
Biotin-conjugated PEGylated porphyrin self-assembled nanoparticles co-targeting mitochondria and lysosomes for advanced chemo-photodynamic combination therapy†
Baskaran
Purushothaman
,
Jinhyeok
Choi
,
Solji
Park
,
Jeongmin
Lee
,
Annie Agnes Suganya
Samson
,
Sera
Hong
and
Joon Myong
Song
 *
*
College of Pharmacy, Seoul National University, Seoul 08826, South Korea. E-mail: jmsong@snu.ac.kr; Fax: +82 2 871 2238; Tel: +82 2 880 7841
First published on 19th November 2018
Abstract
The combination of chemotherapy and photodynamic therapy (chemo–PDT) has been suggested as an alternative therapy for drug-resistant cancers. In this study, biotin-conjugated PEGylated photosensitizer (PS) self-assembled nanoparticles (meso-tetraphenylporphyrin (TPP)–PEG–biotin SANs) were prepared via a self-assembly process to serve as nanocarriers for chemo-drugs as well as PSs. Electron microscopy results reveal the spherical shape of the nanoparticles (NPs). In the NPs, conjugated biotin plays a key role in selective tumor targeting. In vitro cellular experiments revealed the rapid cellular uptake of the TPP–PEG–biotin conjugates by MCF-7 cells that overexpress the biotin receptor, and verified that the conjugates were much more effective PSs than TPPS used as control in the cytotoxicity test. Interestingly, subcellular localization studies showed that the conjugates and their self-assembled NPs were localized mainly in mitochondria and partially in lysosomes, whereas TPPS was localized only in lysosomes. With the exclusive localization in mitochondria, high-content cell based assay showed that the TPP–PEG–biotin SANs induced rapid mitochondrial membrane potential transition (MPT), leading to cellular apoptosis. The chemo-drug doxorubicin (DOX) was successfully encapsulated in the TPP–PEG–biotin SANs (DOX@TPP–PEG–biotin) and had synergistic effects with enhanced cytotoxicity after PDT action. Collectively, the DOX@TPP–PEG–biotin SANs have promising potential as an effective anticancer agent in targeted combination therapy.
1. Introduction
Breast cancer is the second leading cause of death in women in many of the developing countries. In 2018, the American Cancer Society estimated that about 266![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120 new cases of breast cancer will be diagnosed and about 40920 women will die from breast cancer in the United States. Chemotherapy, as one of the anticancer therapies, plays an important role in improving survival among breast cancer patients. However, multidrug resistance (MDR), whether inherent or acquired, greatly threatens clinical outcomes and patients’ lives.1 About 40% of all breast cancer patients suffer local recurrence or distant metastasis after chemotherapy treatment; they are closely correlated with poor chemosensitivity. Also, chemo-drugs are not able to distinguish normal cells from cancer cells, resulting in serious side effects due to the lack of selectivity. An advanced therapy with enhanced therapeutic efficiency and safety is necessary to overcome MDR and minimize severe side effects.2
120 new cases of breast cancer will be diagnosed and about 40920 women will die from breast cancer in the United States. Chemotherapy, as one of the anticancer therapies, plays an important role in improving survival among breast cancer patients. However, multidrug resistance (MDR), whether inherent or acquired, greatly threatens clinical outcomes and patients’ lives.1 About 40% of all breast cancer patients suffer local recurrence or distant metastasis after chemotherapy treatment; they are closely correlated with poor chemosensitivity. Also, chemo-drugs are not able to distinguish normal cells from cancer cells, resulting in serious side effects due to the lack of selectivity. An advanced therapy with enhanced therapeutic efficiency and safety is necessary to overcome MDR and minimize severe side effects.2
In recent years, combination therapy (chemo-photodynamic) has been suggested as an alternative strategy to reverse MDR and minimize side effects through different mechanisms of action, enhancing anticancer efficacy.3–6 Photodynamic therapy (PDT) is a widely used therapy in clinics to treat various cancers and is approved by the United States Federal Drug Administration (USFDA).7 PDT is light-activated cancer therapy, and it involves a PS, a light source, and intracellular molecular oxygen. In the process of PDT, light irradiation of the PS at a specific wavelength in the presence of molecular oxygen produces highly cytotoxic reactive oxygen species (ROS), especially reactive singlet oxygen (1O2). These species live only for a short time (<0.1 ms) in cells and interact with lipid membranes and DNA, causing cellular apoptosis and/or necrosis in targeted sites. Thus, due to these light-activated properties, the PS can selectively attack cancer cells by ROS generation, thereby not damaging normal cells and minimizing side effects in PDT.8,9
However, porphyrin derivatives, which are representative PDT agents, have some limitations in the development of anticancer agents, including low bioavailability, poor water solubility, and low selectivity. To overcome these obstacles, various nanoparticle-based drug delivery systems (NDDSs) have been developed in recent years.10,11 Self-assembly of NPs is one of the technological advancements in the DDS of anticancer agents.12–14 After synthesizing chemicals bearing good water solubility by conjugating a hydrophilic molecule to a PS, self-assembled NPs can be formed by the chemicals following several processes. Furthermore, the processes can include encapsulation of chemo-drugs such as doxorubicin (DOX) and paclitaxel (PTX), which can be used for chemo-photodynamic combination therapy.3 Despite its potential in PDT–chemo therapy, it may not seem to be efficient for self-assembled NPs to accumulate in the tumor region for the target-specific delivery of chemo-drugs.
To solve this issue, conjugation of the targeting agent has been utilized for targeted NDDSs. It is widely reported that many cancer cells overexpress tumor-specific receptors, showing the potential for the development of targeted NDDSs. Some ligands specifically bind to the surfaces of these cancer cells via corresponding receptors and mediate endocytosis of NPs in the cancer cells. The folate receptor has been studied by different groups, and remarkable advancements have been made to date.15,16 However, the biotin receptor has not been well explored as a cancer biomarker for the targeting agent in combination therapy. The growth promoter biotin (vitamin H or B-7) content in cancer cells is considerably higher than that in normal cells.17 Recently, it has been reported that the biotin receptor is overexpressed more than the folate receptor in many cancer cells.18 Therefore, we choose biotin as the cancer targeting agent for the combination therapy.17,19
The self-assembled NPs of PS conjugates are expected to present anticancer efficacy minimizing side effects by target-specific delivery of the chemodrug, rendering the synergistic anticancer effect with PDT action. In addition to this, the PS does not only directly kill cancer cells by generating intracellular ROS but also indirectly attacks the cancer cells, disrupting tumor microvasculature, which induces the depletion of oxygen levels and nutrients in the tumor region. For this reason, blood vessels in the tumor microenvironment become more permeable as a result of the vascular damage caused by PDT, and subsequently, more chemo-drugs encapsulated through self-assembly can be delivered to the tumor site passing readily through the vessels. Therefore, chemo-photodynamic therapy with the self-assembled NPs based on PSs has been suggested as a remarkable cancer treatment.
In this study, we chose the chlorophyll derivative TPP as the PDT agent to develop novel self-assembled NPs for combination therapy. Because of the solubility issues of TPP, we subsequently chose polyethylene glycol (PEG) polymer, which is a hydrophilic component for the conjugation of TPP. The reason for this selection is not only its hydrophilicity, leading to PEGylated TPP having a good aqueous solubility, but also its known antifouling properties. Thus, it provides steric bulk that minimizes the interactions of TPP with proteins or other biomolecules in the blood serum in order to prevent rapid clearance of the PSs, which are commonly found in drug molecules without PEG.20 However, polymer–drug (PEG–drug) conjugation is a common methodology in drug administration for enhancing efficacy;21,22 the formation of nanostructures from PEG–drug conjugates without the aid of nanocarriers or other components of nano-assembly has rarely been reported. Encouraged by the recent progress in the self-assembly of NPs for combination therapy,23 we report here the self-assembly of PEG-conjugated TPP with biotin (TPP–PEG–biotin). The biotin conjugated as the targeting agent can bind to the breast cancer cell line MCF-7, which has been shown to overexpress the biotin receptor. DOX is used as the chemo-drug model for the combination therapy. In this work, the coupling reaction with TPP-amine through an acid–polymer linker (HOOC–PEG–biotin) allowed the conjugation of TPP–PEG–biotin, and the self-assembly of TPP–PEG–biotin with DOX encapsulation was achieved. Consequently, the prepared DOX@TPP–PEG–biotin self-assembled NPs were expected to exhibit great potential in the treatment of cancer, which overexpresses the biotin receptor, with synergistic effects in combination therapy.
2. Results
2.1. Synthesis and characterization of TPP–PEG–biotin self-assembled nanoparticles
The synthetic scheme for the preparation of TPP–PEG–biotin SANs is illustrated in Scheme 1. First, the TPP–PEG–biotin chemical was synthesized from the starting material TPP-NH2. Briefly, the TPP was synthesized according to a previously reported procedure.24 Then, the TPP was nitrated using sodium nitrite in the presence of dichloromethane solvent for 3 minutes to yield mononitro-TPP (TPP-NO2).25 The reduction reaction of TPP-NO2 resulted in amine-TPP (TPP-NH2). Then 1 mmol of biotin–PEG–COOH was activated by 1 mmol of EDCI and DMAP for 3 h and then 1 mmol of TPP-NH2 in DCM was slowly added to the reaction vessel. Then the reaction mixture was mechanically stirred for 24 h at room temperature, and the crude material was purified by column chromatography. The pure TPP–PEG–biotin was dried completely and then characterized by UV-vis, 1H NMR, and MALDI-TOF mass spectroscopy. After confirmation of the TPP–PEG–biotin chemical, the TPP–PEG–biotin species were self-assembled using a sonication method. First, the TPP–PEG–biotin chemical was dissolved in chloroform with a vortex mixer to get a clear solution. Then, the organic solvent was slowly evaporated with a stream of nitrogen gas and then dried completely under vacuum to get a film layer. The film layer was hydrated with 90% of phosphate buffered saline (PBS, pH 7.4) and 10% of DMSO solution. The solution was sonicated for 15 minutes using a Cole-Parmer ultrasonicator. Uniform sized self-assembled TPP–PEG–biotin nanoparticles were obtained by centrifugation using an Amicon Ultra centrifugal tube. The self-assembled TPP–PEG–biotin nanoparticles were characterized by UV-vis absorption spectroscopy, scanning electron microscopy (SEM) (Fig. S12, ESI†), transmission electron microscopy (TEM), and 1H NMR spectroscopy. The UV-vis absorption and emission spectra of TPP–PEG–biotin, TPP–PEG–biotin SANs and DOX loaded TPP–PEG–biotin are shown in Fig. 1a and b. The UV-vis spectra of the TPP–PEG–biotin SANs and DOX loaded TPP–PEG–biotin clearly showed that there is little red shift in the Soret band (421 nm → 450 nm) and Q-bands (522 nm → 526 nm) in the absorption spectra compared to the TPP–PEG–biotin and its SANs, confirming that the DOX was loaded into the TPP–PEG–biotin SANs. The results in Fig. 1c–f showed the size and shape of the TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs. The TEM and dynamic light scattering (DLS) results confirmed that the TPP–PEG–biotin SANs were 150 ± 25 nm, and they were spherical in shape. However, after DOX loading, the size of the particles increased little to 175 ± 25 nm. The zeta potentials of the TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs were +21.76 ± 1.88 mV and +22.16 ± 1.51 mV. The 1H NMR spectrum of the TPP–PEG–biotin SANs was recorded in CDCl3 solvent. The characteristic signal at −2.81 ppm represents the NH protons of the pyrrole ring in the TPP unit, while that of the aromatic protons of the TPP unit shows around 7.49–8.83 ppm. The peak for the –OCH2 protons at 3.62 ppm represents the PEG-unit in the TPP–PEG–biotin SANs. The two –CH protons of the biotin unit show a peak around 4.51–4.54 ppm. The signal at 2.54 ppm represents the –SCH2 proton and the signal at 3.0 ppm represents the –SCH proton in the biotin ring. The peaks for the –CH2 protons of the biotin unit show around 1.10–2.06 ppm. The peaks at 6.57 ppm and 6.59 ppm represent the NH protons of the biotin unit. The results of 1H- and 13C-NMR and MALDI-TOF MS analysis are shown in Fig. S1–S11 (ESI†). The transmittance variance of the TPP–PEG–biotin SANs was monitored for three days in the present study to explore the serum stability of the self-assembled nanoparticles. The stability of the SANs was analyzed under physiological conditions using 10% phosphate-buffered saline (PBS) alone, and Dulbecco's modified Eagle's medium (DMEM) containing 50% (v/v) fetal bovine serum (FBS) and 10% FBS incubated for 3 days at 37 °C (Fig. 2d). The results show that the self-assembled nanoparticles hardly changed the transmittance for 3 days, confirming that there was no particle aggregation during incubation with PBS and FBS. | ||
| Scheme 1 Synthesis of TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs. | ||
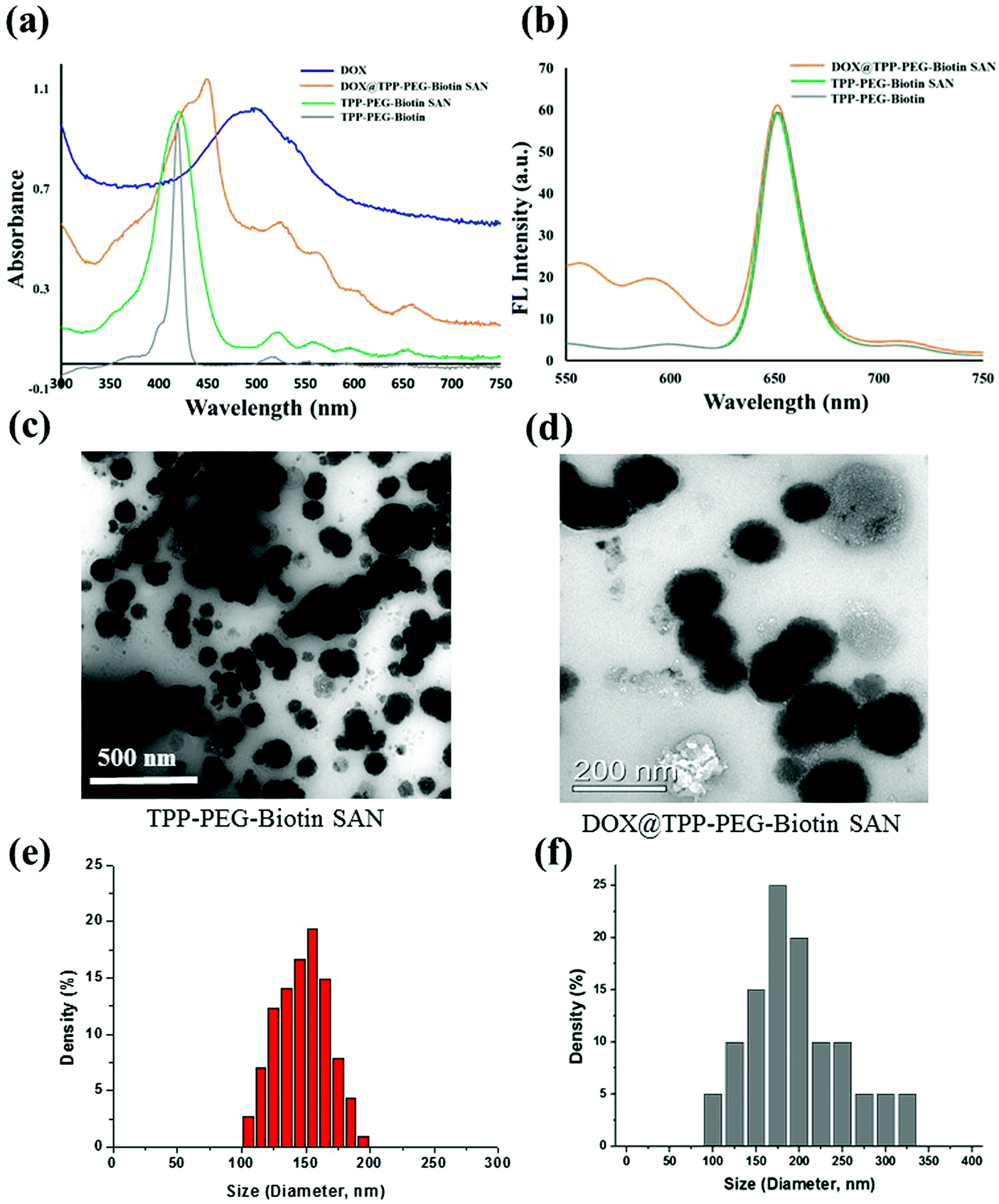 | ||
| Fig. 1 Characterization of the TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs. (a and b) UV-vis absorption and emission spectra of TPP–PEG–biotin, TPP–PEG–biotin SANs, and DOX@TPP–PEG–biotin SANs dissolved in CHCl3. (c and d) TEM images of the TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin nanoparticles. (e and f) Particle sizes of the TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs. | ||
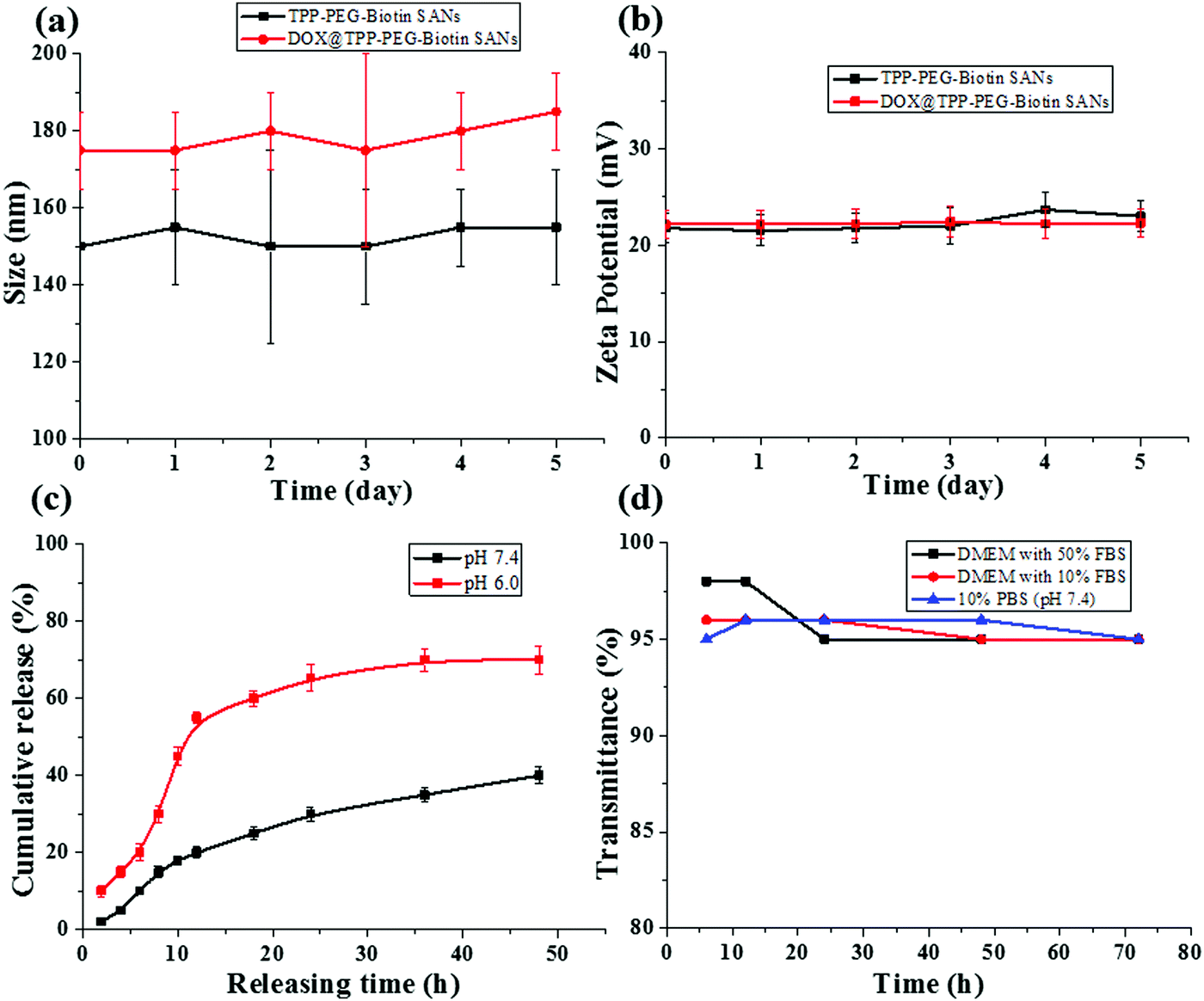 | ||
| Fig. 2 In vitro (a) size and (b) charge stability measurements of TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs with PBS (pH 7.4) at 37 °C. (c) In vitro DOX release from the DOX loaded TPP–PEG–biotin SANs at 37 °C under different PBS buffer (pH 7.4 and pH 6.0) conditions. (d) The variation in transmittance with incubation time of TPP–PEG–biotin SANs with 10% phosphate-buffered saline (PBS), and Dulbecco's modified Eagle's medium (DMEM) containing 50% (v/v) FBS and 10% FBS incubated for 3 days at 37 °C. The data are expressed as means ± SD (n = 3). | ||
2.2. In vitro drug release and stability of the nanoparticles
The release pattern of DOX from the DOX@TPP–PEG–biotin self-assembled nanoparticles was evaluated by the dialysis method. In this experiment, the released sample was measured at different time points using HPLC. Fig. 2c shows the release of DOX from the self-assembled nanoparticles at 37 °C in different PBS media (pH 7.4 and pH 6.0), indicating desirable stability of the DOX@TPP–PEG–biotin SANs. After 24 h, the DOX released was only 30.0 ± 1.8% in the pH 7.4 PBS medium compared to that in the pH 6.0 PBS medium (65.4 ± 2.0%). These results indicate that under acidic conditions the DOX was released into the cellular medium. The sizes and zeta potentials in Fig. 2a and b showed no aggregation behavior at room temperature for five days at 37 °C, indicating that the self-assembled nanoparticles prepared in this study showed adequate stability.2.3. Cytotoxicity tests
The cytotoxicity of PSs and free DOX in the MCF-7 breast cancer cell line was evaluated by MTT assay. Fig. 3 shows the results of the MTT assay performed at different concentrations of free DOX, TPPS, TPP–PEG–biotin, TPP–PEG–biotin SANs, and DOX@TPP–PEG–biotin SANs under dark and light conditions. Under the dark conditions, the PSs did not show any decrease in the cell viability of MCF-7 (Fig. 3a), except for the DOX-loaded TPP–PEG–biotin SANs (Fig. 3c) due to the DOX transported into the cells. The PSs were irradiated using a 660 nm diode laser at 30 mW for 20 min under the light conditions. All PSs irradiated at 660 nm significantly decreased the cell viability of MCF-7 in a concentration-dependent manner due to the light-activated properties of the PS (Fig. 3b and c). The calculated IC50 values of free DOX, TPPS, TPP–PEG–biotin, TPP–PEG–biotin SANs, and DOX-loaded TPP–PEG–biotin SANs are shown in Table 1. Interestingly, the IC50 value of TPP–PEG–biotin was determined as 3.31 μM, indicating the reduction of the IC50 value compared with that of TPPS. This result reveals that TPP–PEG–biotin is a more efficient PS due to the conjugation of biotin as the targeting moiety. However, the TPP–PEG–biotin SANs showed a higher IC50 value than that of TPP–PEG–biotin. The DOX-loaded TPP–PEG–biotin SANs were not only observed to have the lowest IC50 value owing to the synergistic effects under the light conditions but were also observed to decrease the cell viability under the dark conditions. This lowest IC50 value of DOX@TPP-PEG-biotin SANs under light condition is in agreement with the targeting ability of TPP-PEG-biotin SANs (Fig. 4).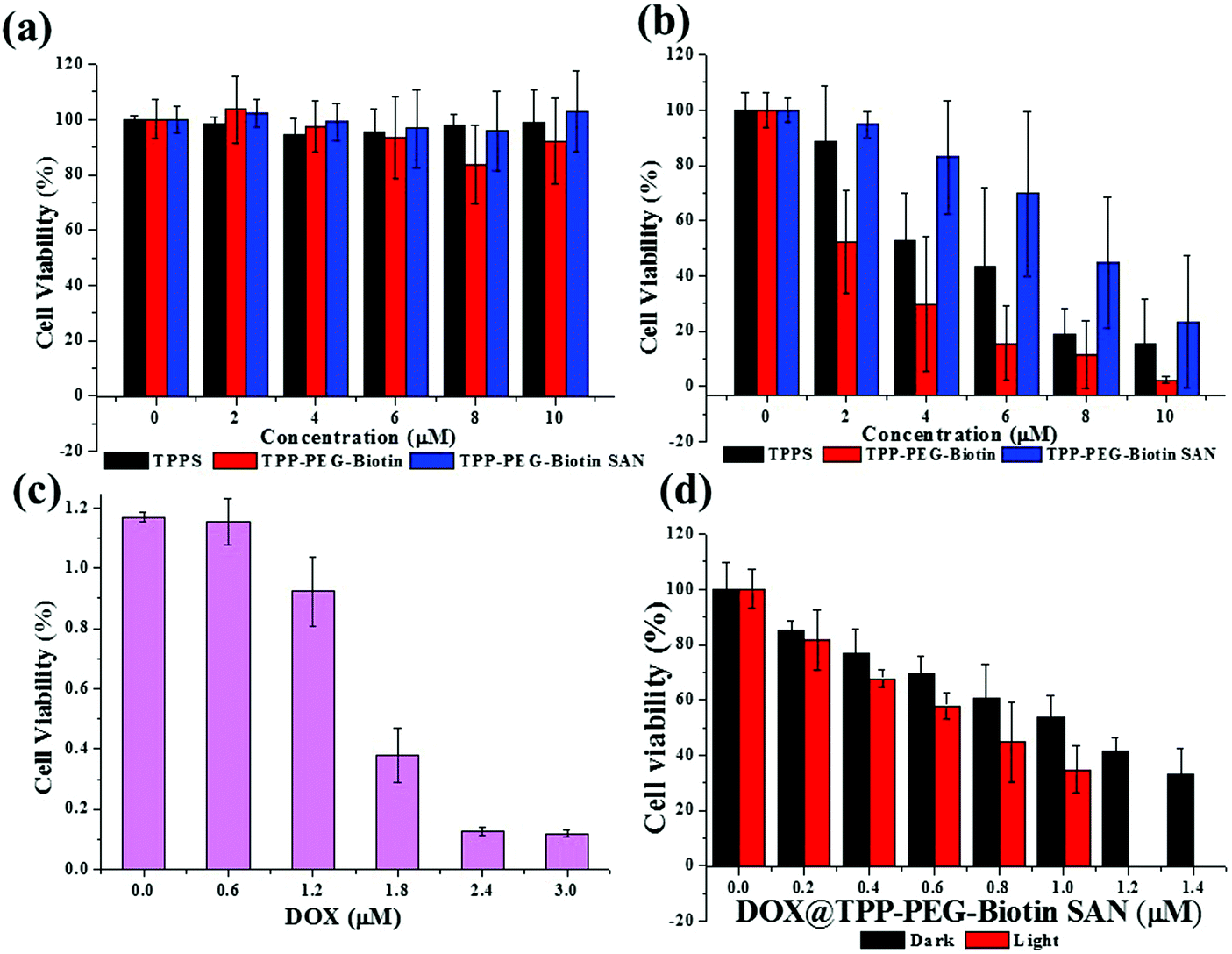 | ||
| Fig. 3 Cytotoxicity test. The cell viability of the MCF-7 cells treated with PSs. TPPS, TPP–PEG–biotin, and TPP–PEG–biotin SANs show negligible cytotoxicity (a) under dark conditions, whereas they show significant cell death (b) under light conditions. (c) The cell viability of doxorubicin (DOX). (d) DOX@TPP–PEG–biotin SANs under the dark or light conditions. Light dose: 660 nm, 30 mW cm−2, 20 min. The data are expressed as means ± SD, n = 6 independent experiments. | ||
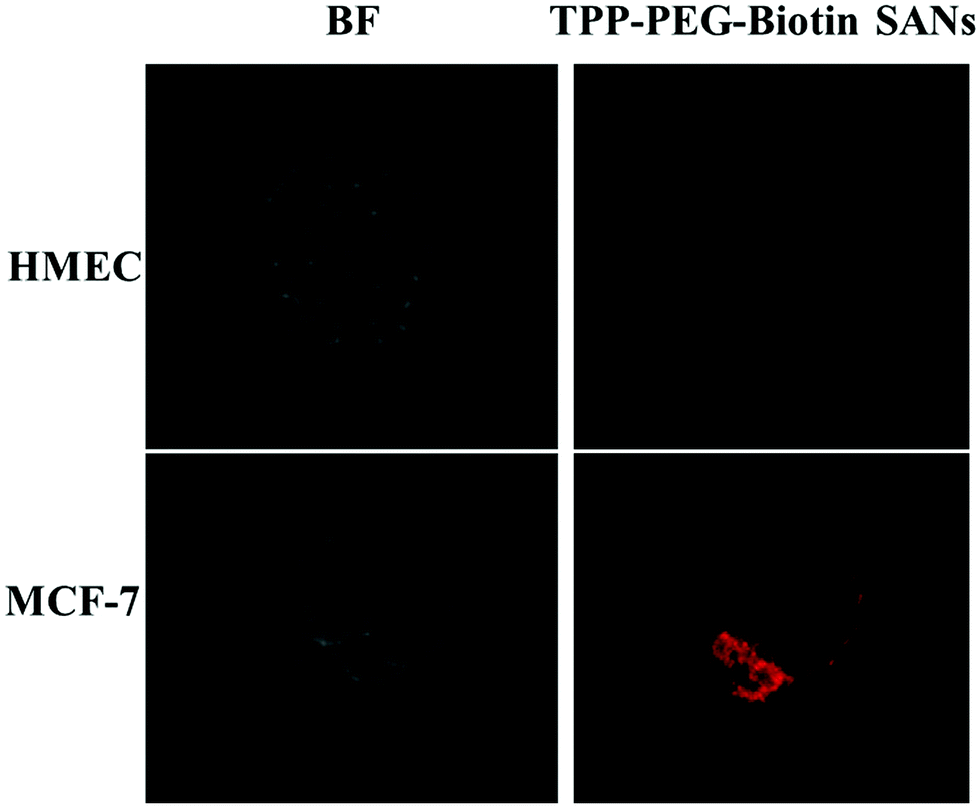 | ||
| Fig. 4 Targeting ability of TPP–PEG–biotin SANs. The confocal microscopy images of a normal cell line (HMEC) and a cancer cell line (MCF-7) treated with 5.0 μM of TPP–PEG–biotin SANs in PBS (total incubation period of 30 min). The left side panels show bright field images and right side panels show fluorescence field images. | ||
2.4. Intracellular ROS generation detection
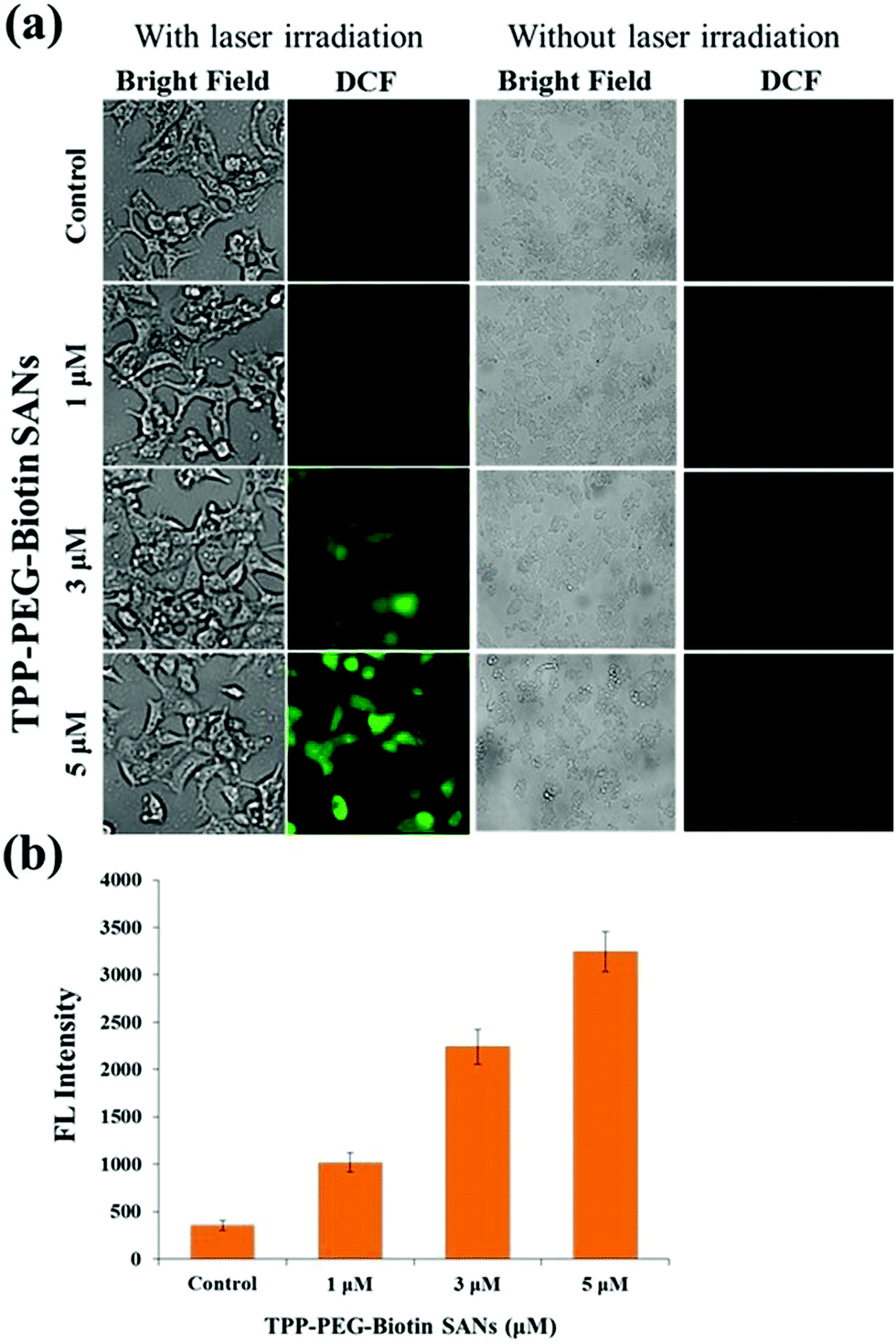
The light activation of PSs at specific wavelengths in cells produces intracellular ROS, presenting cytotoxicity to cancer cells. Intracellular ROS generated by TPP–PEG–biotin SANs was measured using DCFDA. The ROS reacts with DCFDA and produces DCF due to the deacetylation of DCFDA. Fig. 5a shows the bright field images and the fluorescence images of DCF in MCF-7 treated with TPP–PEG–biotin SANs at varying concentrations (1 μM, 3 μM, and 5 μM). The ROS generation in MCF-7 cells treated with TPP–PEG–biotin SANs under light and dark conditions was clearly detected by fluorescence microscopy. Fig. 5b presents the quantitative analysis for the fluorescence intensity of DCF, and it reveals that intracellular ROS production is properly induced by TPP–PEG–biotin SANs in a concentration-dependent manner under laser irradiation conditions.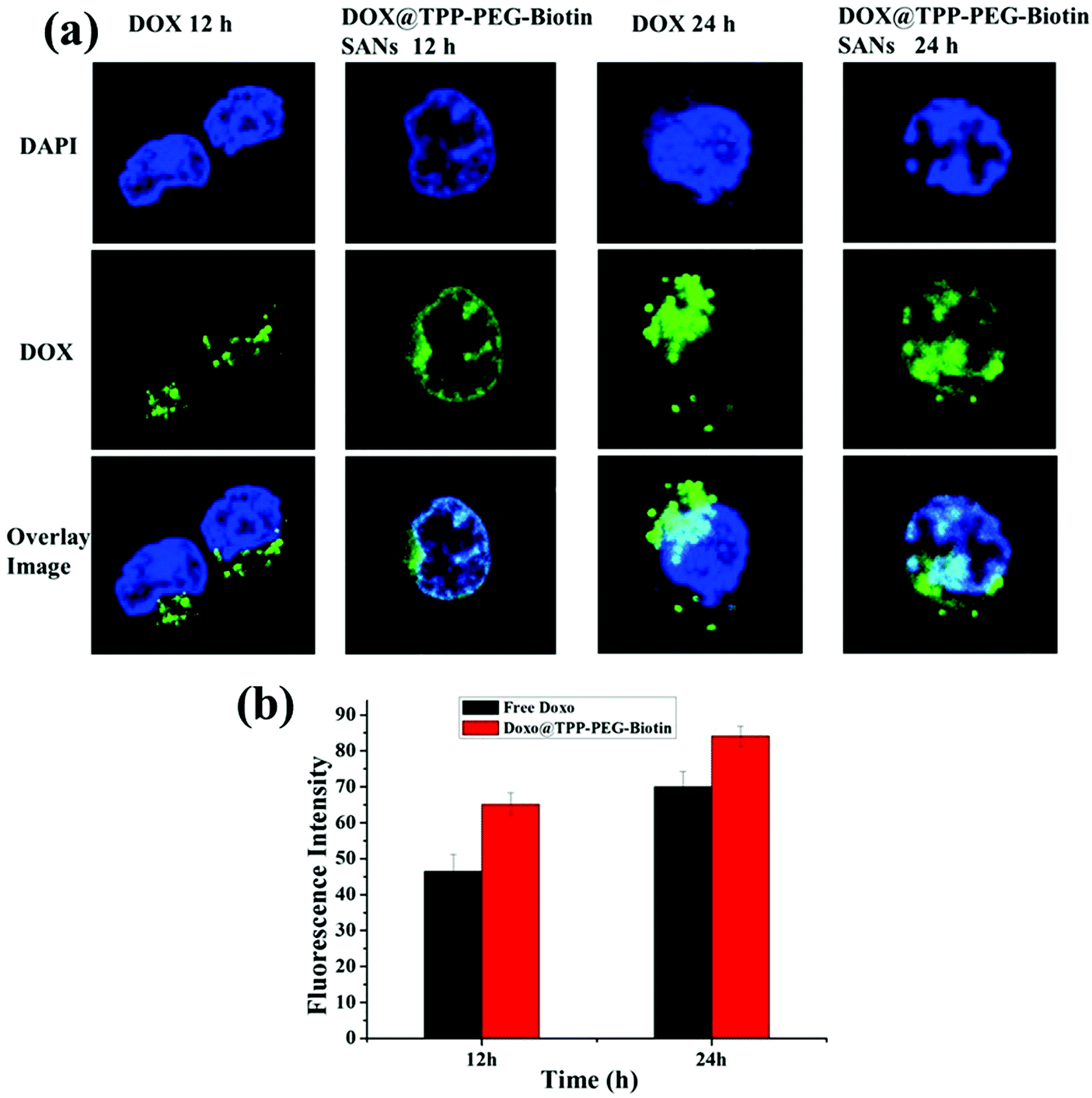 | ||
| Fig. 5 The intracellular DOX release study of DOX@TPP–PEG–biotin SANs was assessed by confocal fluorescence microscopy. (a) Confocal fluorescence images of free DOX and DOX loaded TPP–PEG–biotin SANs. (b) The quantitative analysis for the fluorescence intensity of DOX release was measured using the software MetaVue™. | ||
2.5. In vitro drug loading and intracellular drug release
The anticancer drug doxorubicin was used as a model drug for the combination therapy. DOX loaded TPP–PEG–biotin SANs were prepared by a simple sonication method. Typically, 2 mmol of TPP–PEG–biotin and DOX was mixed with PBS (pH 7.4) and sonicated for 15 min. Then the solution was incubated at room temperature for 24 h under dark conditions. The molar ratio between DOX and TPP–PEG–biotin was 1:5. The DOX was removed by centrifugation using an Amicon Ultra-15 centrifugal filter tube (3 kDa), and the supernatant was measured by UV-vis spectroscopy. The drug loading content was obtained from the standard curve obtained at a series of DOX concentrations. The DOX loading content of DOX@TPP–PEG–biotin was calculated as 25.03%.
The intracellular DOX drug release/delivery behavior of DOX@TPP–PEG–biotin SANs was assessed using confocal laser fluorescence microscopy (CLFM) images (Fig. 6a). MCF-7 cells were incubated with free DOX and DOX@TPP–PEG–biotin SANs at 37 °C, and then the DOX release profiles of DOX@TPP–PEG–biotin SANs at different time intervals (12 h and 24 h) were evaluated by CLFM image analysis. The CLFM image results of DOX@TPP–PEG–biotin SANs at 24 h showed a stronger fluorescence intensity of DOX than the 12 h time release. The free DOX was slowly distributed/delivered in MCF-7 cells compared with the free DOX loaded into TPP–PEG–biotin self-assembled nanoparticles. Fig. 6b presents the quantitative analysis for the fluorescence intensity of DOX release. The results demonstrated that the DOX was efficiently released from the DOX@TPP–PEG–biotin SANs in MCF-7 cells.
 | ||
| Fig. 6 Intracellular ROS detection. The ROS production by TPP–PEG–biotin SANs in MCF-7 was analyzed by fluorescence microscopy. (a) Green fluorescence images of DCF that is deacetylated from DCFDA represent intracellular ROS levels in MCF-7 treated with TPP–PEG–biotin SANs with and without laser irradiation. (b) The mean fluorescence intensity of DCF was quantitatively measured using the software MetaVue™. The graph shows a steady increase in the intracellular ROS level as the concentration of treated TPP–PEG–biotin SANs increases. The data are expressed as means ± SD, n = 3 independent experiments. | ||
2.6. Subcellular localization
As the lifetime of the intracellular ROS produced by PDT treatment is very short, it is difficult for the ROS to move to any other region within the cells. Hence, it is important to study where PSs mainly accumulate in cells. The intracellular localization of TPPS, TPP–PEG–biotin, and TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs was investigated by confocal microscopy using organelle-specific fluorescent probes. Fig. 7a confirms that TPPS is localized in the lysosomes in MCF-7, as previously reported.26 The fluorescence images of TPPS and LysoTracker Green overlapped to a large extent. It demonstrates that TPPS is mainly localized in the lysosomes. In contrast, it is found that TPP–PEG–biotin, TPP–PEG–biotin SANs, and DOX@TPP–PEG–biotin SANs are only partially localized in the lysosomes, which is confirmed by the slight overlap with LysoTracker Green. Interestingly, Fig. 7b shows that TPP–PEG–biotin and TPP–PEG–biotin SANs are mainly localized in the mitochondria. The fluorescence of TPP–PEG–biotin and TPP–PEG–biotin SANs overlapped perfectly with that of MitoTracker Green, but did not merge with that of ER-Tracker (Fig. S13, ESI†). In Fig. 6c, the localization assay of DOX@TPP–PEG–biotin SANs shows that they were mainly localized in mitochondria, and partially in lysosomes and nuclei due to the DOX content in the SANs. The results lead to the inference that TPP–PEG–biotin, TPP–PEG–biotin SANs, and DOX@TPP–PEG–biotin SANs may induce cell death in MCF-7 via cell death mechanisms different from the TPPS mechanisms.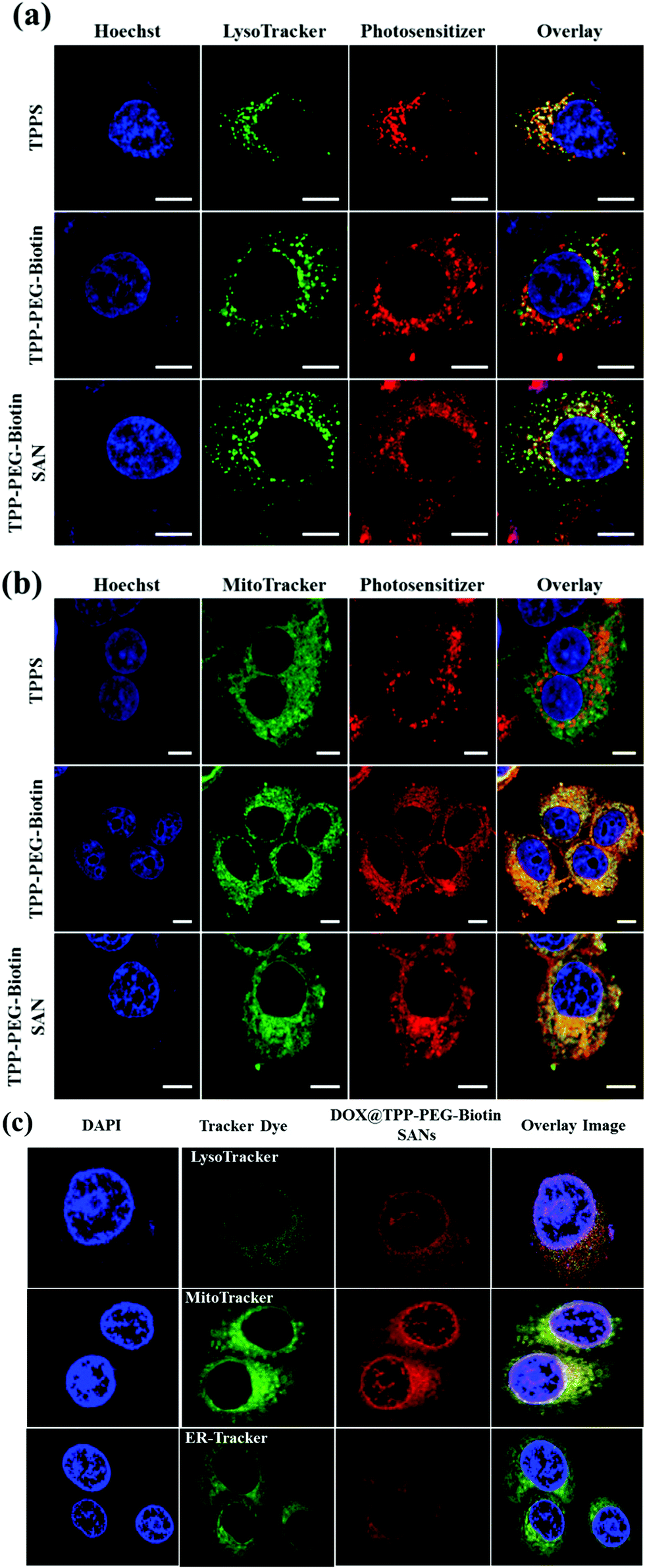 | ||
| Fig. 7 Subcellular localization of PSs. The confocal fluorescence microscopy images of MCF-7 cells treated with 20 μM of TPPS, TPP–PEG–biotin, and TPP–PEG–biotin SANs show their intracellular distribution via staining with organelle-specific fluorescence probes. Image sets (a) and (b) contain Hoechst, LysoTracker, Photosensitizer, and Overlay images. (c) The confocal fluorescence microscopy images of MCF-7 cells treated with 20 μM of DOX@TPP–PEG–biotin SANs. The excitation and emission wavelengths (nm) of the used organelle-specific fluorescent probes: Hoechst 350/461 (λex/λem), Lyso-Tracker 504/511, and Mito-Tracker 490/516. Scale bar, 10 μm. | ||
2.7. Cellular uptake
The cellular uptake of PSs by cancer cells is one of the most important components for good anticancer efficacy. The cellular uptake of PSs by MCF-7 was evaluated by flow cytometry by measuring their fluorescence inside the cells. Fig. 8 shows the FACS analysis data for MCF-7 cells treated with 1.5 μM DOX, and 5 μM concentrations of TPPS, TPP–PEG–biotin, and TPP–PEG–biotin SANs for 30 min, 60 min, and 120 min, respectively. The fluorescence intensities of all the PSs increased in strength with the increase in incubation time. Interestingly, the result of FACS analysis shows that the cellular uptake of TPP–PEG–biotin is rapid compared to that of TPPS. It suggests that the biotin conjugate, as the targeting moiety, promotes the cellular uptake of the TPP–PEG–biotin in MCF-7 cells that overexpress the biotin receptor. The cellular uptake of TPP–PEG–biotin SANs, however, was slower compared to that of its chemical form, TPP–PEG–biotin.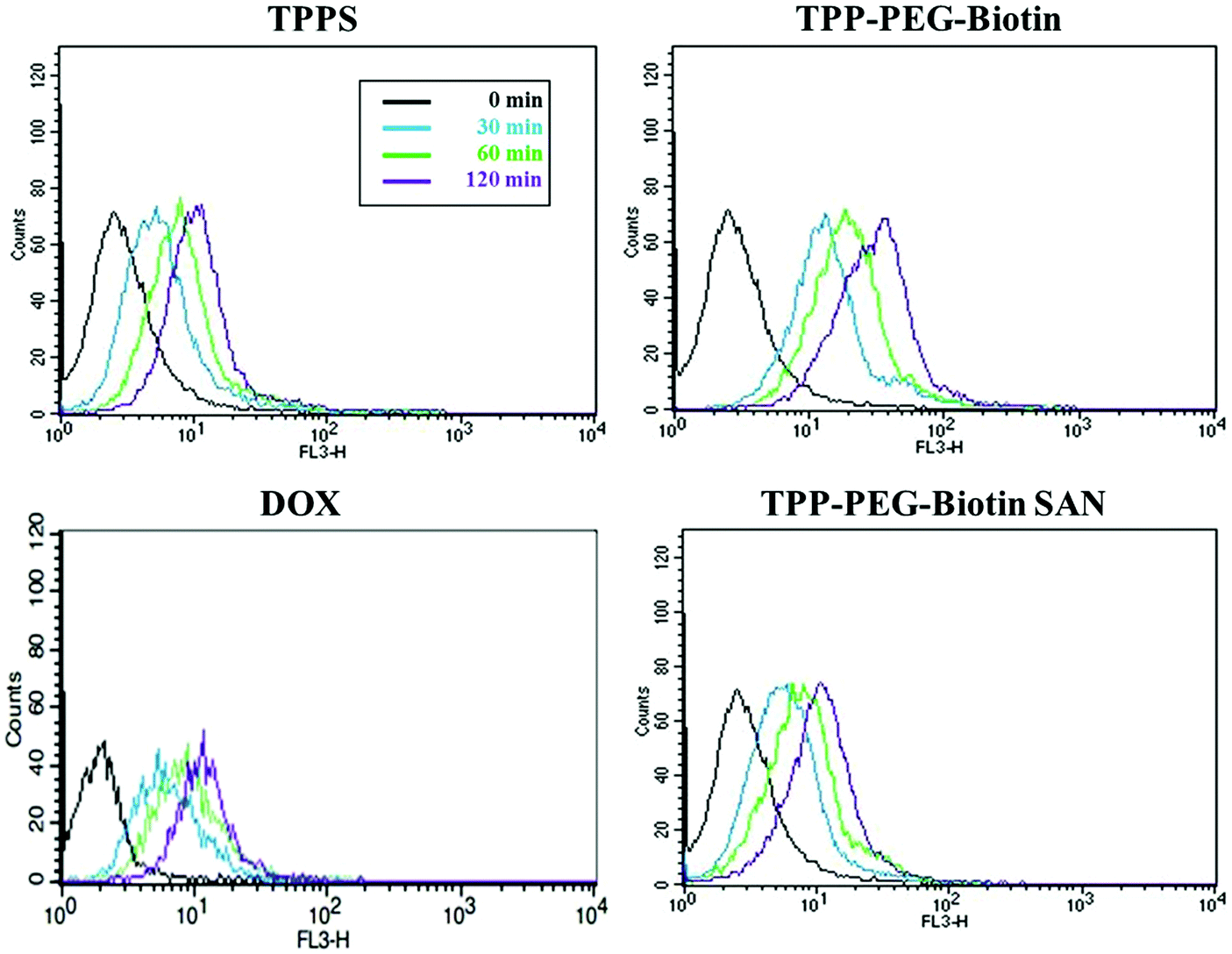 | ||
| Fig. 8 Cellular uptake of PSs and DOX. FACS analysis for MCF-7 treated with 1.5 μM of DOX, 5 μM of TPPS, TPP–PEG–biotin, and TPP–PEG–biotin SANs was performed. The fluorescence emitted from the cells was measured at 30 min, 60 min, and 2 h after PS treatment. The curved graphs obtained using a flow cytometer represent the increasing uptake of PSs as the incubation time increases in MCF-7 cells. | ||
2.8. High-content cell death dynamics
Fig. 9 and 10 present the cell death dynamics of MCF-7 cells treated with TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs. Mitochondrial membrane disruption, the increase of the cytosolic Ca2+ amount, and caspase-3 activation were simultaneously monitored. The fluorescence intensity of the calcein-AM that accumulated in the mitochondria rapidly decreased at 1 μM concentration of TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs, presenting mitochondrial membrane permeability transition (MPT) due to damage to mitochondria caused by ROS. The amount of cytosolic Ca2+ also sharply increased as the concentration of TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs increased with the rapid induction of MPT. On the other hand, the activation of caspase-3 showed a steady increase in MCF-7 treated with TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs in a concentration-dependent manner. However, the fluorescence intensity of the cytosolic Ca2+ amount increased, and the caspase-3 level decreased without light irradiation conditions (Fig. 10c and d). This may be because the chemodrug DOX was loaded into the TPP–PEG–biotin SANs.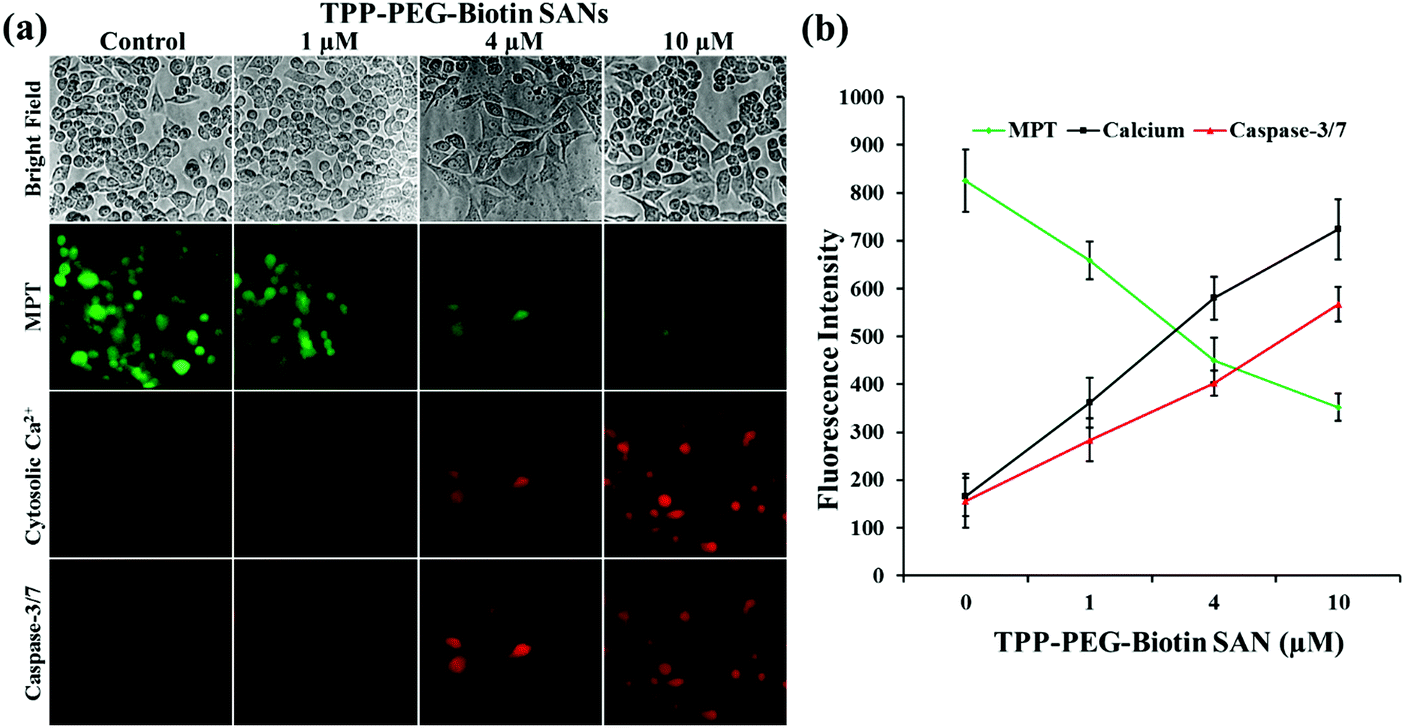 | ||
| Fig. 9 High-content screening assay. The high-content images were obtained using a fluorescence microscope equipped with AOTF. (a) Image sets of MPT, cytosolic Ca2+, caspase-3/7 show their fluorescence images for MCF-7 cells treated with TPP–PEG–biotin SANs, representing cell death dynamics. The mean fluorescence intensity was analyzed by the software MetaVue™, and plotted. (b) The graph indicates the fluorescence intensities as a function of TPP–PEG–biotin SAN concentration. The data are expressed as means ± SD. n = 3 independent experiments. | ||
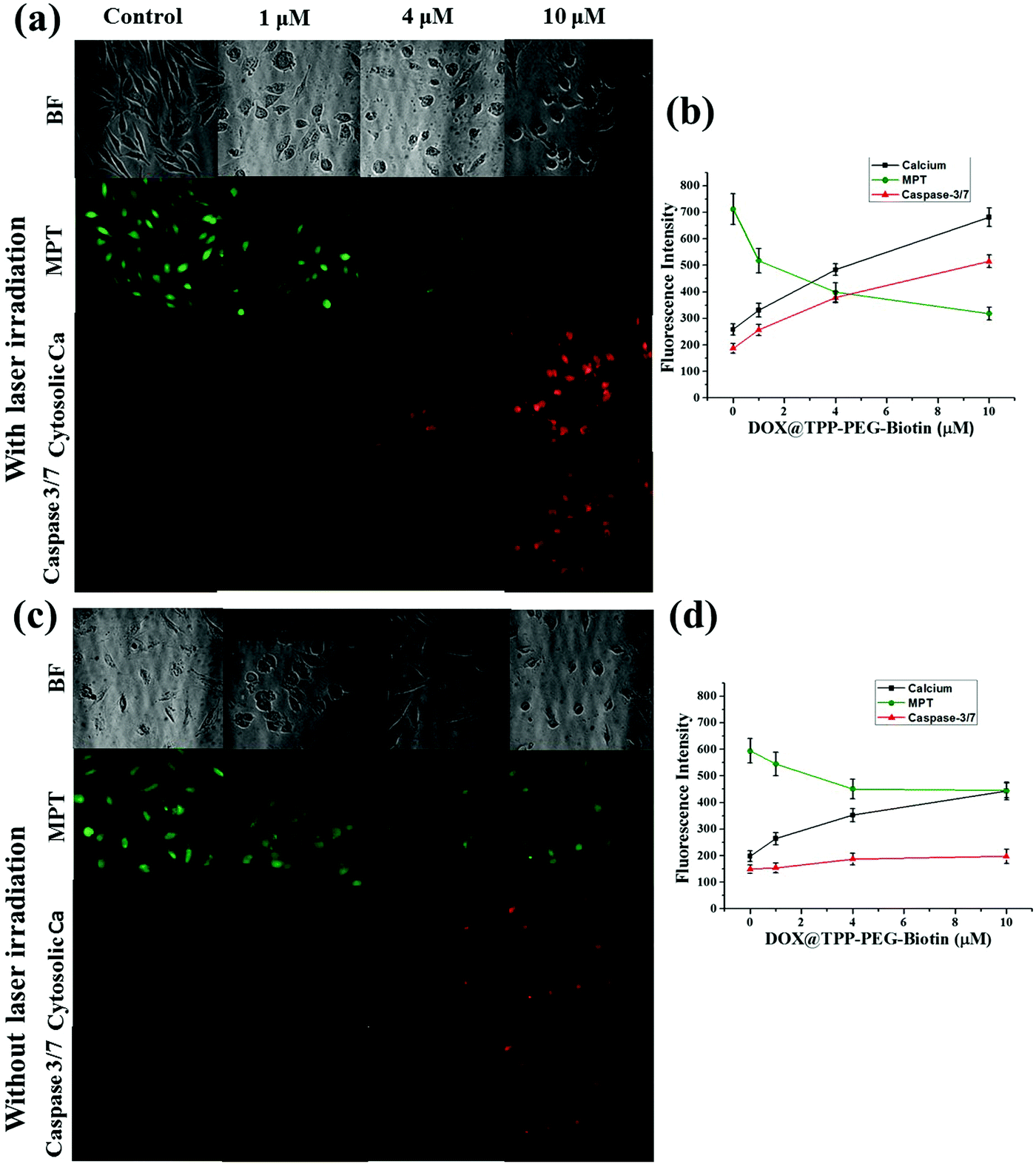 | ||
| Fig. 10 High-content screening assay for DOX@TPP–PEG–biotin SANs with and without light irradiation. The high-content images were obtained using a fluorescence microscope equipped with AOTF. Image sets of MPT, and cytosolic Ca2+ show their fluorescence images for MCF-7 cells treated with TPP–PEG–biotin SANs, representing cell death dynamics with laser irradiation (a and b) and without laser irradiation (c and d). (b and d) The graphs indicate the fluorescence intensities as a function of DOX@TPP–PEG–biotin SAN concentration. The data are expressed as means ± SD. n = 3 independent experiments. The mean fluorescence intensity was analyzed by the software MetaVue™, and plotted. | ||
3. Discussion
Recently, adjuvant chemotherapy with other therapies has been the major anticancer therapy for breast cancer treatment. The chemotherapy contributes to enhanced survival rates; however, approximately 40% of patients treated with chemo-drugs relapsed because of MDR. To overcome MDR, various nanoparticle-based drug delivery systems (NDDSs) have been developed recently.27 Here, we developed a new self-assembled nanoparticle based on a PS for DDSs applied to chemo-drugs. Recently, PDT has also emerged as a useful therapy in cancer treatment. PDT can selectively kill cancer cells, without affecting adjacent normal cells, by light irradiation of the targeted lesion, increasing anticancer efficacy and minimizing side effects.28The combination of chemotherapy and PDT has proven to be the superior approach in the treatment of cancers with drug-resistance by inducing synergistic effects. PDT induces oxidation of endocytic membranes with light irradiation, leading to the release of chemo-drugs from the endosomes to the cytoplasm, and this is crucial in preventing MDR. For the combination therapy, a single nanosystem has been adopted, as the vehicles encapsulate both the chemo-drug and the PS. Nonetheless, there were numerous obstacles including instability, drug leakage, and poor loading efficiency due to the steric hindrance between the chemo-drugs and PSs. Cont et al. reported that co-encapsulation of docetaxel (chemo-drug) and zinc-phthalocyanine (PS) gave rise to a sudden reduction in the entrapment efficiency and loading capacity.29 Therefore, it has been necessary to develop an advanced nanosystem for chemo-photodynamic combination therapy. Tong et al. studied an alternative strategy for combination therapy that adopts near infrared (NIR) light-mediated upconversion nanoparticles@mesoporous silica (UCNPs@mSiO2) as the nanocarriers. Then, TiO2 was used as a PS, and a DOX-UCNPs@mSiO2/TiO2-TC nanocomposite was constructed with synergistic anticancer effects.30 Feng et al. tried tumor-homing and penetrating peptide-functionalized PS-conjugated PEG–PLA NPs for combination therapy and exploited them for encapsulation of paclitaxel (PTX). Established PTX-loaded NPs that have synergistic effects for killing cancer cells were found.3
Because of the distinct photophysical properties of porphyrin, a wide variety of porphyrin derivatives have been synthesized and studied as effective PSs in PDT. Porphyrins can generate intracellular ROS (mainly 1O2) followed by excitation under light exposure (more than 630 nm). In PDT, the major challenge is the depth of tissue penetration by the light source. In the case of porphyrins, the wavelength of the light source could be relatively longer than other PSs; therefore, it is feasible that the light source reaches deeper sites in tissues, damaging diseased tissues by inducing PDT action. In numerous porphyrin derivatives, TPPS has largely been known as a good PS, presenting high anticancer efficacy in PDT. Although TPPS has good water solubility and high anticancer efficacy, it may not ensure selective accumulation in specific diseased tissue in the absence of any targeting moiety.
In this work, we have designed the self-assembly of a PS-conjugated amphiphilic biotinylated polymer (TPP–PEG–biotin) as the nanocarrier for DOX, showing high affinity to the biotin receptor that is commonly overexpressed in cancer cells. PEG polymers have been chosen as new hydrophilic groups conjugated to the hydrophobic TPP. Biotin has been utilized as a targeting moiety for MCF-7 cancer cells. Since vigorous proliferating cancer cells need to take in abundant nutrients for their growth, they tend to overexpress diverse nutrient receptors including the biotin receptor. The biotin receptor has high affinity to biotin and has been reported to be overexpressed in several cancer cell lines, such as MCF-7, SkBr3, HeLa, and HepG2.31 On the basis of this concept, it can be easily assumed that biotinylated PSs can selectively target tumors that overexpress the biotin receptor. In addition, the biocompatibility of TPP–PEG–biotin conjugates may help in their prolonged circulation in blood.
The cytotoxicity of the PSs is activated by light that comes from ROS production in cells reacting with the intracellular oxygen molecule. To evaluate the cytotoxicity of the PSs in MCF-7 breast cancer cell lines, MTT assay was conducted (Table 1). Notably, the IC50 values of the TPP–PEG–biotin chemical and TPP–PEG–biotin SANs have been shown to be much lower than that of the control drug TPPS. However, there is no cytotoxic effect in MCF-7 cells that are not exposed to light. Interestingly, the IC50 value of TPP–PEG–biotin under light irradiation decreased significantly by about 35% compared with that of TPPS. This result demonstrates that TPP–PEG–biotin is more effective in the PSs targeting the MCF-7 cell lines that overexpress the biotin receptor. It was observed, however, that the IC50 value of TPP–PEG–biotin SANs is relatively increased in comparison with the TPP–PEG–biotin chemical. The cellular uptake and subcellular localization of the PSs have been investigated in MCF-7 cells to demonstrate the mechanism of cytotoxicity. The results showed the same tendency with cytotoxicity; the uptake of TPP–PEG–biotin was much faster than that of TPPS, and the uptake of the TPP–PEG–biotin SANs was found to be slower compared to that of its chemical form. This may be a result of the increased size of the spherical TPP–PEG–biotin SANs. In the same context, it can be assumed that the cytotoxicity of the TPP–PEG–biotin SANs is diminished upon light irradiation because of the reduced uptake efficiency. However, overall, the cytotoxicity of DOX@TPP–PEG–biotin SANs showed a low IC50 value (0.71 μM) compared to those of free DOX and TPPS.
In order to evaluate the targeting ability of TPP–PEG–biotin SANs in living systems, the biotin-receptor positive cell line MCF-7 and biotin-receptor negative cell line HMEC were used (Fig. 4). The biotin positive cell line and normal cell line were treated with 5 μM of TPP–PEG–biotin SANs. The CLFM image results reveal that a strong fluorescence intensity was observed in the biotin-receptor positive cell line (MCF-7) than the biotin-receptor negative cell line under similar experimental conditions. The results confirm that the TPP–PEG–biotin SANs were taken up by the biotin positive cell line compared to the normal cell line through receptor mediated endocytosis due to the biotin targeting moiety.
TPP–PEG–biotin SANs have also been evaluated for ROS generation in MCF-7 using DCFDA. As in the case of the cytotoxicity test, TPP–PEG–biotin SANs produced intracellular ROS as observed in the DCF fluorescence mounting in a concentration-dependent manner with and without laser irradiation conditions. As shown in Fig. 5a, TPP–PEG–biotin SANs do not produce ROS fluorescence intensity without laser irradiation conditions. However, the laser irradiated group showed higher ROS fluorescence intensity (Fig. 5b). It revealed that TPP–PEG–biotin SANs can still play a role as the PS in targeted anticancer therapy. This intracellular ROS assay also affirms that the TPP–PEG–biotin was preferentially taken up by the MCF-7 cells (biotin receptor positive cell line) in a concentration dependent manner.
In this study, the most interesting result is that the subcellular localization of TPPS is different from that of TPP–PEG–biotin, TPP–PEG–biotin SANs, and DOX@TPP–PEG–biotin SANs. The intracellular distribution of PSs in cancer cells is very important for determining the cell death mechanism by which the PSs induce apoptosis in cancer cells upon light irradiation. The results of confocal microscopy using organelle-specific fluorescent probes showed that TPPS was localized in lysosomes as previously reported,26 whereas TPP–PEG–biotin and its SANs are predominantly localized in mitochondria and partially in lysosomes. We suggest that PEG–biotin substitution may play a crucial role in selective localization in mitochondria, although the mechanism of selective accumulation of TPP–PEG–biotin and its SANs is not completely understood yet. There has been no report that the PEG polymer or biotin alone targets certain subcellular organelles. In localization studies, the anionic porphyrin TPPS has been confirmed to exclusively accumulate in lysosomes, and this is in accordance with previous studies. Rangasamy et al. designed 5,10,15,20-tetrakis(7-sulfonatobenzo[b]-thiophene)porphyrins (SBTPs), and the SBTPs were found to be mainly localized in mitochondria even though they are negatively charged like TPPS.26 Wilson et al. reported that negatively charged photofrins bind to mitochondria.32 On the basis of this evidence, we assumed that the PEG–biotin moiety in TPP–PEG–biotin and its SANs would be a determining factor in targeting mitochondria. We studied the subcellular localization of DOX loaded TPP–PEG–biotin SANs in MCF-7 cells. The results concluded that the DOX@TPP–PEG–biotin SANs localized in lysosomes and mitochondria and partially in the nucleus. The DOX in the nanoparticles plays a major role in localizing into the nucleus (Fig. 7c). It is highly reasonable to expect that the cell death mechanism in PDT would change in response to the altered intracellular localization of TPP–PEG–biotin SANs. To investigate the cell death mechanism, high-content cell death screening (HCS) has been exploited in the MCF-7 cell lines treated with TPP–PEG–biotin SANs upon light irradiation. After PDT action, cancer cells die by apoptosis or necrosis, the more common mode of cell death being apoptosis. It has already been suggested that PSs localized in mitochondria could induce apoptotic cell death rapidly by PDT action. Disruption of the mitochondrial membrane provokes mitochondrial MPT, breaking the calcium ion homeostasis and increasing the intracellular amount of Ca2+. Subsequently, caspase-3 can be activated, which leads to apoptosis. In our previous study, it was found that the PSs localized mainly in mitochondria initially lead to MPT followed by an increase in the intracellular amount of Ca2+, and they activate caspase-3 in turn. In this study, HCS results indicate that TPP–PEG–biotin SANs exhibit a similar action to the above-mentioned PSs during induction of cell death (Fig. 9 and 10). Yet, there is a difference between them: that is, an increase in the intracellular Ca2+ amount takes place at the same time as the induction of MPT. Considering that the TPP–PEG–biotin SANs merely accumulate in lysosomes, it might make sense that the cytosolic Ca2+ amount is enhanced early due to the release of calcium ions from the lysosome whose membrane is disrupted by PDT action.
For chemo-photodynamic combination therapy, doxorubicin (DOX) was selected as the chemotherapy drug, and it was successfully encapsulated by TPP–PEG–biotin SANs. Encapsulation of DOX inside TPP–PEG–biotin SANs was most efficiently conducted when the molar ratio between DOX and TPP–PEG–biotin was 1:5. In the cytotoxicity test, the DOX-loaded TPP–PEG–biotin SANs presented cytotoxicity in MCF-7 even without light irradiation because of the anticancer effect of transported DOX and higher cytotoxicity with light irradiation due to the PDT action of TPP–PEG–biotin SANs, revealing synergistic effects in combination therapy. The DOX transported into the cancer cells selectively accumulates in the nucleus, damaging DNA via π–π interaction. That is, the DOX-loaded TPP–PEG–biotin SANs can transport DOX into the cancer cells that overexpress the biotin receptor, synergistically enhancing anticancer efficacy with PDT action via the cell death mechanism that is determined by targeting multiple subcellular organelles including the nucleus and mitochondria, and partially the lysosomes. The DOX delivery of DOX@TPP–PEG–biotin SANs was studied using the biotin receptor positive cell line (MCF-7). The CLFM image shows that the DOX was released into the MCF-7 cells in a time dependent manner. The HCS assay of DOX@TPP–PEG–biotin SANs was also conducted under light and dark conditions. The DOX loaded nanoparticles produced more amounts of cytosolic calcium and caspase 3 under light irradiation conditions, and lower amounts of cytosolic calcium and caspase 3 without light irradiation conditions, thus leading to more apoptosis than that under the dark conditions. This HCS result is in agreement with the cell viability of DOX loaded TPP–PEG–biotin SANs (IC50 0.71 μM). In the present work, only in vitro studies have been performed for DOX-loaded TPP–PEG–biotin SANs; therefore, further in vivo studies have to be conducted for clinical application. It is expected that the DOX@TPP–PEG–biotin SANs selectively accumulate in the tumor region that overexpresses the biotin receptor and shows high anticancer efficacy in chemo-photodynamic combination therapy. Furthermore, since TPP–PEG–biotin SANs are hydrophilic PSs, they could also disrupt the vasculature in the tumor microenvironment binding to serum albumin by PDT action. As a result, more DOX-loaded TPP–PEG–biotin SANs can reach the tumor region, leading to higher anticancer efficacy.
4. Conclusions
In this study, we successfully synthesized hydrophilic PSs TPP–PEG–biotin and DOX@TPP–PEG–biotin SANs with high biocompatibility that are capable of selectively targeting cancer cells that overexpress the biotin receptor. In addition, TPP–PEG–biotin and its SANs were found to have higher cytotoxicity upon light irradiation with rapid cellular uptake in MCF-7 cells compared to TPPS. When in vitro PDT activity was studied, it was revealed that the DOX@TPP–PEG–biotin SANs had synergistic anticancer effects in MCF-7 cells. The intracellular localization assay confirmed that TPP–PEG–biotin and its self-assembled nanoparticle targeting subcellular organelles including mitochondria and lysosomes. Taken together, the DOX@TPP–PEG–biotin SANs with synergistic anticancer effects and tumor-targeting potential may prove to be a promising anticancer agent in chemo-photodynamic combination therapy.5. Experimental
5.1. Synthetic materials and methods
All chemicals and reagents purchased from Sigma Aldrich, Korea, and TCI Chemicals and used without further purification. Silica gel 60 (Merck, HF-254, 0.25 mm) was used for column chromatography. Absorption spectra were recorded in 1 cm quartz cuvettes using an Evolution™ 60 UV-Visible Spectrophotometer (Thermo Fisher Scientific, USA). Emission spectra were obtained on a Jasco-FP 6500 spectrofluorometer. 1H and 13C NMR spectra were recorded in deuterated chloroform or dimethyl sulfoxide (Cambridge Isotope Labs), containing 0.05% v/v TMS as an internal standard, using Bruker AVANCE500 and JEOL 400 spectrometers. The abbreviations for the peak multiplicities are as follows: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). Chemical shifts are reported in values (ppm) relative to internal TMS, and J values are reported in Hz. A MALDI-TOF mass spectrum was obtained with a MALDI TOF-TOF 5800 System (AB Sciex, USA).5.2. Synthesis of 5,10,15,20-tetraphenylporphyrin (1)
TPP synthesis was performed as per a previously published procedure.24 Briefly, benzaldehyde and freshly distilled pyrrole were slowly added to refluxing propionic acid. The reaction mixture was stirred at 130 °C for 30 min. Then the reaction mixture was cooled at room temperature, and the solid was slowly filtered and washed with a methanol:acetone (1:1) mixture. The purple color solid was washed again with hot water several times to remove adsorbed propionic acid. The solid was dried in a desiccator for one day, then finally dried in a vacuum pump to get pure TPP. Yield: 200 mg. 1H NMR (500 MHz, chloroform-d) δ 8.84 (s, 3H), 8.21 (dt, J = 6.3, 1.7 Hz, 3H), 7.74 (qd, J = 8.8, 7.9, 3.9 Hz, 5H), −2.78 (s, 1H). 13C NMR (126 MHz, chloroform-d) δ 142.17, 134.54, 127.69, 126.66, 120.12. MALDI-TOF MS: m/z 615.4 [M]+.
5.3. Synthesis of 5-(4-nitrophenyl)-10,15,20-triphenylporphyrin (2)
10 ml of trifluoroacetic acid was taken in a RB flask and 5,10,15,20-tetraphenylporphyrin (200 mg, 0.32 mmol) was added slowly at room temperature, and stirred to dissolve the solid. Then sodium nitrite (40 mg, 0.58 mmol) was added. The reaction mixture was stirred at room temperature for 3 minutes. After 3 minutes, the reaction mixture was immediately poured into ice cold water and extracted with DCM. The DCM layer was washed with sodium bicarbonate and water three times. The collected DCM layer was dried with sodium sulfate. The solvent was removed using a rotary evaporator. The crude material was purified by silica gel column chromatography using ethyl acetate:hexane as a mobile phase. Yield: 230 mg. 1H NMR (500 MHz, chloroform-d) δ 8.88 (d, J = 4.7 Hz, 1H), 8.85 (s, 2H), 8.83 (s, 1H), 8.72 (d, J = 4.8 Hz, 1H), 8.62 (d, J = 8.3 Hz, 1H), 8.38 (d, J = 8.3 Hz, 1H), 8.23–8.17 (m, 3H), 7.82–7.69 (m, 5H), −2.79 (s, 1H). 13C NMR (126 MHz, chloroform-d) δ 141.89 (d, J = 4.6 Hz), 135.11, 134.52, 127.87, 126.74 (d, J = 3.8 Hz), 121.84. MALDI-TOF MS: m/z 662.4 [M + 3]+.5.4. Synthesis of 4-(10,15,20-triphenylporphyrin-5-yl)aniline (3)
5-(4-Nitrophenyl)-10,15,20-triphenylporphyrin (195 mg, 0.29 mmol) was added to concentrated hydrochloric acid (15 ml) in a RB flask. After stirring for 5 min, a clear solution was obtained, then stannous chloride dihydrate (400 mg, 1.77 mmol) was added. The reaction mixture was refluxed at 65 °C for 1 h under a N2 atmosphere. The reaction mixture was cooled to room temperature, then slowly added to ice solution and neutralized with ammonium hydroxide. The pink color solution obtained was extracted with DCM (2× 100 ml). The solution was distilled under a rotary evaporator to get the solid. Yield: 150 mg. 1H NMR (500 MHz, DMSO-d6) δ 8.97 (d, J = 4.8 Hz, 2H), 8.83 (s, 2H), 8.80 (s, 6H), 8.22 (td, J = 6.3, 5.5, 2.8 Hz, 9H), 7.90–7.78 (m, 16H), 7.01 (d, J = 8.0 Hz, 2H), 5.61 (s, 2H), −2.86 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 148.68, 141.33, 135.56, 134.19, 128.24, 128.01, 126.98, 126.95, 121.94, 120.01, 119.73, 119.26, 112.52. MALDI-TOF MS: m/z 630.3 [M]+.5.5. Synthesis of TPP–PEG–biotin (4)
The TPP–PEG–biotin chemical was synthesized by the acid–amine coupling reaction between biotin–PEG–COOH and TPP-amine. Briefly, 1 mmol of biotin–PEG–COOH, DMAP, EDCI and 3 ml of CH2Cl2 were added to a glass vial and the reaction mixture stirred for 3 h at 0 °C. Then 1 mmol of TPP-NH2 was added and the reaction mixture was stirred continuously for 36 h at room temperature under a N2 atmosphere. The unconjugated TPP-NH2 and the EDCI adduct formed were removed by column chromatography using silica gel. Finally, the pure TPP–PEG–biotin chemical was dried completely using a lyophilizer and characterized by spectroscopy methods. MALDI-TOF MS: m/z 4169.3 [M]+.5.6. Preparation of TPP–PEG–biotin self-assembled nanoparticles (5)
The NPs were prepared according to a previously published procedure.33 Briefly, 50 mg of TPP–PEG–biotin was dissolved in 5 ml of chloroform, dried under a flow of nitrogen gas, and dried further under vacuum to get a film layer. For nanoparticle synthesis, the TPP–PEG–biotin film layer was hydrated with 900 μl of PBS (pH 7.4) and 100 μl of DMSO. Then the solution mixture was sonicated for 15 min, and uniform sized self-assembled TPP–PEG–biotin NPs were obtained slowly. The NPs were collected by centrifugation at 15000 rpm using a TJ-25 centrifuge (Beckman Coulter). The characterization of TPP–PEG–biotin SANs was done by TEM and SEM. The stock was diluted with PBS (pH 7.4) and used for in vitro experiments.
5.7. Preparation of DOX@TPP–PEG–biotin self-assembled nanoparticles (6)
A TPP–PEG–biotin (2 mmol) film layer was hydrated with PBS (pH 7.4) containing 20 μl of DOX (1 mg ml−1). After sonication for 15 min, the solution was aged for 24 h, and DOX@TPP–PEG–biotin SANs were finally formed. Free DOX was removed by centrifugation at 15000 rpm using an Amicon Ultra-15 centrifugal filter tube (3 kDa, Merck Millipore, South Korea). The encapsulation efficiency and drug loading efficiency were calculated using the following equations:| Encapsulation efficiency (%) = (amount of DOX drug in NP solution/total weight of drug added initially) × 100. |
| Drug loading percentage (%) = [amount of DOX drug in NP solution/(amount of DOX drug in NP solution + amount of TPP–PEG–biotin added initially)] × 100. |
5.8. In vitro release
Drug release profiles were studied in vitro by the dialysis method as reported previously.34 Briefly, the release profiles of the chemodrug DOX from the DOX@TPP–PEG–biotin self-assembled nanoparticles were obtained using phosphate buffered saline (PBS, pH 7.4 and pH 6.0). An as prepared DOX loaded nanoparticle solution (5 ml) was introduced into a dialysis membrane bag (MWCO 3500 Da) and then subsequently immersed into 50 ml of buffer solutions. The samples was agitated with a shaking water bath at 37 °C and 100 rpm. Then released samples (1 ml) were taken at different time intervals (2, 4, 6, 8, 10, 12, 18, 24, 38, and 48 hours) and replaced with an equal volume of fresh buffer solution. Then the determination of DOX release was monitored using high performance liquid chromatography (HPLC, Hewlett Packard, 1100 Series). All the experiments were conducted in triplicate with standard deviations in the dark.5.9. In vitro stability of self-assembled nanoparticles
The in vitro size and charge stability of TPP–PEG–biotin and DOX@TPP–PEG–biotin SANs were analyzed using dynamic light scattering (DLS) and electrophoretic light scattering (ELS) spectrometers. Briefly, the nanoparticle solution was mixed with 2 ml of PBS (pH 7.4) and incubated at 37 °C. Then, the nanoparticle solution was withdrawn at different time intervals (0, 1, 2, 3, 4, and 5 days) and analyzed using DLS.5.10. Cell culture
The human breast cancer cell line MCF-7 was obtained from the Korean Cell Line Bank (KCLB, Korea). The normal human primary mammary epithelial cell line HMEC (ATCC PCS-600-010) was obtained from the American Type Culture Collection (ATCC). The cell line was grown in Dulbecco's modified Eagle's medium (DMEM; GIBCO, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS; GIBCO, USA) and 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin (GIBCO, USA) at 37 °C in a 5% CO2 atmosphere.5.11. Cytotoxicity assay
Approximately 5.0 × 104 MCF-7 cells were seeded into a 96-well plate and incubated at 37 °C in 5% CO2 overnight. The cells were treated with different concentrations of PSs (0 to 12 μM) and incubated overnight. Subsequently, each well was irradiated using a 660 nm diode laser at 30 mW for 20 min. After irradiation, cells were incubated under the same conditions overnight as earlier. The medium containing the PSs was replaced with 100 μl of 0.5 mg ml−1 MTT solution (Thiazolyl Blue Tetrazolium Bromide; Sigma-Aldrich, USA) and incubated at 37 °C in a 5% CO2 atmosphere under dark conditions for 3 h. After removal of 75 μl of MTT solution, 75 μl of DMSO (Sigma-Aldrich, USA) was added to each well to dissolve insoluble formazan crystals formed by reduction of MTT. The absorbance of formazan/DMSO solution at 540 nm was measured using a multiplate reader (Gemini XS, Molecular Devices, USA). The relative cell viability was analyzed based on the measured absorbance.5.12. Subcellular localization assay
About 1.0 × 106 MCF-7 cells were plated onto a cover slip in a 6-well plate and incubated for 24 h at 37 °C in 5% CO2. The cells were treated with various PSs (TPPS, TPP–PEG–biotin, TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin) at a concentration of 20 μM for 3 h (8 h for TPPS). Then, the treated solution was removed, and the cells were washed with 1× PBS. After washing, the cells were fixed with 4% paraformaldehyde solution (Abel Bio, Korea) for 15 min at room temperature. Subsequently, each well was washed with 1× PBS, and incubated with 0.2% saponin solution (Sigma-Aldrich, USA) for 10 min at room temperature to permeabilize the cell membrane of MCF-7, ensuring the staining of intracellular organelles. The cells were once again washed using 1× PBS, and stained with organelle-specific fluorescent probes such as Hoechst 33342, LysoTracker, MitoTracker, and ER-Tracker (Invitrogen, USA) for 20 min in the dark. Stained cells were washed with 1× PBS, and the cover slip was mounted onto a glass slide using a fluorescence mounting solution (Dako, USA). The cells were then observed and imaged using a confocal microscope (TCS SP8, Leica, Germany).5.13. Evaluation of the intracellular uptake by flow cytometry
Briefly, about 1.0 × 106 MCF-7 cells were plated onto a 6-well plate and incubated at 37 °C in 5% CO2 overnight. Then, the cells were treated with 1.5 μM DOX, and 5 μM TPPS, TPP–PEG–biotin, and TPP–PEG–biotin SANs for different times (30, 60, and 120 min) at 37 °C. After treatment, the cells were detached from the plate using trypsin (GIBCO, USA) for 3 min at 37 °C. The detached cells were washed with 1× PBS and centrifuged at 230 × g for 3 min. After centrifugation, the supernatant solution was removed and the cells were once again washed with 1× PBS followed by centrifugation for 3 min. The obtained cell pellet was resuspended in 500 μl of 1× PBS. Then, the fluorescence intensity of the PSs in MCF-7 was measured by flow cytometric analysis using an FACSCalibur™ (BD Biosciences, USA).5.14. Intracellular drug release
The MCF-7 cells were seeded into a confocal dish (SPL Life Sciences) at a density of 104 cells per dish in 1.0 ml of complete RPMI containing 10% fetal bovine serum, supplemented with 50 IU ml−1 penicillin and 50 IU ml−1 streptomycin. After incubation for 24 h, the culture media were withdrawn and culture media containing DOX@TPP–PEG–biotin and free DOX were supplemented. Subsequently, each dish was washed with 1× PBS. For staining the nuclei, the cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI) for 5 minutes. Stained cells were washed again with 1× PBS. Then, the cells were imaged using a confocal microscope (TCS SP8, Leica, Germany).5.15. Intracellular reactive oxygen species generation detection
The intracellular ROS detection experiment followed the manufacturer's protocol of a DCFDA kit. About 1.0 × 105 MCF-7 cells were seeded into a 48-well plate and incubated for 24 h at 37 °C in 5% CO2. The incubated cells were treated with TPP–PEG–biotin self-assembled nanoparticles at different concentrations (1 μM, 3 μM, and 5 μM) for 4 h at 37 °C under dark and light conditions. After treatment, the cells were washed with 1× PBS and treated with 20 μM 2′,7′-dichlorofluorescin diacetate (DCFDA; Abcam, UK) solution for 30 min under dark conditions. The cells were then irradiated using a 660 nm diode laser at 30 mW for 10 min. The DCFDA solution was removed and the cells were washed with 1× PBS. Lastly, the cells were observed and imaged using a fluorescence microscope (IX73, Olympus, Japan). The excitation and emission wavelengths were 495 nm/529 nm, respectively.5.16. High-content screening assay
MCF-7 cells were plated onto a 12-well plate and incubated at 37 °C in 5% CO2. The cells were treated with TPP–PEG–biotin SANs and DOX@TPP–PEG–biotin SANs at different concentrations (1 μM, 4 μM, and 10 μM) for 24 h. Subsequently, the cells were subjected to irradiation with a 660 nm diode laser at 30 mW for 30 min. After being thoroughly washed with 1× PBS, the cells were immediately treated with a calcium indicator orange solution (Invitrogen, USA) and incubated for 45 min under dark conditions at room temperature. The cells were washed with 1× PBS and treated with calcein acetoxymethyl ester (calcein-AM) solution contained in a MitoProbe Transition Pore Assay kit (Invitrogen, USA). Then, the cells were treated with a Magic Red™ caspase-3/7 substrate solution (ImmunoChemistry Technologies, USA) for 6 min under dark conditions at 37 °C. In turn, the cells were treated with CoCl2 and incubated for 10 min under dark conditions at 37 °C. Finally, the stained cells were washed with 1× PBS, and then imaged using an acousto-optic tunable filter (AOTF; TEAF10-0.45-0.7-s, Brimrose Corporation, USA) with excitation at 488 nm.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Ministry of Education, Science and Technology (MEST) (2016R1A4A1010796 and 2018M3A7B4071235). We are grateful to the Research Institute of Pharmaceutical Sciences at Seoul National University for providing experimental equipment and in vitro facility.References
- R. Krishna and L. D. Mayer, Eur. J. Pharm. Sci., 2000, 11, 265–283 CrossRef CAS.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714 CrossRef CAS PubMed.
- X. Feng, D. Jiang, T. Kang, J. Yao, Y. Jing, T. Jiang, J. Feng, Q. Zhu, Q. Song and N. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 17817–17832 CrossRef CAS PubMed.
- W. Zhang, J. Shen, H. Su, G. Mu, J.-H. Sun, C.-P. Tan, X.-J. Liang, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2016, 8, 13332–13340 CrossRef CAS PubMed.
- L. Xu, M. Zhao, Y. Yang, Y. Liang, C. Sun, W. Gao, S. Li, B. He and Y. Pu, J. Mater. Chem. B, 2017, 5, 9157–9164 RSC.
- L. Huang, G. Wei, X. Sun, Y. Jiang, Z. Huang, Y. Huang, Y. Shen, X. Xu, Y. Liao and C. Zhao, Eur. J. Med. Chem., 2018, 151, 294–303 CrossRef CAS.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene and D. Kessel, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- B. Q. Spring, I. Rizvi, N. Xu and T. Hasan, Photochem. Photobiol. Sci., 2015, 14, 1476–1491 RSC.
- E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2014, 115, 327–394 CrossRef.
- T. A. Debele, S. Peng and H.-C. Tsai, Int. J. Mol. Sci., 2015, 16, 22094–22136 CrossRef CAS.
- C. He, D. Liu and W. Lin, ACS Nano, 2015, 9, 991–1003 CrossRef CAS.
- Y. Lee, S. Lee, D. Y. Lee, B. Yu, W. Miao and S. Jon, Angew. Chem., Int. Ed., 2016, 55, 10676–10680 CrossRef CAS.
- X. Wang, Q. Zhang, J. Zhao and J. Dai, J. Mater. Chem. B, 2013, 1, 4637–4643 RSC.
- C. P. Leamon, Curr. Opin. Invest. Drugs, 2008, 9, 1277–1286 CAS.
- Y.-P. Zeng, S.-L. Luo, Z.-Y. Yang, J.-W. Huang, H. Li, C. Liu, W.-D. Wang and R. Li, J. Mater. Chem. B, 2016, 4, 2190–2198 RSC.
- W. X. Ren, J. Han, S. Uhm, Y. J. Jang, C. Kang, J.-H. Kim and J. S. Kim, Chem. Commun., 2015, 51, 10403–10418 RSC.
- G. Russell-Jones, K. McTavish, J. McEwan, J. Rice and D. Nowotnik, J. Inorg. Biochem., 2004, 98, 1625–1633 CrossRef CAS PubMed.
- S. Maiti and P. Paira, Eur. J. Med. Chem., 2018, 145, 206–223 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS PubMed.
- G. Pasut and F. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- C. Y.-S. Chung, S.-K. Fung, K.-C. Tong, P.-K. Wan, C.-N. Lok, Y. Huang, T. Chen and C.-M. Che, Chem. Sci., 2017, 8, 1942–1953 RSC.
- R. Zhang, R. Xing, T. Jiao, K. Ma, C. Chen, G. Ma and X. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 13262–13269 CrossRef CAS.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- R. Luguya, L. Jaquinod, F. R. Fronczek, M. G. H. Vicente and K. M. Smith, Tetrahedron, 2004, 60, 2757–2763 CrossRef CAS.
- S. Rangasamy, H. Ju, S. Um, D.-C. Oh and J. M. Song, J. Med. Chem., 2015, 58, 6864–6874 CrossRef CAS PubMed.
- Y. Dai, Z. Yang, S. Cheng, Z. Wang, R. Zhang, G. Zhu, Z. Wang, B. C. Yung, R. Tian, O. Jacobson, C. Xu, Q. Ni, J. Song, X. Sun, G. Niu and X. Chen, Adv. Mater., 2018, 30, 1704877 CrossRef.
- S. S. Jalde, A. K. Chauhan, J. H. Lee, P. K. Chaturvedi, J.-S. Park and Y.-W. Kim, Eur. J. Med. Chem., 2018, 147, 66–76 CrossRef CAS.
- C. Conte, F. Ungaro, G. Maglio, P. Tirino, G. Siracusano, M. Sciortino, N. Leone, G. Palma, A. Barbieri and C. Arra, J. Controlled Release, 2013, 167, 40–52 CrossRef CAS.
- Y. Chen, F. Zhang, Q. Wang, R. Tong, H. Lin and F. Qu, Dalton Trans., 2017, 46, 14293–14300 RSC.
- J. G. Vineberg, E. S. Zuniga, A. Kamath, Y.-J. Chen, J. D. Seitz and I. Ojima, J. Med. Chem., 2014, 57, 5777–5791 CrossRef CAS PubMed.
- B. Wilson, M. Olivo and G. Singh, Photochem. Photobiol., 1997, 65, 166–176 CrossRef CAS PubMed.
- Y. Lee, S. Lee, D. Y. Lee, B. Yu, W. Miao and S. Jon, Angew. Chem., 2016, 128, 10834–10838 CrossRef.
- P. Ju, J. Hu, F. Li, Y. Cao, L. Li, D. Shi, Y. Hao, M. Zhang, J. He and P. Ni, J. Mater. Chem. B, 2018, 6, 7263–7273 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb01923a |
| This journal is © The Royal Society of Chemistry 2019 |