
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh frequency, calibration-free molecular measurements in situ in the living body†
Hui
Li‡
 a,
Shaoguang
Li‡
a,
Jun
Dai
b,
Chengcheng
Li
a,
Man
Zhu
a,
Hongxing
Li
a,
Xiaoding
Lou
a,
Fan
Xia
*a and
Kevin W.
Plaxco
*cd
a,
Shaoguang
Li‡
a,
Jun
Dai
b,
Chengcheng
Li
a,
Man
Zhu
a,
Hongxing
Li
a,
Xiaoding
Lou
a,
Fan
Xia
*a and
Kevin W.
Plaxco
*cd
aEngineering Research Center of Nano-Geomaterials of Ministry of Education, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, China. E-mail: xiafan@cug.edu.cn
bDepartment of Obstetrics and Gynecology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430074, China
cDepartment of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, California 93106, USA. E-mail: kwp@chem. ucsb.edu
dCenter for Bioengineering, University of California Santa Barbara, Santa Barbara, California 93106, USA
First published on 6th November 2019
Abstract
Abolition of the need for end-users to perform sensor calibration proved key to the widespread use of home-glucose monitors. Motivated by this observation here we have adapted electrochemical aptamer-based (E-AB) sensors, a sensing technology that is far more general than the glucose monitor, to the problem of performing calibration-free in vivo measurements of molecules other than glucose. Specifically, we first demonstrate the ability of E-AB sensors to achieve the accurate and precise measurement of cocaine, ATP and kanamycin in vitro in undiluted whole blood, achieving clinically relevant accuracy (better than ±20%) in this sample matrix without the need to calibrate individual sensors. We then demonstrate similar, calibration-free accuracy (±30%) for ATP and kanamycin measurements with sensors placed in situ in the jugular veins of live rats over multi-hour measurements runs that achieve time resolution of seconds and concentration precision of a few micromolar.
Introduction
The “home glucose meter,” which is used millions of times a day to measure glucose in finger-prick blood samples, achieves clinically relevant accuracy via highly reproducible fabrication (achieving just 3–5% variation from sensor to sensor in a given batch1), thus enabling the calibration of a few sensors in each batch to replace the need to calibrate each individual sensor.2 The alleviation of end-user calibration has not yet, however, proven possible for in vivo glucose sensors (i.e., “continuous glucose monitors”).2–6 In part this is due to physiology-driven changes in the relationship between interstitial glucose levels (i.e., the measurement site) and plasma glucose levels (the measure of interest). But it is also due to the complex manufacturing process associated with these sensors, which results in greater sensor-to-sensor variation (typically on the order of 10%, as tested in vitro).1 Because of this, each individual in vivo glucose sensor must be calibrated by the end-user. For example, the FreeStyle Navigator observes a 10 h equilibration period, followed by four in vivo calibrations over the following 5 days. The Dexcom STS requires a 1 h equilibration followed by twice daily calibrations over the ensuing daily wear. This necessary calibration step is both cumbersome and introduces opportunities for user error, which can lead in turn to inappropriate clinical action.In order to ease the adaptation of biosensors to clinical practice we present here a means of achieving calibration-free operation for electrochemical aptamer-based (E-AB) sensors, a broad class of in vivo biosensors that, because they are independent of the chemical or enzymatic reactivity of their targets, are quite versatile.7–14 E-AB sensors are comprised of a redox-reporter-modified, target-recognizing oligonucleotide “probe” that is deposited on an interrogating electrode via the formation of a self-assembled monolayer. Due to binding-induced changes in probe conformation, in the environment of the redox reporter, or in probe flexibility (e.g., due to the steric bulk of the target) the presence of target alters the kinetics with which electrons exchange to/from the redox reporter, producing an easily measurable change in current when the sensor is interrogated, for example, using square wave voltammetry.14,15 This reagentless, reversible, single-step read-out renders E-AB sensors selective enough that they perform well when challenged in vitro in undiluted blood serum.16–18 Indeed, in recent years we have adapted E-AB sensors to the problem of achieving high-frequency molecular measurements in situ in the living body,19–22 an advance that required correction19 or elimination20,21 the baseline drift seen under these challenging conditions. The resulting in vivo sensors now support seconds (or even sub-second21) resolved, multi-hour measurements when placed intravenously in live rats.19–22
The above-described success aside, all prior in vivo E-AB sensors suffered from the same hurdle as in vivo glucose monitors: the need to calibrate each individual sensor to correct for significant, sensor-to-sensor fabrication variation. Specifically, due to difficult-to-avoid variation in electrode surface area and, less so, aptamer packing density, the absolute currents produced by (currently hand-fabricated) E-AB sensors vary by up to several fold from one sensor to the next. Historically we have solved this problem by calibrating each individual in vivo sensor against a sample of known target concentration, most often using a sample of which the target concentration is known to be zero [e.g., ref. 19–22]. Previously, however, Ellington and co-workers have shown that a dual-reporter approach can be employed to render this class of sensors calibration free when they are deployed in vitro in a simple buffer.23 Building on their work, we demonstrate here adaptation of the approach to the much more challenging task of performing calibration-free measurements both in vitro in undiluted whole blood and in situ in the living body.
Results and discussion
Due to variations in the microscopic surface area of the electrode and in aptamer packing density the number of aptamers on an E-AB sensor, and thus the absolute currents they produce, differ significantly from one sensor to the next (Fig. 1C). To correct this we employ here a “dual reporter” approach, similar to that reported by Ellington and co-workers for use in vitro,23 that uses the signal from a second redox reporter as a reference to obviate the need to calibrate each individual sensor. Here the “sensing” reporter (methylene blue; MB) is placed on the distal terminus of the DNA chain and thus the current it produces is strongly altered by the conformational change associated with target recognition. A second, “reference” reporter (anthraquinone; AQ) is placed instead near the surface attachment site, a position that responds differently to the presence of the target. This allows the second reporter to serve as an internal reference with which to correct for sensor-to-sensor variability (Fig. 1A and B).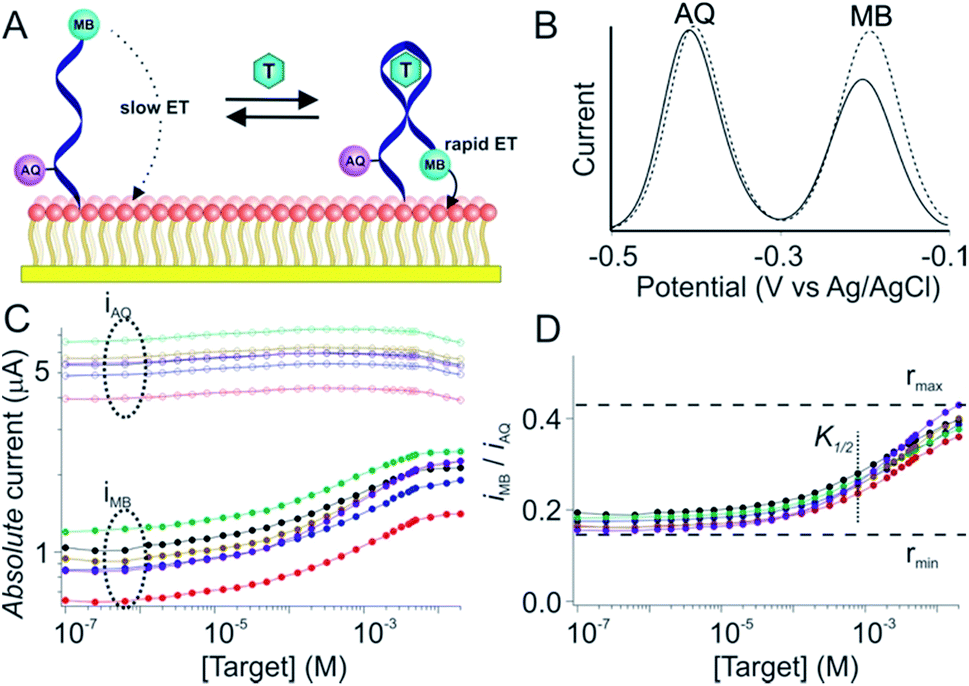 | ||
| Fig. 1 The dual-reporter approach to calibration-free E-AB sensing. (A) E-AB sensors are comprised of a redox-reporter-modified, target (T in the figure)-recognizing aptamer “probe” that is deposited on an interrogating electrode via the formation of a self-assembled monolayer. Target binding alters the kinetics with which electrons exchange to and from an attached redox reporter (here methylene blue; MB), producing an easily measurable change in current when the sensor is interrogated using square wave voltammetry. In this work we have modified the aptamer with a second reporter, anthraquinone (AQ), at a second site on the aptamer such that it responds differently to the presence of target, providing an internal reference for use in correcting sensor-to-sensor fabrication variation. (B) As shown here for a cocaine-detecting E-AB sensor, MB and AQ report at distinct, non-overlapping redox potentials, and thus can be interrogated independently of one another. (C) Due to variation in electrode surface area and the density with which their aptamer probes are packed there is significant sensor-to-sensor variation in the absolute currents produced by these reporters. (D) The ratio of their currents, however, is quite reproducible from one sensor to the next, thus providing a route to accurate, calibration-free operation when the sensors are deployed, as shown here, in vitro. In this paper, however, we have adapted the dual-reporter approach to the much more challenging task of performing calibration-free measurements in situ in the living body. | ||
The dual-reporter approach achieves excellent sensor-to-sensor reproducibility and good measurement accuracy when deployed in vitro. Specifically, despite ∼2-fold sensor-to-sensor variation in absolute current from both reporters (Fig. 1C), the ratio of the signals output by the two reporters is quite reproducible from one sensor to the next (±20%; Fig. 1D). To use this to achieve calibration-free operation we first titrated a training set of sensors and fit the resulting concentration–response profiles to the Hill equation (eqn (1)) to obtain the relevant parameters for each class of sensors (i.e., for all sensors employing a given aptamer probe under a given set of solvent conditions).
 | (1) |
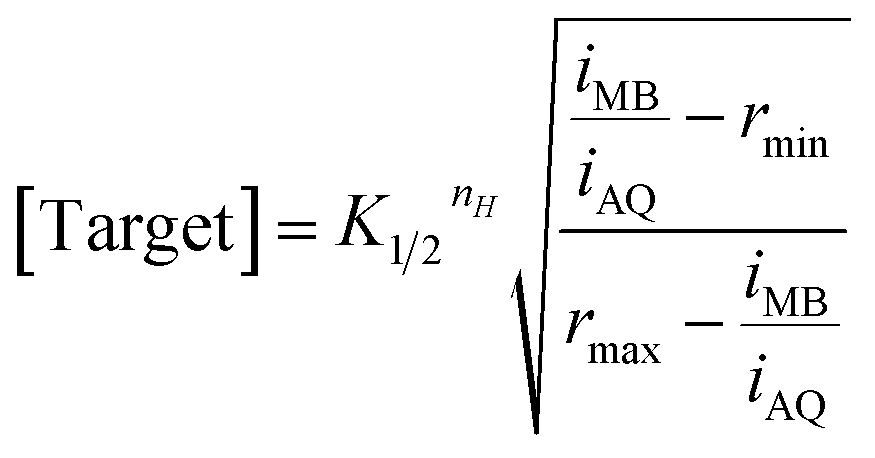 | (2) |
As our first test of the dual-reporter approach we applied it to a cocaine-detecting E-AB sensor.25,26 To do so we first challenged a training set of cocaine sensors in flowing, undiluted whole blood using an in vitro system meant to mimic circulation in the vasculature.24 As expected, while the currents produced by their reporters vary dramatically from sensor to sensor, their ratio is, again, quite reproducible (Fig. 2A, 1C and D). Using the data from this training set we defined rmin, rmax, nH and K1/2 for sensors in this class (Fig. 2B). Applying these parameters to a set of cocaine sensors outside of the training set we recover cocaine concentration estimates that are within 20% of the actual (spiked) concentration of the drug in whole blood over the range from 0.1 to 10 mM (Fig. 2C; black symbols), a level of accuracy comparable to that achieved using the traditional calibration approach on the same sensors (Fig. 2C, blue symbols).
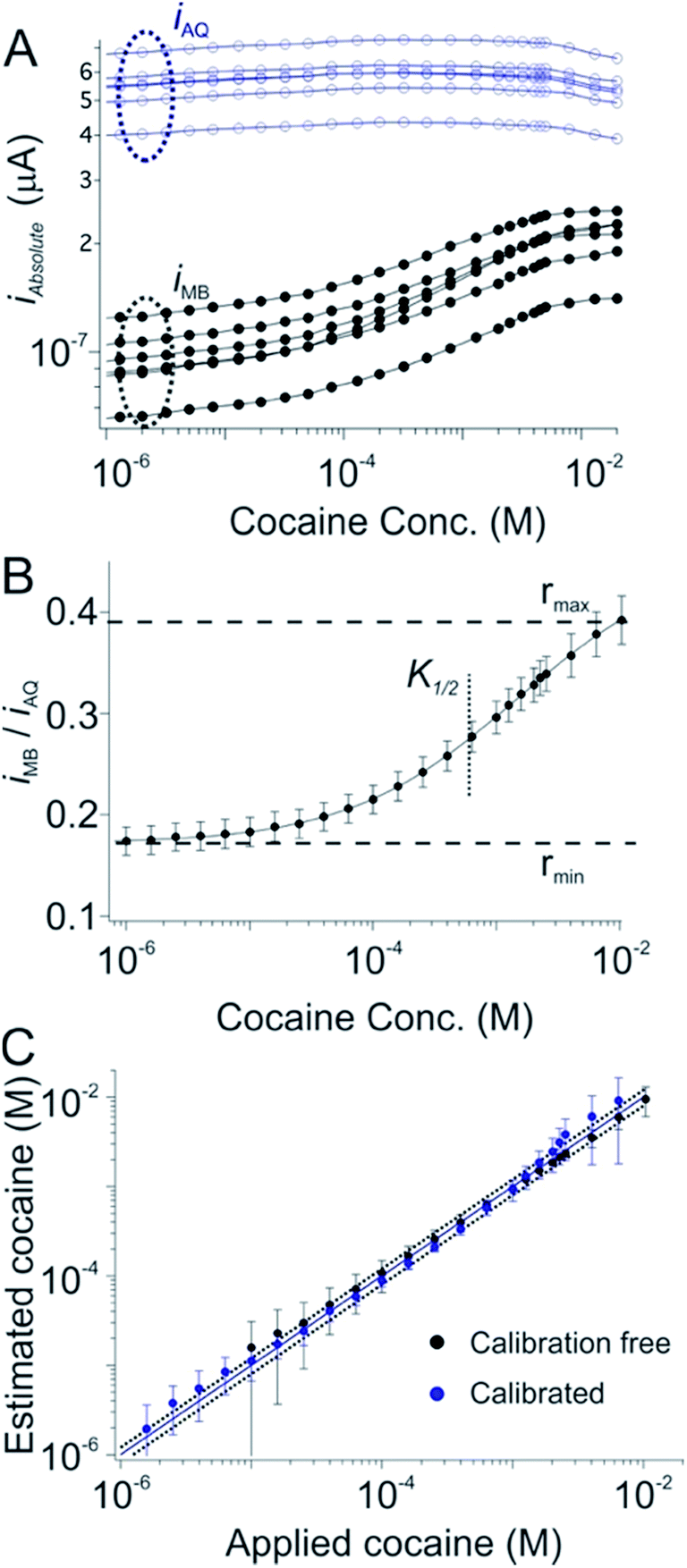 | ||
| Fig. 2 A dual-reporter, calibration-free sensor for cocaine. (A) We first challenged a set of dual-reporter, cocaine-detecting sensors with their target in whole blood, observing significant variation in both iMB and iAQ, the peak currents produced by the two reporters. (B) In contrast, the ratio iMB/iAQ is quite reproducible from sensor to sensor. Using these data as our training set we derived the parameters rmin, rmax, nH and K1/2 for this class of sensors; (C) applying these parameters to a set of sensors outside of our original training set illustrates our ability to reproducibly estimate cocaine concentration without the need to calibrate each individual sensor (the error bars indicate the standard deviations of measurements collected using multiple, independently fabricated, out-of-training-set sensors, thus illustrating reproducibility). Specifically, this dual-reporter calibration free approach produces cocaine concentration estimates (here in undiluted whole blood) within 20% of the actual (spiked) concentration of the drug over a two order of magnitude concentration range (black symbols, see individual sensors in Fig. S1†). This is quite similar to the accuracy we achieve by calibrating the same sensors in a sample of known (here zero) target concentration (blue symbols). This level of accuracy is also similar to that of commercial glucose sensors.4,5 Specifically, both approaches achieved limits of detection of ∼10 μM, defined as the concentration of target that produces a signal of iMB/iAQ three times the average noise level of a signal of iMB_0/iAQ_0 obtained in the absence of target. | ||
Dual-reporter, calibration-free operation also works well for sensors employing other aptamers. To show this we first employed a sensor against adenosine triphosphate (ATP). Once again, while the absolute currents produced by the reporters vary significantly from sensor to sensor (Fig. 3A), the ratio of these outputs is quite reproducible (Fig. 3B). As expected, after using a training set to define rmax, rmin, nH and K1/2 for this sensor class we obtain excellent measurement accuracy when we apply these parameters to new sensors outside of the training set. Specifically, the estimated target concentrations (in whole blood) we obtained are accurate to within 20% of the spiked concentrations across an approximate 30-fold concentration range (Fig. 3C, black), a level of accuracy quite similar to that achieved using the traditional calibration approach (Fig. 3C, blue symbols).
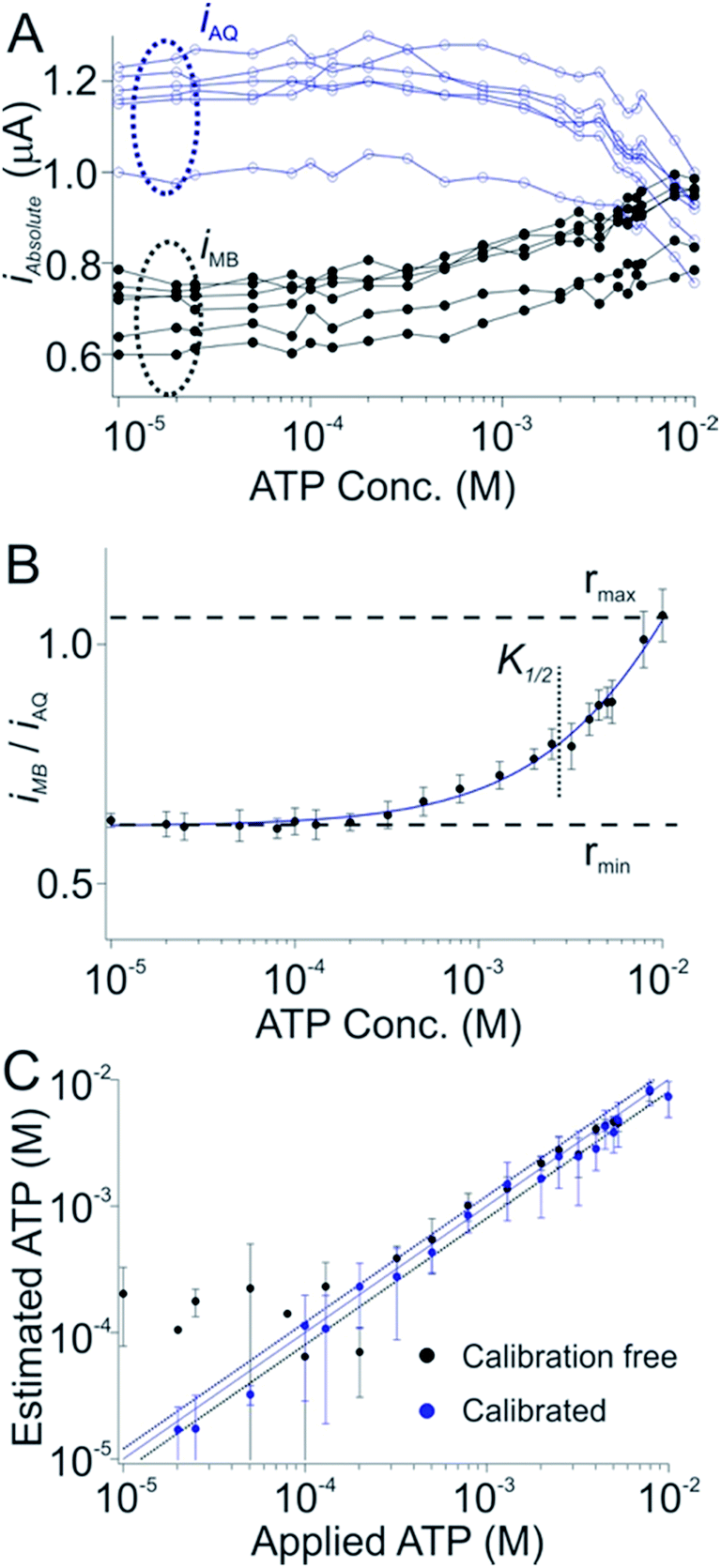 | ||
| Fig. 3 A dual-reporter, calibration-free sensor for ATP. (A) Challenging a training set of dual-reporter, ATP-detecting sensors with their target in undiluted whole blood, we again see significant variation in the absolute currents produced by the two reporters. (B) The ratio of their currents, however, is again quite reproducible from one sensor to the next. We employed this data set to derive the parameters rmin, rmax, nH and K1/2 for this class of sensors. (C) Applying these parameters to perform calibration-free measurements to a new, out-of-training-set group of sensors we reproducibly achieved ATP concentration estimates (in undiluted whole blood) within 20% of the actual (spiked) concentration across an approximate 30-fold concentration range (black symbols, see individual sensors in Fig. S2†), a level of accuracy we achieved by calibrating the same sensors in a sample of known (here zero) target concentration (blue symbols). Specifically, both approaches achieved limits of detection of ∼300 μM, defined as the concentration of target that produces a signal of iMB/iAQ three times the average noise level of a signal of iMB_0/iAQ_0 obtained in the absence of target. | ||
As a third test of the generality of the dual-reporter approach we adapted it to an E-AB sensor for the detection of the aminoglycoside antibiotics. In this case the target-induced signal change observed from the AQ reporter was (counter to our expectations) larger than that produced by the MB reporter (Fig. 4A), presumably due to binding-induced changes in environments of the two reporters rather than via the binding-induced conformational change illustrated above (Fig. 1A). Nevertheless, when we fit the ratio iAQ/iMB, to eqn (1) to obtain rmax, rmin, nH and K1/2, and apply these parameters to a set of sensors outside of out training set we still achieve measurement accuracy of better than ±20% across an approximate 15-fold concentration range (Fig. 4C, black) in results that closely match the accuracy of our traditional calibration approach on the same sensors (Fig. 4C, blue).
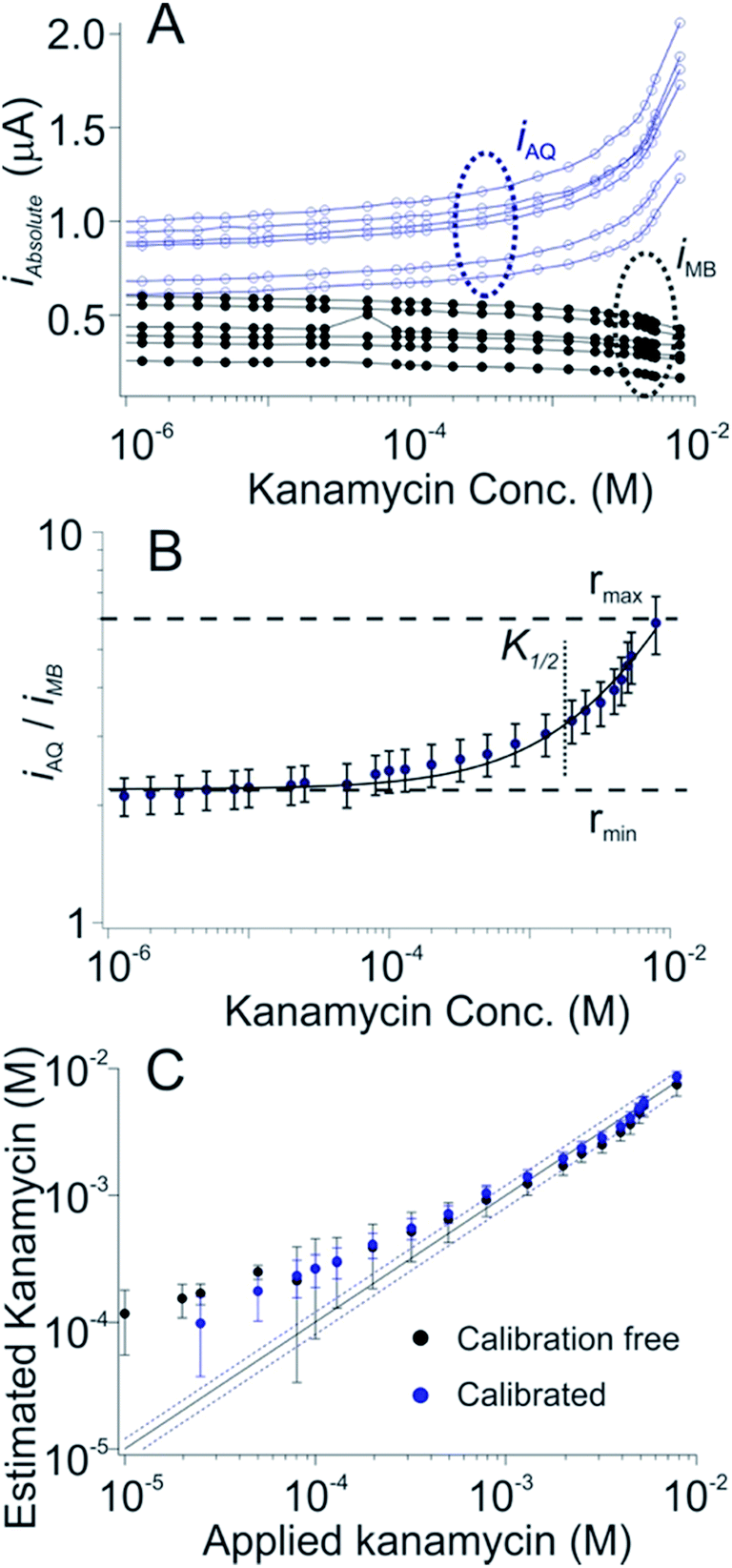 | ||
| Fig. 4 A dual-reporter, calibration-free sensor for kanamycin. (A) We first challenged a set of dual-reporter, aminoglycoside-detecting sensors with increasing concentrations of the antibiotic kanamycin in whole blood, observing once again significant variation in the absolute currents produced by the two reporters; (B) the ratio of their two currents, however, is quite reproducible. (C) Applying the dual-reporter calibration free approach we reproducibly achieved kanamycin concentration estimates within 20% of the actual (spiked) concentration of the drug across an approximate 15-fold concentration range (black symbols, see individual sensors in Fig. S3†), a level of accuracy we achieved by calibrating the same sensors in a sample of known (here zero) target concentration (blue symbols). Specifically, both approaches achieved limits of detection of ∼200 μM, defined as the concentration of target that produces a signal of iMB/iAQ three times the average noise level of a signal of iMB_0/iAQ_0 obtained in the absence of target. | ||
Dual-reporter calibration-free measurements provide a convenient, single-step approach to the measurement of specific molecules in whole blood in vitro (ex vivo). But can this approach also be applied to the problem of performing measurements in vivo? A problem with such measurements is that E-AB sensors exhibit significant baseline drift when placed in situ in the living body.19,20,27 We have previously shown, however, that replacement of their hydroxyl-terminated self-assembled monolayer with a more adsorption-resistant phosphatidylcholine monolayer largely eliminates this drift, thus supporting multi-hour measurements even under these challenging conditions.15 Here we have found that this same monolayer supports dual-reporter calibration-free operation (Fig. 5) suggesting that such sensors could support calibration-free in vivo measurements.
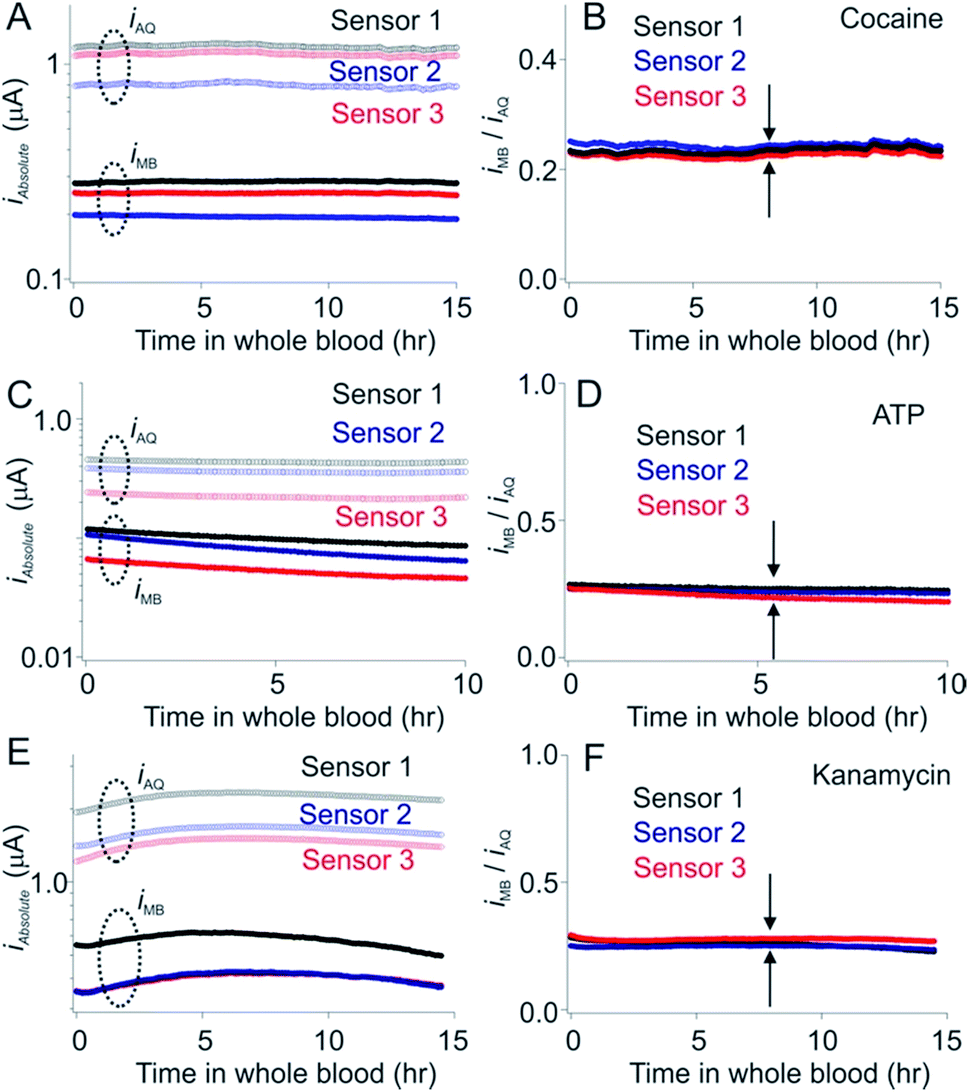 | ||
| Fig. 5 Phosphatidyl choline self-assembled monolayers minimize the drift seen in whole blood in vitro. E-AB sensors fabricated using hydroxyl-terminated monolayers drift significantly (typically of order 10 to 15% per hour) when deployed directly in undiluted whole blood.15,25. (Left hand column) As shown here, however, sensors fabricated using a phosphatidyl choline-terminated monolayer are much more resistant to this drift. Shown are the absolute currents produced by both the MB and AQ reporters on our cocaine, ATP and aminoglycoside sensors. (Right hand column) The ratio of the output currents likewise resists drift (less than 0.5% per hour). | ||
To test our ability to achieve calibration-free in vivo measurements we emplaced sensors for the detection of either ATP or kanamycin in the external jugular veins of anesthetized Sprague–Dawley rats. Upon injecting the relevant target into the opposite external jugular vein and applying the dual-reporter calibration-free approach to the sensor outputs produced by these in vivo tests, we achieved seconds-resolved, micromolar-precision molecular measurements following one or two injections of the target molecule over several hours of continuous monitoring (Fig. 6). The resulting plots of concentration versus time presents consecutive spikes corresponding to each of the injections performed, producing in the case of kanamycin pharmacokinetic values closely consistent with previous observations.19 To better define the accuracy of the calibration approach in vivo we compared these results with data obtained by calibrating the same sensor in vivo prior to the first target injection (i.e., at zero target concentration). As desired, the target concentration estimates produced by the calibration-free approach remained within 30% of those obtained using calibration on the same sensor throughout the duration of the experiment.
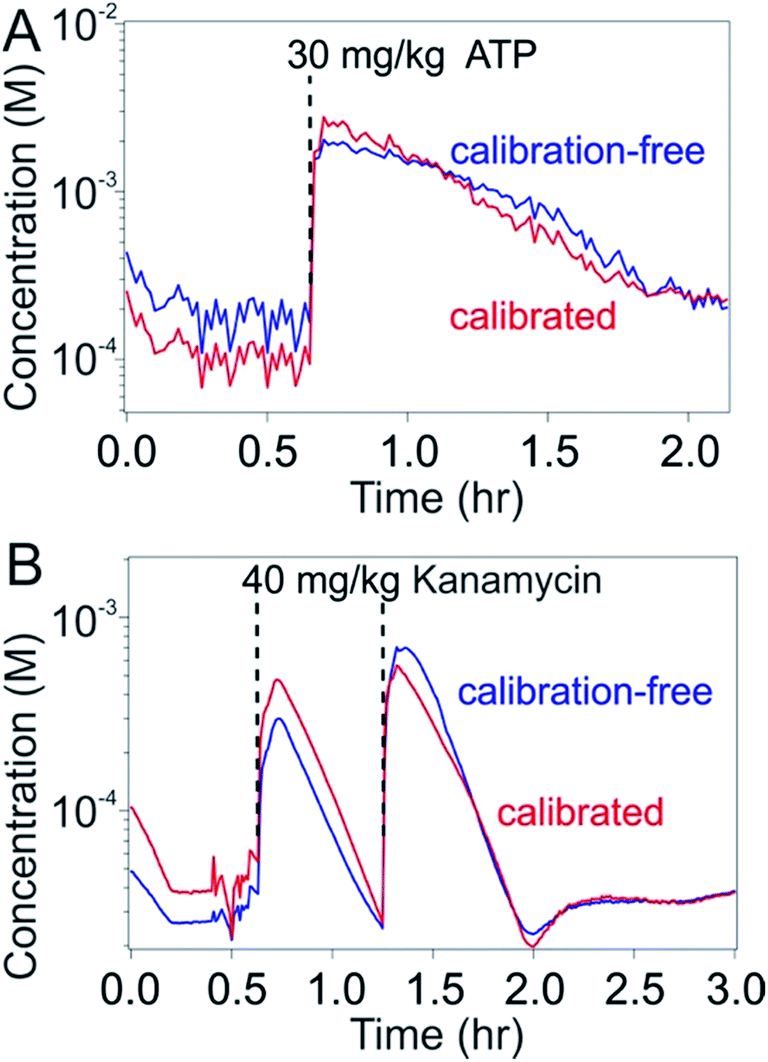 | ||
| Fig. 6 The calibration-free, high-frequency measurement of specific molecules in situ in the living body. Here we have employed phosphatidylcholine-modified, dual-reporter E-AB sensors emplaced in situ in the jugular veins of live rats to perform seconds-resolved measurements of plasma (top) ATP (a single intravenous injection of 30 mg kg−1) and (bottom) kanamycin (two sequential injections of 40 mg kg−1) levels using both calibrated (red) and calibration-free (blue) approaches applied to the same sensor. The calibration-free approach produces concentration estimates within 30% of those obtained via calibration over the duration of the measurement runs. | ||
Conclusions
Here we demonstrate high-frequency molecular measurements obtained in real time using uncalibrated E-AB sensors placed in situ in the living body. Specifically, by coupling dual-reporter calibration-free operation with drift-eliminating surface passivation we achieved clinically accurate (±30%) measurements in situ in the veins of live animals for measurements runs lasting hours and achieved time resolution of seconds and concentration precision of a few micromolar. Moreover, the approach we have taken to this end is relatively general, as the specificity of E-AB sensors can be switched via the simple expedient of replacing its aptamer. Indeed, the approach we have employed may be more general even than E-AB sensors, as it may also be adaptable other electrochemical sensing architectures employing binding-induced changes in electron transfer kinetics.28–32 Given this we believe that the described approach may broadly improve the utility of in vivo molecular measurements.Conflicts of interest
One author (KWP) has a financial interest in and serves on the scientific advisory boards of three companies attempting to commercialize E-AB sensors.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21804121, 21801231, 21525523, 21722507, 21574048, 21874121), the National Basic Research Program of China (973 Program, 2015CB932600), the National Key R&D Program of China (2017YFA0208000), National Institutes of Health (Grant R01GM125789), and the Fundamental Research Funds (CUG170665, CUG170668) for the Central Universities, China University of Geosciences (Wuhan). We would like to acknowledge Drs Philippe Dauphin-Ducharme and Gabriel Ortega for the development of the MATLAB script used to analyze square wave voltammetric (SWV) data.References
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS PubMed.
- J. J. Shin, K. R. Holtzclaw, N. D. Dangui, J. S. Kanderian, J. J. Mastrototaro, P. I. Hong, US Patent 7,029, 444, 2006.
- B. Feldman, R. Brazg, S. Schwartz and R. Weinstein, Diabetes Technol. Ther., 2003, 5, 769–779 CrossRef CAS PubMed.
- D. C. Klonoff, Diabetes Spectr., 2014, 27, 174–179 CrossRef PubMed.
- https://www.dexcom.com/get-startedcgm?utm_source=adwords%26utm_campaign=b%26sfc=70133000001LlpgAAC%26gclid=CO3fwIK419MCFRSDfgod2nwEiw .
- E. Waltz, Nat. Biotechnol., 2017, 35, 11–15 CrossRef CAS PubMed.
- A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43, 496–505 CrossRef CAS PubMed.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Angew. Chem., Int. Ed., 2005, 44, 5456–5459 CrossRef CAS PubMed.
- B. S. Ferguson, D. A. Hoggarth, D. Maliniak, K. Ploense, R. J. White, N. Woodward, K. Hsieh, A. J. Bonham, M. Eisenstein, T. E. Kippin, K. W. Plaxco and H. T. Soh, Sci. Transl. Med., 2013, 5, 213ra165 CrossRef PubMed.
- A. Idili, N. Arroyo-Curras, K. L. Ploense, A. T. Csordas, M. Kuwahara, T. E. Kippin and K. W. Plaxco, Chem. Sci., 2019, 10, 8164–8170 RSC.
- Y. Wu, B. Midinov and R. J. White, ACS Sens., 2019, 4, 498–503 CrossRef CAS PubMed.
- J.-X. Liu, N. Bao, X. Luo and S.-N. Ding, ACS Omega, 2018, 3, 8595–8604 CrossRef CAS PubMed.
- N. Arroyo-Curraś, P. Dauphin-Ducharme, G. Ortega, K. L. Ploense, T. E. Kippin and K. W. Plaxco, ACS Sens., 2018, 3, 360–366 CrossRef PubMed.
- X. Zuo, S. Song, J. Zhang, D. Pan, L. Wang and C. Fan, J. Am. Chem. Soc., 2007, 129, 1042–1043 CrossRef CAS PubMed.
- Y. Liu, J. Yan, M. C. Howland, T. Kwa and A. Revzin, Anal. Chem., 2011, 83, 8286–8292 CrossRef CAS PubMed.
- E. E. Ferapontova, E. M. Olsen and K. V. Gothelf, J. Am. Chem. Soc., 2008, 130, 4256–4258 CrossRef CAS PubMed.
- A. A. Lubin, R. Y. Lai, B. R. Baker, A. J. Heeger and K. W. Plaxco, Anal. Chem., 2006, 78, 5671–5677 CrossRef CAS PubMed.
- A. Vallee-Belisle, F. Ricci, T. Uzawa, F. Xia and K. W. Plaxco, J. Am. Chem. Soc., 2012, 134, 15197–15200 CrossRef CAS PubMed.
- N. Arroyo-Curraś, J. Somerson, P. A. Vieira, K. L. Ploense, T. E. Kippin and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 645–650 CrossRef PubMed.
- H. Li, P. Dauphin-Ducharme, N. Arroyo-Curras, C. H. Tran, P. A. Vieira, S. Li, C. Shin, J. Somerson, T. E. Kippin and K. W. Plaxco, Angew. Chem., Int. Ed., 2017, 56, 7492–7495 CrossRef CAS PubMed.
- N. Arroyo-Curras, P. Dauphin-Ducharme, G. Ortega, K. Ploense, T. Kippin and K. W. Plaxco, ACS Sens., 2018, 3, 360–366 CrossRef CAS PubMed.
- N. Arroyo-Currás, K. Scida, K. L. Ploense, T. E. Kippin and K. W. Plaxco, Anal. Chem., 2017, 89, 12185–12191 CrossRef PubMed.
- Y. Du, B. J. Lim, B. Li, Y. S. Jiang, J. L. Sessler and A. D. Ellington, Anal. Chem., 2014, 86, 8010–8016 CrossRef CAS PubMed.
- H. Li, P. Dauphin-Ducharme, G. Ortega and K. W. Plaxco, J. Am. Chem. Soc., 2017, 139, 11207–11213 CrossRef CAS PubMed.
- B. R. Baker, R. Y. Lai, M. S. Wood, E. H. Doctor, A. J. Heeger and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 3138–3139 CrossRef CAS PubMed.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2001, 123, 4928–4931 CrossRef CAS PubMed.
- H. Li, N. Arroyo-Curraś, D. Kang, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2016, 138, 15809–15812 CrossRef CAS PubMed.
- E. Prasad, B. W. Knettle and R. A. Flowers, J. Am. Chem. Soc., 2004, 126, 6891–6894 CrossRef CAS PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- K. Plumb and H.-B. Kraatz, Bioconjugate Chem., 2003, 14, 601–606 CrossRef CAS PubMed.
- G. A. Orlowski, S. Chowdhury and H.-B. Kraatz, Langmuir, 2007, 23, 12765–12770 CrossRef CAS PubMed.
- D. J. Feld, H.-T. Hsu, A. L. Eckermann and T. J. Meade, Langmuir, 2012, 28, 939–949 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04434e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |