with hydroxylamine for up to one hour (ESI Fig. S1†). By using the established MesG/hydroxylamine assay which also relies on hydroxylamine quenching but detects released PPi,20 kinetic constants were determined for the substrates L-Phe, D-Phe, L-Trp, L-Tyr, L-Leu, L-Met, and L-Val (ESI Fig. S2†). The kcat/KM's for these substrates cover a range of five orders of magnitude. MesG/hydroxylamine and PPi exchange assay yielded overall consistent results (ESI Table S1 and ESI Fig. S3†). Deviations could arise from intrinsic chemical preferences of the assay reactions – attack of hydroxylamine on the aminoacyl adenylate in one case and of pyrophosphate in the other. We conclude that hydroxamate formation is an informative parameter for the characterization of adenylation reactions.A prerequisite of hydroxamate quantification by LC-MS/MS are standards for optimizing and calibrating hydroxamate detection. Hydroxamates of proteinogenic and three nonproteinogenic amino acids (phenyl-glycine, β-phenylalanine and pipecolic acid) were synthesized from corresponding methyl esters by treatment with hydroxylamine. Asparagine, glutamine and ornithine hydroxamates could not be obtained, presumably due to intramolecular cyclization and instability. Hydroxamate standards are available from the authors upon reasonable request. Hydrophilic interaction chromatography on a BEH-amide phase provided efficient separation of the highly polar compounds which showed little retention on reversed phase columns. Specific fragmentations of hydroxamates were detected by ESI-MS/MS on a Xevo TQ-S micro (Waters), with limits of quantitation in the range of 3 to 400 nM and a dynamic range of at least three orders of magnitude (ESI Table S2†). Detection of serine hydroxamate was hampered by isobaric, coeluting compounds present in the assay mixture. Ile and Leu hydroxamates coelute but were differentiated by addition of deuterium labelled Leu-d7. Similarly, addition of deuterium labelled isotopes allowed mass-differentiation of enantiomeric pairs of Phe and Val.
For HAMA, we performed reactions similar to the MesG/hydroxylamine assay but in the presence of a 1 mM substrate mixture containing all amino acids of interest. To avoid substrate depletion, reactions were run up to 10% conversion of the most active substrate. Under these conditions, the amounts of hydroxamates determined by LC-MS/MS should be proportional to the corresponding specificity constant kcat/KM of the amino acid substrate. Since the activity of TycA is ca. 103-fold larger for L-Phe (kcat/KM = 1600 mM−1 min−1; ESI Table S1†) than for the best alternative substrates, a second reaction without L-Phe was conducted with longer reaction time to bring the less active substrates into the quantifiable range. The hydroxamate profile obtained from two reactions and two chromatographic runs yielded results consistent with full saturation kinetics recorded with the MesG/hydroxylamine assay (Fig. 2A). While the detailed kinetic analysis with PPi exchange and release assays is tedious and time consuming, a complete specificity profile with HAMA can be completed in less than one hour.
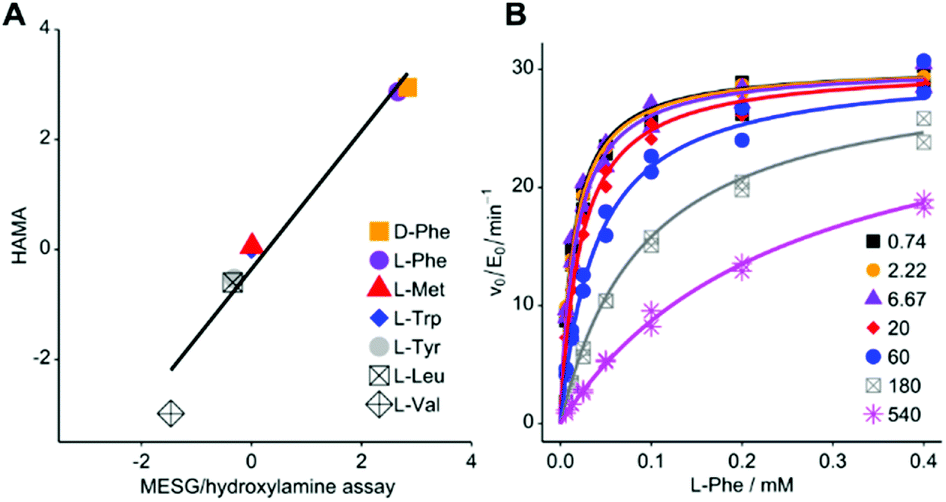 | ||
| Fig. 2 (A) Rapid LC-MS/MS quantification of hydroxamates formed by Phe-activating NRPS module TycA in a competition reaction yields specificity data equivalent to saturation kinetics recorded with the MESG/hydroxylamine assay (ESI Table S1†; R2 = 0.959; slope = 1.26 ± 0.12). Logarithms of hydroxamate concentrations obtained by HAMA are plotted against log(kcat/KM * mM min) values obtained with the MesG/hydroxylamine assay. All activities are relative to Trp. Reported activity for L-Ile (3.4 mM−1 min−1)29 could not be detected in either assay format. (B) PheHA is a weak competitive inhibitor of TycA (Ki = 30.3 ± 1.4 μM). The inset shows PheHA concentrations in μM. Each point was measured as technical duplicate. | ||
Based on the subtle, two atom difference between amino acids and amino acid hydroxamates, we hypothesized that hydroxamates would act as competitive inhibitors of adenylation. Indeed, we found that L-Phe hydroxamate (PheHA) is a weak competitive inhibitor of TycA with an inhibition constant (Ki = 30 μM) similar to the Michaelis constant (KM = 20 μM) of L-Phe (Fig. 2B and ESI Fig. S2†). Notably, competitive inhibition is not expected to skew specificity profiles, since the preference of the free enzyme remains unaltered. Formation of the enzyme–inhibitor complex only diminishes the free enzyme concentration, and hence, the overall rate. As predicted, ratios of hydroxamates remained constant over the course of the reaction (ESI Fig. S4†). Determination of kcat values for individual substrates is not the purpose of HAMA but if necessary, recording the time course of hydroxamate formation (ESI Fig. S5†) will ensure linear, initial velocity conditions without inhibition.
After validating HAMA on TycA, we proceeded to demonstrate its general applicability with a panel of NRPS modules encoding various known specificities. For this purpose, four modules from the gramicidin S cluster (GrsB1CAT [L-Pro], GrsB2CAT [L-Val], GrsB3CAT [L-Orn or L-Lys], GrsB4CATTe [L-Leu])34 and three modules from the surfactin A cluster (SrfA-A1CAT [L-Glu], SrfA-B2CAT [L-Asp], SrfA-CCATTe [L-Leu])35 were expressed in Escherichia coli, purified via nickel affinity chromatography and assayed with a mixture of 1 mM proteinogenic amino acids (Fig. 3A). The published specificities were correctly identified by HAMA while promiscuous side activities were minimal. Additionally, one uncharacterised AT domain from the jessenipeptin biosynthetic cluster from Pseudomonas aeruginosa QS1027,36 supposed to activate L-Thr (JesA1AT), was expressed and assayed, and the specificity profile clearly matched the expectation (Fig. 3A).
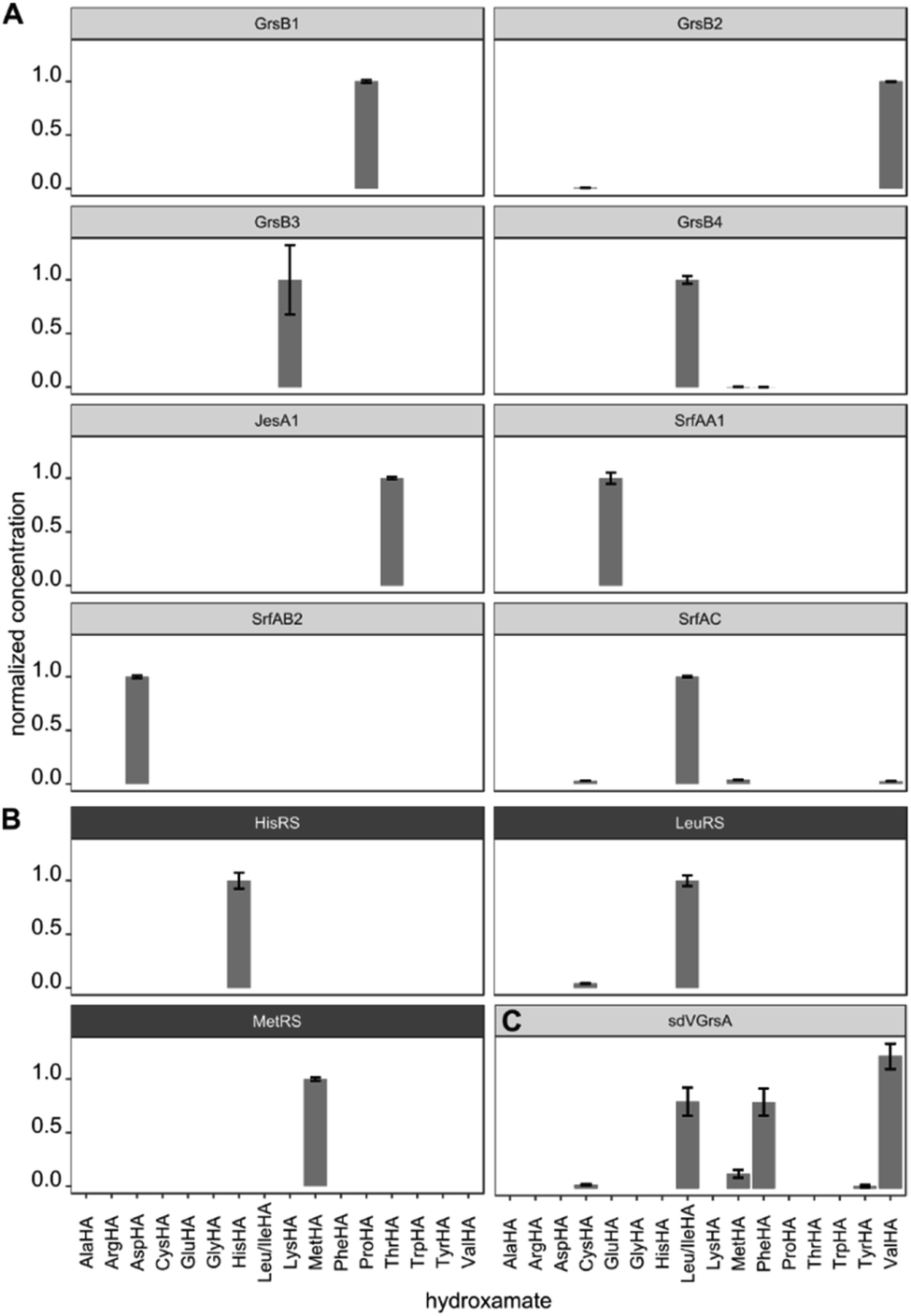 | ||
| Fig. 3 (A) Specificity profiles of several NRPS modules and (B) aaRSs have been determined using the HAMA assay. (C) Low activity of the engineered NRPS module sdVGrsA30 is detectable. Error bars indicate the standard deviation from three technical replicates. | ||
In ribosomal protein synthesis, aaRSs are key enzymes which have been thoroughly engineered aiming for expansion of the genetic code with unnatural amino acids.3,4 To demonstrate the potential of HAMA for the analysis of this enzyme class, we expressed and profiled three aaRSs from E. coli (MetG, LeuS, HisS; Fig. 3B). Again, all three specificities were correctly identified and almost no side-activities detected, as expected for highly proficient enzymes from primary metabolism.
Production of large NRPS proteins in a pure form is notoriously difficult, but a meaningful specificity profile was also obtained by HAMA using an enzyme preparation (NRPS module JesA1) of low purity (Fig. 3A and ESI Fig. S6†). The small fraction of expressed enzyme generated enough hydroxamate to deduce specificity and the large quantity of unknown contaminants did not interfere with the assay. Possible contaminants that unavoidably disturb adenylation assays are other carboxylate activating enzymes present in every cell, such as aaRSs, precluding measurements of adenylation activity in cellular lysates. In our hands, a single purification step via nickel affinity chromatography effectively eliminated background activity arising from these enzymes.
In enzyme engineering, low activities are often encountered, which present an analytical challenge. We tested the applicability of HAMA on sdVGrsAATE, a chimeric and poorly active NRPS module with imperfect L-Val specificity (kcat/KM[Val] = 0.3 mM−1 min−1) grafted by subdomain-swapping into L-Phe specific GrsA.30 Despite the low activity, determination of the sdVGrsA specificity profile succeeded. Reported side-activities for L-Phe and L-Leu next to the designed L-Val activity were confirmed (Fig. 3C).
In order to improve activity of sdVGrsA, we performed a brief directed evolution experiment37 and characterized selected mutants with HAMA. First, mutants were assessed based on the rate of Val-Pro diketopiperazine formation in a dimodular system together with GrsB1.30 Three rounds of mutagenesis were targeted to the interface between the grafted subdomain and the surrounding protein. Subdomain residues were reverted to the identity of the corresponding residue in GrsA at 5 positions at the A–T domain interface (1st round) and at 12 positions in the hydrophobic core of the A domain (2nd round). In the 3rd round, beneficial mutations were combined, yielding sdVGrsA-STAP (D306S, N334T, S338A, A356P) which produced 6.2-fold more peptide than sdVGrsA after 3 h at 37 °C. However, a HAMA profile of sdVGrsA-STAP revealed no improvement in substrate specificity compared to the broadly specific starting point sdVGrsA (Fig. 4). Another mutant, sdVGrsA-MSTP (G243M, D306S, N334T, A356P), showed 2-fold higher preference for Val (72%) at only 3-fold higher activity than sdVGrsA. Here, peptide yields in combination with comprehensive HAMA profiles allow to balance activity-specificity trade-offs and to find the most promising pathway for directed evolution.
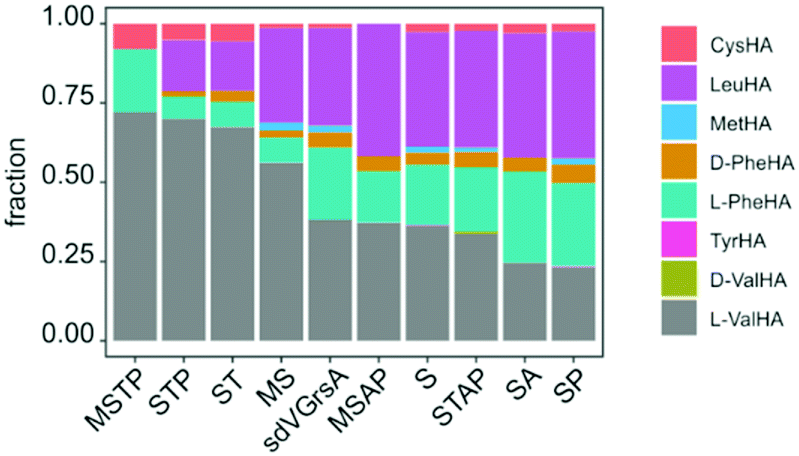 | ||
| Fig. 4 Specificity profiles of sdVGrsA variants obtained in a directed evolution experiment, sorted by L-Val specificity (MSTP: G243M, D306S, N334T, A356P; STP: D306S, N334T, A356P; ST: D306S, N334T; MS: G243M, D306S; MSAP: G243M, D306S, S338A, A356P; S: D306S; STAP: D306S, N334T, S338A, A356P; SA: D306S, S338A; SP: D306S, A356P) were measured at 33 °C and 3 h. | ||
Since HAMA resolves different products by mass, multimodular NRPSs encoding multiple adenylation activities on one protein can be measured. Heterologously expressed, four-modular GrsB from the gramicidin S synthetase was profiled (Fig. 5). Three out of the expected four hydroxamates (L-Pro, L-Val, L-Orn, L-Leu) were detected together with side activities, in particular for L-Lys which seems to be a good surrogate of L-Orn in GrsB3. L-Orn hydroxamate could not be quantified because synthesis of the standard failed, presumably due to its proclivity to cyclize. Indeed, Lys-containing gramicidin S analogues have been detected in the natural producer by mass spectrometry.38 It should be noted that a rigorous prediction of intracellular NRPS activities has to take the intracellular amino acid concentrations into account, and these vary from low micromolar to low millimolar.39
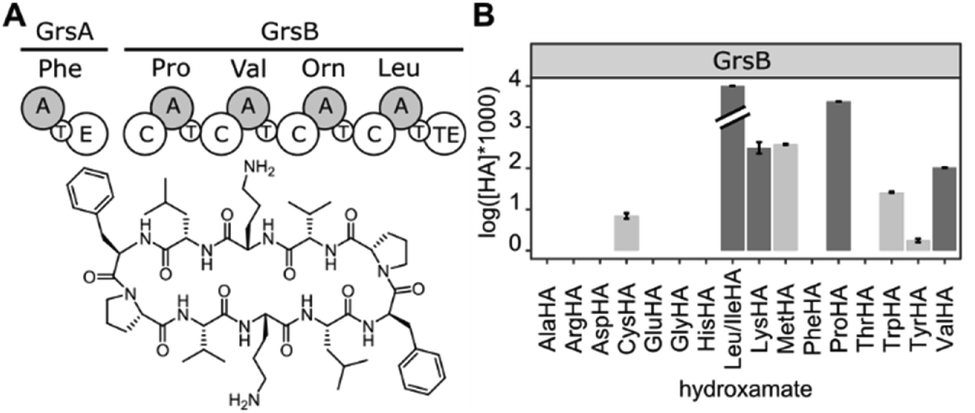 | ||
| Fig. 5 (A) The tetramodular NRPS GrsB is part of the gramicidin S synthetase. (B) HAMA profile of GrsB. Turnover for Leu was markedly faster than for the other substrates and exceeded 10% conversion. Promiscuous activities are shown in light grey. | ||
Conclusions
In summary, we have demonstrated HAMA's utility in substrate profiling of nonribosomal A domains and aaRSs coming from various origins and encoding various specificities. HAMA delivers detailed specificity profiles under competition conditions in a short time and with minimal experimental effort. Being superior to previous assays in several aspects, HAMA has potential to serve as a reliable standard tool in the engineering and discovery of adenylating enzymes. When the goal is to verify the link between genes and natural products in NRPS discovery, HAMA can provide valuable data because it discerns product structures via MS fragmentation. Previous MS methods observed acylated natural product synthetases or fragments ejected from acylated prosthetic groups.40 In contrast to these methods, HAMA calibrated with chemical standards has allowed accurate and highly sensitive quantification of weak, promiscuous activities. Structural information about the products not only alleviates problems caused by common sample contaminants such as amino acids, enzymes, or phosphate but also allows to resolve activities of multimodular NRPSs (Fig. 5). In the age of exploding sequence databases, substrate predictions for NRPSs obtained through HAMA could contribute to the deorphanization of biosynthetic pathways. By resolving multimodular activities, HAMA could also help to better understand the context dependence observed for NRPS substrate preference.24,41 Efficient substrate profiling will especially benefit screening and directed evolution efforts addressing the specificity of adenylating enzymes. In summary, HAMA offers a new analytical tool to several fields of research where adenylate-forming enzymes play important roles and will potentially expedite the development of tailored proteins and life-saving drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Daniel Braga for performing high-resolution MS measurements, to Heike Heinecke and Luka Raguž for help with NMR measurements, and to Oliver Waldmann, Philipp Stephan, and Hsin-Mei Huang for help with protein purification (all HKI Jena). We thank Linda Schlittenbauer (Waters GmbH) for advice on LC-MS/MS method development. This work has been supported by the Daimler und Benz Stiftung (HK). We further acknowledge donation of Pseudomonas jessenii genomic DNA from the Stallforth Lab (HKI Jena), and donation of plasmid pTrc99a_GrsBMtoL from David Niquille and Donald Hilvert (ETH Zurich) under an MTA.Notes and references
- F. Kudo, A. Miyanaga and T. Eguchi, J. Ind. Microbiol. Biotechnol., 2018, 46, 515–536 CrossRef PubMed.
- S. Schmelz and J. H. Naismith, Curr. Opin. Struct. Biol., 2009, 19, 666–671 CrossRef CAS PubMed.
- C. Fan, J. M. L. Ho, N. Chirathivat, D. Söll and Y. S. Wang, ChemBioChem, 2014, 15, 1805–1809 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 35, 1210–1228 RSC.
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC.
- H. Neumann, K. Wang, L. Davis, M. Garcia-Alai and J. W. Chin, Nature, 2010, 464, 441–444 CrossRef CAS PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS PubMed.
- M. Petchey, A. Cuetos, B. Rowlinson, S. Dannevald, A. Frese, P. W. Sutton, S. Lovelock, R. C. Lloyd, I. J. S. Fairlamb and G. Grogan, Angew. Chem., Int. Ed., 2018, 57, 11584–11588 CrossRef CAS PubMed.
- R. Hara, K. Hirai, S. Suzuki and K. Kino, Sci. Rep., 2018, 8, 2950 CrossRef PubMed.
- A. Stanišić and H. Kries, ChemBioChem, 2019, 20, 1347–1356 CrossRef PubMed.
- M. P. Stulberg and G. D. Novelli, Methods Enzymol., 1962, 5, 703–707 CAS.
- L. G. Otten, M. L. Schaffer, B. R. M. Villiers, T. Stachelhaus and F. Hollfelder, Biotechnol. J., 2007, 2, 232–240 CrossRef CAS PubMed.
- V. V. Phelan, Y. Du, J. A. McLean and B. O. Bachmann, Chem. Biol., 2009, 16, 473–478 CrossRef CAS PubMed.
- T. Kittilä, M. Schoppet and M. J. Cryle, ChemBioChem, 2016, 17, 576–584 CrossRef PubMed.
- H. Katano, H. Watanabe, M. Takakuwa, C. Maruyama and Y. Hamano, Anal. Sci., 2013, 29, 1095–1098 CrossRef CAS PubMed.
- H. Katano, R. Tanaka, C. Maruyama and Y. Hamano, Anal. Biochem., 2012, 421, 308–312 CrossRef CAS PubMed.
- T. J. McQuade, A. D. Shallop, A. Sheoran, J. E. Delproposto, O. V. Tsodikov and S. Garneau-Tsodikova, Anal. Biochem., 2009, 386, 244–250 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. Leigh Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- D. J. Wilson and C. C. Aldrich, Anal. Biochem., 2010, 404, 56–63 CrossRef CAS PubMed.
- R. Hara, R. Suzuki and K. Kino, Anal. Biochem., 2015, 477, 89–91 CrossRef CAS PubMed.
- S. D. Copley, Curr. Opin. Struct. Biol., 2017, 47, 167–175 CrossRef CAS PubMed.
- H. Kaljunen, S. H. H. Schiefelbein, D. Stummer, S. Kozak, R. Meijers, G. Christiansen and A. Rentmeister, Angew. Chem., Int. Ed., 2015, 54, 8833–8836 CrossRef CAS PubMed.
- S. Meyer, J. C. Kehr, A. Mainz, D. Dehm, D. Petras, R. D. Süssmuth and E. Dittmann, Cell Chem. Biol., 2016, 23, 462–471 CrossRef CAS PubMed.
- J. W. Han, E. Y. Kim, J. M. Lee, Y. S. Kim, E. Bang and B. S. Kim, Biotechnol. Lett., 2012, 34, 1327–1334 CrossRef CAS PubMed.
- M. Zhu, L. Wang and J. He, ACS Chem. Biol., 2019, 14, 256–265 CrossRef CAS PubMed.
- F. Ishikawa, A. Miyanaga, H. Kitayama, S. Nakamura, I. Nakanishi, F. Kudo, T. Eguchi and G. Tanabe, Angew. Chem., Int. Ed., 2019, 58, 6906–6910 CrossRef CAS PubMed.
- K. Throckmorton, V. Vinnik, R. Chawdhury, T. Cook, M. G. Chevrette, C. Maranas, B. Pfleger and M. G. Thomas, ACS Chem. Biol., 2019, 14(9), 2044–2054 CrossRef PubMed.
- B. Villiers and F. Hollfelder, Chem. Biol., 2011, 18, 1290–1299 CrossRef CAS PubMed.
- H. Kries, D. L. Niquille and D. Hilvert, Chem. Biol., 2015, 22, 640–648 CrossRef CAS PubMed.
- B. R. M. Villiers and F. Hollfelder, ChemBioChem, 2009, 10, 671–682 CrossRef CAS PubMed.
- H. Bisswanger, Enzyme Kinetics, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- B. P. Duckworth, D. J. Wilson and C. C. Aldrich, Measurement of Nonribosomal Peptide Synthetase Adenylation Domain Activity Using a Continuous Hydroxylamine Release Assay, Springer New York, New York, NY, 2016, vol. 1401 Search PubMed.
- J. Kratzschmar, M. Krause and M. A. Marahiel, J. Bacteriol., 1989, 171, 5422–5429 CrossRef CAS PubMed.
- B. Kluge, J. Vater, J. Salnikow and K. Eckart, FEBS Lett., 1988, 231, 107–110 CrossRef CAS PubMed.
- J. Arp, S. Götze, R. Mukherji, D. J. Mattern, M. García-Altares, M. Klapper, D. A. Brock, A. A. Brakhage, J. E. Strassmann, D. C. Queller, B. Bardl, K. Willing, G. Peschel and P. Stallforth, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3758–3763 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- K. Hori and T. Kurotsu, J. Biochem., 1997, 122, 606–615 CrossRef CAS PubMed.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Nat. Chem. Biol., 2009, 5, 593–599 CrossRef CAS PubMed.
- P. C. Dorrestein, J. Blackhall, P. D. Straight, M. A. Fischbach, S. Garneau-Tsodikova, D. J. Edwards, S. McLaughlin, M. Lin, W. H. Gerwick, R. Kolter, C. T. Walsh and N. L. Kelleher, Biochemistry, 2006, 45, 1537–1546 CrossRef CAS PubMed.
- A. Degen, F. Mayerthaler, H. D. Mootz and B. Di Ventura, Sci. Rep., 2019, 9, 5119 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04222a |
| This journal is © The Royal Society of Chemistry 2019 |