
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchical MoS2–carbon porous nanorods towards atomic interfacial engineering for high-performance lithium storage†
Zhenyou
Li
 *abg,
Alexander
Ottmann
a,
Qing
Sun
ac,
Anne K.
Kast
f,
Kai
Wang
h,
Ting
Zhang
b,
Hans-Peter
Meyer
e,
Claudia
Backes
d,
Christian
Kübel
ghi,
Rasmus R.
Schröder
cf,
Junhui
Xiang
b,
Yana
Vaynzof
ac and
Rüdiger
Klingeler
ac
*abg,
Alexander
Ottmann
a,
Qing
Sun
ac,
Anne K.
Kast
f,
Kai
Wang
h,
Ting
Zhang
b,
Hans-Peter
Meyer
e,
Claudia
Backes
d,
Christian
Kübel
ghi,
Rasmus R.
Schröder
cf,
Junhui
Xiang
b,
Yana
Vaynzof
ac and
Rüdiger
Klingeler
ac
aKirchhoff Institute of Physics, Heidelberg University, INF 227, 69120 Heidelberg, Germany. E-mail: zhenyou.li@kit.edu
bCollege of Materials Science and Opto-Electronic Technology, University of Chinese Academy of Sciences, Yuquan Road 19A, Beijing, 100049, China
cCentre for Advanced Materials (CAM), Heidelberg University, INF 225, 69120 Heidelberg, Germany
dInstitute of Physical Chemistry, Heidelberg University, INF 253, 69120 Heidelberg, Germany
eInstitute of Earth Sciences, Heidelberg University, INF 236, D-69120 Heidelberg, Germany
fBioQuant, Cryo Electron Microscopy, Heidelberg University, INF 267, 69120 Heidelberg, Germany
gHelmholtz Institute Ulm (HIU), Helmholtzstraße 11, D-89081 Ulm, Germany
hInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
iKarlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 6th March 2019
Abstract
Hierarchical nanostructures have attracted considerable attention for rechargeable battery systems since they combine the benefits of size effects induced by nanoscaling with the integrity of bulk materials. Despite significant progress, the hierarchical structures reported so far are designed only down to the nanoscale. To improve the battery performance, downsizing the designed building blocks of the hierarchical structure to smaller scales (molecular or even atomic level) is essential. This novel concept has been realized in a MoS2/C composite system, where MoS2 and N-doped carbon molecular layers are alternately stacked to form nanosheet building blocks, which are further assembled into a porous nanorod structure. This hierarchical heterostructure converts the guiding principle of sub-nanoscale engineering into practice, aiming at increasing the interfaces between MoS2 and carbon towards the largest possible molecular contact level. The resultant MoS2/N-doped carbon porous nanorods (MoS2/NC-PNR) electrode exhibits outstanding performances in lithium-ion batteries including high initial discharge capacity of ∼1300 mA h g−1, cycling stability for 700 cycles and excellent rate performance (443 mA h g−1 at 10C). The outstanding performance of the MoS2/NC-PNR superstructure illustrates the enormous potential of the hierarchically designed 2D compounds from molecular layer level, which could be extended to other layered materials.
Introduction
Molybdenum disulfide (MoS2) is one of the most stable and multifunctional members of the transition metal dichalcogenide family and hence promises a wide range of applications, including hydrogen evolution,1,2 solar cells3,4 and rechargeable batteries.5–8 Its tuneable layered structure and high theoretical lithium storage capacity of 670 mA h g−1 render it a great candidate as anode material in Li-ion batteries (LIBs).9,10 However, the semi-conductive nature and large volume changes during the conversion reaction process, which cause disintegration of the structure, lead to very limited cycling stability and poor rate capability.11 Furthermore, the involved redox couple Li2S/S suffers from the shuttle effect of polysulfides, which causes the loss of active material, significantly reducing the specific capacities as well.12Rational design of nanoscale, hierarchical MoS2 composite structures has been proven to be an effective route to tackle the abovementioned problems.13–16 A hierarchical structure both exploits the size effects of the bare nanoscaled building blocks and maintains the integrity of the bulk material to some extent. On the one hand, the Li+ diffusion pathways are shortened due to the size effect. On the other hand, a relatively loose arrangement of the individual building blocks can facilitate electrochemical reactions by providing increased contact area between active material and electrolyte.17 In this context, an adequate backbone structure is needed to guarantee the stability of the composite by buffering mechanical strain during electrochemical cycling. In case of MoS2-based composites, usually carbon layers are introduced to increase the overall conductivity and the adsorption rate of polysulfides, and to further alleviate the volume changes.18 The resultant composite materials usually show enhanced electrochemical performance. For example, Yu et al.19 reported a general method to synthesize multiscale-ordered metal–sulphide/carbon structures, which exhibit stable dis-/charge capacities for up to 300 cycles. MoS2@C nanotubes have been produced by Qian's group20via the Kirkendall effect, featuring 90% capacity retention after 300 cycles at 0.5C. Lou et al.21 have synthesised uniform MoS2 hollow spheres with a high specific capacity of 1100 mA h g−1 at 500 mA g−1 (∼0.75C) and excellent cycling stability. Very recently, Świerczek's group has reported epitaxial growth of carbon-sheathed MoS2 on CNTs which displays long-term stable capacities of ∼900 mA h g−1 at 1 A g−1 (∼1.5C) for 500 cycles.22
However, most of the previously reported hierarchical structures are constructed from the nanoscale building blocks. Since electrochemical processes are electron-driven redox reactions on the atomic level, it seems extremely beneficial to optimize the design of hierarchical MoS2 composite structures down to the single molecular layer scale. And building this highly ordered superstructure requires carefully engineering the MoS2:C interfaces. Recent findings23 suggest that the performance of MoS2-based anode materials can be substantially improved by maximizing the molecular layer contact area between MoS2 and carbon. In fact, increasing the MoS2:C heterointerfaces has also been a guiding principle to us for developing composite MoS2/C nanostructures. According to our previous studies, a better cycling stability and rate capability have been achieved when change from point-to-surface contacts24 to a surface-to-surface architecture25 (sketched in Fig. 1a and b). Rigorous advancement of this strategy to maximize the battery performance of MoS2-based electrodes implies a layer-by-layer contact as illustrated in Fig. 1c. Such kind of molecular layer stacked structures are expected to largely enhance the multi-function of carbon species mentioned above. Moreover, the intercalation of carbon layers between the MoS2 ones presumably provides expanded interlayer distances and hence facilitates Li+ diffusion so that layer-by-layer stacked composite materials exhibit extraordinarily better performance than its single components and larger scale mixtures, respectively.26,27 By exfoliation and reassembly, top-down methods are common ways to synthesis the layer-by-layer structures, but difficult to further build into hierarchical structures.23 So far, constructing this hierarchical superstructure is still challenging.
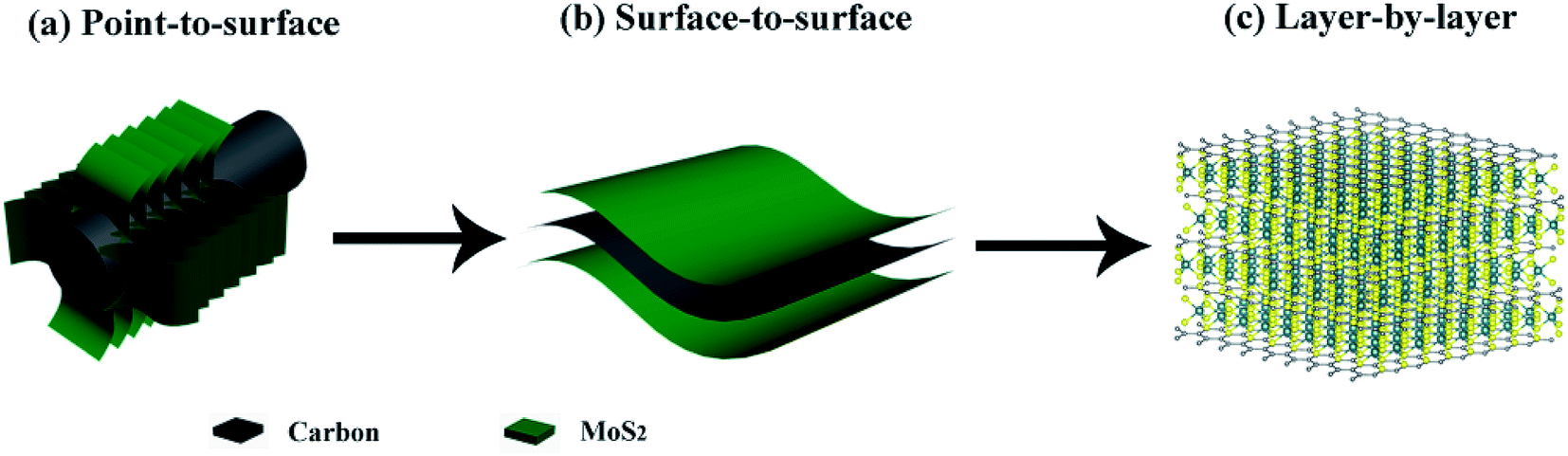 | ||
| Fig. 1 Interfacial engineering of MoS2:C: (a) point-to-surface (1D); (b) surface-to-surface (2D); (c) layer-by-layer (3D) heterointerfaces. | ||
In this work, we apply a facile bottom-up method to synthesize MoS2/N-doped carbon porous nanorods (MoS2/NC-PNR), targeting the hierarchical superstructure to achieve high performance lithium storage. In the reported material, similar hexagonal structures enable alternating stacking of MoS2 molecular layers and N-doped carbon layers, which promotes the largest possible contact interfaces between the active material (MoS2 layers) and the buffer layers (carbon). In addition, such ordered stacking yields an expanded interlayer distance as compared to bulk hexagonal MoS2 (2H-MoS2). In this architecture, the superior properties of both MoS2 and N-doped carbon are fully maintained while a synergetic effect due to a molecular layer level homogeneous distribution is introduced, potentially leading to ultra-fast electron and ion transfer.28 Furthermore, the interoverlapped MoS2/C nanosheets serve as building blocks to construct a hierarchical rod-like structure which is robust enough to endure the repeated de-/lithiation processes and offers a large number of electrochemically active sites as well. These merits endow MoS2/NC-PNR as anode material in LIB with greatly enhanced lithium storage performance. Importantly, this materials design strategy has great potential to be extended to other 2D material systems, thereby realizing high performance hierarchical materials for other energy related applications.
Results and discussion
Synthesis of the MoS2/NC-PNR
As illustrated in Fig. 2, the MoS2/NC-PNR superstructure was prepared via a modified template method. Firstly, α-MoO3 nanorods with around several μm long and 200 nm in diameter (see SEM images in Fig. S1a†) were mixed into a dopamine (DOPA)/TRIS solution to form a Mo(VI) – DOPA complex (MDC), which served as self-sacrifice template. The formation of this complex is indicated by the initially colourless DOPA/TRIS solution changing to a light brown suspension, immediately after the addition of the α-MoO3 nanorods (see Fig. S1c†). During complexation of the MDC, the morphology (Fig. S1b†) and orthorhombic crystal structure (Fig. S1d†) of the α-MoO3 nanorod template are maintained. However, the UV-vis spectra of α-MoO3, DOPA and MDC reveal a different electronic structure in MDC compared to α-MoO3 due to different chemical environments. In Fig. 3a, a broad absorption peak at about 315 nm, which corresponds to bulk-type molybdate species,29,30 is observed in the α-MoO3 spectrum but disappeared in the MDC spectrum. In the latter spectrum, an absorption peak at 280 nm presents in the same position as that of the DOPA spectrum, which originates from the La–Lb coincident transition of DOPA molecules31 due to an excess amount of DOPA in the complexation suspension. High-resolution N 1s spectra of DOPA (Fig. 3b) and MDC (Fig. 3c), collected using X-ray photoemission spectroscopy (XPS), show the presence of different amines in the compounds. In case of DOPA, two species at binding energies of 401.8 eV and 399.2 eV contribute to the signal and are assigned to primary and secondary amine, respectively.32 Although the latter indicates the polymerization of DOPA to a certain degree, the main peak at 401.8 eV confirms that the dominant species is monomeric. In the N 1s spectrum of MDC, four separate peaks at 401.7 eV, 399.5 eV, 398.6 eV, and 396.5 eV are observed, which are attributed to primary, secondary and tertiary amine, and molybdenum (Mo 3p3/2), respectively. The large share of tertiary amine, which is associated with tautomeric species of the intermediates, can be explained by electron donation from DOPA to Mo(VI), resulting in reduced electron density in the DOPA part.33 These results confirm the formation of the MDC complex as an essential intermediate to finally synthesize the layer-by-layer structures.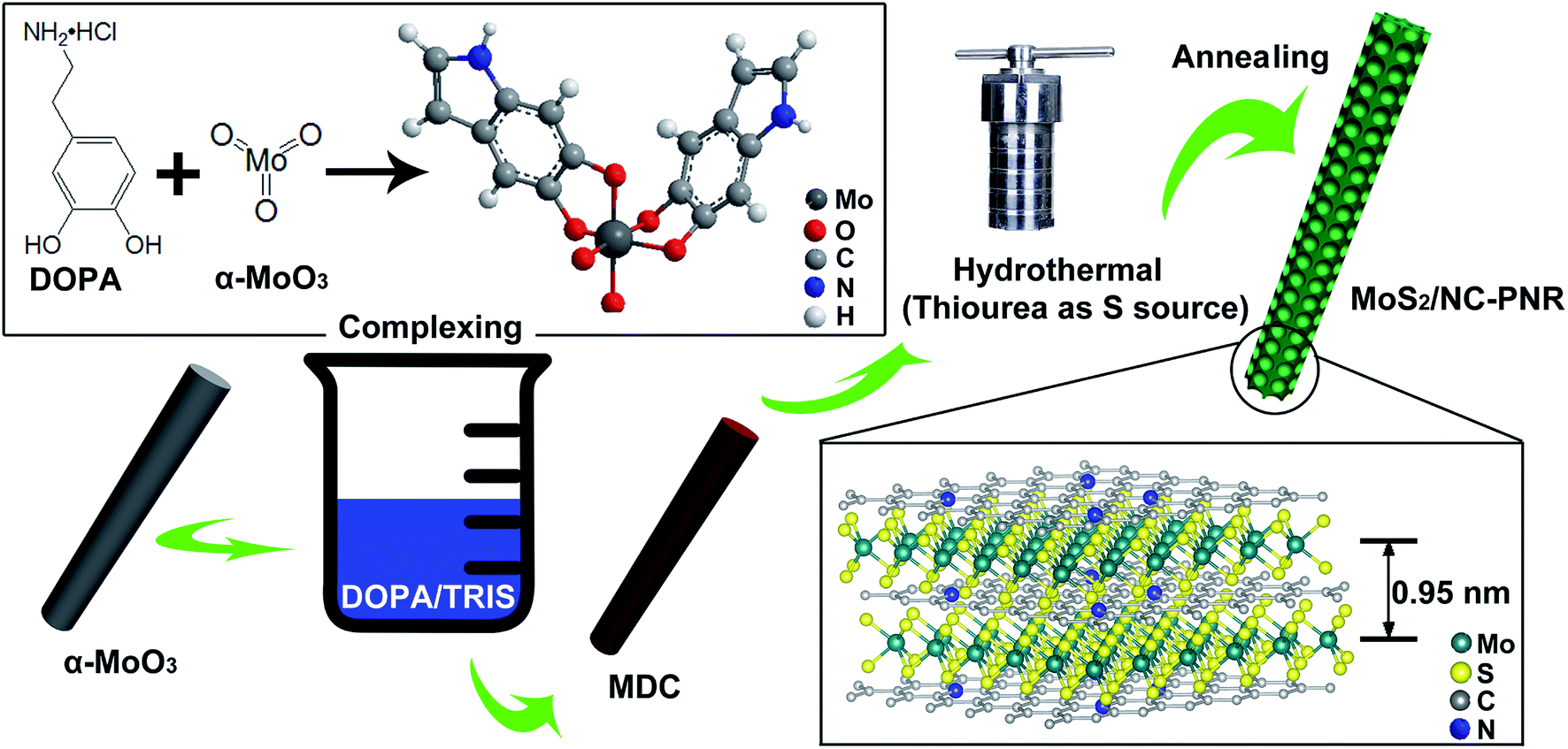 | ||
| Fig. 2 Schematic illustration of MoS2/NC-PNR formation. | ||
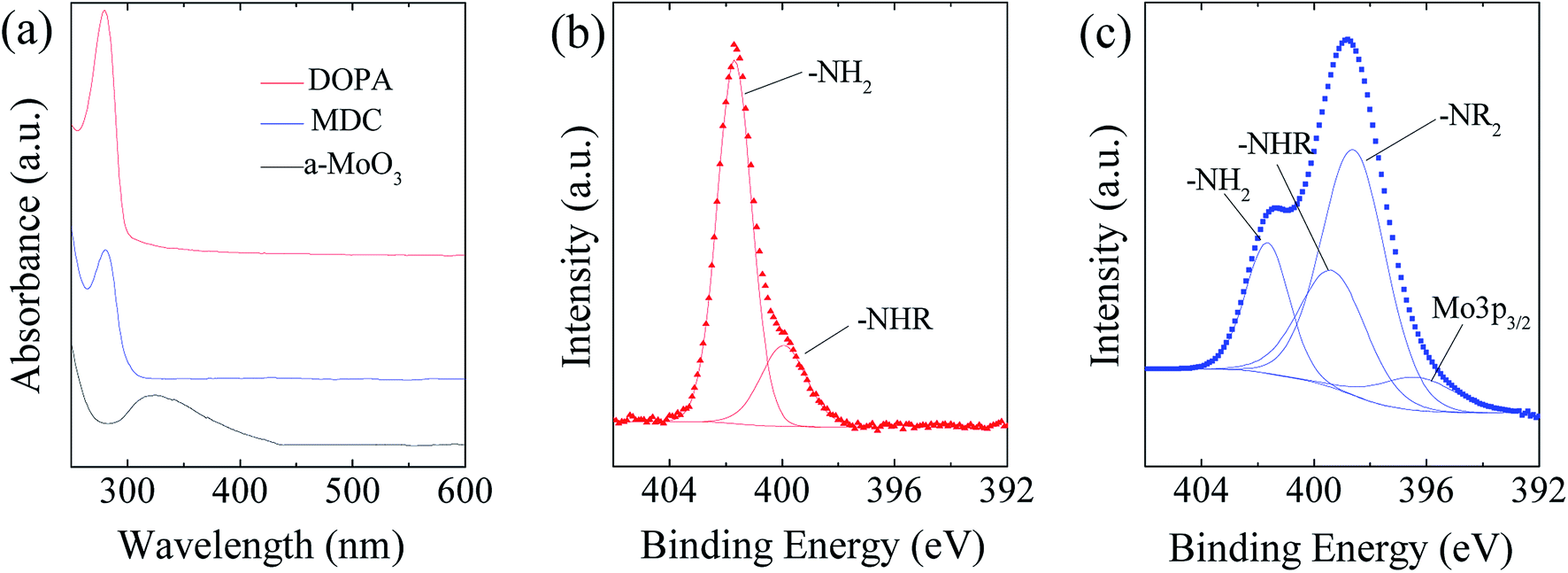 | ||
| Fig. 3 (a) UV-vis spectra of α-MoO3, DOPA and MDC; spectra are offset for clarity. (b and c) XPS N 1s spectra of DOPA and MDC, respectively. –NH2, –NHR and –NR2 indicate primary, secondary and tertiary amines. | ||
In order to obtain the final product, the MDC complex was hydrothermally treated with thiourea as the sulfur source followed by an annealing procedure. In the early stage, self-polymerization of DOPA is supposed to take place with the emission of NH3 from thiourea which forms a mild basic environment (pH = 10.2 after hydrothermal reaction).34 In the meantime, Mo(VI) in the MDC complex is reduced to MoS2 with the help of H2S, growing in situ on the surface of polydopamine (PDA). Thus, the hydrothermal reaction yields MoS2/polydopamine porous nanorods (MoS2/PDA-PNR), which were then sintered at 900 °C under Ar atmosphere to prepare the final product MoS2/NC-PNR. Altogether, this procedure results in the formation of a layer-by-layer MoS2/NC structure while on the larger length scale the porous nanorod morphology is maintained.
Characterization of the interoverlapped structure
Analysis of the chemical composition of the resultant MoS2/NC-PNR superstructure was carried out by means of XPS, displayed in Fig. 4a and S2.† The survey spectrum (Fig. S2a†) shows distinct peaks of Mo, S, C, N and O in which the molar ratio of S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo equals 2.40 ± 0.01. The discrepancy to the stoichiometry of MoS2 can be explained by surface defects of the nanocomposite.35 A high-resolution scan of the Mo 3d range (Fig. 4a left) shows a doublet at 229.2 eV and 232.7 eV, which is attributed to the Mo4+ in MoS2. A minor contribution from MoOx is also observed. The S 2p spectrum (Fig. S2b†) consists of a single doublet at binding energies of 162.3 eV and 163.6 eV, which relates to the S 2p3/2 and 2p1/2 orbitals of S2−.36 In the C 1s spectrum (Fig. 4a mid), a dominant C
:Mo equals 2.40 ± 0.01. The discrepancy to the stoichiometry of MoS2 can be explained by surface defects of the nanocomposite.35 A high-resolution scan of the Mo 3d range (Fig. 4a left) shows a doublet at 229.2 eV and 232.7 eV, which is attributed to the Mo4+ in MoS2. A minor contribution from MoOx is also observed. The S 2p spectrum (Fig. S2b†) consists of a single doublet at binding energies of 162.3 eV and 163.6 eV, which relates to the S 2p3/2 and 2p1/2 orbitals of S2−.36 In the C 1s spectrum (Fig. 4a mid), a dominant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C signal at 284.5 eV and traces of a CO one at 287.4 eV are observed, indicating low oxygen content in the carbon layers. Furthermore, the C-N species at 285.8 eV accounts for 14.1% of all carbon, revealing the N-rich nature of the carbon component.37 The N 1s spectrum (Fig. 4a right) is composed of a Mo 2p3/2 peak and three different N species, i.e., graphitic, pyrrolic, and pyridinic, suggesting a successful doping of N into the carbon layer.38 The Raman shift spectrum of MoS2/NC-PNR in Fig. 4b further confirms the chemical composition of the hierarchical superstructure. At lower wave numbers, the peaks present at 383.43 and 404.71 cm−1 correspond to the in-plane E12g and out of plane A1g mode of MoS2, respectively.39 It is worth noting that the A1g peak is more pronounced than the E12g peak, indicating a more edge-terminated and less layered structure.40 Actually, the frequency difference of the two peaks Δk = 21.28 cm−1 is close to the value of single layer MoS2 reported in the literature39 (20.2–21.2 cm−1), suggesting that single layer MoS2 is featured in the MoS2/NC-PNR composite. At higher wave numbers, characteristic peaks at 1369 and 1598 cm−1 correspond to disordered carbon (D-band) and ordered graphitic carbon (G-band), respectively, with a peak intensity ratio of approximately 1:1. The pronounced D-band indicates a defect rich nature of the N-doped carbon layers.
C signal at 284.5 eV and traces of a CO one at 287.4 eV are observed, indicating low oxygen content in the carbon layers. Furthermore, the C-N species at 285.8 eV accounts for 14.1% of all carbon, revealing the N-rich nature of the carbon component.37 The N 1s spectrum (Fig. 4a right) is composed of a Mo 2p3/2 peak and three different N species, i.e., graphitic, pyrrolic, and pyridinic, suggesting a successful doping of N into the carbon layer.38 The Raman shift spectrum of MoS2/NC-PNR in Fig. 4b further confirms the chemical composition of the hierarchical superstructure. At lower wave numbers, the peaks present at 383.43 and 404.71 cm−1 correspond to the in-plane E12g and out of plane A1g mode of MoS2, respectively.39 It is worth noting that the A1g peak is more pronounced than the E12g peak, indicating a more edge-terminated and less layered structure.40 Actually, the frequency difference of the two peaks Δk = 21.28 cm−1 is close to the value of single layer MoS2 reported in the literature39 (20.2–21.2 cm−1), suggesting that single layer MoS2 is featured in the MoS2/NC-PNR composite. At higher wave numbers, characteristic peaks at 1369 and 1598 cm−1 correspond to disordered carbon (D-band) and ordered graphitic carbon (G-band), respectively, with a peak intensity ratio of approximately 1:1. The pronounced D-band indicates a defect rich nature of the N-doped carbon layers.
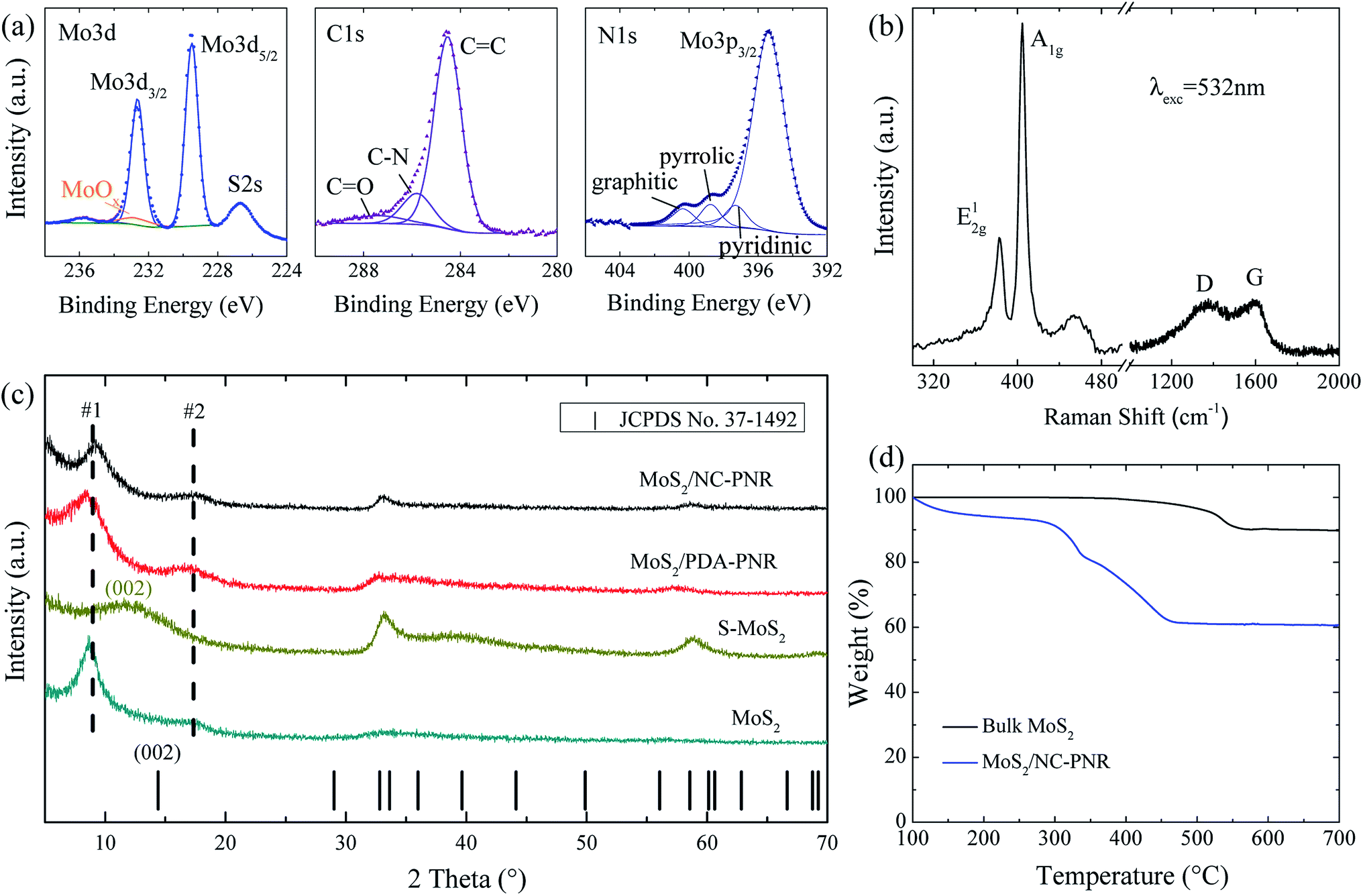 | ||
| Fig. 4 (a) XPS Mo 3d, C 1s and N 1s spectrum of MoS2/NC-PNR; (b) Raman spectrum of the MoS2/NC-PNR excited at 532 nm; (c) XRD patterns of MoS2/NC-PNR, MoS2/PDA-PNR, S-MoS2 and MoS2. Vertical ticks display the standard Bragg peaks of MoS2 (JCPDS no. 37-1492); (d) TGA curve of MoS2/NC-PNR and of bulk MoS2. | ||
The TGA data (Fig. 4d) of both MoS2/NC-PNR and bulk MoS2 illustrate oxidation of MoS2 to MoO3, accompanied by a theoretical mass loss of 10.1 wt%. The bigger total mass loss in case of MoS2/NC-PNR confirms a significant carbon content, which is determined to 25.8 wt%, assuming complete C combustion. In addition, the onset of oxidation at lower temperatures compared to bulk MoS2 further indicates the defect rich nature of the MoS2/NC-PNR superstructure. XRD patterns of both MoS2/PDA-PNR and MoS2/NC-PNR as well as of pure MoS2 samples are displayed in Fig. 4c. Instead of one Bragg peak below 20° in case of standard 2H-MoS2 (JCPDS no. 37-1492), two new broad peaks appear, corresponding to d spacings of 9.7 Å (#1) and 5.1 Å (#2), respectively, for both MoS2/PDA-PNR and MoS2/NC-PNR. Taking the interlayer distances of standard MoS2 (6.2 Å) and graphitic carbon (3.4 Å)23,40 into account, the combined “crystallographic layer thickness” is expected to be 9.6 Å, which agrees with the d spacing of 9.7 Å (#1) determined from XRD. Therefore, the more pronounced peak #1 at 9.1° can be assigned to the (002) reflection of MoS2, whose interlayer distance is enlarged by the interoverlapped N-doped carbon layers, while the peak at 17.4° (#2) might belong to a higher order diffraction along the c-direction. Few recent works1,41 also reported hydrothermally synthesized MoS2 nanostructures with an expanded interlayer distance of 9.5 Å, which was ascribed to either amine insertion between the MoS2 layers or oxygen incorporation under hydrothermal conditions. However, these structures are only stable below 260 °C. At high temperatures, the expanded structure will shrink. In contrast, the MoS2/PDA-PNR is thermodynamically stable as the interlayer expansion is maintained upon sintering at 900 °C (MoS2/NC-PNR), as evidenced by very similar XRD patterns in Fig. 4c. But this is not the case for the pure MoS2 sample, where dopamine was not added to the reaction solution, which hence does not show the characteristic Bragg peak #1 anymore after sintering (S-MoS2, Fig. 4c). In addition, with the large interlayer distance and the N-doped carbon layers in between, the MoS2 layers are expected to exhibit monolayer behavior, which is revealed by the Raman spectra. In summary, a MoS2/NC-PNR superstructure was confirmed by both crystallographic and spectroscopic methods. Additional morphological analysis also supports this conclusion, which will be discussed in detail in the following part. The resulting superstructure offers a novel way of assembling materials in a chosen sequence by precise arrangement of the building blocks within molecular layer scale.42 Owing to the similar hexagonal layered structure of the single components MoS2 and carbon, even weak van der Waals forces are sufficient to stabilize the stacked structure, which maximizes the molecular contact area and consequently the synergetic effects.23
Characterization of the hierarchical structure
From the structural point of view, MoS2/NC-PNR is more advanced than a simple interoverlapped structure because it exhibits an additional hierarchical arrangement. The structural organisation of MoS2/NC-PNR at different length scales is presented in Fig. 5, including a schematic summary in Fig. 5g. By virtue of the α-MoO3 template, MoS2/NC-PNR features a rod-like morphology on the micrometer scale. The SEM/TEM images in Fig. 5a and b show that the rods are still several μm long, but increased in diameter from 200 nm to around 600 nm. The magnified images in Fig. 5c and d reveal that the rods are constructed by homogeneous nanosheets of around 20 nm thick. The MoS2/NC nanosheets crosslink to form a 3D porous structure with a pore size of 100–200 nm, resulting in a high specific surface area of 26.9 ± 0.7 m2 g−1 as compared to 6.97 ± 0.03 m2 g−1 of the pure S-MoS2 nanosheet agglomerates (see Fig. S3†). HR-TEM tilt-series were recorded and reconstructed to further confirm the nanosheets constructed rod-like structure. In a tilt-series movie (Movie S1†), one can clearly see that the lines of the stacking appear and disappear depending on how the sample is oriented. This is highlighted in Fig. 6 which shows selected frames from the movie. The red and white arrows indicate the same parts of the nanorod as it is being tilted. The red arrow points to an area, which shows the lines of the stacking for the negative tilt-angles. While for the sample tilted in such a manner at 35° and 61°, no stacking lines were observed, indicating that the projection goes straight through the layers. The tilt-series shows that the nanorod indeed consists of the proposed nanosheets building blocks (Fig. 5). By tilting the sample, the largest interlayer distance of the red arrow marked area, which corresponds to the actual interlayer distance of the MoS2/NC-PNR, was determined. The corresponding HR-TEM image (Fig. 5e) suggests that the MoS2/NC nanosheets consist of smaller subunits. The lattice fringes exhibit an ordered layer structure with 3–5 layers. Fig. 5f presents the line scan of the white arrow in Fig. 5e, from which the interlayer distance is determined to be 0.96 ± 0.16 nm. This value is in accordance with the d spacing value of the XRD peak #1 (Fig. 4c), confirming the alternately stacked superstructure. It is worth noting that the hierarchical porous nanorod structure cannot be realized without the addition of DOPA during the synthesis. Abundant N-doped carbon introduced by the carbonized self-polymerized DOPA acts as additional backbone for the superstructure, as evidenced by the non-layered parts in the HR-/TEM images in Fig. 5. On the contrary, S-MoS2 synthesized by the same procedure but without adding DOPA does not maintain the nanorod morphology during hydrothermal treatment, but forms blossom-like agglomerates of MoS2 nanosheets (see Fig. S4†).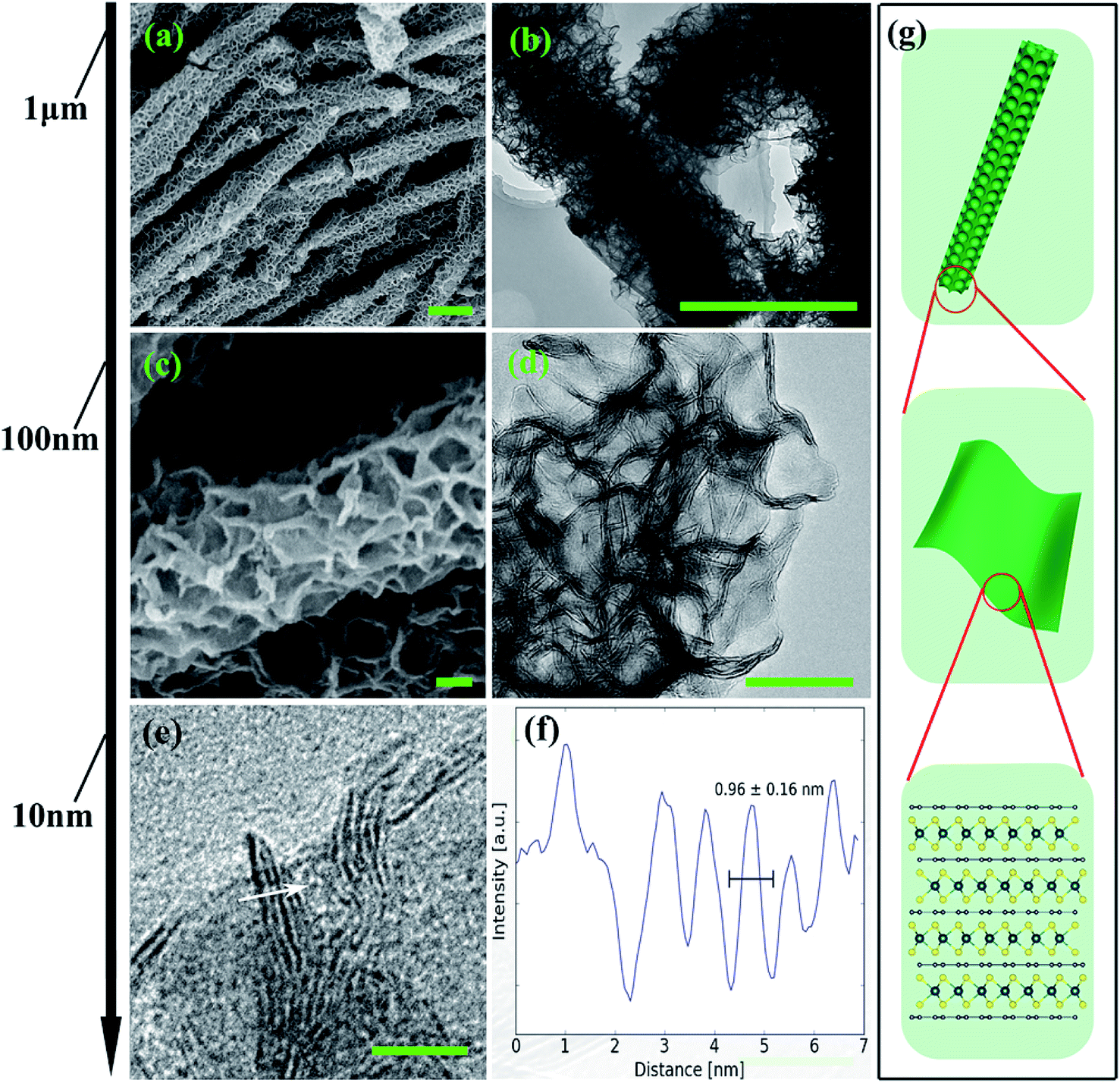 | ||
| Fig. 5 Structural organisation of MoS2/NC-PNR on several length scales: (a, c) SEM images (scale bar 1 μm), (b, d) TEM images (scale bar 100 nm), (e) HR-TEM images (scale bar 10 nm), (f) line scan of the white arrow in (e) and (g) schematic illustration of the hierarchical structure. | ||
 | ||
| Fig. 6 HR-TEM images: selected frames from a movie (Movie S1†) showing a tilt series. The arrows indicate the same part of the nanorod as it is being tilted. While −46° to 15° show the lines of the stacking, indicated by the red arrow, the sample is tilted in such a manner at 35° and 61°, that the projection goes straight through the layers (scale bar: 50 nm). | ||
High-performance lithium storage
The structural advance endows the MoS2/NC-PNR with excellent lithium storage performance, which was studied by cyclic voltammetry (CV) and galvanostatic cycling in the voltage range of 0.01–3.0 V vs. Li/Li+. The CV curves of MoS2/NC-PNR at 0.1 mV s−1 are presented in Fig. 7a. Two pronounced reduction peaks at 1.01 V and 0.65 V during the initial cathodic scan are typical features of MoS2-based electrode materials, corresponding to the Li+ intercalation into MoS2 interlayer space and the subsequent conversion reaction of LixMoS2 to metallic Mo and Li2S, respectively.43,44 Due to the irreversible nature of the conversion reaction, both peaks vanish during the following cycles while a broad reduction peak appears around 1.92 V from the 2nd cycle on, which corresponds to the stepwise reduction of S to S2−.24 It belongs to the reversible redox pair Li2S/S with the related oxidation peak at 2.25 V in the first anodic scan, where Li2S is oxidized to elemental S.45 The Li2S/S redox pair shows very good cycling stability, except a slight decrease in intensity and a moderate shift of the oxidation peak up to 2.31 V in cycle 10. Upon continued cycling, further electrochemical activity is identified by means of several reductive and oxidative features in the voltage ranges of 0.2–1.6 V (cathodic) and 1.1–1.9 V (anodic). They may, i.e., originate from the de-/lithiation of an amorphous Mo/Li2S matrix, including the re-formation of MoSx. The N-doped carbon in the superstructure can also accommodate Li+, which is indicated by a distinct stable reduction feature at 0.01 V.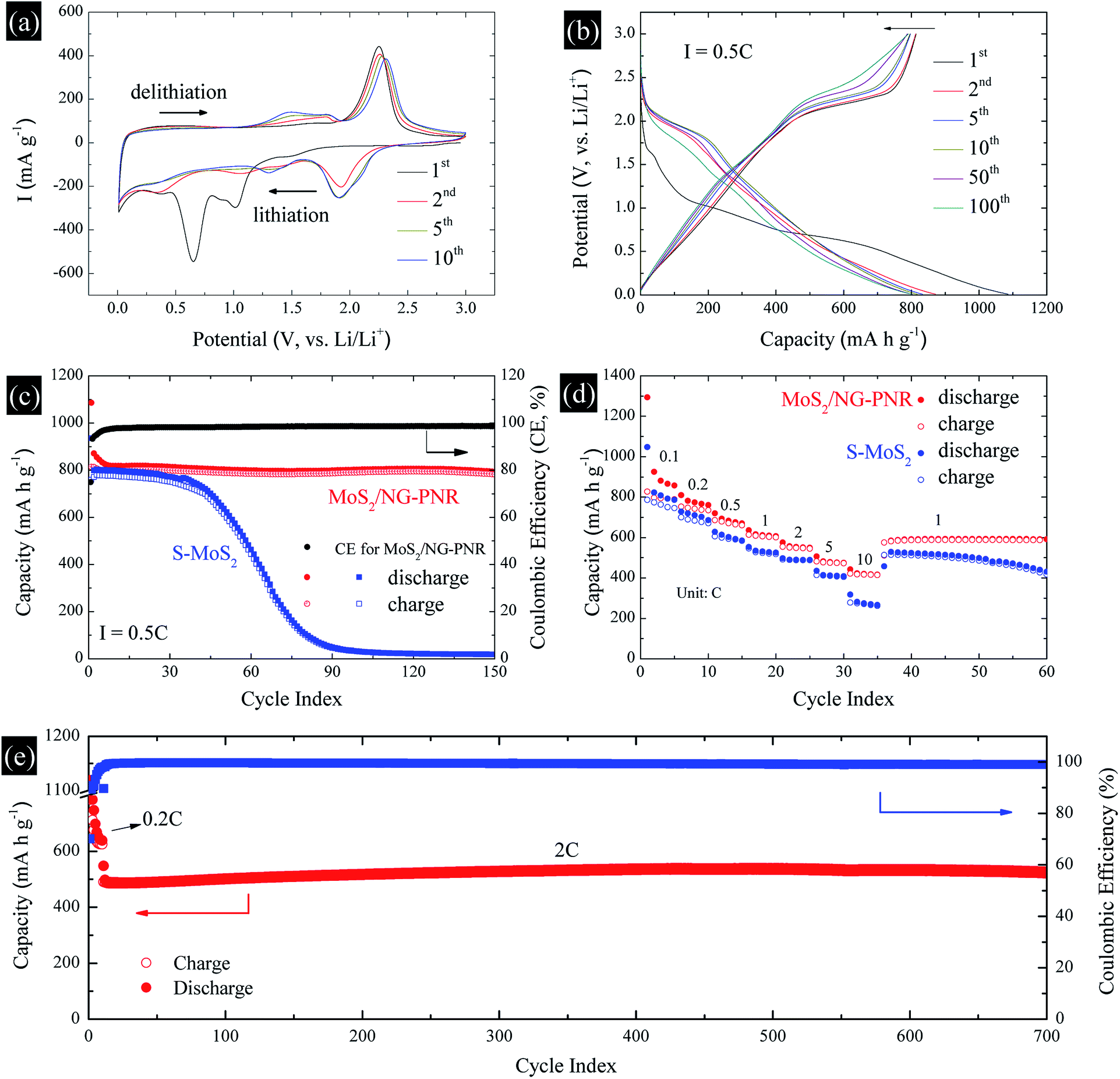 | ||
| Fig. 7 Electrochemical performance of MoS2/NC-PNR: (a) CV, (b) GCPL measurements at specific cycles, (c) cycling stability at 0.5C and the comparison with S-MoS2, (d) rate capability and (e) long-term cycling stability at 2C (activated at 0.2C for 10 cycles). | ||
The charge–discharge profile of a MoS2/NC-PNR-based electrode upon galvanostatic cycling at a current density of 0.5C (1C = 670 mA g−1) is shown in Fig. 7b. In agreement with the CV curves, there are two plateaus at ∼1.0 and ∼0.7 V during the first discharge process referring to the different lithium storage processes, while only one prominent plateau appears at ∼2.2 V during the first charge process. From the second cycle on, the dominant plateaus at 2.0/2.25 V as well as the slopes below 2.0 V, corresponding to the Li2S/S redox pair, show quite reversible behavior, which is superior to usually reported Li–S batteries.46 The observed high reversibility may be promoted by the N-doped carbon layers and by the appearance of conductive Mo nanoparticles.12 The stacked N-doped carbon layers could offer physical confinement to the active species and provide more electroactive sites for the strong chemisorption of lithium polysulfides. The Mo nanoparticles derived from the MoS2 reduction boost the electrochemical reaction, further hindering the polysulfide shuttle effect. As a result, the corresponding cycling performance in Fig. 7c presents a stable discharge capacity of ∼800 mA h g−1 for up to 150 cycles after initial decay from 1086 mA h g−1. The initial capacity loss is mainly attributed to the partially irreversible conversion reaction and the formation of solid-electrolyte interphase (SEI).47 The durable cycling capability of the hierarchical superstructure is highlighted by the comparison with a S-MoS2 electrode, whose dis-/charge capacities drop noticeably after 40 cycles before stabilizing at around underwhelming 30 mA h g−1. The poor cycling stability of S-MoS2 can be attributed to structural changes of the active MoS2 material as well as the shuttle effect of in situ formed (poly)sulfide from the 2nd cycle on.48 However, the MoS2/NC-PNR superstructure design specifically aims at maintaining the high specific capacities by maximizing the interfacial contact area between active material and interoverlapped carbon, which may buffer the mechanical strain to the largest extend possible.23 In the SEM images of electrodes after 150 cycles (Fig. S5†), the rod-like morphology of MoS2/NC-PNR is partially preserved, while in case of the S-MoS2 material only irregular nanoparticles remained. Further post mortem characterizations of the MoS2/NC-PNR electrode demonstrate the buffer function of the N-doped carbon layers to the amorphous products. In Fig. S6a,† the XRD patterns of the MoS2/NC-PNR electrode after cycling suggest completely non-crystallized feature of the MoS2 structure. The HAADF-STEM image of the cycled electrode and the corresponding SAED patterns in Fig. S7a–c† also present a disordered structure of the Mo species. In the SAED patterns, the only weak diffraction rings can be indexed as disordered graphitic carbon. The EDX together with the STEM images (Fig. S7d–f†) indicate a homogeneous Mo distribution in the carbon matrix. All the results highlight the significant role of carbon in maintaining the structural integrity of the electrode. Therefore, the MoS2/NC-PNR electrode exhibits an ultra-long cycling lifetime of 700 cycles at a current of 2C, delivering a stable capacity of ∼520 mA h g−1 as shown in Fig. 7e. In addition, the N-doped carbon layers compensate the intrinsic low conductivity of MoS2 in MoS2/NC-PNR, allowing faster electron transfer during the dis-/charge process.49 As a consequence, the MoS2/NC-PNR electrode shows much improved rate capability as compared to the S-MoS2 electrode (Fig. 7d). The former exhibits an initial discharge capacity of 1294 mA h g−1 at 0.1C, stabilizing at 810, 720, 636, 575, and 507 mA h g−1 upon increasing the current density to 0.2, 0.5, 1, 2, and 5C, respectively. All of the values are higher than that of the latter. Especially at high current density of 10C, the MoS2/NC-PNR electrode presents a large reversible capacity of 443 mA h g−1, which is twice the value of the S-MoS2 electrode.
In order to clarify the enhanced electrochemical kinetics of the MoS2/NC-PNR electrode, further CV measurements and electrochemical impedance spectroscopy (EIS) have been carried out. The CV curves at different scan rates (see Fig. 8a,) show similar shape, which indicates excellent adaptability even at large scan rates. The peak current (I) and the scan rate (v) obey a power law I = aνb, from which both the capacitive and the diffusion controlled contributions of the charge storage process can be evaluated.50 Fitting the data in Fig. 8b yields that the parameter b equals to 1.02, 0.86, 0.77 and 0.88 for the cathodic and anodic peaks (C1, C2, A1, A2), respectively. The values suggest that capacitive processes (mainly the redox pseudocapacitive contribution) are dominant in the associated electrochemical reactions. The exact (pseudo)capacitive and diffusion controlled contributions can be assessed by using the equation I = k1ν + k2ν1/2 (see Fig. S8†).51 In Fig. 8c, the (pseudo)capacitive contribution is proportional to the scan rate, reaching 85% at 2 mV s−1. The high (pseudo)capacitive contribution suggests fast redox reactions in the MoS2/NC-PNR electrode, which is straightforwardly attributed to the effective contact between MoS2 and N-doped carbon as well as the porous nanorods architecture. This conclusion is further supported by EIS. In the Nyquist plots obtained at open circuit voltage (OCV) in Fig. 8d, both MoS2/NC-PNR- and S-MoS2-based electrodes show a depressed semi-circle in the high to medium frequency range as well as a sloping behavior in the low frequency range. The diameter of the semicircle, which correlates with the charge transfer resistance (R2 in the corresponding equivalent circuit as shown in the inset of Fig. 8d), is only half the value in MoS2/NC-PNR (183 ± 2 Ω) compared to S-MoS2 (342 ± 1 Ω). Upon cycling, R2 decreases abruptly in the MoS2/NC-PNR electrode (Fig. S9†) which can be attributed to irreversible structural changes during the initial lithiation. Interestingly, the MoS2/NC-PNR electrode exhibits few changes in subsequent cycles, which also holds for the low frequency domain, revealing a stable and reversible de-/lithiation process. These results imply not only improved conductivity of the pristine MoS2/NC-PNR compound, but also more efficient ion transport pathways through the N-doped carbon layers of the superstructure.
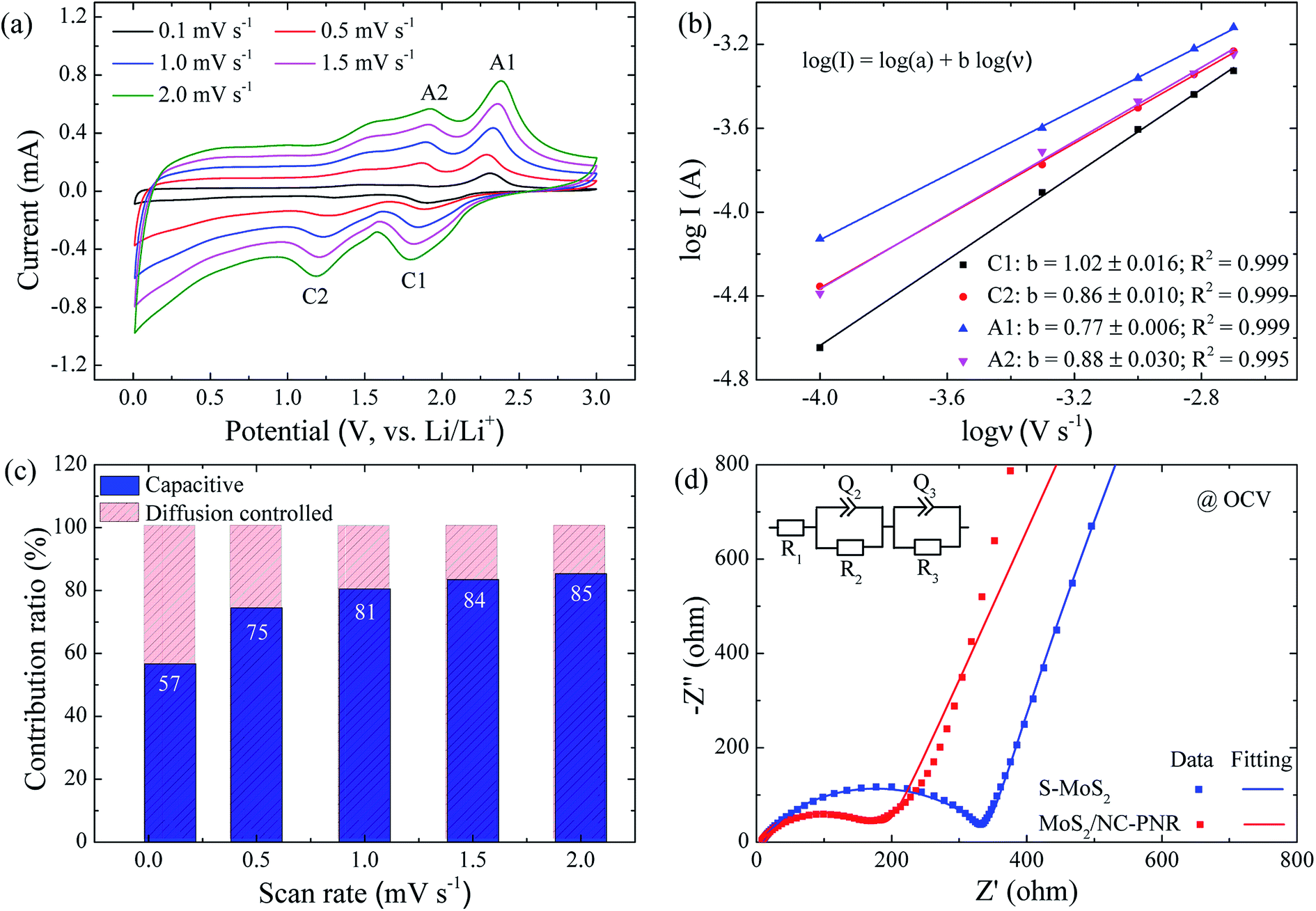 | ||
| Fig. 8 Kinetic analysis of MoS2/NC-PNR as LIB anode: (a) CV curves at different scan rate; (b) current response versus scan rate (logI vs. logv) plots at each redox peak; (c) capacitive and diffusion controlled contributions to the electrochemical processes at corresponding scan rate; the number labels the percentile capacitive contributions; (d) Nyquist plots and the fitting curves of MoS2/NC-PNR and S-MoS2 electrodes. The inset shows the equivalent circuit. | ||
The abovementioned results clearly imply that the hierarchical MoS2/C composite with well-designed molecular contact interface ensures an enhanced electrochemical performance. This conclusion is further confirmed by comparing the present results with our previous work.25 In ref. 27, we have reported C@MoS2@C sandwiched hollow spheres with a surface-to-surface contact between MoS2 and carbon layers. Interestingly, the carbon content in C@MoS2@C sandwiched hollow spheres and the MoS2/NC-PNR superstructures is almost identical (∼26%). With the same chemical composition, the major differences between the two materials are the interfacial contact and morphological architecture. Due to its much more effective interfacial contact (layer-by-layer contact), the MoS2/NC-PNR electrode exhibits well enhanced electrochemical performance as compared to the C@MoS2@C electrode including higher capacities of ∼800 mA h g−1vs. ∼640 mA h g−1 at 0.5C; longer cycling stability up to 700 cycles at 2C vs. 200 cycles at 1C; and a better rate capability of 507 mA h g−1vs. 382 mA h g−1 at 5C. Actually, these values are comparable or even better than most of recently published MoS2-based hierarchical structures as seen in Table S1† while our approach employs an advantageous environmentally friendly synthetic route and exploits a low cost, easy access carbon source. In general, the reported performances show that the rational structure design of nanocomposites is indispensable to their application potential and the present work provides a route to realize such a hierarchical structure down to the molecular layer level.
Conclusions
In summary, we report on a hierarchically structured nanocomposite with optimized atomic interfacial contact for application in lithium ion batteries. The MoS2/NC-PNR superstructure has been prepared by means of a one-pot synthesis approach. The alternating stacking of the MoS2 layers with intercalated N-doped carbon layers provides a maximum specific contact area between the active material and the buffer layers, effectively alleviating the loss of active material due to pulverization of MoS2 or dissolution of polysulfides. In addition, the buffer layers also act as a conductive matrix, enabling faster Li-ion and electron transfer during the de-/lithiation processes. At a larger length scale, the rod-like porous structure offers large specific surface area and abundant active sites for electrochemical reactions which also contributes to the dominant capacitive charge storage behaviour. As a result, the interfacially engineered MoS2/NC-PNR superstructure exhibits a high specific capacity of 1294 mA h g−1 at 0.1C, stable cycling performance up to 700 cycles at 2C, and a high rate capability of 443 mA h g−1 at 10C. The ordered MoS2/NC-PNR superstructure with well-defined interfaces is capable of buffering huge mechanical strain and offering efficient electron and ion transport pathways, which indicates a new way of rationally designing other hierarchical functional materials.Experimental methods
Synthesis of α-MoO3 nanorods
α-MoO3 nanorods were prepared through a hydrothermal method according to the literature.52 1000 mg ammonium molybdate tetrahydrate (AMT, Sigma-Aldrich, 81.0–83.0% MoO3 basis) was dissolved in 34 mL deionized water. 6 mL nitric acid (65 wt%) was added dropwise before stirring the AMT solution for 15 min. After stirring for another 15 min, the mixture was transferred to a 50 mL Teflon autoclave, heating at 180 °C for 24 h. The milky white precipitate was washed and collected for further use.Synthesis of MoS2/NC-PNR superstructure and S-MoS2
MoS2/NC-PNR was produced by adding α-MoO3 nanorods into a dopamine/Tris solution and then stirring for 24 h. Then the precipitate was centrifuged and added into a thiourea solution under vigorous stirring. The homogeneously dispersed suspension was hydrothermally treated at 200 °C for 24 h. After the reaction, the black precipitate was washed with distilled water and ethanol for at least 3 times before vacuum drying at 80 °C. Finally, the black powder was sintered at 900 °C for 3 h under Ar atmosphere. For comparison, pure MoS2 nanosheets were synthesized by using the same α-MoO3 nanorods and same sulfur source at the same hydrothermal conditions and subsequent thermal treatment. The pure MoS2 samples before and after sintering are labeled as MoS2 and S-MoS2, respectively.Characterization
The morphology and microstructure of the sample was characterized by scanning electron microscopy (SEM, ZEISS Leo 1530) and transmission electron microscopy (TEM, JEM 2100F). TEM tilt-series images were acquired using a ZEISS Libra 200 MC Cs-Corr DMU Kronos with 200 kV acceleration voltage at highest magnification (pixel size: 0.578 Å) from −68° to + 64° in 1° steps. 5 images were acquired at each step with exposure times of 1000 ms, which were later aligned and summed to increase resolution. The tilt-series was then aligned using the software provided by the IMOD package.53–55 For TEM measurement, the sample (powder) was dispersed in ethanol:water (2:1) mixture at 3 mg mL−1. The dispersion was ultrasonicated for 30 minutes and then centrifuged at 14800 rpm for 60 seconds. 1 μL of the material that had collected at the bottom of the tube was then mixed with 30 μL of water. 2.5 μL of this mixture was dropped onto a copper TEM grid with carbon-coated Formvar film. Before the sample could dry, 2.5 μL of a mixture of water and gold fiducials (6 nm in size), used for image alignment, was dropped onto both sides of the TEM grid. After waiting for one minute, excess liquid was carefully removed with filter paper.
X-Ray powder diffraction (XRD) was performed in Bragg–Brentano geometry (Bruker-AXS D8 ADVANCE ECO) applying Cu-Kα1 radiation (λ = 1.54056 Å). The step size Δ2θ was 0.02° with a scan speed of 0.5° min−1. X-ray photoemission spectroscopy (XPS) was carried out in an ESCALAB 250Xi ultra-high vacuum system with an Al Kα radiation source (hν = 1486.6 eV), 900 μm spot size and 20 eV pass energy. Nitrogen physisorption measurements were performed at 77 K with a Gemini V (Micro-meritics, Norcross, GA) after degassing the sample at 120 °C for 2 h. Brunauer–Emmett–Teller (BET) analysis from the amount of N2 absorbed at various relative vapor pressures (six points 0.05 < P/P0 < 0.3, nitrogen molecular cross-sectional area = 0.162 nm2) was used to determine the surface area. Barrett–Joyner–Halenda (BJH) analysis was done on the adsorption isotherms in order to obtain the pore-size distribution. Raman spectroscopy was performed on bulk powder samples using a Renishaw InVia microscope with 532 nm excitation lasers in air under ambient conditions. The laser power was kept below 100 μW to avoid decomposition. Measurements on minimum five different spots were performed and showed the samples to be highly homogeneous under these measurement conditions. The spectra were baseline corrected and averaged after acquisition. UV-vis absorption spectra were measured with a Jascon UV-660 spectrophotometer in the range from 250 to 600 nm. The absorption of the substrate was subtracted as baseline correction. Thermogravimetric analysis was performed by using a TGA/DSC1 STARe System (Mettler Toledo) at a heating rate of 10 °C min−1 in air.
Electrochemical measurement
Electrochemical studies were carried out using Swagelok-type cells.56 For both the pure MoS2 and the MoS2/NC-PNR electrodes, the active material, carbon black (SuperP, Timcal) and polyvinylidene fluoride (PVDF, Sigma-Aldrich, 99%) binder were mixed in anhydrous 1-methyl-2-pyrrolidinone (NMP, Sigma-Aldrich, 99%) with a weight ratio of 7:2:1. The slurry was pasted on a circular Cu plate (∼10 mm in diameter) with a mass loading of 1 mg cm−2, dried overnight under vacuum at 80 °C and pressed. The two-electrode Swagelok-type cells were assembled in an Ar atmosphere glove box using lithium foil as counter electrode and 1 M LiPF6 in a 1:1 mixture of ethylene carbonate and dimethyl carbonate as liquid electrolyte (Merck LP30). Cyclic voltammetry and galvanostatic cycling of the cells were performed at 25 °C between 0.01 and 3.0 V versus Li/Li+ at various scan/current rates using a VMP3 multichannel potentiostat (Bio-Logic SAS). Electrochemical impedance spectroscopy (EIS) was carried out also by using the VMP3 multichannel potentiostat in the frequency range of 100 kHz to 0.1 Hz. The EIS data were analyzed with the help of the Z Fit function of the EC-Lab software (Bio-Logic).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank I. Glass for experimental support. Financial support by the CleanTech-Initiative of the Baden-Württemberg-Stiftung (Project CT3 Nanostorage) and by the IMPRS-QD is gratefully acknowledged. Z. L. acknowledges financial support by the Chinese Scholarship Council, the Excellence Initiative of the German Federal Government, and by the Götze foundation.References
- Y. J. Tang, Y. Wang, X. L. Wang, S. L. Li, W. Huang, L. Z. Dong, C. H. Liu, Y. F. Li and Y. Q. Lan, Adv. Energy Mater., 2016, 6, 1600116 CrossRef.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- M.-L. Tsai, S.-H. Su, J.-K. Chang, D.-S. Tsai, C.-H. Chen, C.-I. Wu, L.-J. Li, L.-J. Chen and J.-H. He, ACS Nano, 2014, 8, 8317–8322 CrossRef CAS PubMed.
- X. Gu, W. Cui, H. Li, Z. Wu, Z. Zeng, S.-T. Lee, H. Zhang and B. Sun, Adv. Energy Mater., 2013, 3, 1262–1268 CrossRef CAS.
- P. T. Dirlam, J. Park, A. G. Simmonds, K. Domanik, C. B. Arrington, J. L. Schaefer, V. P. Oleshko, T. S. Kleine, K. Char, R. S. Glass, C. L. Soles, C. Kim, N. Pinna, Y.-E. Sung and J. Pyun, ACS Appl. Mater. Interfaces, 2016, 8, 13437–13448 CrossRef CAS PubMed.
- S. H. Choi, Y. N. Ko, J.-K. Lee and Y. C. Kang, Adv. Funct. Mater., 2015, 25, 1780–1788 CrossRef CAS.
- C. Zhu, X. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., Int. Ed., 2014, 53, 2152–2156 CrossRef CAS PubMed.
- X. Chen, Small Methods, 2017, 1, 1600029 CrossRef.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- Z. Hu, Q. N. Liu, W. Y. Sun, W. J. Li, Z. L. Tao, S. L. Chou, J. Chen and S. X. Dou, Inorg. Chem. Front., 2016, 3, 532–535 RSC.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- X. Fang, X. Guo, Y. Mao, C. Hua, L. Shen, Y. Hu, Z. Wang, F. Wu and L. Chen, Chem.–Asian J., 2012, 7, 1013–1017 CrossRef CAS PubMed.
- Y. M. Chen, X. Y. Yu, Z. Li, U. Paik and X. W. Lou, Sci. Adv., 2016, 2, e1600021 CrossRef PubMed.
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. Lou, Energy Environ. Sci., 2014, 7, 3302–3306 RSC.
- F. Gong, L. Peng, H. Liu, Y. Zhang, D. Jia, S. Fang, F. Li and D. Li, J. Mater. Chem. A, 2018, 6, 18498–18507 RSC.
- W. Wang, P. Yang, Z. Jian, H. Li, Y. Xing and S. Zhang, J. Mater. Chem. A, 2018, 6, 13797–13805 RSC.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H.-W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- P.-p. Wang, H. Sun, Y. Ji, W. Li and X. Wang, Adv. Mater., 2014, 26, 964–969 CrossRef CAS PubMed.
- C. Wu, J. Maier and Y. Yu, Adv. Mater., 2016, 28, 174–180 CrossRef CAS PubMed.
- X. Zhang, X. Li, J. Liang, Y. Zhu and Y. Qian, Small, 2016, 12, 2484–2491 CrossRef CAS PubMed.
- Y. Wang, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2016, 55, 7423–7426 CrossRef CAS PubMed.
- Z. Zhang, H. Zhao, Y. Teng, X. Chang, Q. Xia, Z. Li, J. Fang, Z. Du and K. Świerczek, Adv. Energy Mater., 2018, 8, 1700174 CrossRef.
- H. Jiang, D. Ren, H. Wang, Y. Hu, S. Guo, H. Yuan, P. Hu, L. Zhang and C. Li, Adv. Mater., 2015, 27, 3582 CrossRef.
- Z. Li, A. Ottmann, E. Thauer, C. Neef, H. Sai, Q. Sun, K. Cendrowski, H.-P. Meyer, Y. Vaynzof, E. Mijowska, J. Xiang and R. Klingeler, RSC Adv., 2016, 6, 76084–76092 RSC.
- Z. Li, A. Ottmann, T. Zhang, Q. Sun, H.-P. Meyer, Y. Vaynzof, J. Xiang and R. Klingeler, J. Mater. Chem. A, 2017, 5, 3987–3994 RSC.
- G. Lian, C.-C. Tuan, L. Li, S. Jiao, K.-S. Moon, Q. Wang, D. Cui and C.-P. Wong, Nano Lett., 2017, 17, 1365–1370 CrossRef CAS PubMed.
- L. Li, B. Song, L. Maurer, Z. Lin, G. Lian, C.-C. Tuan, K.-S. Moon and C.-P. Wong, Nano Energy, 2016, 21, 276–294 CrossRef CAS.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- A. Manivel, G.-J. Lee, C.-Y. Chen, J.-H. Chen, S.-H. Ma, T.-L. Horng and J. J. Wu, Mater. Res. Bull., 2015, 62, 184–191 CrossRef CAS.
- H. Yang, X. Li, A. Wang, Y. Wang and Y. Chen, Chin. J. Catal., 2014, 35, 140–147 CrossRef CAS.
- E. Karabulut, T. Pettersson, M. Ankerfors and L. Wågberg, ACS Nano, 2012, 6, 4731–4739 CrossRef CAS PubMed.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- C. Zhao, J. Kong, L. Yang, X. Yao, S. L. Phua and X. Lu, Chem. Commun., 2014, 50, 9672–9675 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- K.-K. Liu, W. Zhang, Y.-H. Lee, Y.-C. Lin, M.-T. Chang, C.-Y. Su, C.-S. Chang, H. Li, Y. Shi, H. Zhang, C.-S. Lai and L.-J. Li, Nano Lett., 2012, 12, 1538–1544 CrossRef CAS PubMed.
- X.-D. Zhu, K.-X. Wang, D.-J. Yan, S.-R. Le, R.-J. Ma, K.-N. Sun and Y.-T. Liu, Chem. Commun., 2015, 51, 11888–11891 RSC.
- Y. Zhang, K. Fugane, T. Mori, L. Niu and J. Ye, J. Mater. Chem., 2012, 22, 6575–6580 RSC.
- K. Chang, D. Geng, X. Li, J. Yang, Y. Tang, M. Cai, R. Li and X. Sun, Adv. Energy Mater., 2013, 3, 839–844 CrossRef CAS.
- Y. Yu, C. Li, Y. Liu, L. Su, Y. Zhang and L. Cao, Sci. Rep., 2013, 3, 1866 CrossRef PubMed.
- M.-R. Gao, M. K. Y. Chan and Y. Sun, Nat. Commun., 2015, 6, 7493 CrossRef PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- F. Xiong, Z. Cai, L. Qu, P. Zhang, Z. Yuan, O. K. Asare, W. Xu, C. Lin and L. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 12625–12630 CrossRef CAS PubMed.
- L. Wang, Z. Xu, W. Wang and X. Bai, J. Am. Chem. Soc., 2014, 136, 6693–6697 CrossRef CAS PubMed.
- Z. Wan, J. Shao, J. Yun, H. Zheng, T. Gao, M. Shen, Q. Qu and H. Zheng, Small, 2014, 10, 4975–4981 CrossRef CAS PubMed.
- N. He, L. Zhong, M. Xiao, S. Wang, D. Han and Y. Meng, Sci. Rep., 2016, 6, 33871 CrossRef CAS PubMed.
- Z. Zhu, Y. Tang, Z. Lv, J. Wei, Y. Zhang, R. Wang, W. Zhang, H. Xia, M. Ge and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 3656–3660 CrossRef CAS PubMed.
- J.-Y. Hwang, H. M. Kim, S.-K. Lee, J.-H. Lee, A. Abouimrane, M. A. Khaleel, I. Belharouak, A. Manthiram and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501480 CrossRef.
- X.-Y. Yu, H. Hu, Y. Wang, H. Chen and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 7395–7398 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518 CrossRef CAS PubMed.
- X. Wang, D. Chen, Z. Yang, X. Zhang, C. Wang, J. Chen, X. Zhang and M. Xue, Adv. Mater., 2016, 28, 8645–8650 CrossRef CAS PubMed.
- X. W. Lou and H. C. Zeng, Chem. Mater., 2002, 14, 4781–4789 CrossRef CAS.
- J. R. Kremer, D. N. Mastronarde and J. R. McIntosh, J. Struct. Biol., 1996, 116, 71–76 CrossRef CAS PubMed.
- D. N. Mastronarde, J. Struct. Biol., 1997, 120, 343–352 CrossRef CAS.
- D. N. Mastronarde and S. R. Held, J. Struct. Biol., 2017, 197, 102–113 CrossRef PubMed.
- C. Neef, C. Jahne, H. P. Meyer and R. Klingeler, Langmuir, 2013, 29, 8054–8060 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta12293h |
| This journal is © The Royal Society of Chemistry 2019 |