
Enhanced water oxidation reaction kinetics on a BiVO4 photoanode by surface modification with Ni4O4 cubane†
Bin
Gao
a,
Tao
Wang
 *a,
Xiaoli
Fan
a,
Hao
Gong
a,
Peng
Li
a,
Yaya
Feng
a,
Xianli
Huang
a,
Jianping
He
*a and
Jinhua
Ye
*b
*a,
Xiaoli
Fan
a,
Hao
Gong
a,
Peng
Li
a,
Yaya
Feng
a,
Xianli
Huang
a,
Jianping
He
*a and
Jinhua
Ye
*b
aCollege of Materials Science and Technology, Jiangsu Key Laboratory of Electrochemical Energy Storage Technologies, Nanjing University of Aeronautics and Astronautics, Nanjing 210016, PR China. E-mail: wangtao0729@nuaa.edu.cn; jianph@nuaa.edu.cn; Fax: +86 25 52112626; Tel: +86 25 52112900
bInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: Jinhua.YE@nims.go.jp; Fax: +81 29 8604958; Tel: +81 29 8592646
First published on 20th November 2018
Abstract
Transition-metal–organic molecular catalysts have been regarded as novel, promising and high-efficiency water oxidation cocatalysts for photoelectrochemistry. Here, molecular Ni4O4 cubane was prepared and combined with a BiVO4 nanoplate photoanode to generate a water oxidation cocatalyst. Intensity modulated photocurrent spectroscopy tests verify that the Ni4O4 cubane catalyst not only facilitates the kinetics of water oxidation, but also suppresses the recombination of carriers at the electrolyte/photoanode interface. As expected, the BiVO4 nanoplates modified with Ni4O4 cubane show superior activity (3.9 mA cm−2 at 1.23 V vs. RHE), with a more than twice increased activity compared to the BiVO4 nanoplates. Meanwhile, the onset potential also generates a 350 mV cathodic shift. In order to prevent desorption of the molecular catalyst due to the weak binding between the semiconductor and molecular catalyst, an Al2O3 adsorbed layer is constructed on the surface of the nanoplates. Consequently, Ni4O4/Al2O3/BiVO4 shows a significant improvement in PEC stability.
Introduction
Photoelectrochemical (PEC) water splitting into hydrogen and oxygen is considered as a promising technology to transform solar energy into steady chemical energy.1–3 In a PEC system, the photoanode, as the site of the oxygen evolution reaction (OER), plays a key role in the efficient conversion of solar radiation.4–6 Thus, many semiconductor materials have been investigated as photoanodes, such as TiO2,7–9 WO3,10,11 Fe2O3,12,13 BiVO4,14–16etc. However, due to some intrinsic defects of semiconductor photoanodes such as poor water oxidation kinetics and high electron–hole recombination,17,18 the OER process always shows high overpotential and low reaction efficiency of surface holes, causing low conversion efficiency of solar radiation. To solve these problems, many strategies have been adopted such as surface modification,19,20 element doping,21–23 loading of co-catalysts24–26 and construction of heterojunctions.27,28Among these strategies, loading of OER catalysts has been regarded as an effective approach to enhance the OER reaction efficiency. Due to the poor water oxidation kinetics of semiconductor photoanodes, plenty of photo-generated holes are enriched on the semiconductor/electrolyte interface, which further cause the recombination of photo-generated carriers.29,30 The introduction of OER catalysts can lower the activation energy and accelerate the reaction kinetics of the water oxidation reaction, so that the surface holes are rapidly consumed by water and the recombination of photo-generated carriers is restricted.31,32 In general, water oxidation catalysts (WOCs) are transition-metal–inorganic compounds like Co3O4, Co–Pi, and NiOOH.33–38 However, inorganic heterogeneous WOCs still exhibit some defects limiting their development, such as the high interface barrier between the semiconductor and catalyst, blocking light absorption.
Recently, transition-metal–organic WOCs have drawn much attention from many researchers.39,40 Compared to inorganic heterogeneous WOCs, organic molecular WOCs have distinct molecular orbitals and almost no effect on the light absorption of the semiconductor.41,42 What's more, organic molecular WOCs show good catalytic activity and accelerate the OER kinetics at the photoanode surface. Thus, seeking novel, economical and efficient molecular WOCs have been one of the most popular research fields. Wu et al. obtained a cobalt porphyrin complex-modified BiVO4 photoanode with almost a 450 mV negative shift at 0.5 mA cm−2 and a double enhancement of photocurrent density at 1.23 V vs. RHE compared to bare BiVO4.43 Kandiel et al. synthesized a Co/3-aminopropionic acid-hematite photoanode with a 3.8-fold higher photocurrent at 1.23 V vs. RHE and a 290 mV cathodic shift of the onset potential compared to the pristine hematite photoanode.44 Li et al. achieved a photocurrent density of 5 mA cm−2 at 1.23 V vs. RHE for molecular Co4O4 cubane catalyst-modified BiVO4, which is a six-fold enhancement over that for unmodified BiVO4.45 However, there is almost no research reported on nickel based molecular WOCs for PEC water splitting. As an abundant element, it has been proved that the inorganic compounds of nickel exhibit excellent catalytic activity for the oxygen evolution reaction.46–49 Therefore, nickel-based organic molecules have promising potential as WOCs in theory. Meanwhile, the biologically inspired “cubelike” metal–organics, which are inspired by the key catalyst of photosynthesis, have attracted researchers' attention. A series of “cubelike” metal–organics has been synthesized and their catalytic activity for water oxidation has been confirmed.50–52 Therefore, nickel-based “cubelike” organic molecules have promising potential as WOCs in theory. In addition, there are few reports about the mechanism of molecular catalysts in a PEC system, which has led to the random selection of a molecular catalyst.
In this study, a BiVO4 nanoplate photoanode is synthesised by a hydrothermal method, which acts as the light absorber and the hole-provider, and then molecular Ni4O4 cubane is synthesized via a liquid-phase reaction and combined with the BiVO4 photoanode which acts as the water oxidation catalyst to improve the PEC performance. As a result, the molecular catalyst-modified BiVO4 photoanode exhibits an enhanced photocurrent density of 3.9 mA cm−2 at 1.23 V vs. RHE under AM 1.5 G solar illumination, which is more than 2-fold higher than that of the bare BiVO4 photoanode. What's more, the onset potential of the OER is negatively shifted by 350 mV relative to the unmodified photoanode. The IMPS test reveals that Ni4O4 cubane not only facilitates the kinetics of water oxidation, but also suppresses the recombination of carriers on the surface of the photoanode. Meanwhile, to overcome the poor PEC stability of molecular catalysts, an Al2O3 adsorbed layer is constructed on the surface of the BiVO4 nanoplate, which can adsorb more Ni4O4 cubane catalysts and promote the PEC stability. The composite photoanode is tested for 2 h with a residual photocurrent of 80%. As a result, the composite photoanode exhibits dramatic PEC performance and stability.
Experimental section
Synthesis of the Ni4O4 cubane molecule
The synthesis of the Ni4O4 cubane molecule was performed according to the previous literature.53 First, 10 mmol NiCl2·6H2O was added to 20 ml of MeOH solution with 10 mmol 2-hydroxybenzaldehyde and 15 mmol triethylamine, and a green precipitate appeared immediately. Then, the mixture was vigorously stirred for two hours. Finally, the green product was collected by suction filtration, washed with methanol and H2O, and dried at room temperature in air.Synthesis of the BiVO4 photoanode
The BiVO4 photoanode was synthesized by a seed layer-assisted hydrothermal method based on our previous work.54The BiVO4 seed layer was prepared on FTO conductive glass by the spin-coating method, with bismuth nitrate as the bismuth source, ammonium metavanadate as the vanadium source and ethylene diamine tetraacetic acid (EDTA) as the complexing agent. In the solution preparation process, ammonia water was used to adjust the pH to 10.
In the hydrothermal process, first, 0.2 mmol bismuth nitrate as the bismuth source and 0.2 mmol EDTA as the complexing agent were dispersed in 30 ml of distilled water. Then, 0.75 ml of a 2 M NaOH solution was added to adjust the pH and the mixture was rapidly stirred until the powder was dissolved completely. Next, 0.2 mmol ammonium metavanadate as the vanadium source was dissolved into the previous solution under stirring until it became clear and transparent. Finally, the obtained solution was poured into a Teflon-lined autoclave with the BiVO4 seed layer substrate leaning against the autoclave wall and the seed layer side facing down. The hydrothermal process was carried out at 180 °C for 6 h to obtain the BiVO4 photoanode. In the end, the BiVO4 film photoanode was calcined at 500 °C for 4 h.
Synthesis of the Ni4O4/BiVO4 and Ni4O4/Al2O3/BiVO4 photoanode
Ni4O4/BiVO4: The prepared BiVO4 photoanode was immersed into the Ni4O4 cubane-saturated aqueous solution at 80 °C for 1 h. Finally, the photoanode was rinsed with acetonitrile, ethanol and water.Ni4O4/Al2O3/BiVO4: The prepared BiVO4 photoanode was immersed into 30 ml of isopropanol with 0.5 ml of aluminum tri-sec-butoxide at 60 °C for 0.5 h. Then, the photoanode was heated up to 200 °C for 1 h. Finally, the Al2O3/BiVO4 photoanode was immersed into the Ni4O4 cubane-saturated aqueous solution at 80 °C for 1 h.
Characterization
The FT-IR spectra were obtained on a Perkin-Elmer Spectrum BX FT-IR spectrometer as a KBr pellet. X-ray power diffraction analysis was performed on a D8 Advance diffractometer (Bruker, Germany) with Cu Kα radiation (λ = 0.154056 nm). Scanning electron microscopy (SEM) images were collected on a Hitachi Co. S4800 field emission scanning electron microscope. A transmission electron microscope (JEOL 2100F) was used to collect high-resolution transmission electron microscopy (HRTEM) images and energy-dispersive X-ray spectroscopy (EDS) was carried out in STEM mode. Diffuse reflectance ultraviolet-visible (DR UV-vis) spectra were recorded on a Shimadzu UV-3600 spectrophotometer. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALab220i-XL spectrometer.Electrochemical and photoelectrochemical measurements
A typical three-electrode system was used to conduct electrochemical and photoelectrochemical tests. In the three-electrode system, a platinum plate electrode and a saturated calomel electrode were used as the counter electrode and reference electrode, respectively. Phosphate buffer (0.2 M, pH = 7) was used as the electrolyte. A 300 W xenon lamp with an AM 1.5 G filter (Perfect, PLS-SXE300C, China) was used as the light source and the light intensity was adjusted to 100 mW cm−2. A Zahner IM6 electrochemical workstation was used to test the electrochemical and photoelectrochemical performance. Electrochemical impedance spectra were recorded at 1.23 V vs. RHE with an amplitude of 10 mV from 0.01 Hz to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz. Intensity modulated photocurrent spectra were measured under a light intensity of 100 mW cm−2 from 0.01 Hz to 100000 Hz. I–t curves and incident photon-to-current efficiencies were obtained at 1.23 V vs. RHE. O2 gas evolved from the PEC system was detected using a gas chromatograph (Techcomp GC 7900, China), and a syringe was used to transfer 50 μL from a sealed electrolytic cell to the GC instrument every 30 min.
000 Hz. Intensity modulated photocurrent spectra were measured under a light intensity of 100 mW cm−2 from 0.01 Hz to 100000 Hz. I–t curves and incident photon-to-current efficiencies were obtained at 1.23 V vs. RHE. O2 gas evolved from the PEC system was detected using a gas chromatograph (Techcomp GC 7900, China), and a syringe was used to transfer 50 μL from a sealed electrolytic cell to the GC instrument every 30 min.
Results and discussion
Characterization of the Ni4O4/BiVO4 composite photoanode
The Ni4O4 cubane catalyst was synthesized following the previous literature,53 and characterized by FTIR. The broad peak at 3426 cm−1 shown in Fig. 1b is attributed to the OH group. The peaks at 1631, 1531 and 1465 cm−1 are assigned to the benzene skeleton vibration. The alcoholic C–O stretching mode occurs at 1049 cm−1 with a strong peak. The peak at 1153 cm−1 is ascribed to the ν(C–Ophen) stretching mode. In addition, some weak peaks appearing in the range 3000–2800 cm−1 are caused by the aromatic and aliphatic C–H stretching. The BiVO4 photoanode was prepared based on the method reported by us and characterized by SEM (Fig. 2a), XRD (Fig. S1†) and HRTEM (Fig. 2c).54 The SEM image shows that the photoanode is composed of nanoplate arrays with a thickness of approximately 200 nm. The XRD spectrum indicates that it has a well-crystallized monoclinic BiVO4 structure, in accord with JCPDS Card 44-0081 (Fig. S1†). As shown in Fig. 2c, the HRTEM image of BiVO4 exhibits legible lattice fringes of 0.309 nm, which match well with the (121) lattice plane of monoclinic scheelite BiVO4. Also, the XRD pattern of Ni4O4/BiVO4 is recorded and is no different from that of BiVO4, indicating that molecular cubane is amorphous. The selected-area electron diffraction (SAED) pattern of BiVO4 confirms that the nanoplate is a single crystal. The SEM image of the BiVO4 photoanode after modification by the Ni4O4 cubane catalyst is shown in Fig. 2b, which shows the unchanged nanoplate structure with the ultrathin Ni4O4 cubane layer adsorbed on the surface. According to the TEM image shown in Fig. 2e, the BiVO4 nanoplate is uniformly coated with a layer of the Ni4O4 cubane catalyst with a thickness of about 10 nm. The catalyst layer adheres to the surface of BiVO4 closely, which favors the transfer of carriers. The EDS mapping images are shown in Fig. 2f, and the mapping image of the Ni element matches well with the SEM image shown in Fig. 2b. Moreover, the image of Ni mapping verifies the uniform distribution of Ni4O4 cubane on the surface of the BiVO4 nanoplates.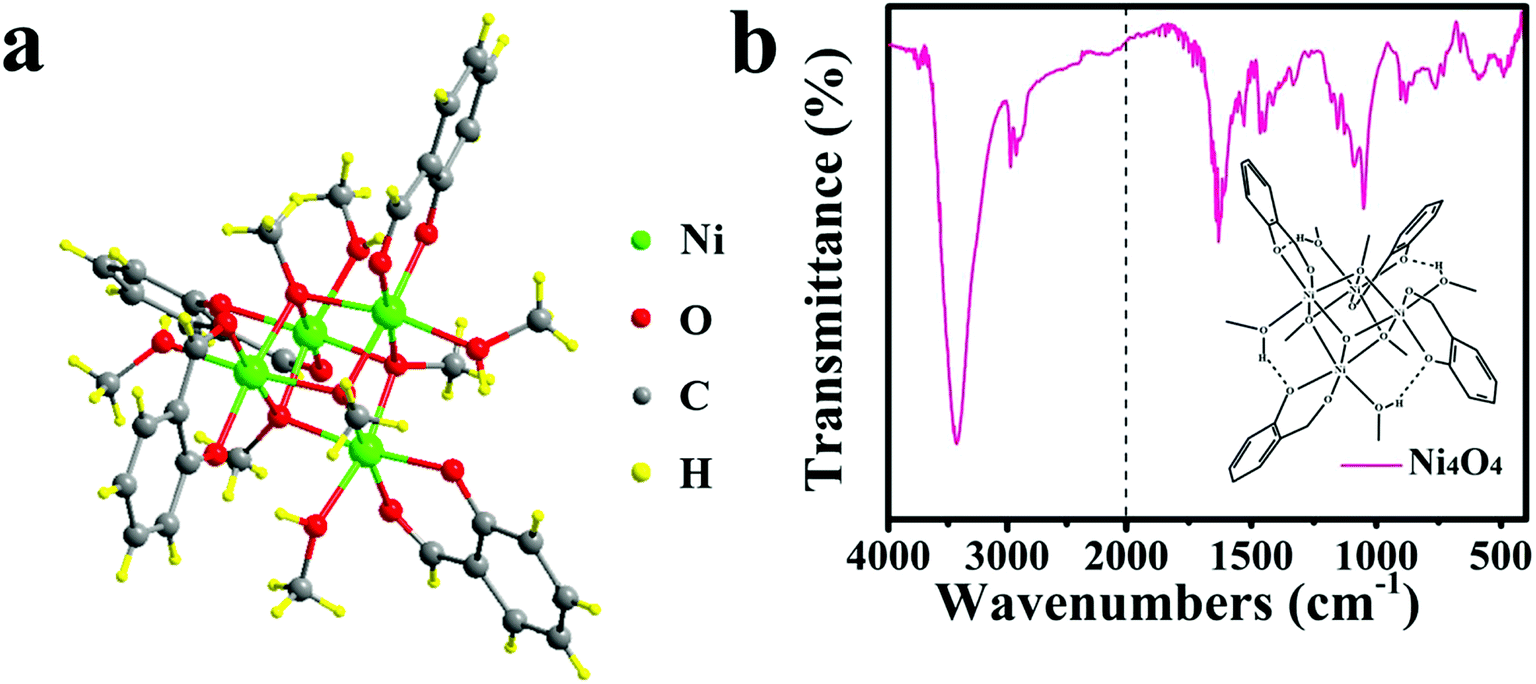 | ||
| Fig. 1 (a) Ball-and-stick representation and (b) FTIR of Ni4O4 cubane catalyst. | ||
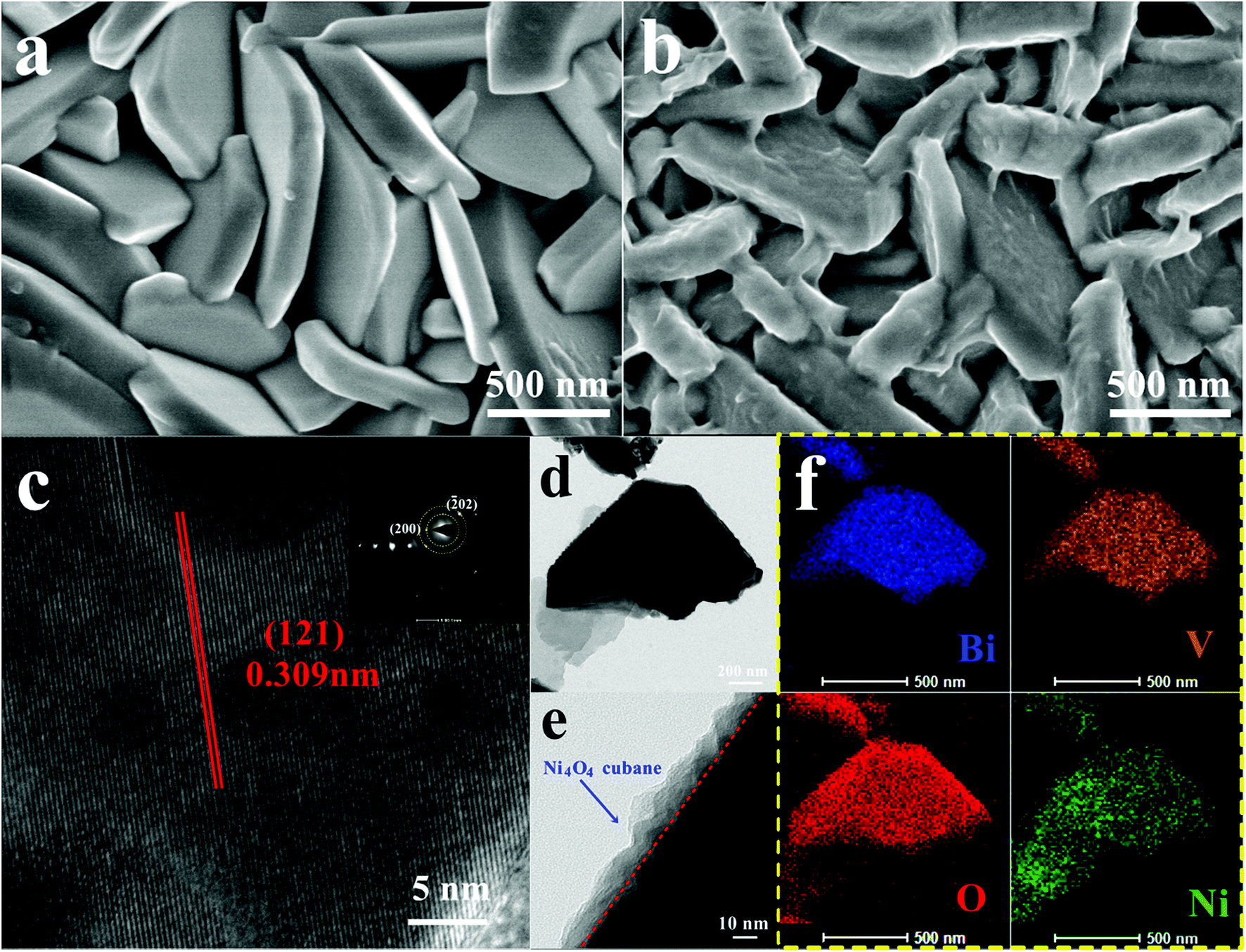 | ||
| Fig. 2 SEM images of BiVO4 (a) and Ni4O4/BiVO4 (b), (c) HRTEM image of BiVO4, and TEM (d and e) and EDS mapping (f) images of Ni4O4/BiVO4. | ||
In order to analyze the surface chemical states of the modified BiVO4 photoanode, XPS tests were performed. As shown in Fig. 3d, the characteristic peaks of Ni2+ were detected, indicating the existence of Ni4O4 cubane on the BiVO4 surface. In addition, as shown in Fig. 3e, the O 1s XPS spectrum of BiVO4 can be deconvoluted into two peaks at 531.0 eV and 529.7 eV, corresponding to the dangling oxygen and the lattice oxygen from V–O, respectively. After modification by Ni4O4 cubane, new peaks at 532.9 eV and 532.1 eV appear and are attributed to C–O and Ni–O from Ni4O4 cubane, respectively. Moreover, because Ni4O4 cubane covers the surface of BiVO4, the O 1s peaks at 531.0 eV and 529.7 eV, which originate from BiVO4, become weak. In addition, the XPS spectra of Ni(OH)2 were recorded and shown in Fig. S2.† The binding energy of the O 1s peak is different for the composite photoanode, indicating that the Ni element in the Ni4O4/BiVO4 photoanode exists in the form of Ni4O4 cubane rather than Ni(OH)2. Besides, the Ni4O4/BiVO4 photoanode after annealing at 400 °C was tested by XPS. The high-resolution spectrum of O 1s is shown in Fig. S3,† and it's obvious that the peak assigned to C–O vanished and a new peak appeared which was attributed to Ni–O, indicating that the Ni element in the Ni4O4/BiVO4 photoanode exists in the form of Ni4O4 cubane rather than NiO. The optical properties of the samples were investigated via UV-vis spectra. As shown in Fig. S4a,† the pristine BiVO4 has an absorption edge of approximately 510 nm, which corresponds to the theoretical value of monoclinic BiVO4. Tauc plots are obtained from the UV-vis absorption spectrum and show similar band gaps of 2.4 eV, indicating that the deposition of Ni4O4 cubane does not change the band structure of the photoanode (Fig. S4b†). Meanwhile, the Ni4O4/BiVO4 photoanode shows a similar UV-vis absorption spectrum, indicating that the loading of Ni4O4 cubane does not change the optical absorption of the photoanode and does not block the light absorption of BiVO4.
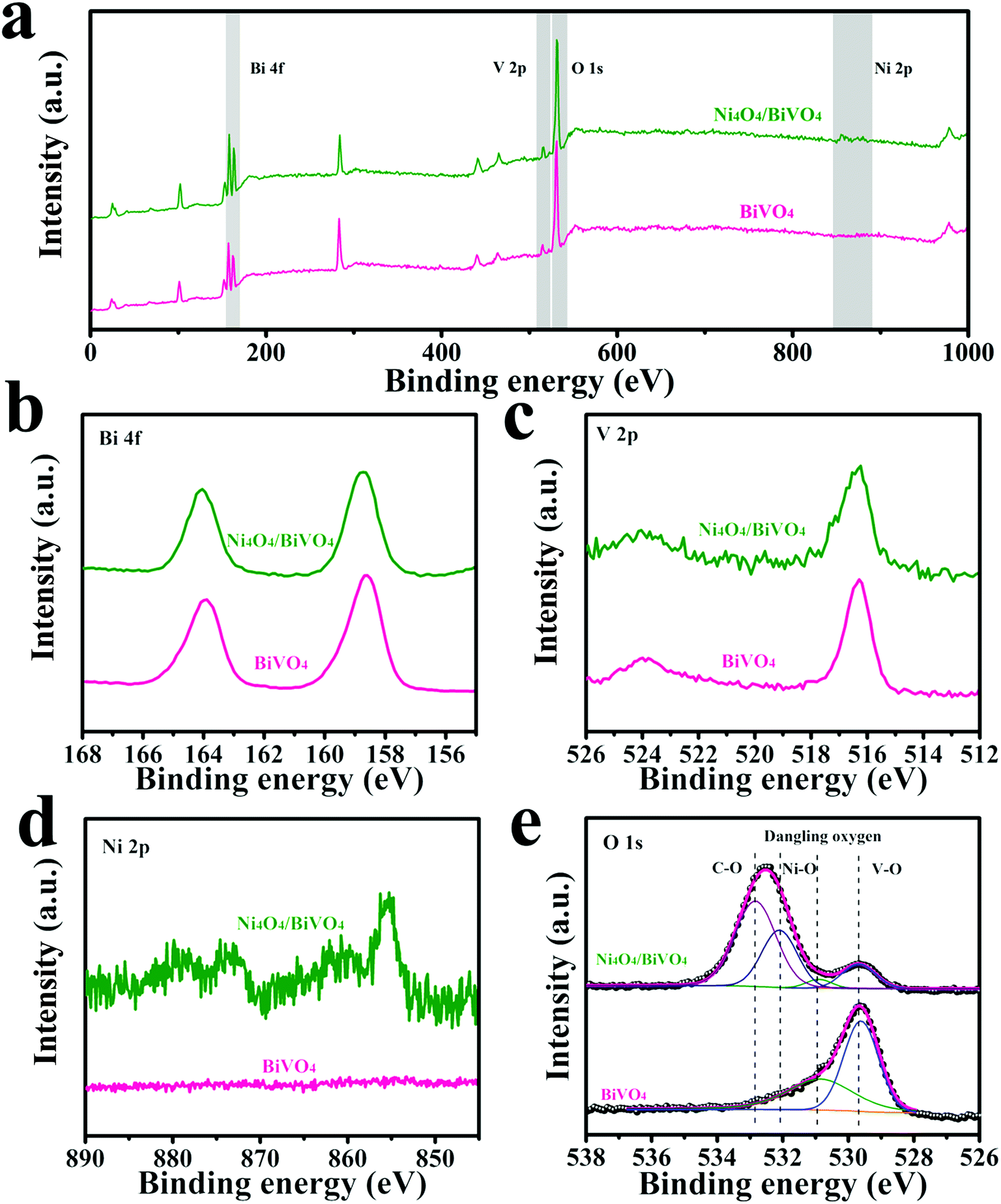 | ||
| Fig. 3 XPS survey spectra (a) and high-resolution spectra of Bi 4f (b), V 2p (c), Ni 2p (d), and O 1s (e) of BiVO4 and Ni4O4/BiVO4. | ||
Performance of the Ni4O4/BiVO4 composite photoanode
In order to investigate the contribution of the Ni4O4 cubane catalyst to the photoelectrochemical performance, the BiVO4 photoanode modified with the Ni4O4 cubane catalyst was tested in a water splitting setup and compared to the pristine BiVO4 photoanode. The water oxidation activity is directly reflected in the linear sweep voltammetry (LSV) curves. As shown in Fig. 4a, the pristine BiVO4 exhibited a photocurrent density of 1.5 mA cm−2 at 1.23 V vs. RHE and an onset potential of 0.7 V vs. RHE. The high photocurrent density is due to the unique single crystal structure and specific crystal orientation, which are beneficial for transfer and separation of photo-generated carriers.55,56 After modification by the Ni4O4 cubane catalyst, the photoanode showed an obviously increased photocurrent at all potentials with a photocurrent of 3.9 mA cm−2 at 1.23 V vs. RHE, more than 2-fold higher than that of pristine BiVO4, and a more negative onset potential of 0.35 V vs. RHE. Moreover, the Ni4O4/BiVO4 photoanode shows a much higher photocurrent than the NiOOH modified BiVO4 photoanode, which indicates the superiority of the molecular catalyst (Fig. S5†). Compared with reported conventional cocatalysts, Ni4O4 cubane exhibits excellent cocatalytic ability for the water oxidation reaction (Table S1†). The enhanced photocurrent and more negative onset potential indicate that the Ni4O4 cubane catalyst can effectively promote the PEC performance of the photoanode. Moreover, the photocurrent transients of the photoanodes were measured to investigate the recombination at the photoanode surface. In Fig. 4b, it's clear that the photocurrent of Ni4O4/BiVO4 was more than 2-fold higher than that of BiVO4. In addition, the photocurrent of pristine BiVO4 showed a sharp spike after turning on the light, which was attributed to the fact that the photo-generated holes were accumulated at the electrolyte/semiconductor interface and then consumed by recombination with photo-generated electrons. However, this transient spike is suppressed in the presence of the Ni4O4 cubane catalyst, indicating that there is a reduction of recombination processes in the photoanode surface, which is due to the fast water oxidation reaction kinetics of the Ni4O4 cubane catalyst. The applied bias photon-to-current efficiencies (ABPE) were obtained from the LSV curves and the ABPE of Ni4O4/BiVO4 reached a maximum of 0.91% at 0.8 V vs. RHE (Fig. 4c). The solar conversion efficiency was investigated using the incident photon-to-current efficiency (IPCE) and shown in Fig. 4d. It's clear that the Ni4O4/BiVO4 photoanode exhibits an enhanced IPCE compared to BiVO4 from the ultraviolet to the visible light region. Since the optical absorptivity did not change after the modification by Ni4O4 cubane, the enhanced IPCE may be attributed to the increased separation efficiency and faster kinetics of the water oxidation reaction.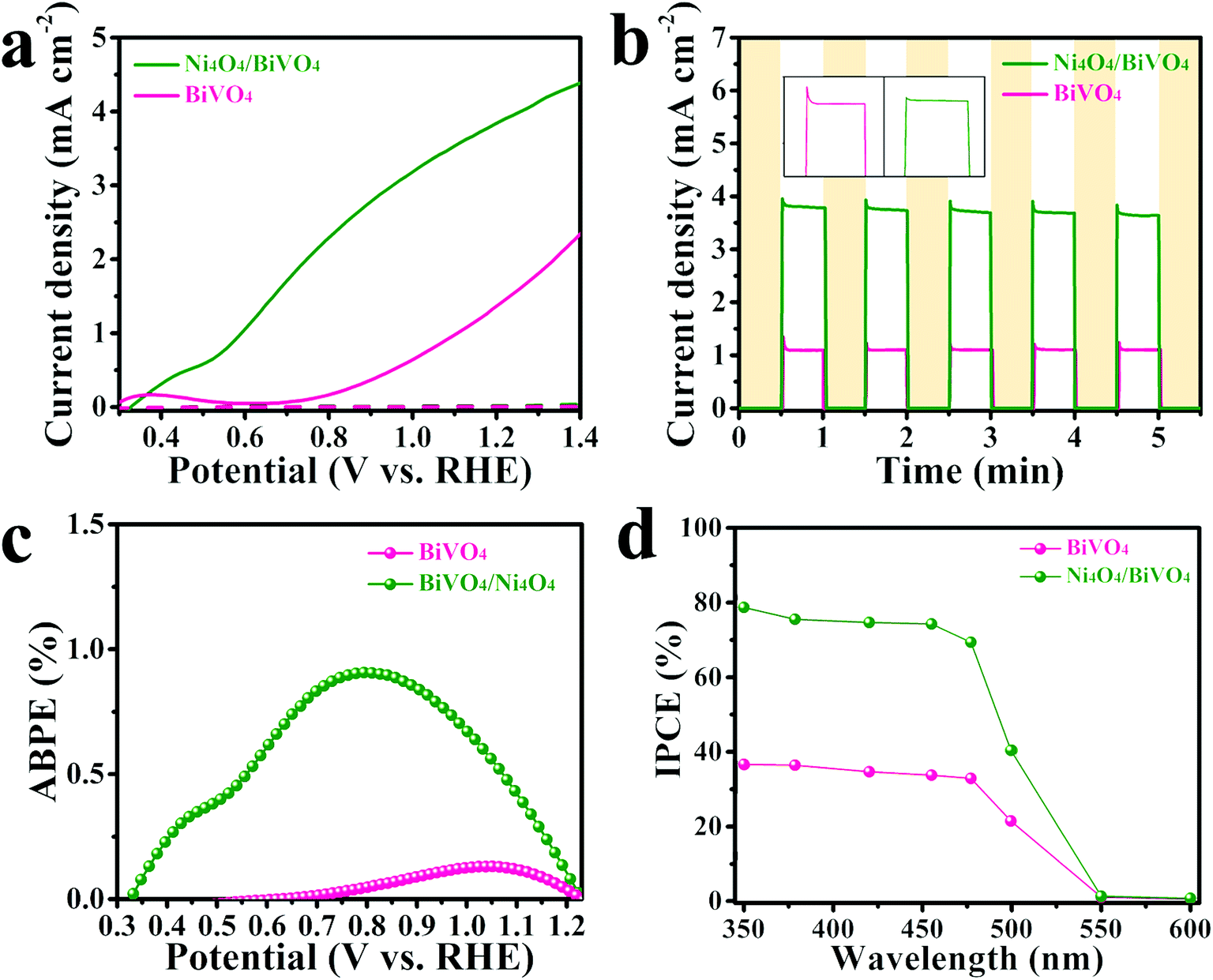 | ||
| Fig. 4 (a) LSV curves, (b) I–t curves, (c) applied bias photon-to-current efficiencies (ABPE) and (d) incident photon-to-current efficiencies (IPCE) of the BiVO4 and Ni4O4/BiVO4 photoanodes. | ||
Discussion on mechanism
In order to investigate the transfer of carriers in the PEC system, electrochemical impedance spectroscopy (EIS) was carried out on the samples. The EIS spectra are shown in Fig. 5a, and both the curves exhibit a typical resistance arc. It's obvious that the resistance arc of Ni4O4/BiVO4 has a smaller radius compared with that of BiVO4, which indicates better carrier mobility. The adsorption of Ni4O4 cubane changes the properties of the electrolyte/semiconductor interface, but not the intrinsic properties of BiVO4. The better carrier mobility may be attributed to the better hole transfer efficiency or reduced recombination of carriers at the electrolyte/semiconductor interface. The hole transfer efficiency was investigated using the ratio of the sulfite-oxidation current to the water-oxidation current (Fig. 5b). Because of the fast oxidation kinetics of sulfite, the Jwater/Jsurface ratio was identified as the yield of holes that participate in the water-oxidation reaction at the electrolyte/semiconductor interface (ηsurface). As shown in Fig. 5c, after modification by Ni4O4 cubane, the ηsurface of the photoanode exhibits a dramatic increase compared to that of the pure BiVO4 photoanode. The ηsurface of Ni4O4/BiVO4 reaches 78% at 1.23 V (vs. RHE), while that of the BiVO4 photoanode only reaches 30%. It's clear that the modification of Ni4O4 cubane can form an interface which is beneficial to the water-oxidation reaction between the electrolyte and semiconductor. To further investigate the catalytic properties of Ni4O4 cubane and BiVO4 for the water oxidation reaction, the electrocatalytic oxygen evolution reaction curves were obtained via the rotating disk electrode method and are shown in Fig. 5d. Ni4O4 exhibited an electrocatalytic OER performance preferable to that of BiVO4, as evidenced by the lower overpotential with the higher current density. The results reveal that Ni4O4 cubane has a more efficient water oxidation activity, so that the modified photoanode exhibited a more negative onset potential for the oxygen evolution reaction. Thus, the composite photoanode with Ni4O4 cubane as the cocatalyst has faster kinetics of the water oxidation reaction compared with pristine BiVO4, which resulted in a more effective utilization of the surface reaching holes.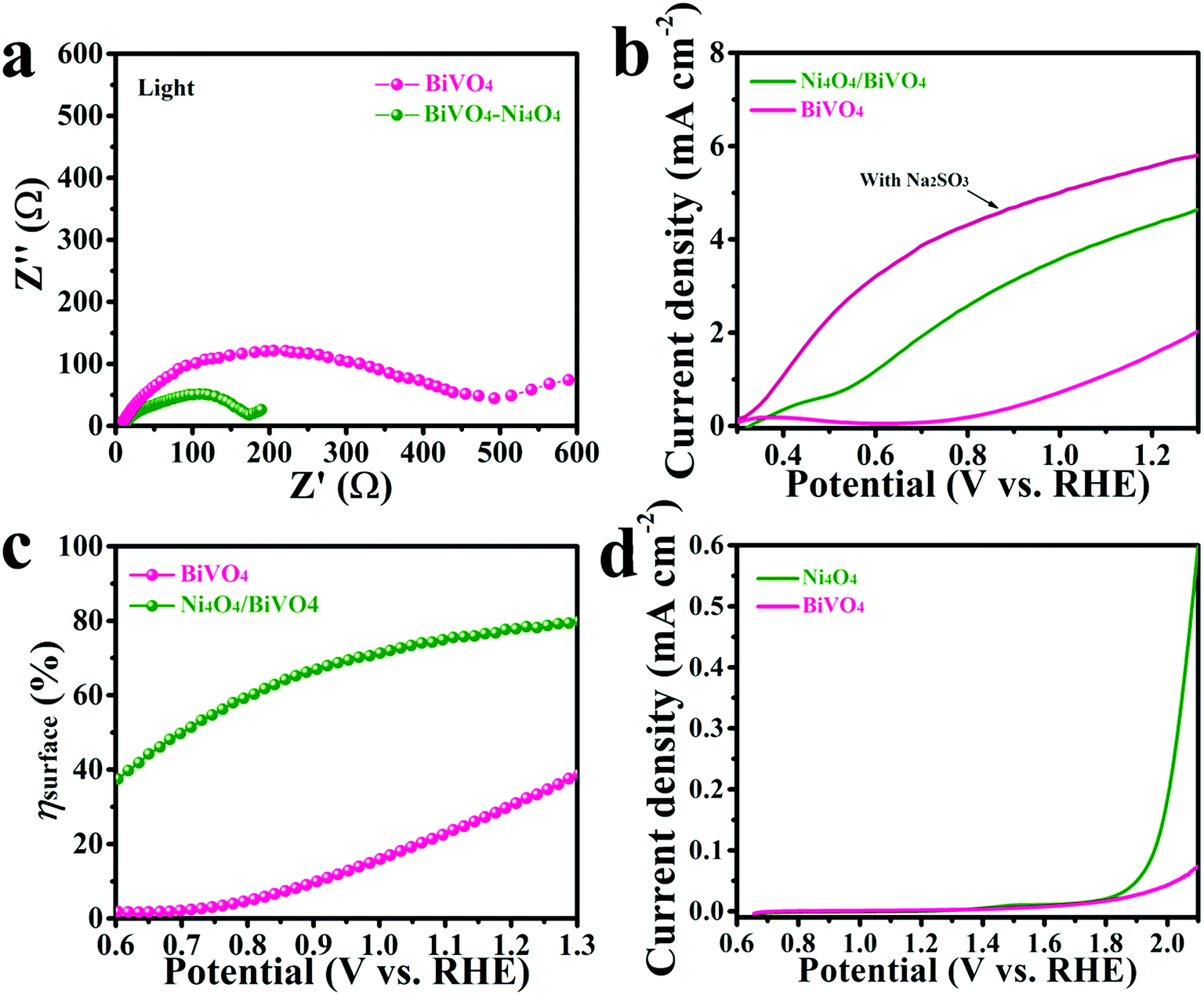 | ||
| Fig. 5 EIS spectra (a), sulfite and water oxidation current curves (b) and surface hole reaction efficiency (ηsurface) (c) of the BiVO4 and Ni4O4/BiVO4 photoanodes and (d) oxygen evolution reaction curves of BiVO4 and Ni4O4 cubane obtained by the rotating disk electrode method at 1600 rpm. | ||
To investigate the water-oxidation reaction behavior at the electrolyte/semiconductor interface in detail, intensity modulated photocurrent spectroscopy (IMPS) was performed. On the surface of the photoanode, the surface-reaching holes could generate two kinds of current, charge transfer current and surface recombination current. The obtained photocurrent is the combined contribution of the two kinds of current. On the premise that a small perturbation of light intensity changes the charge density but not the degree of band bending, by applying sinusoidal modulated light on the photoanode, the frequency response of the photocurrent to the modulated light intensity is measured and information about hole transfer and recombination can be obtained.57–61 As shown in Fig. 6a and b, the Nyquist plots are composed of two arcs. The arc in the fourth quadrant is due to RC attenuation, and the other arc in the first quadrant can be attributed to the change of surface recombination in relation to the modulated light source. The upper arc has two X-intercepts at the low frequency range and high frequency range, respectively. The X-intercept at the high frequency range indicates that the surface recombination velocity cannot match the constantly changing surface hole concentration along with light intensity. At this point, the surface recombination can be ignored and the measured photocurrent is equal to the hole current, which was caused by the surface-reaching hole flux. At the other X-intercept, the change in light intensity is too slow to cause the change in the surface hole concentration in this state. At this time, the behavior of surface recombination is similar to that observed when the photoanode is under steady illumination and the obtained photocurrent is exactly equal to the water-oxidation current. Thus, information on transfer and surface recombination can be obtained. As a result, after normalization processing, the X-intercept at the low frequency range is equal to kt/(kt + kr), where kt and kr are the rate constants of charge transfer and surface recombination, respectively. The sum of kt and kr is determined by the angular frequency of the peak of the Nyquist plot. According to the above discussion, the rate constants of transfer and surface recombination are obtained by analyzing the IMPS data and shown in Fig. 6c and d. For the BiVO4 photoanode, the rate constant of transfer gradually increases with the bias voltage increase. This is owing to the fact that the degree of band bending gradually increases and photo-generated holes can overcome the energy barrier to react with water more easily. Meanwhile, due to being driven by the electric field, the carriers separate faster and recombination is suppressed, so that the rate constant of recombination decreases gradually. However, the Ni4O4/BiVO4 photoanode exhibits high kt and low kr from the beginning, indicating that the fast transfer rate of holes can be reached and the recombination is suppressed at a low potential. Obviously, Ni4O4 cubane can facilitate the water oxidation reaction kinetics and suppress the recombination at the electrolyte/semiconductor interface. In addition, based on the analysis of IMPS data, the hole current, photocurrent and recombination current could be acquired. The hole current is the current generated by surface-reaching holes, which could be divided into photocurrent and recombination current. The photocurrent and recombination current represents the water-oxidation current and current consumed by recombination, respectively. At the high-frequency intercept, the recombination of surface-reaching holes could be ignored and the measured current is the hole current. At the low-frequency intercept, the frequency of light intensity is too low to change the surface state of the photoanode, so that the measured current is the steady photocurrent. According to the fitting results (Fig. 6e), the BiVO4 photoanode has a high recombination current which increased first and then decreased. At a low potential, the photo-generated holes accumulate at the surface and recombine with photo-generated electrons due to the slow water oxidation reaction kinetics. With the increase of potential, recombination is suppressed and Jrecom decreases gradually. Fig. 6f shows the fitting results of the Ni4O4/BiVO4 photoanode, where a negligible recombination current is observed, indicating that the recombination is effectively suppressed at the surface of the photoanode in the whole range of potential. Thus, the Ni4O4 cubane cocatalyst can effectively accelerate the water oxidation reaction kinetics and suppress the recombination of carriers at the electrolyte/semiconductor interface, which results in the dramatic increase in the photocurrent of the Ni4O4/BiVO4 photoanode. As shown in Fig. 7, under illumination, BiVO4 could absorb energy to generate electrons and holes. The photo-generated electrons migrated to the conductive substrate and the holes migrated to the surface of BiVO4, driven by the electric field. The surface-reaching holes can accumulate and recombine on the surface due to the sluggish water oxidation kinetics. When the photoanode was modified by Ni4O4 cubane WOCs, the photo-generated holes migrated to the cocatalyst layer and the recombination of surface-reaching carriers was restricted. The holes could participate in the water oxidation reaction rapidly due to the fast water oxidation kinetics and low overpotential of Ni4O4 cubane, as confirmed by the negatively shifted onset potential and increased photocurrent.
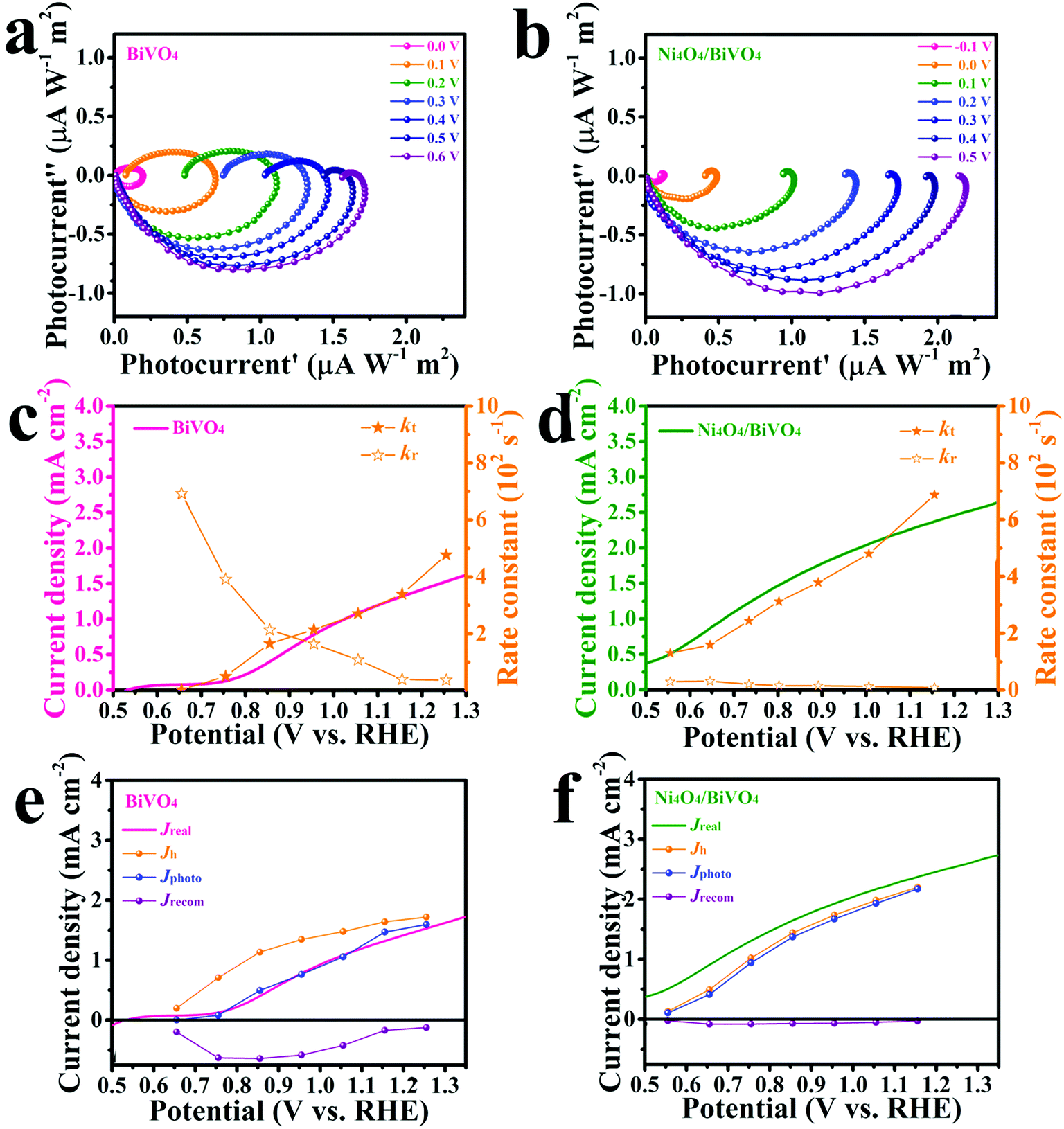 | ||
| Fig. 6 The IMPS Nyquist plots (the potentials are relative to SCE) and rate constants and theoretical currents extracted from the IMPS data at different applied potentials of the BiVO4 photoanode (a, c, and e) and Ni4O4/BiVO4 photoanode (b, d, and f). | ||
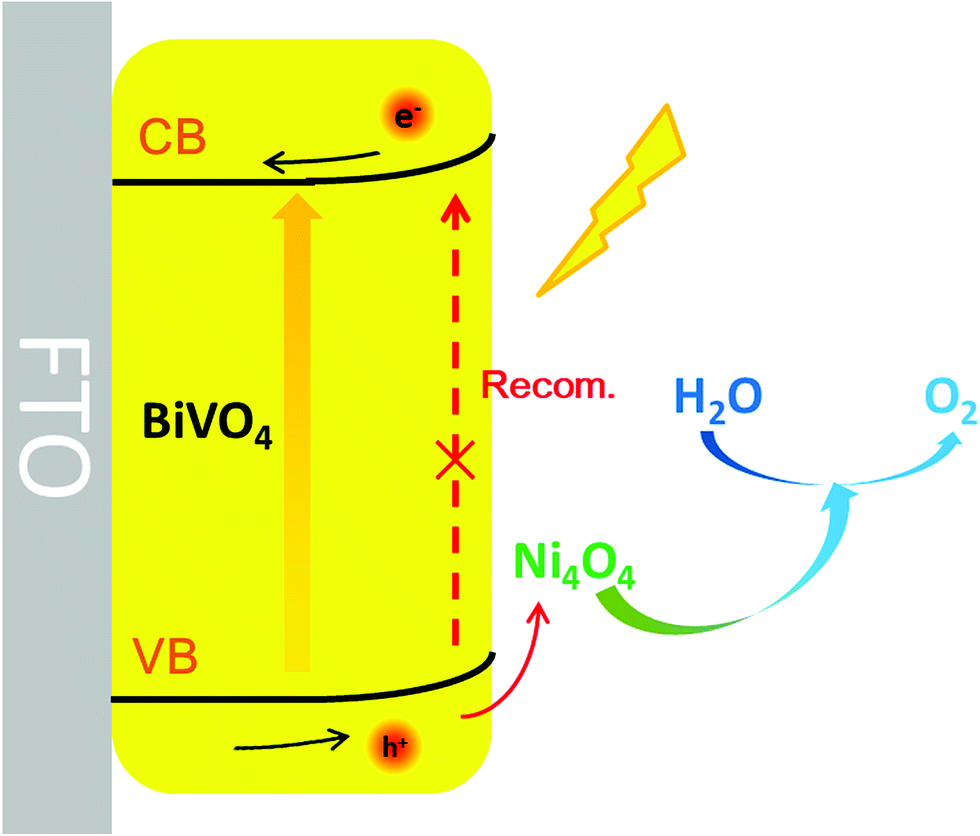 | ||
| Fig. 7 Schematic representation of the integrated BiVO4 photoanode system. | ||
In addition, some researchers have reported that the metal–organic molecular catalysts would release dissociative metal ions to form metallic oxides in the test and the metallic oxide would be the real catalytic active site. In order to verify that the active cocatalyst is Ni4O4 cubane rather than nickel oxide, bpy (2,2′-bipyridine) as the molecular probe was used to identify the real catalytic species. If the dissociative nickel ion is released from Ni4O4 cubane, bpy would chelate with the nickel ion and prevent the formation of nickel oxide. Thus, the PEC performance is tested with the presence of bpy in the electrolyte and the results are shown in Fig. S6.† After adding bpy into the electrolyte, the onset potential and photocurrent of the OER curves barely changed, indicating that there are no free nickel ions during the process of water oxidation. To further investigate the chemical stability of Ni4O4 cubane in the water-oxidation reaction, the XPS spectra are also recorded after testing and shown in Fig. S7.† The characteristic peaks of Ni 2p and O 1s didn't shift after testing, indicating that the chemical states of Ni and O are not altered. In particular, the peaks of O 1s scarcely changed before and after testing and do not match with the characteristic peaks of nickel oxide. This is further evidence that Ni4O4 cubane doesn't form nickel oxide in the PEC process.
PEC stability of the composite photoanode
Finally, the PEC stability of the Ni4O4/BiVO4 photoanode is investigated by chronoamperometry. However, the stability of the photocurrent is unsatisfactory. As shown in Fig. 8d, at the bias voltage of 1.23 V vs. RHE, the photocurrent of Ni4O4/BiVO4 rapidly decays and decreases to 60% after 1 h of the PEC test. According to the SEM image of the photoanode after being tested for 1 h shown in Fig. S8,† a majority of Ni4O4 cubane has been released from the surface of the BiVO4 nanoplates. Thus, desorption of Ni4O4 cubane is the primary cause of performance degradation. The desorption of molecular catalysts is a universal problem in a molecular WOC/semiconductor combined system; thus their long-term durability isn't always ideal. To solve the stability problem, an Al2O3 adsorbed layer is deposited onto the surface of the BiVO4 nanoplates as shown in Fig. 8c. The SEM image is shown in Fig. 8a, and it's clear that the Al2O3 adsorbed layer is closely and uniformly adsorbed on the surface of the nanoplates. The thickness of the Al2O3 adsorbed layer is measured via TEM, and shown in Fig. S9a.† Compared with the smooth BiVO4 crystal (Fig. S10†), Al2O3/BiVO4 exhibits a loose layer on the surface of the crystal. It's clear that the 20 nm Al2O3 is closely attached to the BiVO4 nanoplates. The HRTEM image shown in Fig. S9c† indicates that the Al2O3 on the surface is amorphous. The chemical states of the Al2O3 layer were also investigated using the XPS test, and it was verified that the adsorbed layer is composed of Al2O3 (Fig. S11†). The UV-vis spectrum and Tauc plots indicate that the deposition of the Al2O3 layer does not change the light absorption and band gap (Fig. S12†). In addition, the FTIR spectroscopy results verified that the surface of the modified photoanode had more hydroxyl groups compared with that of the pristine photoanode, which could combine with Ni4O4 cubane to form hydrogen (Fig. S13†). After constructing the Al2O3 adsorbed layer, the identical conditions are applied to load Ni4O4 cubane onto the Al2O3/BiVO4 photoanode. As shown in Fig. 8b, the Ni4O4/Al2O3/BiVO4 photoanode exhibits a thicker Ni4O4 cubane adsorbed layer compared to the Ni4O4/BiVO4 photoanode shown in Fig. 2b. The TEM image shown in Fig. S9b† shows the 40 nm plicated layer on the surface of the BiVO4 nanoplate. The increased adsorbing capacity is also certified by inductively coupled plasma-atomic emission spectrometry (ICP). By adding the Al2O3 adsorbed layer, the adsorption amount of Ni4O4 cubane increases from 0.1734 μmol cm−2 to 0.324 mol cm−2 (Fig. S14†). Obviously, the construction of the Al2O3 layer significantly promotes the adsorbing capacity of Ni4O4 onto BiVO4. The PEC properties of Al2O3/BiVO4 are shown in Fig. S15,† and it's clear that there is no visible difference between the photoanode with and without the Al2O3 layer. The unchanged performance may be due to the fact that the loose Al2O3 layer neither hinders water molecules migrating to surface of BiVO4 nor acts as a water oxidation cocatalyst. In addition, the flat-band potential of BiVO4 and Al2O3/BiVO4 was obtained using Mott–Schottky plots shown in Fig. S16.† It's clear that there is no difference in the flat-band potential between BiVO4 and Al2O3/BiVO4, indicating that the band structure was unaltered after the deposition of the Al2O3 layer. Moreover, the Ni4O4/BiVO4 photoanode and Ni4O4/Al2O3/BiVO4 exhibit similar PEC properties, although the content of Ni4O4 cubane in the former is far lower that in the latter. In addition, according to the ICP results, the ethanol solution with Ni4O4 cubane was added dropwise onto the BiVO4 photoanode with single and five-fold quantity compared with the catalyst loading amount of Ni4O4/BiVO4 (Ni4O4/BiVO4-d-1 and Ni4O4/BiVO4-d-2). As shown in Fig. S17,† Ni4O4/BiVO4-d-1 showed similar light absorption to the Ni4O4/BiVO4 photoanode, indicating that the deposition of Ni4O4 cubane is not enough to affect light absorption. Meanwhile, the photocurrent showed a slight decrease, which may be due to the non-uniform distribution of Ni4O4 cubane on the BiVO4 nanoplate. However, after the five-fold addition of Ni4O4 cubane dropwise, the absorption spectrum of the photoanode changed obviously, and the photocurrent decreased. This result indicated that overmuch Ni4O4 cubane could affect the light absorption and reduce the photocurrent. Therefore, the deposition weight of the Ni4O4 layer on the photoanode with the Al2O3 layer was not enough to affect light absorption and also PEC performance. Finally, the Ni4O4/Al2O3/BiVO4 photoanode is placed in a PEC system to test its stability. As shown in Fig. 8d, the composite photoanode exhibits excellent stability with an attenuation of 20% after two hours. Furthermore, the SEM image of the post-test composite photoanode shown in Fig. S18† shows that there is still a mass of Ni4O4 cubane on the surface of the BiVO4 nanoplates. Thus, the Al2O3 adsorbed layer could combine more Ni4O4 cubane and effectively keep it from being desorbed, so that a satisfactory PEC stability is obtained. Also, the long-term test for 6 hours was performed and it was verified that the Ni4O4/Al2O3/BiVO4 photoanode had excellent stability compared to the Ni4O4/BiVO4 photoanode (Fig. S19†). In order to prove that the excellent photocurrent is due to O2 evolution rather than self-oxidation or impurities in the electrolyte, the O2 production was monitored under 0.6 V (vs. RHE) bias and shown in Fig. 9. The actual measured result matched well the theoretical result and the faradaic efficiency was calculated to be 96.3%, indicating that the photocurrent resulted from oxygen evolution (Table S2†).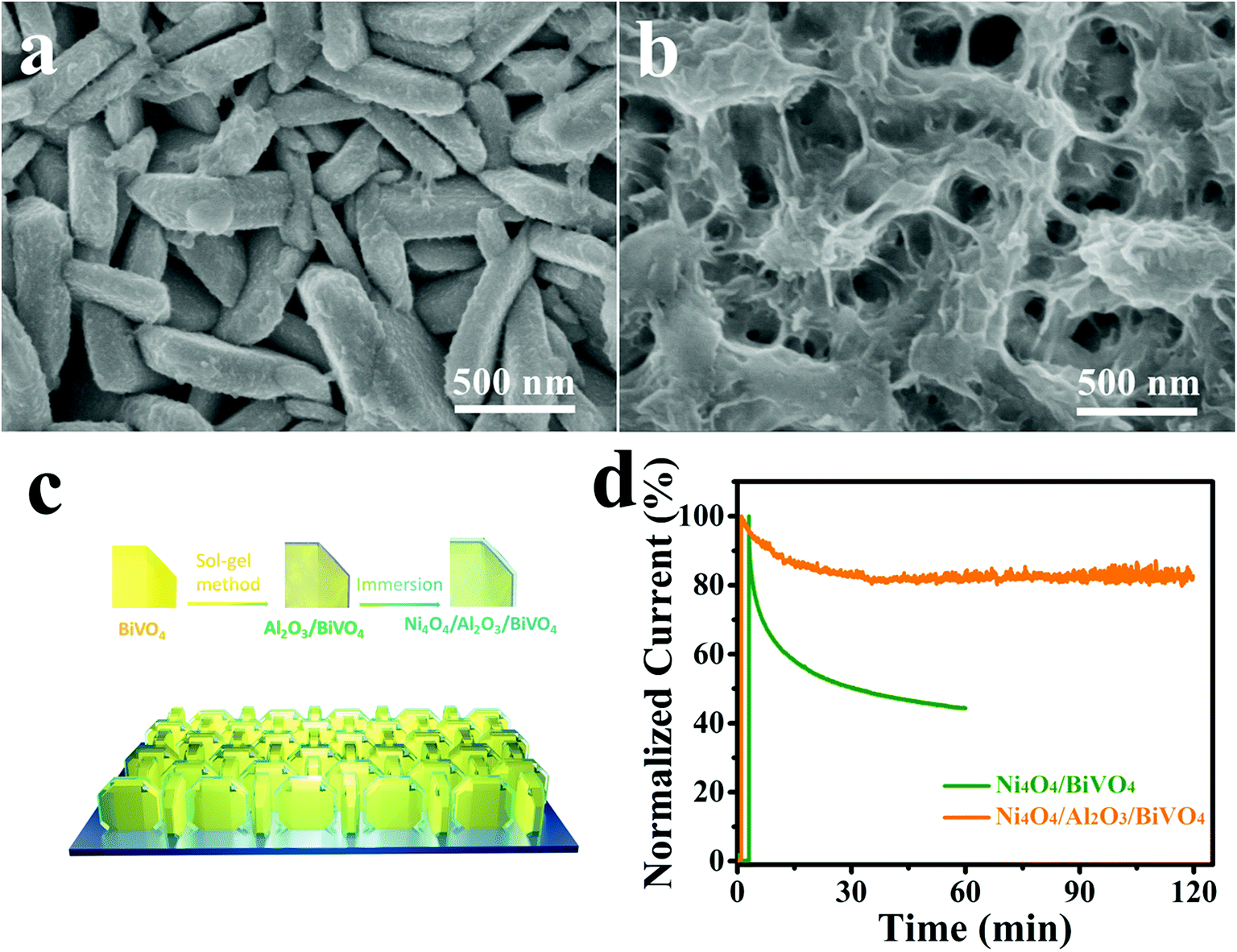 | ||
| Fig. 8 The SEM images of the Al2O3/BiVO4 (a) and Ni4O4/Al2O3/BiVO4 (b) photoanodes. (c) Schematic of the Ni4O4/Al2O3/BiVO4 photoanode. (d) I–t curve of the Ni4O4/BiVO4 and Ni4O4/Al2O3/BiVO4 photoanodes. | ||
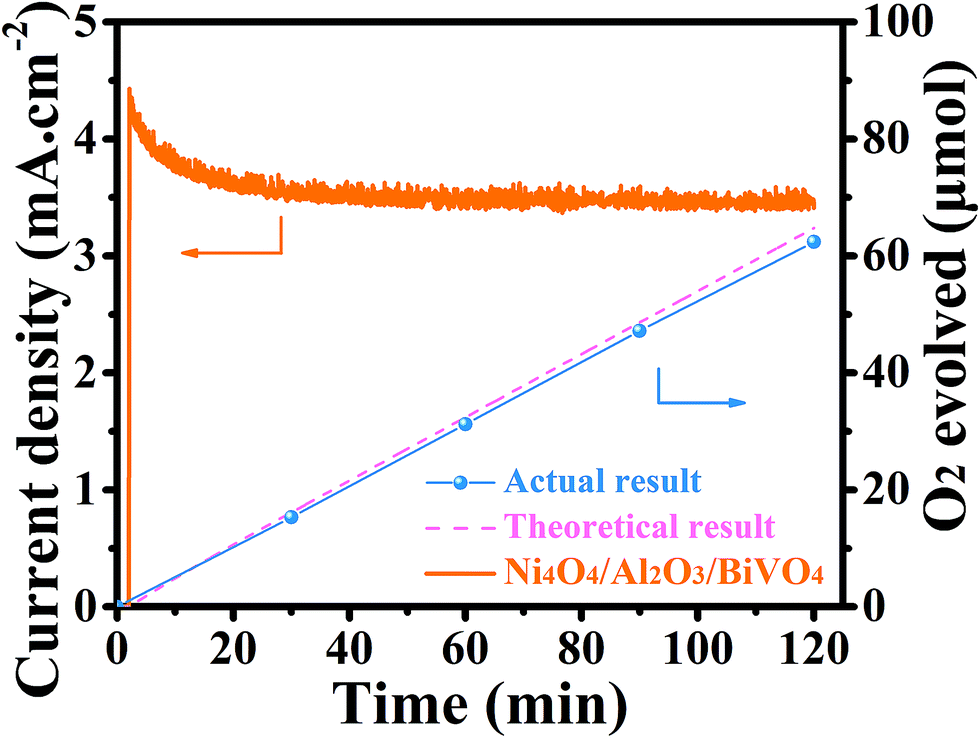 | ||
| Fig. 9 O2 evolution at 0.6 V (vs. RHE); the dashed line indicates the theoretical result from measured photocurrents. | ||
Conclusions
In conclusion, earth-abundant and low-cost Ni4O4 cubane was synthesized and combined with a BiVO4 photoanode as a water-oxidation catalyst. The composite photoanode exhibits an increased photocurrent and a more negative onset potential than the pristine BiVO4 photoanode. The study of the mechanism shows that the Ni4O4 cubane cocatalyst not only accelerated the water-oxidation kinetics but also suppressed the recombination of photo-generated carriers at the interface of the electrolyte/semiconductor. In addition, the introduction of an Al2O3 adsorbed layer on the BiVO4 photoanode could efficiently adsorb more cocatalyst resulting in the excellent PEC stability. These results provide a promising strategy to achieve high-efficiency water oxidation by combining a molecular cocatalyst with a semiconductor photoanode.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by the National Natural Science Foundation of China (51602153 and 11575084), the Natural Science Foundation of Jiangsu Province (BK20160795) and the Fundamental Research Funds for the Central Universities (No. NE2018104) and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 238–245 CrossRef.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- S. Kment, F. Riboni, S. Pausova, L. Wang, L. Wang, H. Han, Z. Hubicka, J. Krysa, P. Schmuki and R. Zboril, Chem. Soc. Rev., 2017, 46, 3716–3769 RSC.
- Y. Yu, Z. Zhang, X. Yin, A. Kvit, Q. Liao, Z. Kang, X. Yan, Y. Zhang and X. Wang, Nat. Energy, 2017, 2, 17045 CrossRef CAS.
- K. H. Ye, Z. Wang, J. Gu, S. Xiao, Y. Yuan, Y. Zhu, Y. Zhang, W. Mai and S. Yang, Energy Environ. Sci., 2017, 10, 772–779 RSC.
- D. Bae, B. Seger, P. C. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC.
- H. Gong, T. Wang, H. Xue, X. Fan, B. Gao, H. Zhang, L. Shi, J. He and J. Ye, Energy Storage Materials, 2018, 13, 49–56 CrossRef.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef PubMed.
- X. Fan, T. Wang, B. Gao, H. Gong, H. Xue, H. Guo, L. Song, W. Xia, X. Huang and J. He, Langmuir, 2016, 32, 13322–13332 CrossRef CAS PubMed.
- W. Yu, J. Chen, T. Shang, L. Chen, L. Gu and T. Peng, Appl. Catal., B, 2017, 219, 693–704 CrossRef CAS.
- X. Fan, B. Gao, T. Wang, X. Huang, H. Gong, H. Xue, H. Guo, L. Song, W. Xia and J. He, Appl. Catal., A, 2016, 528, 52–58 CrossRef CAS.
- D. Cao, W. Luo, J. Feng, X. Zhao, Z. Li and Z. Zou, Energy Environ. Sci., 2014, 7, 752–759 RSC.
- X. She, J. Wu, H. Xu, J. Zhong, Y. Wang, Y. Song, K. Nie, Y. Liu, Y. Yang and M. T. F. Rodrigues, Adv. Energy Mater., 2017, 7, 1700025 CrossRef.
- J. Xie, C. Guo, P. Yang, X. Wang, D. Liu and C. M. Li, Nano Energy, 2016, 31, 28–36 CrossRef.
- S. Wang, P. Chen, J. H. Yun, Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 8500–8504 CrossRef CAS PubMed.
- S. Wang, P. Chen, Y. Bai, J. H. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef PubMed.
- X. Ding, L. Zhang, Y. Wang, A. Liu and Y. Gao, Coord. Chem. Rev., 2018, 357, 130–143 CrossRef CAS.
- N. Wu, Nanoscale, 2018, 10, 2679–2696 RSC.
- T. Li, J. He, B. Peña and C. P. Berlinguette, Angew. Chem., Int. Ed., 2016, 55, 1769–1772 CrossRef CAS PubMed.
- B. Trzesniewski and W. A. Smith, J. Mater. Chem. A, 2016, 4, 2919–2926 RSC.
- A. T. Oliveira, M. Rodriguez, T. S. Andrade, H. E. D. Souza, J. D. Ardisson, H. S. Oliveira, L. C. Oliveira, E. Lorençon, A. C. Silva, L. L. Nascimento, A. O. T. Patrocínio and M. C. Pereira, Sol. RRL, 2018, 2, 180089 Search PubMed.
- X. Zhong, H. He, M. Yang, G. Ke, Z. Y. Zhao, F. Dong, B. Wang, Y. Chen, X. Shi and Y. Zhou, J. Mater. Chem. A, 2018, 6, 10456–10465 RSC.
- B. Gao, T. Wang, X. Fan, H. Gong, H. Guo, W. Xia, Y. Feng, X. Huang and J. He, Inorg. Chem. Front., 2017, 4, 898–906 RSC.
- S. Wang, T. He, J. H. Yun, Y. Hu, M. Xiao, A. Du and L. Wang, Adv. Funct. Mater., 2018, 8, 1802685 CrossRef.
- L. Wang, D. Mitoraj, S. Turner, O. V. Khavryuchenko, T. Jacob, R. K. Hocking and R. Beranek, ACS Catal., 2017, 7, 4759–4767 CrossRef CAS.
- B. Zhang, L. Wang, Y. Zhang, Y. Ding and Y. Bi, Angew. Chem., Int. Ed., 2018, 57, 2248–2252 CrossRef CAS PubMed.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- M. Zhu, Z. Sun, M. Fujitsuka and T. Majima, Angew. Chem., Int. Ed., 2018, 57, 2160–2164 CrossRef CAS PubMed.
- T. W. Kim and K. S. Choi, Science, 2014, 45, 990–994 CrossRef PubMed.
- H. Park, H. Kim, G. Moon and W. Choi, Energy Environ. Sci., 2015, 9, 411–433 RSC.
- P. Kuang, L. Zhang, B. Cheng and J. Yu, Appl. Catal., B, 2017, 218, 570–580 CrossRef CAS.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- W. Liu, H. Liu, L. Dang, H. Zhang, X. Wu, B. Yang, Z. Li, X. Zhang, L. Lei and S. Jin, Adv. Funct. Mater., 2017, 27, 1603904 CrossRef.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
- X. Chang, T. Wang, Z. Peng, J. Zhang, A. Li and J. Gong, J. Am. Chem. Soc., 2015, 137, 8356–8359 CrossRef CAS PubMed.
- S. Hernández, G. Gerardi, K. Bejtka, A. Fina and N. Russo, Appl. Catal., B, 2016, 190, 66–74 CrossRef.
- H. Gong, H. Xue, T. Wang, H. Guo, X. Fan, L. Song, W. Xia and J. He, ACS Appl. Mater. Interfaces, 2016, 8, 18060–18068 CrossRef CAS PubMed.
- W. Xia, J. Li, T. Wang, L. Song, H. Guo, H. Gong, C. Jiang, B. Gao and J. He, Chem. Commun., 2018, 54, 1623–1626 RSC.
- Y. Liu, Y. Jiang, F. Li, F. Yu, W. Jiang and L. Xia, J. Mater. Chem. A, 2018, 6, 10761–10768 RSC.
- S. Nakashima, R. Negishi and H. Tada, Chem. Commun., 2016, 52, 3665–3668 RSC.
- H. Y. Jiang, P. Zhou, Y. Wang, R. Duan, C. Chen, W. Song and J. Zhao, Adv. Mater., 2016, 28, 9776–9781 CrossRef CAS PubMed.
- T. Rosser, M. A. Gross, Y. H. Lai and E. Reisner, Chem. Sci., 2016, 7, 4024–4035 RSC.
- B. Liu, J. Li, H. L. Wu, W. Q. Liu, X. Jiang, Z. J. Li, B. Chen, C. H. Tung and L. Z. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 18577–18583 CrossRef CAS PubMed.
- A. Y. Ahmed, M. G. Ahmed and T. A. Kandiel, J. Phys. Chem. C, 2016, 120, 23415–23420 CrossRef CAS.
- Y. Wang, F. Li, X. Zhou, F. Yu, J. Du, L. Bai and L. Sun, Angew. Chem., Int. Ed., 2017, 56, 6911–6915 CrossRef CAS PubMed.
- K. Dang, X. Chang, T. Wang and J. Gong, Nanoscale, 2017, 9, 16133–16137 RSC.
- H. Kai, T. Kreuger, B. Mei and G. Mul, ACS Catal., 2017, 7, 1610–1614 CrossRef PubMed.
- Q. Liu, F. Cao, F. Wu, H. Lu and L. Li, Adv. Mater. Interfaces, 2016, 3, 1600256 CrossRef.
- X. Mu, Q. Wen, G. Ou, D. Yuemin, P. He, M. Zhong, H. Zhu, H. Wu, S. Yang and Y. Liu, Nano Energy, 2018, 51, 83–90 CrossRef CAS.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS PubMed.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335–7338 CrossRef CAS PubMed.
- G. Zahariou, Inorg. Chem., 2017, 56, 6105–6113 CrossRef CAS PubMed.
- H. A. Rudbari, F. Lloret, M. Khorshidifard, G. Bruno and M. Julve, RSC Adv., 2016, 6, 7189–7194 RSC.
- B. Gao, T. Wang, X. Fan, H. Gong, X. Meng, P. Li, Y. Feng, X. Huang, J. He and J. Ye, Sol. RRL, 2018, 2, 1800102 CrossRef.
- C. W. Kim, Y. S. Son, M. J. Kang, D. Y. Kim and Y. S. Kang, Adv. Energy Mater., 2016, 6, 1501754 CrossRef.
- Y. Zhu, J. Ren, X. Yang, G. Chang, Y. Bu, G. Wei, W. Han and D. Yang, J. Mater. Chem. A, 2017, 5, 9952–9959 RSC.
- A. Tsyganok, D. Klotz, K. D. Malviya, A. Rothschild and D. A. Grave, ACS Catal., 2018, 8, 2754–2759 CrossRef CAS.
- L. M. Peter, J. Solid State Electrochem., 2013, 17, 315–326 CrossRef CAS.
- L. M. Peter, K. G. Wijayantha and A. A. Tahir, Faraday Discuss., 2012, 155, 309–322 RSC.
- S. Xiao, H. Chen, Z. Yang, X. Long, Z. Wang, Z. Zhu, Y. Qu and S. Yang, J. Phys. Chem. C, 2015, 199, 23350–23357 CrossRef.
- J. E. Thorne, J. Jang, E. Liu and D. Wang, Chem. Sci., 2016, 7, 3347–3354 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta09404g |
| This journal is © The Royal Society of Chemistry 2019 |