DOI:
10.1039/C9SC04522H
(Edge Article)
Chem. Sci., 2019,
10, 9446-9453
Chemical communication in spatially organized protocell colonies and protocell/living cell micro-arrays†
Received
9th September 2019
, Accepted 11th September 2019
First published on 18th September 2019
Abstract
Micro-arrays of discrete or hemifused giant unilamellar lipid vesicles (GUVs) with controllable spatial geometries, lattice dimensions, trapped occupancies and compositions are prepared by acoustic standing wave patterning, and employed as platforms to implement chemical signaling in GUV colonies and protocell/living cell consortia. The methodology offers an alternative approach to GUV micro-array fabrication and provides new opportunities in protocell research and bottom-up synthetic biology.
Introduction
Giant unilamellar lipid vesicles (>1 μm, GUVs)1 have been used widely as a model cell system in biophysical research to investigate membrane mechanical properties,2 shape transformations,3 phase separation,4 budding and division,5,6 fusion,7–9 permeability,10 and protein/lipid bilayer interactions.11 GUVs have also been exploited as enzyme-based microreactors12 and for the design and construction of lipid-based protocell models exhibiting diverse functions.5,6,12–18 Significantly, microarrays of single GUVs of uniform size for potential use in high-throughput biochemical and chemical analysis have been developed using microcontact printing19,20 or microfluidics.21,22 Herein, we demonstrate a novel approach to the direct fabrication of GUV micro-arrays based on the spontaneous trapping of lipid vesicles in an acoustic (ultrasonic) standing wave pressure field. Although multi-dimensional trapping of micro-particles,23,24 liquid droplets,25 cells23,24,26 and organisms23,24 in water have been achieved using acoustic radiation forces, GUV micro-arrays have not previously been acoustically trapped due to the lack of acoustic contrast between the lipid vesicles and the surrounding aqueous continuous phase. We circumvent this problem by using a sucrose/glucose density gradient across the vesicle membrane and engineer the acoustic standing wave to generate one dimensional (1D) or two dimensional (2D) reconfigurable micro-arrays of GUVs with controllable spatial geometries (linear, square, rectangular, triangular) and lattice spacings. We demonstrate that micro-arrays consisting of single or multiple GUVs trapped at each zero-pressure point (node) of the acoustic pressure field can be fabricated by controlling the number of GUVs added to the acoustic trapping device and exploit the co-location of multiple GUVs to produce spatially arranged colonies27 of hemifused GUVs.
We use the micro-arrays of acoustically trapped GUVs for the spatial positioning and signaling of enzyme reactions. By trapping colonies of heterogeneous GUV populations containing either glucose oxidase (GOx) or horseradish peroxidase (HRP) we produce spatially distributed communities of synthetic protocells capable of localized enzyme-mediated chemical signaling triggered by a pore-forming peptide (melittin) inserted into the lipid membrane. We employ a similar strategy for the localization and killing of cancer cells28 by co-trapping clusters of GOx-containing GUVs and HepG2 cells. Finally, by co-trapping clusters of isopropyl β-D-thiogalactoside (IPTG)-containing GUVs and bacterial cells (Escherichia coli) at the acoustic pressure nodes in the presence of melittin, we demonstrate a protocell/living cell transmitter/receiver arrangement for bacterial gene expression.
Results and discussion
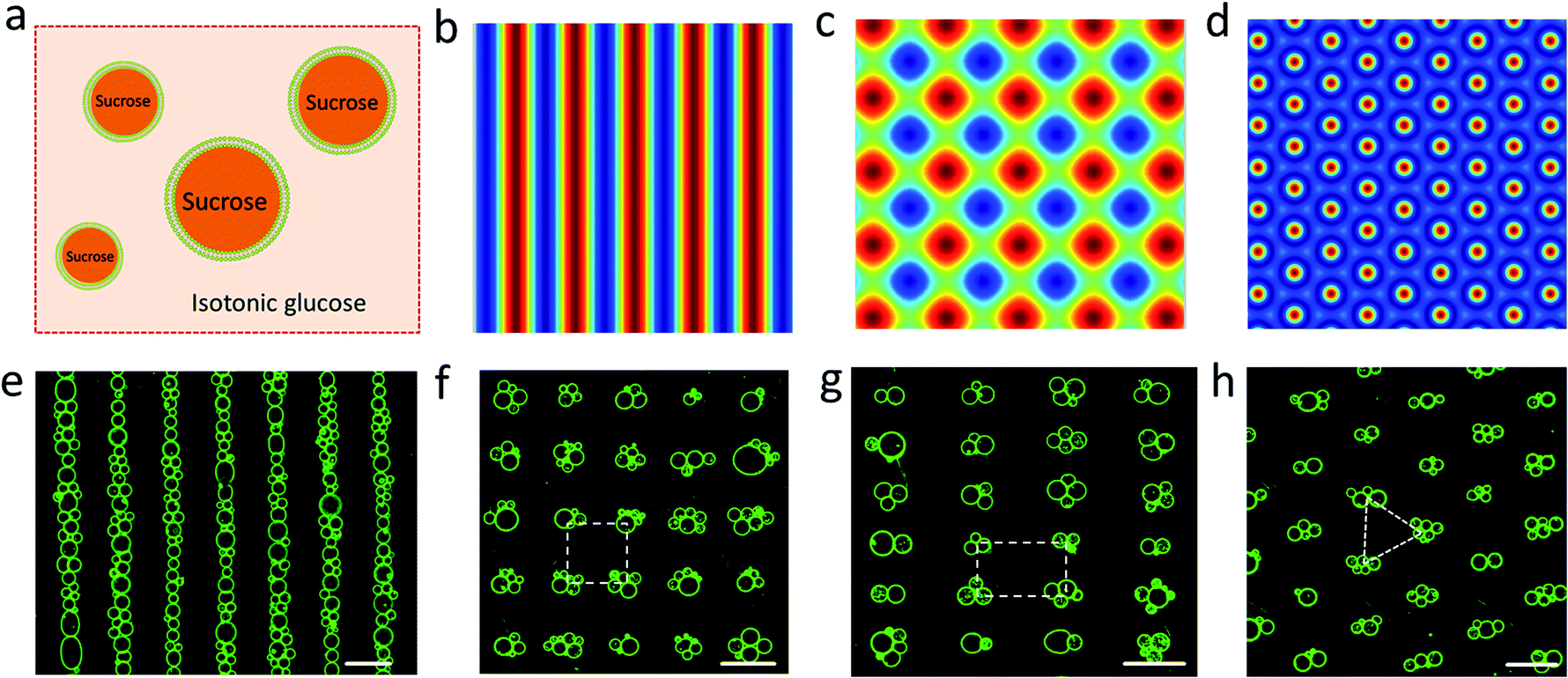
Spherically shaped GUVs typically 25 ± 14 μm in diameter were prepared from 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) using an electroformation method (Fig. S1†).29 We used sucrose-containing GUVs dispersed in an isotonic glucose solution (>25 mM) to generate a persistent density gradient (>0.5%) with sufficient acoustic contrast for efficient and reproducible trapping in an ultrasonic standing wave pressure field (Fig. 1a, S2 and S3†). The protocells were trapped typically within 15 min to produce 1D rows, 2D square grids or triangular micro-arrays by engineering the acoustic pressure fields accordingly (Fig. 1b–h and S4†). In each case, the DOPC vesicles remained structurally intact and non-fused in the acoustic pressure field, and the centre-to-centre line spacings could be readily modulated by changing the operational frequencies (Fig. 1f, g and S5, S6†). Moreover, reversible dynamic reconfiguration of the GUV arrays from vertical to horizontal alignments or from 1D to 2D arrangements could be achieved within ca. 4 min by on/off switching of the corresponding pairs of piezoelectric transducers (Fig. S7 and Movie S1†). In general, switching off the acoustic field resulted in re-dispersal of the GUVs into the bulk solution typically within 5–10 min (Fig. S8†).
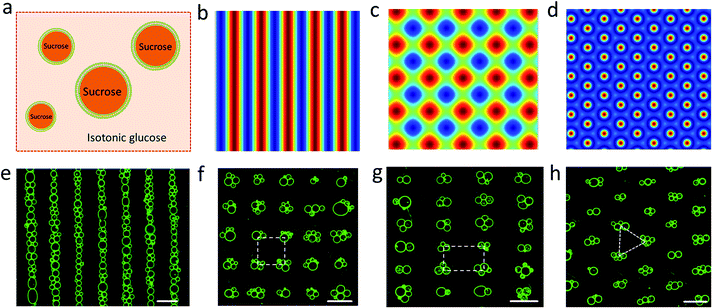 |
| | Fig. 1 (a) Schematic of sucrose-containing GUVs in isotonic glucose solution used for acoustic trapping. (b–d) Simulation of the acoustic pressure distribution in the acoustic trapping device for one pair of piezoelectric transducers (PZTs) (b), two pairs of PZTs with a square arrangement (c), and three pairs of PZTs with hexagonal arrangement (d); high pressure (blue, antinodes), low pressure (red, nodes). (e–h) Representative fluorescence microscopy images of micro-arrays of green GUVs (5% NBD-PE) produced in a 1D acoustic pressure field (6.71 MHz, 10 V) (e); 2D field (6.69/6.71 MHz, 10 V) (f); 2D field (5.06/9.13 MHz, 10 V) (g), and 2D field (6.70/6.71/6.72 MHz, 10 V) (h). Centre-to-centre line spacings: ca. 110 μm (e), 110 × 110 μm (inter-vesicle distance, ca. 50 μm) (f), 146 × 81 μm (g). All scale bars are 100 μm. | |
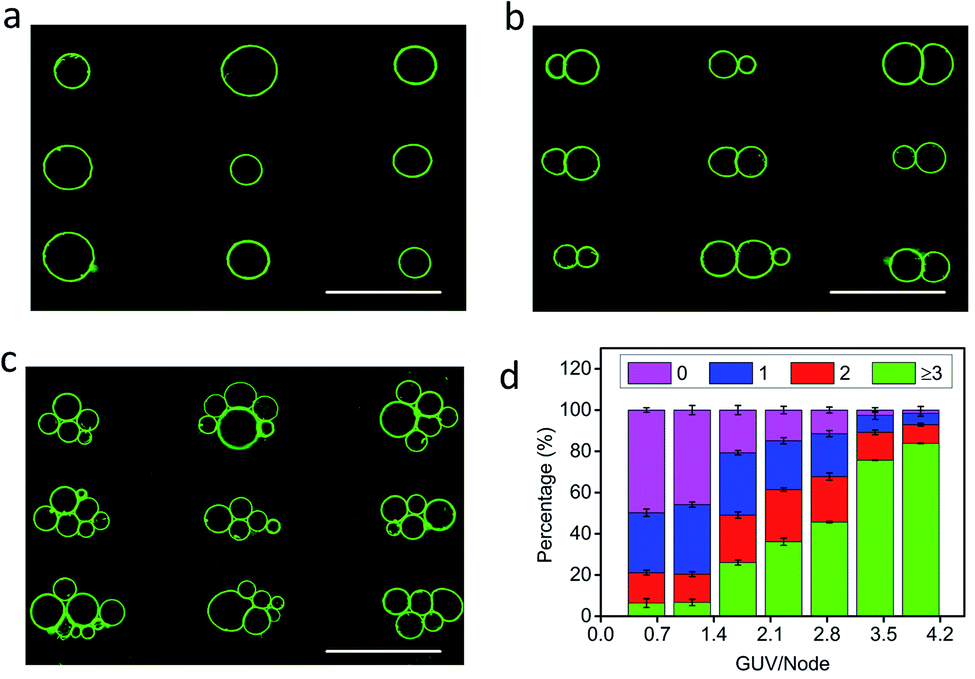
The level of protocell occupancy at each acoustic pressure node was adjusted by changing the GUV/node number ratio (RG/N) (Fig. 2a–c and S9†). When RG/N was below 1.2, ca. 33% of the nodes were occupied by single vesicles with the remaining sites being unoccupied (Fig. 2a and S9a†). The occurrence of two vesicles per node reached a maximum value (ca. 25%) when the RG/N ratio was ca. 2.2 (Fig. 2b and S9b†), and multiple numbers of vesicles per node (ca. 76%) were observed at RG/N values above 3.4 (Fig. 2c and S9c†). Clusters of multiple GUVs were predominantly trapped at RG/N > 3.4 (Fig. 2d), enabling the co-location of mixed protocell populations at each acoustic pressure node (Fig. S10†). We used this approach as a step towards the periodic arrangement of spatially separated colonies of protocells capable of structural integration and functional activation. For this, we exploited local contact points between the GUVs as foci for the Ca2+-induced hemifusion of the co-localized protocells in the presence of the acoustic field (Fig. S11†). Hemifusion of the vesicles resulted in mixing of the membrane lipids (Fig. S12†), while no lipid exchange was observed in the absence of Ca2+ ions (Fig. S13†). Moreover, in comparison to the non-fused GUVs, the hemifused GUV clusters remained spatially fixed at the lattice positions when the acoustic pressure field was switched off. This allowed us to prepare arrays using a two-step patterning procedure that contained co-located clusters of non-fused and hemifused GUVs that exhibited protocell-selective dynamical responses to changes in the acoustic pressure field (Fig. S14 and Movie S2†).
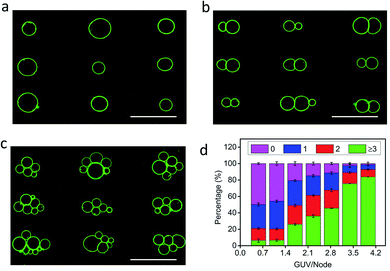 |
| | Fig. 2 (a–c) Representative fluorescence microscopy images of acoustically trapped rectangular micro-arrays consisting of single (a), double (b) and multiple (c) GUVs at each pressure node; samples prepared using GUV/node number ratios (RG/N) of 1.2 (a), 2.2 (b) and 3.4 (c). All scale bars are 100 μm. (d) Plots of the percentage of unoccupied nodes (no GUVs, black) and occupied nodes containing single (red), double (blue) or multiple GUVs (≥3, cyan) against RG/N. In total, three independent replicates were conducted at each GUV/node number ratio (RG/N). In each replicate, 500 nodes were counted. Error bars represent the standard deviation. | |
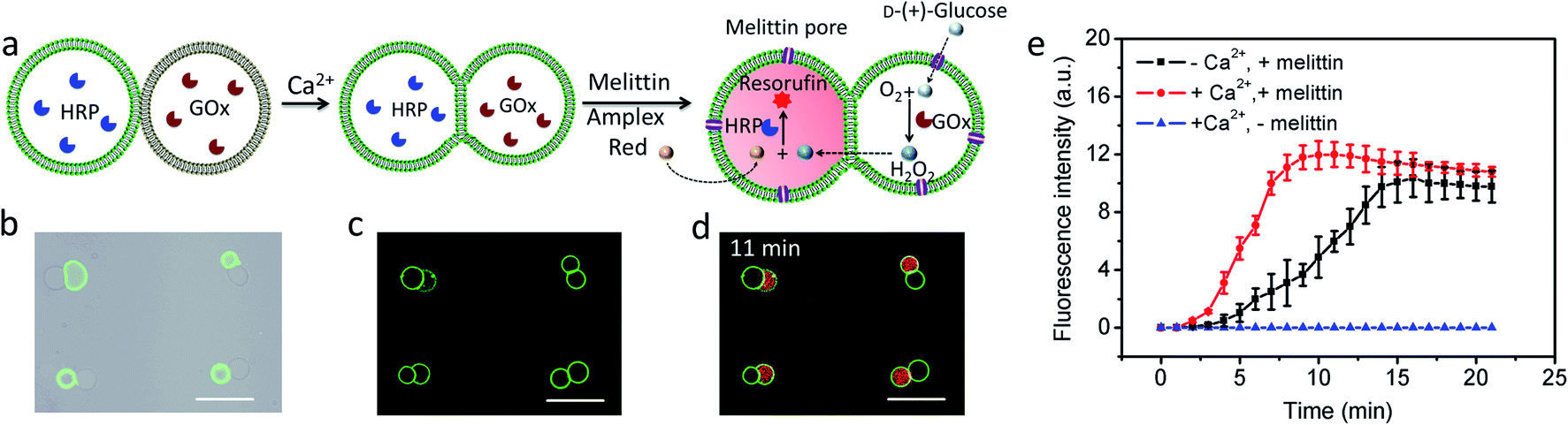
Given the above observations, we exploited the structurally integrated GUVs for the spatial positioning of enzyme reactions within colonies of the hemifused protocells. For a proof of concept, we implemented a simple and well-established signal transduction pathway into an inter-vesicular enzyme GOx/HRP cascade reaction (Fig. 3a).12,30,31 All the GUVs showed similar enzyme concentration, and the encapsulation efficiency was independent on the size of GUVs (Fig. S15†). The cascade was triggered by the spontaneous membrane insertion of a toroidal pore-forming cationic peptide (melittin)32 and concomitant uptake of glucose into the GUVs. A red fluorescence output was detected specifically in the HRP-loaded but not in the co-trapped GOx-containing hemifused GUVs within a few minutes in the presence of melittin, Amplex red and glucose (Fig. 3b–d, and S16†), consistent with facile penetration of glucose through the peptide channels and inter-vesicular transfer of H2O2. No fluorescent output was observed in hemifused colonies of co-trapped GUVs that did not include melittin (Fig. S17†) or both types of enzymes (Fig. S18†), indicating that uptake of glucose through the peptide channels implemented a localized pathway for signal transduction specifically between the different protocells. Similar observations were made for resorufin production in non-fused aggregates of co-trapped GUVs comprising mixed populations of melittin-loaded enzyme-containing vesicles (Fig. S19†). However, the rate of resorufin production measured at a similar enzyme loading, substrate concentration and GUV number ratio per node was considerably lower in the non-fused GUVs compared with the hemifused arrangement (Fig. 3e). We attributed this to increased diffusional constraints associated with the transfer of H2O2 across two separated bilayers for colonies of non-fused GUVs compared with the single bilayer connections present in the hemifused protocells.
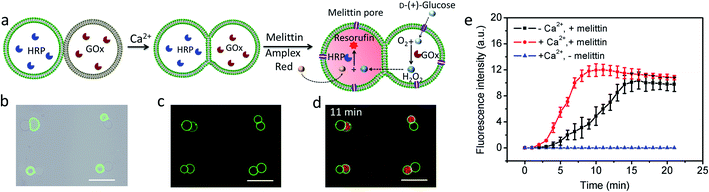 |
| | Fig. 3 (a) Schematic of a hemifused heterogeneous pair of acoustically trapped GUVs containing either HRP or GOx, followed by addition of melittin, Amplex red and glucose. (b) Superimposition of bright field and fluorescence microscopy images of a rectangular micro-array consisting of colonies of co-trapped NBD-PE-labeled HRP-containing GUVs (green fluorescence) and unlabeled GOx-containing GUVs. (c) As for (b), but after Ca2+-induced hemifusion. (d) As for (c), but superimposition of red and green fluorescence image recorded 11 min after addition of melittin and Amplex red. All scale bars are 50 μm. (e) Time-dependent changes in mean fluorescence intensity for GOx/HRP-mediated formation of resorufin in acoustically trapped micro-arrays comprising melittin-functionalized hemifused multiple GUVs (red), melittin-functionalized non-fused multiple GUVs (black) and hemifused GUVs without melittin (blue). The increase of fluorescence intensity at the single GUV level was followed and the error bars represent variations between GUVs during a single experiment. Initial rates; 2.37 ± 0.48 and 0.67 ± 0.16 a.u for hemifused and non-fused GUVs, respectively. | |
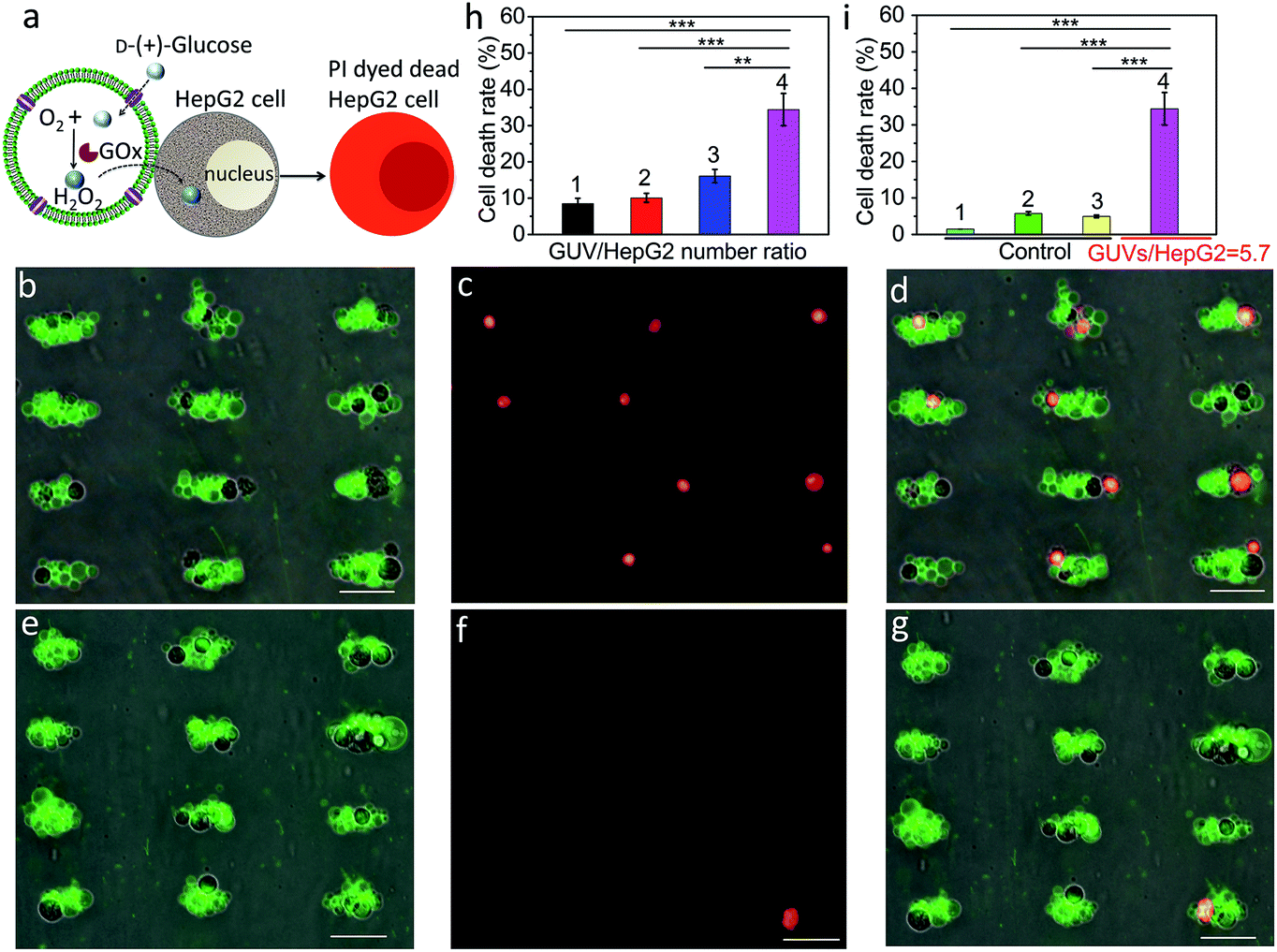
We employed a similar signal-induced process for the localization and killing of cancer cells by co-trapping clusters of melittin-functionalized GOx-containing GUVs and HepG2 cells at the acoustic pressure nodes (Fig. 4a). As shown by optical and fluorescence microscopy, co-localized clusters of NBD-PE labeled melittin/GOx GUVs (green fluorescence) and propidium iodide (PI)-stained HepG2 cells displayed significant numbers of dead cells at the acoustic nodes (Fig. 4b–d) compared to the control experiments in which clusters of GUVs and HepG2 cells were co-cultured in the absence of GOx in the GUVs (Fig. 4e–g), indicating that toxic levels of hydrogen peroxide were transferred into the adjacent cells. Increasing the number of GUVs per node increased the killing efficiency, consistent with the increased flux of hydrogen peroxide under these conditions (Fig. S20†). As a consequence, over 35% of the trapped HepG2 cells were killed in a period of 4 h at a GUV/HepG2 number ratio of 5.7 (Fig. 4h and S21†). Control experiments undertaken in the absence of encapsulated GOx or melittin, or without GUVs showed minimal levels of cell death (Fig. 4i and S22†).
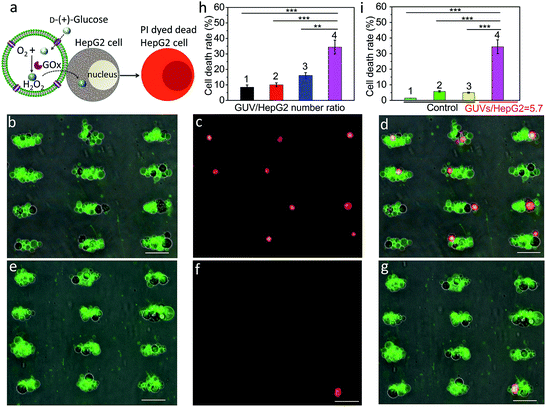 |
| | Fig. 4 (a) Schematic showing H2O2-induced killing of HepG2 cells by co-trapped melittin-functionalized GOx-containing GUVs. (b) Superposition of optical and green fluorescence microscopy images of co-trapped clusters of melittin/GOx-GUVs and HepG2 cells (dark objects) recorded 4 h after addition of glucose (GUV/HepG2 number ratio, 5.7). (c) Red fluorescence microscopy image of (b) after staining with PI to determine the number of dead cancer cells. (d) Superposition of (b) and (c). (e) Superposition of optical and green fluorescence microscopy images of co-trapped clusters of melittin/GUVs and HepG2 cells (dark objects) without GOx encapsulated in the GUVs recorded 4 h after addition of glucose (GUV/HepG2 number ratio, 5.7). (f) Red fluorescence microscopy image of (e) after staining with PI to determine the number of dead cancer cells. (g) Superposition of (e) and (f). Scale bars in (b–g) are 50 μm. (h) Corresponding histogram of percentage of dead HepG2 cells after 4 h for GUV/HepG2 number ratios of 0.5 (1), 1.4 (2), 2.9 (3) and 5.7 (4). Approximately 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were used in each experiment; (n = 3, **p < 0.01, ***p < 0.001). (i) Control experiments undertaken at a GUV/HepG2 number ratio of 5.7; histograms showing low percentage (<6%) of dead cells after 4 h for HepG2 cells alone (1), HepG2/melittin-GUVs (no GOx) (2) and HepG2/GOx-GUVs (no melittin) (3) compared with HepG2/melittin/GOx-GUVs (4); (n = 3, ***p < 0.001). Three independent replicates were conducted in each experimental group and control group. Error bars represent the standard deviation. 000 cells were used in each experiment; (n = 3, **p < 0.01, ***p < 0.001). (i) Control experiments undertaken at a GUV/HepG2 number ratio of 5.7; histograms showing low percentage (<6%) of dead cells after 4 h for HepG2 cells alone (1), HepG2/melittin-GUVs (no GOx) (2) and HepG2/GOx-GUVs (no melittin) (3) compared with HepG2/melittin/GOx-GUVs (4); (n = 3, ***p < 0.001). Three independent replicates were conducted in each experimental group and control group. Error bars represent the standard deviation. | |
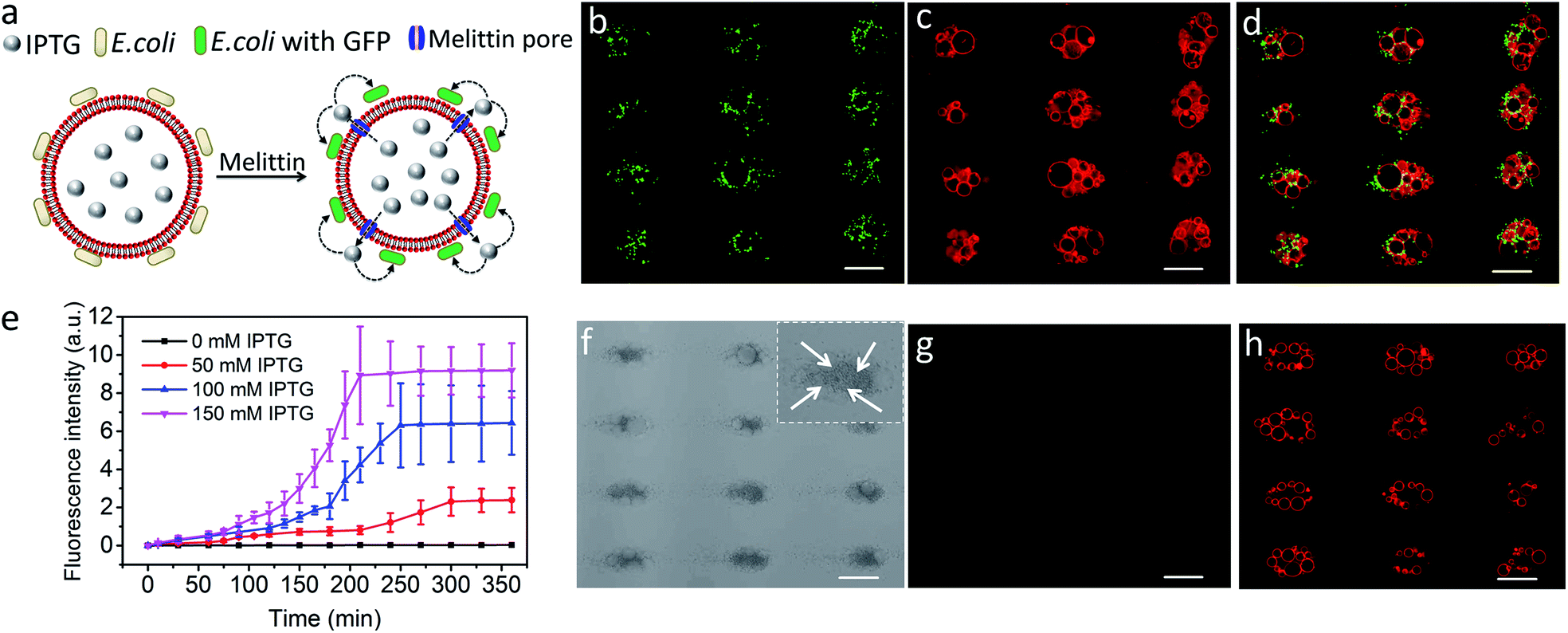
Finally, we explored the possibility of producing integrated protocell/living cell microarrays by co-locating clusters of GUVs and E. coli cells at the acoustic pressure nodes and exploiting the mutual organization for protocell-mediated induction of bacterial gene expression (Fig. 5a).33–36 To achieve this, we encapsulated IPTG in the GUVs prior to acoustic trapping with a population of E. coli BL21(DE3) pLysS cells transformed with a plasmid encoded for green fluorescent protein (GFP) expression under a T7 promoter/lac operator. Melittin was added to initiate membrane-mediated transfer of IPTG from the GUVs to neighbouring E. coli, which in turn resulted in GFP expression in the bacterial cells (Fig. 5b–d and S23–25†). In contrast, control experiments showed that there was no GFP expression in E. coli in the absence of melittin suggesting that there was no passive leakage of IPTG from the GUVs (Fig. 5f–h). Moreover, accumulation of bacteria at the GUV membrane in the acoustic field did not lead to pore formation on the lipid membrane. An IPTG concentration-dependent response was observed in the bacterial cells with maximum levels of GFP expression being observed typically over a period of 200 min for entrapped inducer concentrations of 150 mM (Fig. 5e, S26, and S27†).
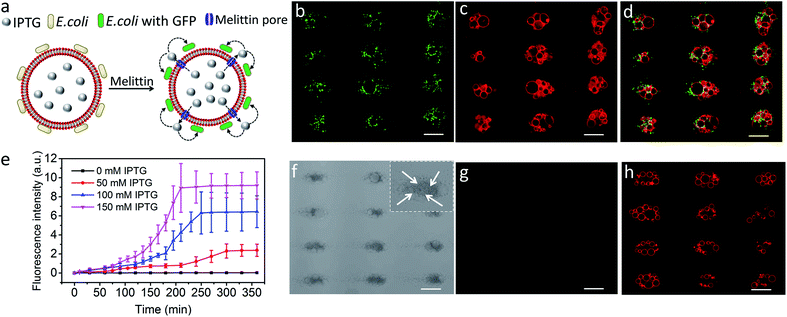 |
| | Fig. 5 (a) Schematic showing IPTG-induced GFP expression in E. coli by co-trapped melittin-functionalized IPTG-containing GUVs. (b–d) Representative fluorescence microscopy images of GFP expression in E. coli cells (green fluorescence) (b), co-trapped GUVs (red fluorescence, 0.5% TR-DHPE) (c) and green/red superimposition (d) of co-trapped clusters of IPTG (150 mM)-containing GUVs and E. coli recorded 210 min after addition of melittin. (e) Time-dependent changes in mean fluorescence intensity for IPTG-induced GFP expression in E. coli by co-trapped melittin-functionalized GUVs containing IPTG with different concentrations. (f–h) Representative bright field image (f) and fluorescence microscopy images recorded at 463 nm excitation (green fluorescence) (g) and 535 nm (red fluorescence, 0.5% TR-DHPE labelled GUVs) (h) of co-localized IPTG containing GUVs and E. coli recorded 6 h without melittin. All scale bars are 50 μm. The darker dots observed at the nodes (white arrows in the inset in (f)) are E. coli cells. Image g indicates no GFP expression in E. coli in the absence melittin. Scale bars are 50 μm. | |
Conclusion
In conclusion, 1D and 2D micro-arrays of GUVs with controllable spatial geometries, lattice spacings and number of vesicles per pressure node are fabricated by acoustic trapping. The methodology is straightforward, adaptable in terms of micro-engineering, and readily amenable to in situ chemical manipulations possibly including non-equilibrium spatiotemporal sensing.30 Spatially arranged clusters of GUVs can be used to produce hemifused colonies of co-trapped vesicles capable of localized enzyme-directed chemical communication via peptide-mediated inter-vesicular diffusion. Similarly, clusters of HepG2 cells and GUVs are employed as platforms for the signal-induced enzyme-mediated killing of co-trapped cancer cells. Additionally, micro-arrays of IPTG-containing GUVs and E. coli cells can be employed as transmitter/receiver platforms for the local induction of GFP expression, suggesting a step towards high throughput chemical signalling between artificial cells and living cells. Our results are relevant for research in bottom-up synthetic biology,19,20,37 including the assembly of synthetic prototissues,38–41 and design of vesicle-mediated chemical communication pathways in protocell/living cell consortia.33,36,42,43 A key advantage of the described methodology is that it provides a quick access to various arrays of GUVs or GUVs/cells, which in principle could be adapted as multi-functional high throughput platforms for chemical and biochemical screening, enzymatic assays and clinical diagnostics. For instance, chemical signal transfer within vesicle colonies could be explored in heterogeneous multi-vesicle arrays to implement cooperative inter-cellular communications. Moreover, arrays of hybrid vesicles and living cells provide opportunities for the rapid screening of diverse cell-lipid vesicle interactions.
Experimental
Materials
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) was obtained from Avanti Polar Lipids; Texas Red-labeled 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine triethylammonium salt (TR-DHPE) and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine triethylammonium salt (NBD-PE) were supplied by Thermal Fisher Scientific; D-(+)-glucose (≥99.5%), chloroform, melittin (≥85% HPLC, Sigma), Amplex red (≥98.0% HPLC), glucose oxidase (GOx) from Aspergillus niger (type VII, ≥100000 units per g solid), horseradish peroxidase (HRP) (≥250 units per g solid), bovine serum albumin (BSA), rhodamine-B, rhodamine-isothiocyanate (RITC), fluorescein-isothiocyanate (FITC) and isopropyl β-D-thiogalactoside (IPTG) were purchased from Sigma Aldrich. Glass slides coated with indium tin oxide (ITO, sheet resistance ≈ 8 to 12 Ω per square, thickness ≈ 160 nm) were produced by Hangzhou Yuhong Technology. Saline solution and sucrose were supplied by Xilong Chemicals. Millipore MilliQ water with a resistivity of 18.0 MΩ cm was used throughout.
Preparation of sucrose-containing GUVs
GUVs were prepared using an electroformation method. In general, a chloroform solution of the lipids (DOPC, 8 μL, 5 mg mL−1) labelled with the fluorescent lipids TR-DHPE 0.5% (red fluorescence) or NBD-PE 5% (green fluorescence) was deposited on two indium tin oxide (ITO) coated glass slides, and then spread gently on the conductive side of ITO. The residual chloroform was removed under a nitrogen flow. The two ITO-coated glasses were separated by a polytetrafluoroethylene (PTFE) spacer to form an electroformation chamber, which was filled with sucrose solution (0–400 mM). An AC electric field (10 Hz, 5 V) was then applied by a signal generator for 2 h to produce a stock dispersion of the sucrose-containing GUVs. The number of GUVs per unit volume (vesicle density) was obtained as follows. 10 μL of a sucrose-containing GUVs stock solution was added to 120 μL of isotonic glucose solution, and placed into the observation chamber of a fluorescence microscope and the GUVs allowed to sediment onto the substrate (10 min). Fluorescence microscopy images were then arbitrarily recorded and the GUV density (ρ) was calculated from ρ = σ/V × AS/AF, where ρ represents the vesicle density, σ the average number of the GUVs in the field of view, V is the volume (10 μL) of added GUVs to the chamber, AS is the total area of the observation chamber, and AF is the area of the field of view. Typically, the value of ρ was 2152 ± 110 per μL, which was consistent with flow cytometry results (BD Accuri C6 flow cytometer).
Enzyme encapsulation in GUVs
0.05 mg mL−1 RITC-HRP or HRP (0.1 U mL−1) were dissolved in a 400 mM aqueous sucrose solution and encapsulated in DOPC GUVs by electroformation. HRP-containing GUVs were washed three times with isotonic glucose to remove external HRP, and monitored by UV/vis spectroscopy (280 nm) and fluorescence microscopy. Alternatively, 0.05 mg mL−1 FITC-GOx or GOx (1 U mL−1) were dissolved in a 400 mM aqueous sucrose solution and encapsulated in DOPC GUVs by electroformation in the presence of dextran (65 mg mL−1). GOx in the external phase was removed using a Sephadex G150 column loaded with sucrose, or by centrifugation and washing with isotonic sucrose solution. Removal of GOx from the external phase was monitored by UV/vis spectroscopy (280 nm) or by using an Amplex red assay of the supernatant.
Operation of the acoustic trapping device
The GUV micro-array was prepared in a custom-built acoustic trapping device with a square arrangement of four piezoelectric transducers (PZT, Noliac, NCE 51, L15 × W2 mm) or a hexagonal arrangement of six PZTs. The orthogonal transducer pairs were wired in series, driven with a sinusoidal voltage (10 V) by a signal generator (Agilent 33220a-001), and connected to an oscilloscope (Agilent DSOX2014A). A bovine serum albumin (BSA)-modified glass coverslip was attached with adhesive to the bottom of the device. Transducer pairs were operated at their resonance frequency (5.06, 6.71 or 9.13 MHz for 1D patterns; 6.69/6.71 MHz for the square pattern; 5.06/9.13 MHz for the rectangular pattern; 6.70/6.71/6.72 MHz for the hexagonal pattern) with corresponding wavelengths of 162, 146 or 110 μm, 221/220 μm, 292/162 μm and 221.96/220.63/220.30 μm, respectively.
Hemifusion of acoustically trapped GUVs
Hemifusion of co-trapped GUVs prepared with different fluorescent labels was induced by addition of aqueous CaCl2 (10 μL, 133 mM) to the acoustic trapping chamber in the presence of an applied acoustic field. Exchange of lipids between the adjacent vesicles was observed after 20 minutes.
Dynamic modulation of GUV array
The GUV array was prepared by adding 30 μL of sucrose containing GUVs (400 mM) to isotonic glucose solution (1 mL, 400 mM). To control dynamic modulation of the GUV array, the two orthogonal pairs of transducers were switched on/off every 30 s. For the dynamic modulation of a hemifused/non-hemifused GUV array, a 2D array of TR-DHPE labelled GUVs (red) was firstly prepared by adding 30 μL of sucrose containing GUVs (400 mM) to isotonic glucose solution (1 mL, 400 mM) in the presence of a 2D acoustic pressure field (5.06/9.13 MHz, 10 V), and then was hemifused by the additional of CaCl2 (10 μL, 133 mM) in the same acoustic pressure field. Then, the supernatant in the acoustic trapping chamber was carefully replaced by glucose solution (400 mM) to remove the Ca2+. The hemifused/non-hemifused hybrid GUV array was obtained by the addition of 20 μL of sucrose containing NBD-PE labelled GUV (green) to the acoustic trapping chamber. To control dynamic modulation of the GUV array, the two orthogonal pairs of transducers were switched on/off about every 60 s.
Enzyme cascade reactions in acoustically trapped GUV micro-arrays
Sucrose-containing GOx-(1 U mL−1) GUVs were co-trapped with sucrose-containing HRP-(0.1 U mL−1)-loaded GUVs in the presence of isotonic glucose (400 mM) and Amplex red (10 μL, 50 μM). A signal transduction pathway between the different GUVs before or after Ca2+-induced hemifusion was initiated by addition of melittin (final concentration, 4 μg mL−1), which inserted into the vesicle bilayers and promoted the glucose influx required for the GOx/HRP enzyme cascade. Location of the individual GOx- and HRP-containing GUVs in the co-localized clusters was determined using a mixture of NBD-PE-labelled HRP-containing GUVs (green fluorescence) and unlabeled GOx-loaded GUVs, and identifying the spatial positions of the different GUVs by a combination of optical and fluorescence microscopies. Formation of resorufin specifically in the HRP-containing GUVs was monitored by fluorescence microscopy (560 nm).
Signal transduction in co-trapped GUV/HepG2 cell assemblies
A slightly modified electroformation method was used to prepare GOx-containing GUVs that were functionalized with melittin prior to acoustic trapping with living HepG2 cells. This was done to prevent deleterious effects on the cell membrane accompanying addition of aqueous melittin to the acoustically trapped GUV/HepG2 assemblies. In addition, dextran was encapsulated along with sucrose and GOx to extend the trapping lifetime of the GUVs, which was otherwise curtailed by efflux of sucrose through the melittin pores in the lipid membrane. Typically, a chloroform solution of DOPC (8 μL, 5 mg mL−1), NBD-PE (5%) and melittin (5 μg mL−1) was deposited on ITO coated glass slides, and a pre-mixed solution of GOx (50 U mL−1), dextran (0.065 g mL−1) and sucrose (300 mM) added, followed by electroformation. The obtained GOx-containing GUV suspension was diluted 3 times in isotonic sucrose (300 mM), and GOx in the supernatant removed by six cycles of centrifugation (7000 rpm, 10 min) and washing. The number density of the GOx-containing GUVs was (5.3 ± 0.3) × 103/μL. To prepare micro-arrays consisting of co-localized clusters of multiple GOx-containing GUVs and living HepG2 cells, different numbers of GUVs (ca. 2.6 × 104, 7.8 × 104, 1.5 × 105 and 2.3 × 105) were added to a stock suspension of HepG2 cells (ca. 5.6 × 104 cells; 1 mL cell culture medium) and the mixed suspension placed in the acoustic chamber with two transducer pairs (5.06/9.13 MHz). After 20 min, 100 μL of 300 mM glucose was added into the acoustic chamber to initiate the generation of H2O2 in the GOx-containing GUVs. The micro-arrays were incubated for 4 h, and propidium iodine (PI) in PBS buffer solution (20 μL, 20 μg mL−1) was then added to the acoustic chamber to stain the dead cells over a period of 20 min. The numbers of dead cells produced after 4 h were determined from fluorescence microscopy images (excitation wavelength, 536 nm).
Optical and fluorescence microscopy
Optical microscopy experiments were carried out on a Leica DMI 3000B optical microscope. Fluorescence imaging was performed using a Leica DFC 310FX set up, and dye molecules were excited by using specific filters with the following excitation (λex) and emission wavelength cut offs (λem); NBD-PE, λex = 463 nm, λem = 536 nm; FITC, λex = 492 nm and λem = 518 nm; TR-DHPE, λex = 582 nm and λem = 601 nm; rhodamine-B, λex = 528 nm and λem = 551 nm; RITC, λex = 570 nm and λem = 590 nm; resorufin, λex = 560 nm and λem = 572 nm; propidium iodide (PI), λex = 536 nm, λem = emission 617 nm. All glass slides used for the imaging were coated with BSA.
HepG2 cell cultures
HepG2 cells were cultured in medium (Dulbecco's Modified Eagle's Medium; 4.5 g L−1 glucose, sodium pyruvate and sodium bicarbonate supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 μg mL−1 streptomycin and 50 μg mL−1 penicillin (all from Sigma-Aldrich)) at 37 °C and 5% CO2 atmosphere. The pre-cultured HepG2 cells were digested by trypsin, which was removed by centrifugation at 1000 rpm for 3 min, and the concentrated cells were re-dispersed in the medium (5 mL). The density of HepG2 cells was 3 × 106 ± 4 × 105/mL. The HepG2 cells were kept at low temperature by placing tubes of the cell suspensions in ice before trapping.
Preparation of E. coli
E. coli BL21(DE3) pLysS transformed with a plasmid-encoding GFP behind a T7 promoter and a lac operator sequence were incubated in Luria–Bertani (LB) liquid medium (including 10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl) for 16 h at 37 °C.
IPTG-induced gene expression of GFP in co-trapped GUV/E. coli arrays
IPTG containing GUVs were electroformed (3.5 V, 10 Hz) in different mixtures of IPTG/sucrose mixtures (50 mM/250 mM, 100 mM/200 mM, 150 mM/150 mM) for 2 h. The obtained IPTG-containing GUV suspension was diluted 3 times in isotonic glucose (300 mM) solution. IPTG in the supernatant was removed by six cycles of centrifugation (4000 rpm, 10 min) and washing by 300 mM glucose. E. coli cells in medium (3 mL) were concentrated by centrifugation (5000 rpm, 15 min) and re-dispersed in 300 mM glucose (400 μL). In all the GUV/E. coli arrays, 20 μL of above-mentioned GUVs glucose solution and 20 μL of above-mentioned E. coli glucose solution were added in 1 mL glucose solution (300 mM) to the acoustic chamber. Signalling between the co-trapped GUVs and E. coli was induced by addition of melittin (final concentration, 4 μg mL−1).
Simulation of the acoustic pressure in the device
The acoustic pressure field in the device was simulated according to a previous report.44 In this simulation, the following assumptions were made: (i) the reflections of the acoustic waves are ignored; (ii) the initial transient period was not considered for the simulation after each opposed transducer pair is excited with a sinusoidal voltage; (iii) the standing wave was treated as the sum of two counter-propagating plane waves and any interference between these two fields averages to zero when each of the transducer pairs were operated at different frequencies. The total acoustic pressure in the devices is then given by the sum of the two standing waves created by the orthogonal pairs:| | 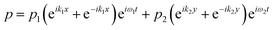 | (1) |
where 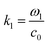 and ω1 is the angular frequency in rad s−1 and c0 is the speed of sound in the host fluid. The numerical subscript denotes the orthogonal pair under consideration, and is required as the transducer pairs were operated at different frequencies. The Cartesian axes are defined by x and y. Transducer pair-1 creates a standing wave in x whereas transducer pair-2 creates a standing wave in y. Gor'kov described the forces as resulting from a potential field, U. Using this approach the acoustic radiation force,
and ω1 is the angular frequency in rad s−1 and c0 is the speed of sound in the host fluid. The numerical subscript denotes the orthogonal pair under consideration, and is required as the transducer pairs were operated at different frequencies. The Cartesian axes are defined by x and y. Transducer pair-1 creates a standing wave in x whereas transducer pair-2 creates a standing wave in y. Gor'kov described the forces as resulting from a potential field, U. Using this approach the acoustic radiation force, ![[F with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0046_20d1.gif) , can be found from
, can be found from| | | = −∇U, | (2) |
| | 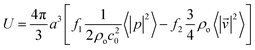 | (3) |
| | 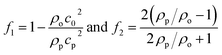 | (4) |
where 〈|p|2〉 and 〈|![[v with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0076_20d1.gif) |2〉 are the mean squared pressure and particle velocity respectively at the object, a is the radius of the spherical object, ρ is the density and the subscripts denote the particle, ‘p’, or host, ‘o’ properties. Note also, for a harmonic sound field,
|2〉 are the mean squared pressure and particle velocity respectively at the object, a is the radius of the spherical object, ρ is the density and the subscripts denote the particle, ‘p’, or host, ‘o’ properties. Note also, for a harmonic sound field, 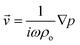 .
.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21773050 and 21929401) and the Natural Science Foundation of Heilongjiang Province for Distinguished Young Scholars (JC2018003). L. T. and S. M. thank BrisSynBio (a BBSRC/EPSRC Synthetic Biology Research Centre; BB/L01386X/1) at the University of Bristol and Engineering and Physical Sciences Research Council (EPSRC, UK; EP/L002957/1) for financial support.
Notes and references
- P. Walde, K. Cosentino, H. Engel and P. Stano, ChemBioChem, 2010, 11, 848–865 CrossRef CAS PubMed.
- L. Lu, W. J. Doak, J. W. Schertzer and P. R. Chiarot, Soft Matter, 2016, 12, 7521–7528 RSC.
- Y. S. Ryu, D. Yoo, N. J. Wittenberg, L. R. Jordan, S. D. Lee, A. N. Parikh and S. H. Oh, J. Am. Chem. Soc., 2015, 137, 8692–8695 CrossRef CAS PubMed.
- K. Karamdad, J. W. Hindley, G. Bolognesi, M. S. Friddin, R. V. Law, N. J. Brooks, O. Ces and Y. Elani, Chem. Sci., 2018, 9, 4851–4858 RSC.
- T. Zhu and J. W. Szostak, J. Am. Chem. Soc., 2009, 131, 5705–5713 CrossRef CAS PubMed.
- M. Andes-Koback and C. D. Keating, J. Am. Chem. Soc., 2011, 133, 9545–9555 CrossRef CAS PubMed.
- T. Robinson, P. E. Verboket, K. Eyer and P. S. Dittrich, Lab Chip, 2014, 14, 2852–2859 RSC.
- T. Tanaka and M. Yamazaki, Langmuir, 2004, 20, 5160–5164 CrossRef CAS PubMed.
- N. Mora, A. Bahreman, H. Valkenier, H. Li, T. H. Sharp, D. N. Sheppard, A. P. Davis and A. Kros, Chem. Sci., 2016, 7, 1768–1772 RSC.
- M. Chabanon, J. C. S. Ho, B. Liedberg, A. N. Parikh and P. Rangamani, Biophys. J., 2017, 112, 1682–1691 CrossRef CAS PubMed.
- H. Pick, A. C. Alves and H. Vogel, Chem. Rev., 2018, 118, 8598–8654 CrossRef CAS PubMed.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 CrossRef CAS PubMed.
- N. N. Deng, M. A. Vibhute, L. Zheng, H. Zhao, M. Yelleswarapu and W. T. S. Huck, J. Am. Chem. Soc., 2018, 140, 7399–7402 CrossRef CAS PubMed.
- R. K. Kumar, X. Yu, A. J. Patil, M. Li and S. Mann, Angew. Chem., Int. Ed., 2011, 50, 9343–9347 CrossRef PubMed.
- W. Zong, S. H. Ma, X. N. Zhang, X. J. Wang, Q. C. Li and X. J. Han, J. Am. Chem. Soc., 2017, 139, 9955–9960 CrossRef CAS PubMed.
- M. Weiss, J. P. Frohnmayer, L. T. Benk, B. Haller, J. Janiesch, T. Heitkamp, M. Börsch, R. B. Lira, R. Dimova, R. Lipowsky, E. Bodenschatz, J. C. Baret, T. Vidakovic-Koch, K. Sundmacher, I. Platzman and J. P. Spatz, Nat. Mater., 2018, 17, 89–96 CrossRef CAS PubMed.
- R. Tomasi, J. Noel, A. Zenati, S. Ristori, F. Rossi, V. Cabuil, F. Kanou and A. Hassan, Chem. Sci., 2014, 5, 1854–1859 RSC.
- A. Bhattacharya, R. J. Brea and N. K. Devaraj, Chem. Sci., 2017, 8, 7912–7922 RSC.
- P. Taylor, C. Xu, P. D. I. Fletcher and V. N. Paunov, Phys. Chem. Chem. Phys., 2003, 5, 4918–4922 RSC.
- Y. Kang, H. S. Wostein and S. Majd, Adv. Mater., 2013, 25, 6834–6838 CrossRef CAS PubMed.
- Y. Kazayama, T. Teshima, T. Osaki, S. Takeuchi and T. Toyota, Anal. Chem., 2016, 88, 1111–1116 CrossRef CAS PubMed.
- N. Yandrapalli and T. Robinson, Lab Chip, 2019, 19, 626–633 RSC.
- X. Ding, S. C. S. Lin, B. Kiraly, H. Yue, S. Li, I. K. Chiang, J. J. Shi, S. J. Benkovic and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11105–11109 CrossRef CAS PubMed.
- B. W. Drinkwater, Lab Chip, 2016, 16, 2360–2375 RSC.
- L. Tian, N. Martin, P. G. Bassindale, A. J. Patil, M. Li, A. Barnes, B. W. Drinkwater and S. Mann, Nat. Commun., 2016, 7, 13068 CrossRef CAS PubMed.
- D. J. Collins, B. Morahan, J. Garcia-Bustos, C. Doerig, M. Plebanski and A. Neild, Nat. Commun., 2015, 6, 8686 CrossRef CAS PubMed.
- P. Carrara, P. Stano and P. L. Luisi, ChemBioChem, 2012, 13, 1497–1502 CrossRef CAS PubMed.
- C. Zhu, E. M. Taipaleenmäki, Y. Zhang, X. Han and B. Städler, Biomater. Sci., 2018, 6, 793–802 RSC.
- H. M. Bi, B. Yang, L. Wang, W. Cao and X. J. Han, J. Mater. Chem. A, 2013, 1, 7125–7130 RSC.
- L. Tian, M. Li, J. B. Liu, A. J. Patil, B. W. Drinkwater and S. Mann, ACS Cent. Sci., 2018, 4, 1551–1558 CrossRef CAS PubMed.
- S. Li, X. Wang, W. Mu and X. Han, Anal. Chem., 2019, 9, 6859–6864 Search PubMed.
- M. Mally, J. Majhenc, S. Svetina and B. Zeks, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1179–1189 CrossRef CAS PubMed.
- R. Lentini, S. P. Santero, F. Chizzolini, D. Cecchi, J. Fontana, M. Marchioretto, C. Bianco, J. L. Terrell, A. C. Spencer, L. Martini, M. Forlin, M. Assfalg, M. D. Serra, W. E. Bentley and S. S. Mansy, Nat. Commun., 2014, 5, 4012 CrossRef CAS PubMed.
- G. Rampioni, F. D'Angelo, M. Messina, A. Zennaro, Y. Kuruma, D. Tofani, L. Leoni and P. Stano, Chem. Commun., 2018, 54, 2090–2093 RSC.
- R. Lentini, N. Y. Martín, M. Forlin, L. Belmonte, J. Fontana, M. Cornella, L. Martini, S. Tamburini, W. E. Bentley, O. Jousson and S. S. Mansy, ACS Cent. Sci., 2017, 3, 117–123 CrossRef CAS PubMed.
- T. Trantidou, L. Dekker, K. Polizzi, O. Ces and Y. Elani, Interface Focus, 2018, 8, 20180024 CrossRef PubMed.
- A. Joesaar, S. Yang, B. Bögels, A. Linden, P. Pieters, B. V. V. S. P. Kumar, N. Dalchau, A. Phillips, S. Mann and T. F. A. de Greef, Nat. Nanotechnol., 2019, 14, 369–378 CrossRef CAS PubMed.
- P. Gobbo, A. J. Patil, M. Li and S. Mann, Nat. Mater., 2018, 17, 1145–1153 CrossRef CAS PubMed.
- M. J. Booth, V. R. Schild, A. D. Graham, S. N. Olof and H. Bayley, Sci. Adv., 2016, 2, e1600056 CrossRef PubMed.
- G. Bolognesi, M. S. Friddin, A. Salehi-Reyhani, N. E. Barlow, N. J. Brooks, O. Ces and Y. Elani, Nat. Commun., 2018, 9, 1882 CrossRef PubMed.
- Y. Elani, A. Gee, R. V. Law and O. Ces, Chem. Sci., 2013, 4, 3332–3338 RSC.
- P. M. Gardner, K. Winzer and B. G. Davis, Nat. Chem., 2009, 1, 377–383 CrossRef CAS PubMed.
- A. X. Lu, H. Oh, J. L. Terrell, W. E. Bentley and S. R. Raghavan, Chem. Sci., 2017, 8, 6893–6903 RSC.
- C. R. P. Courtney, C. K. Ong, B. W. Drinkwater, A. L. Bernassau, P. D. Wilcox and D. R. S. Cumming, Proc. - R. Soc. Edinburgh, Sect. A: Math. Phys. Sci., 2011, 468, 337–360 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04522h |
| ‡ X. J. Wang and L. Tian contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Hang
Du
a,
Mei
Li
b,
Hang
Du
a,
Mei
Li
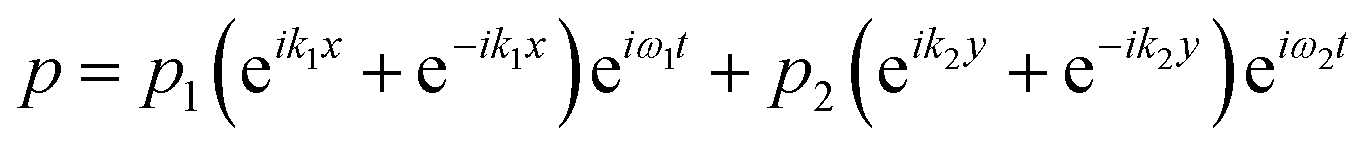
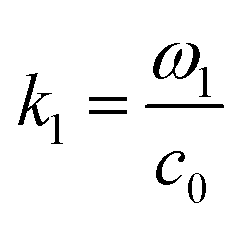 and ω1 is the angular frequency in rad s−1 and c0 is the speed of sound in the host fluid. The numerical subscript denotes the orthogonal pair under consideration, and is required as the transducer pairs were operated at different frequencies. The Cartesian axes are defined by x and y. Transducer pair-1 creates a standing wave in x whereas transducer pair-2 creates a standing wave in y. Gor'kov described the forces as resulting from a potential field, U. Using this approach the acoustic radiation force,
and ω1 is the angular frequency in rad s−1 and c0 is the speed of sound in the host fluid. The numerical subscript denotes the orthogonal pair under consideration, and is required as the transducer pairs were operated at different frequencies. The Cartesian axes are defined by x and y. Transducer pair-1 creates a standing wave in x whereas transducer pair-2 creates a standing wave in y. Gor'kov described the forces as resulting from a potential field, U. Using this approach the acoustic radiation force, 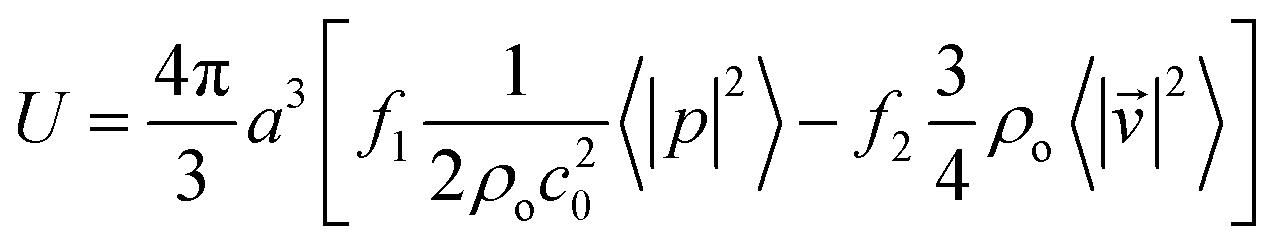
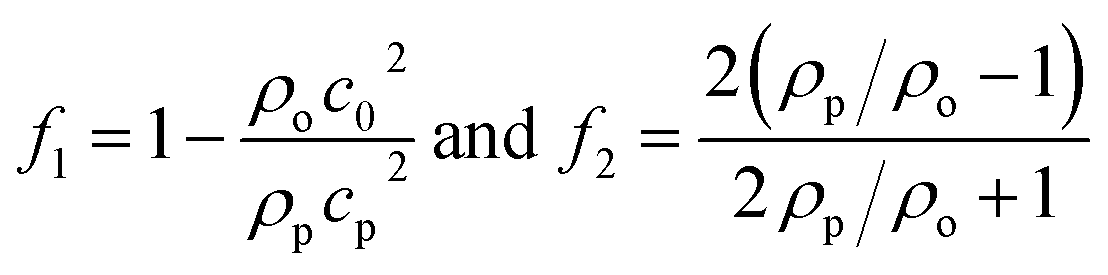
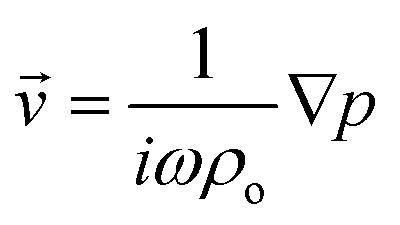 .
.