
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessment of capillary volumetric blood microsampling for the analysis of central nervous system drugs and metabolites†
Michele
Protti‡
 a,
Camilla
Marasca‡
ab,
Marco
Cirrincione
a,
Andrea
Cavalli
bc,
Roberto
Mandrioli
d and
Laura
Mercolini
*a
a,
Camilla
Marasca‡
ab,
Marco
Cirrincione
a,
Andrea
Cavalli
bc,
Roberto
Mandrioli
d and
Laura
Mercolini
*a
aResearch Group of Pharmaco-Toxicological Analysis (PTA Lab), Department of Pharmacy and Biotechnology (FaBiT), Alma Mater Studiorum – University of Bologna, Via Belmeloro 6, 40126 Bologna, Italy. E-mail: laura.mercolini@unibo.it
bItalian Institute of Technology (IIT), Via Morego 30, 16163 Genoa, Italy
cDepartment of Pharmacy and Biotechnology (FaBiT), Alma Mater Studiorum – University of Bologna, Via Belmeloro 6, 40126 Bologna, Italy
dDepartment for Life Quality Studies, Alma Mater Studiorum – University of Bologna, Corso d'Augusto 237, 47921 Rimini, Italy
First published on 4th August 2020
Abstract
Therapeutic drug monitoring (TDM) is an important tool for correlating the administered drug dose to drug and metabolite concentrations in the body and to therapeutic and adverse effects. In the case of treatment with drugs active on the central nervous system (CNS), frequent TDM becomes really useful, especially for patient compliance checking and for therapy optimisation. The selective serotonin reuptake inhibitors (SSRIs) fluoxetine and sertraline, chosen as target compounds for this study, are two antidepressants mainly used for major depression, but also for obsessive-compulsive disorder associated with neurodegenerative diseases and for eating disorders. Microsampling approaches can be used to make TDM patient-friendly, by means of minimally invasive fingerpricking instead of classic invasive venipuncture. In this study, an innovative volumetric microsampling approach based on the use of hemaPEN technology is proposed to simultaneously obtain four identical dried whole blood microsamples by means of a single capillary sampling. The developed strategy shows significant advantages in terms of blood collection and storage, fast and feasible extraction procedure and sensitive LC-MS/MS analysis, also providing satisfactory validation results (extraction yield >81%, RSD <12.0%, and <6.3% loss in analyte stability after 3 months). The proposed methodology has proven to be sound and reliable for application to the TDM of psychiatric patients treated with antidepressant drugs such as fluoxetine and sertraline. The original capillary volumetric microsampling procedure using hemaPEN has been demonstrated to be suitable for the accurate sampling of capillary whole blood, in order to be successfully exploited in self- and home-sampling procedures in future and to pave the way for precision medicine approaches for the treatment of CNS disorders.
Introduction
The use of dry microsampling approaches by finger pricking avoids recourse to the traditional invasive practice of large-volume venipuncture. It also simplifies post-collection processing steps, reduces biohazard risks and increases the feasibility of sample storage and shipment. Due to water loss, dried microsamples can usually be stored at room temperature (RT) without significantly impacting analyte stability.1 In some cases, stability in dried micromatrices at RT is better than that in fluid matrices stored at freezing or sub-freezing temperatures.2 This in turn can lead to cost decrease when compared to refrigerated storage, and to a decrease in overall analysis times, thanks to the increased possibility of implementing high-throughput, automated strategies. The collection of capillary whole blood on filter paper to generate dried blood spots (DBS) is one of the best known alternative dry microsampling strategies, used in screening tests for neonatal metabolic diseases.3 During the past decade, DBS approaches have been successfully applied for the monitoring of different classes of drugs such as antibiotics, antidepressants, antiepileptics, antipsychotics, and immunosuppressants.4–9However, DBS techniques can suffer from lack of sampling accuracy and spot area variability, mainly due to haematocrit (HCT) effects. In order to overcome these issues, other microsampling approaches closely related to DBS have been investigated such as dried plasma spots (DPS) and dried serum spots (DSS), as well as newer possibilities such as volumetric absorptive microsampling (VAMS) and microfluidic-based techniques.10–16
A recently developed technology based on capillary volumetric blood microsampling (hemaPEN®) allows the simultaneous collection of four identical DBS replicates (2.74 μL each) from a single sample, thanks to the presence of four end-to-end EDTA-coated microcapillaries. The pen-like design of this device makes it easy to handle and, with the inclusion of a desiccant, sample integrity is ensured while preventing any contamination. It is a single use and tamper-resistant device, requiring a specific opening tool for the retrieval of DBS samples.17 The HCT independence of blood microsamples collected by means of hemaPEN technology has been studied by quantifying caffeine and its metabolite paraxanthine in dried whole blood.18 Moreover, hemaPEN has been tested in a study on iohexol for kidney function assessment.19
Alternative and more patient-friendly microsampling strategies are particularly attractive in order to promote accurate therapeutic drug monitoring (TDM) practices, i.e. the constant monitoring of drug and metabolite concentrations in haematic samples during pharmacotherapy.
Dry microsampling is especially suitable for the simplification of TDM procedures in delicate populations, such as paediatric patients and those affected by central nervous system (CNS) disorders, and to facilitate self- and home-sampling procedures.20 In fact, TDM plays a central role in achieving the optimisation and personalisation of drug therapy. Clinical decision-making benefits from the knowledge of chemical–clinical correlations between administered dose and drug and metabolite concentrations in biological fluids, and between dose and therapeutic effect but also side effects. Consequently, this practice dramatically increases both therapeutic efficacy and safety, as a key component of personalised and precision medicine regimens.21 Frequent TDM becomes essential for delicate patients under therapy with drugs active on the CNS, especially in the cases of polypharmacy regimens, often prescribed for the treatment of symptoms associated with neurodegenerative diseases.22,23 This is becoming relatively common for antidepressant drugs such as the best-known tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs) and selective serotonin reuptake inhibitors (SSRIs); in this last class, fluoxetine (FLX) and sertraline (SRT) are among the most frequently administered antidepressants.24–27 SSRIs are so-called “second-generation” or “new-generation” antidepressants and their efficacy is similar to that of TCAs and MAOIs, but their safety profile is usually considered to be more favourable.
Within this research work, the chosen target analytes were FLX (N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine, Fig. 1a) together with its active metabolite norfluoxetine (Fig. 1b, NFLX) and SRT ((1S,4S)-4-(3,4-dichlorophenyl)-N-methyl-1,2,3,4-tetrahydronaphthalen-1-amine, Fig. 1c) along with N-desmethylsertraline (NSRT, Fig. 1d), its main active metabolite.28 The commonly accepted plasma therapeutic ranges for the analytes are: FLX + NFLX, 120–500 ng mL−1; SRT, 10–150 ng mL−1; and NSRT, 15–500 ng mL−1.29
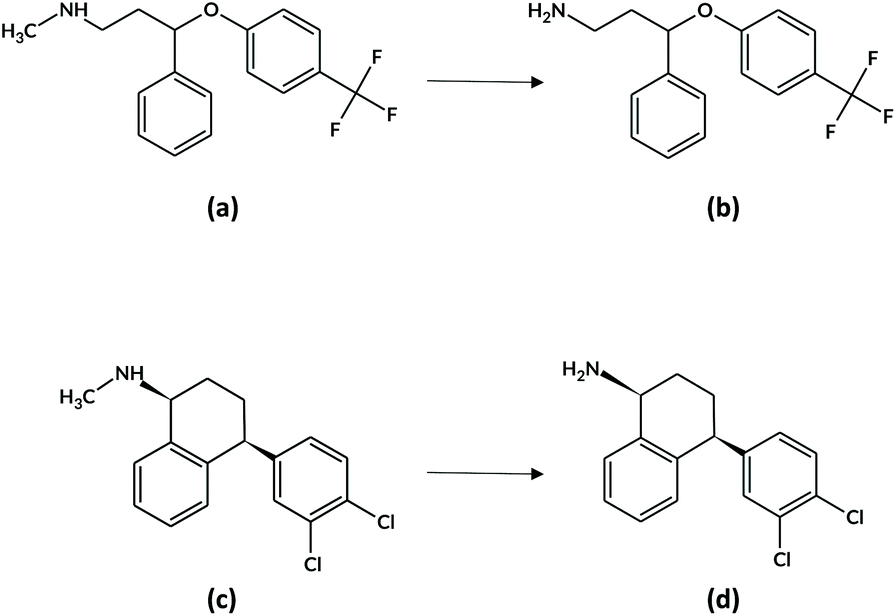 | ||
| Fig. 1 Chemical structures of (a) FLX and its metabolite (b) NFLX, and (c) SRT and its metabolite (d) NSRT. | ||
Microsampling approaches have been exploited for simultaneous determination of FLX and NFLX in DBS using high-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) and fast gas chromatography-tandem mass spectrometry (GC-MS/MS).30,31 Rat PK studies have been performed by means of VAMS technique to collect dried blood samples after FLX administration.32 With regards to SRT, whole blood drops were spotted on alginate and chitosan foams as the sampling media and on commercial cards for comparison.33 Twenty-two antidepressants, including FLX and SRT (without metabolites), have been determined in DBS after collection of post-mortem samples for toxicological purposes.34
To the best of our knowledge, this is the first time that a microsampling approach based on hemaPEN and coupled to LC-MS/MS is used for the TDM of patients undergoing treatment with CNS drugs. The aim of this study is to develop and validate an original method for miniaturised sampling and pretreatment, in order to evaluate hemaPEN applicability for TDM purposes. In perspective, this approach could improve therapy efficacy and allow personalisation within the framework of psychiatric pharmacological regimens, including the treatment of depressive symptoms in neuro-degenerative diseases such as amyotrophic lateral sclerosis, Alzheimer's disease and frontotemporal dementia.
Experimental
Instrumentation, chemicals and chromatographic conditions
FLX hydrochloride, NFLX hydrochloride, SRT hydrochloride and NSRT hydrochloride, all pure powders (>99% purity), were purchased from Sigma-Aldrich (Milan, Italy); internal standard (IS) reference solutions of FLX-D6 oxalate (1 mg mL−1), NFLX-D6 oxalate (1 mg mL−1) and SRT-D3 hydrochloride (100 μg mL−1) were purchased from Cerilliant (Round Rock, TX, US); NSRT-D4 hydrochloride was purchased from LGC Standards (Teddington, Middlesex, UK). HPLC-grade (>99.8%) methanol, acetonitrile (ACN) and 95% formic acid (FA), all pure for analysis, were bought from Sigma-Aldrich. Ultrapure water (18.2 MΩ cm) was obtained by means of a Milli Q apparatus from Millipore (Milford, USA). HemaPEN devices were kindly provided by Trajan Scientific and Medical (Ringwood, Australia). Stock solutions of analytes and ISs were prepared at 1 mg mL−1 in methanol and stored at −20 °C in amber glass vials, and were stable for at least 2 months, as assessed by LC-MS/MS analysis. Working solutions for calibrators and QCs were prepared daily in a mixture of 0.1% FA in acetonitrile and 0.1% FA in water (50/50, v/v).The HPLC-MS/MS system includes a Waters (Milford, MA, USA) Alliance e2695 chromatographic pump equipped with an autosampler and coupled to a Waters Micromass Quattro Micro triple quadrupole mass spectrometer. Chromatographic separation was performed on a Waters Sunfire C18 reversed-phase column (50 × 3.0 mm, 3.5 μm) coupled to a Sunfire C18 guard column, both kept at RT. The mobile phase was a mixture of 0.1% FA in acetonitrile and 0.1% FA in water, flowing at 0.1 mL min−1 with the following gradient composition: 0.0–2.0 min, constant 20% A; 2.1–3.5 min, linear 20%–40% A gradient; 3.6–6.4 min, constant 40% A; 6.5–7.5 min, linear 40%–20% A gradient; and 7.6–8.0 min, constant 20% A to re-equilibrate the column. The injection volume was 10 μL.
Tandem mass spectrometry acquisition was carried out in multiple reaction monitoring (MRM) mode, using an electrospray ionisation source in positive mode (ESI+). For all analytes and ISs, the quasi-molecular [M + H]+ ion was selected and acquired, and two different MRM transitions for the analytes were chosen: the most abundant one for quantitation and the second most abundant one for identity confirmation, while for the ISs a single daughter ion was selected. The optimised parameters were as follows: ion source voltage 3.8 kV, ion source temperature 125 °C, desolvation gas temperature 200 °C, and desolvation gas flow rate 250 L h−1. Nitrogen was used as the desolvation gas, while argon was used as the collision gas. Precursor and product ions, with collision energy, cone voltage and dwell time values for each analyte and ISs were optimised. Waters MassLynx 4.1 software was used to acquire and process the LC-MS/MS data.
Sample collection and pretreatment
For this evaluation study, whole blood for both method validation and for method application was collected in the fluid form, then DBS samples were generated by means of hemaPEN technology. Blank blood samples for method validation were withdrawn by fingerpricking from healthy volunteers, not subjected to any pharmacological treatment, in sterile plastic tubes with EDTA coating. Whole blood samples for method application were instead obtained from in- and out-patients of different Italian psychiatric clinics and hospitals receiving antidepressants as part of their pharmacological treatment (these samples had been already collected for general needs). Spiked whole blood samples (fortified with both analytes and ISs for method validation and with ISs only for method application) were prepared by adding to 90 μL of whole blood 10 μL of a mixture of 0.1% FA in acetonitrile and 0.1% FA in water (50/50, v/v) containing analytes and/or ISs.All hemaPEN DBS samples were prepared by barely touching with the tip of all four capillaries the surface of a drop of blood positioned on a hydrophobic surface, to simulate the droplet generated by fingerpricking. The device was held for 10 seconds in a tilted position to facilitate the filling of capillaries, then flipped over and closed with the dedicated cap to trigger the emptying of the whole capillary volume on the pre-cut disks inside the hemaPEN (Fig. ESI1 of the ESI†). The four obtained 2.74 μL DBS were left to dry at RT for 1 hour inside the device, without additional packaging in the case of immediate pretreatment and analysis. In the case of delayed analysis, after drying the already sampled hemaPENs were stored inside sealed zip-lock bags. Each device was then opened by means of a dedicated cutter tool, then a spot was retrieved, transferred into a centrifuge microtube together with 50 μL of pure methanol and subjected to ultrasound-assisted extraction (UAE) for 30 minutes, followed by centrifugation for 10 min at 3200 rpm at RT. An aliquot of 10 μL of the supernatant was then injected into the LC-MS/MS system.
In order to evaluate the analytical performance of the proposed dry microsampling approach, the results obtained from the hemaPEN samples were compared to those obtained by reference plasma analysis. Both hemaPEN and plasma samples were obtained from fluid whole blood from patients under antidepressant treatment. For the analysis of plasma using fully validated procedures, whole blood samples were centrifuged at 3200 rpm for 15 min in EDTA-coated tubes, then the plasma aliquots were transferred into polypropylene tubes and stored at −20 °C until pretreatment and analysis. Plasma samples were subjected to solid phase extraction (SPE), as described in previously published research papers.35,36 Then, the methanolic extracts were analysed by LC-MS/MS under the previously described conditions.
Method validation
Method validation was carried out following the European Medicines Agency (EMA) and U.S. Food and Drug Administration (FDA) guidelines on bioanalytical method validation also taking into account DBS-specific parameters.37,38For linearity, aliquots of 10 μL of standard solutions containing the target compounds at seven different concentrations and ISs at a constant concentration of 50 ng mL−1, were added to 90 μL of fluid blank whole blood. After hemaPEN collection, the pretreatment procedure previously described was applied to dried spiked samples, which were then analysed by LC-MS/MS (n = 3). The calibration curve was set-up by means of the least-square method, plotting analyte/IS peak area ratios (pure numbers) against the nominal analyte concentrations (expressed as ng mL−1) and the lower limit of quantification (LLOQ) was set at the level of the lowest calibrator.
Extraction yield and precision were evaluated by adding known amounts of the analytes (at three different QC concentrations, corresponding to the LLOQ, an intermediate point and the upper value of the calibration curve) to blank blood samples, pretreated and analysed by LC-MS/MS. The extraction yield was calculated by comparing analyte peak areas with those of standard solutions at the same theoretical concentrations and expressed as percentage recovery. Intra-day and inter-day precision values were obtained by repeating the assays six times on the same day and over six different days, respectively, both expressed as percentage relative standard deviation (RSD%).
For selectivity, blank hemaPEN samples from six different volunteers were collected, pretreated and injected into the LC-MS/MS system, and the chromatogram of the blank samples was compared with the peak area of the LLOQ of each analyte, at their respective retention time. Selectivity was considered acceptable if any interfering peak area was ≤20% of the LLOQ peak area of each analyte (≤5% for ISs).
Matrix effect of DBS generated by means of hemaPEN was assessed on blank sample extracts from three different sources, spiked after extraction by adding known amounts of analytes at three concentrations and ISs at fixed concentration. Then, analyte/IS peak area ratios of each extract were compared with analyte/IS peak area ratios from standard solutions and the resulting percentage variation was calculated. The matrix effect was considered acceptable when it was within ±15% of the nominal value.
Carryover was evaluated by means of solvent blank injection immediately after the extraction of blood samples, spiked with analyte concentrations equivalent to the upper limit of linearity ranges. The acceptance criterion was no analyte peak higher than 20% of LLOQ levels (5% for IS).
Hematocrit effect
To evaluate the possible impact of HCT on analyte recovery and matrix effect, two sets of whole blood samples were prepared with low (0.30), medium (0.50) and high (0.70) HCT levels. The first set of fluid whole blood samples was fortified with the analytes and ISs at three different concentration levels (LLOQ, an intermediate point and the upper value of each calibration curve) before hemaPEN sampling and processing. The second set was left blank and sampled with hemaPEN, while analytes and ISs were added directly into the extraction solvent. Extraction yield (expressed as % recovery) was calculated by comparing peak areas of the set spiked before extraction with those of the corresponding samples spiked after extraction. The recoveries of the low- and high-HCT samples were then compared to the extraction recovery of the medium-HCT samples. Recovery was considered HCT-independent when that of low- and high-HCT hemaPEN samples was within ±15% of that observed for medium-HCT. In order to evaluate HCT impact on matrix effect, the set of samples fortified post-extraction were compared with analyte standard solution analysis at the same nominal concentrations as previously described and the resulting percentage was calculated. The matrix effect was considered independent from HCT when the calculated percentage recovery of low- and high-HCT hemaPEN samples was within ±15% of that observed for medium-HCT.Stability of hemaPEN DBS
Long-term stability (3 months) was assessed by storing DBS samples spiked with the analytes within intact hemaPEN enclosed in sealable zip-lock bags at RT, at two concentration levels (LLOQ and the upper value of each calibration curve). Short-term stability (5 days) was investigated at 50 °C and at the same two concentration levels. The acceptance criterion for stability was analyte losses lower than 15% of the nominal values.HemaPEN misuse testing
The misuse of hemaPEN has been tested in terms of possible undersampling or oversampling, subjecting blank whole blood spiked with analytes at two different concentration levels (LLOQ and the upper value of each calibration curve) to hemaPEN sampling, and applying contact times different from that indicated by the manufacturer. In particular, contact times of 2, 4, 6, and 8 s (for possible undersampling), and 15 and 20 s (for possible oversampling) were tested. The analytical responses (in terms of % recovery) were compared with those obtained by standard 10 s sampling and practical considerations on the quality of the DBS samples were made. The assays were repeated in 6 replicates.Clinical application
Having validated the analytical methodology involving hemaPEN microsampling for the production of volumetrically accurate DBS samples in combination with LC-MS/MS analysis, the procedure was applied to real samples from patients affected by CNS diseases and undergoing therapy with FLX and/or SRT, in order to determine, for TDM purposes, the concentrations of the parent compounds together with their main active metabolites, namely NFLX for FLX and NSRT for SRT. The experiments were performed in compliance with the relevant laws and institutional guidelines, in particular according to D.Lgs. 30/06/2003 n. 196, as amended and complemented by D.Lgs. 10/08/2018, n. 101 and to General Authorisation 15/12/2016, n. 9 issued by the Italian Data Protection Authority. Institutional committee approval of the experiments was not required, since all samples had already been collected for general needs related to the therapy and anonymised immediately after collection and all subjects provided informed consent prior to their participation. Completely anonymised biological samples are considered to have been destroyed according to the above mentioned legislature and guidelines.Since the whole blood samples had already been collected by classic blood withdrawal into tubes containing anticoagulant, the proof of concept presented herein was assessed by sampling whole blood microvolumes using hemaPEN for the generation of DBS samples which were then pre-treated and analysed according to the procedures previously described.
The same patient blood samples in the fluid form were then analysed by means of reference, fully validated procedures (see the ESI,† section EXP1) and the results obtained from hemaPEN analysis were compared with those from the analysis of fluid samples in order to verify the overlapping of the qualitative–quantitative data from microsample analysis.
All the results obtained from real samples were compared by plotting the results from hemaPEN samples versus those from fluid whole blood ones. Then, the least-square method was applied to calculate linearity correlation coefficient and slope of each curve, while Bland Altman plots were built to evaluate biases between results from microsamples and those from whole blood analysis.
For accuracy, analyte standard solutions at three concentrations were added to hemaPEN patient samples whose analyte content was already assessed. The samples were then pretreated and analysed and accuracy was expressed as percentage recovery. Assays were repeated in triplicate and SD was calculated.
Results and discussion
Mass spectrometry analysis
All spectra of the analytes and ISs were acquired using the ESI+ ionisation mode (50–600 m/z) by direct infusion of 1 μg mL−1 solutions at 20 μL min−1, using a mixture of 0.1% FA in acetonitrile and 0.1% FA in water (50/50, v/v) for dilutions, in order to choose the best MS/MS conditions for each analyte and IS. A complete overview of optimised MRM transitions and MS/MS parameters is reported in Table 1.| Analyte/IS | MW | Q1 (m/z) | Q3 (m/z) | Dwell time (s) | Cone voltage (V) | Collision energy (eV) |
|---|---|---|---|---|---|---|
| FLX | 309.33 | 310.13 | 148.1 | 0.300 | 23 | 17 |
| 44.2 | 12 | |||||
| NFLX | 295.31 | 296.11 | 134.9 | 0.300 | 16 | 11 |
| 30.3 | 8 | |||||
| SRT | 306.23 | 307.34 | 275.2 | 0.300 | 20 | 25 |
| 159.1 | 15 | |||||
| NSRT | 292.20 | 293.17 | 159.0 | 0.300 | 18 | 21 |
| 129.1 | 13 | |||||
| FLX-D6 | 315.37 | 316.33 | 154.2 | 0.300 | 18 | 16 |
| NFLX-D6 | 301.34 | 302.33 | 150.1 | 0.300 | 13 | 10 |
| SRT-D3 | 309.25 | 310.30 | 278.3 | 0.300 | 18 | 22 |
| NSRT-D4 | 296.23 | 297.20 | 163.1 | 0.300 | 15 | 19 |
Development of hemaPEN sampling and pretreatment
The whole blood volume absorbed by pre-cut DBS after hemaPEN sampling was investigated by measuring the weight differences between individual, disassembled pre-cut discs and the corresponding DBS samples after hemaPEN sampling (n = 6). Then, the accuracy and precision of sampling volume were calculated. HemaPEN volume testing confirmed the high mean volume accuracy (102%) and precision (RSD = 1.9%) with respect to the capillary volume capacity of 2.74 μL stated by the manufacturer.With regards to the sampling time, it has been studied in terms of possible undersampling or oversampling and will be discussed separately (section Evaluation of hemaPEN misuse).
Gravimetric assays on the time required to dry a set of four 2.74 μL DBS samples within hemaPEN showed that the samples were fully dried within 1 h after closing the device.
For the development of the sample pretreatment protocol, dried blood spotting itself can be considered a form of sample pretreatment, as the analyte-matrix-cellulosic support interactions can be exploited to selectively desorb and extract the analytes from the spot. Several solvent extraction procedures were investigated, by testing different solvents (methanol, acetonitrile, water, ethyl acetate, phosphate buffer solution and their mixtures), solvent volumes (50–1000 μL), extraction times (1–60 min) and extraction methods (vortex-, ultrasound- and microwave-assisted extraction). Methanol provided better results in terms of extraction yield and matrix purification when compared to other solvents and solvent mixtures, with satisfactory results even when using a volume as low as 50 μL, while higher volumes did not lead to significantly better performances. The use of assistive technologies in the extraction step provided increased yields, with better performances using UAE (with respect to vortex-assisted and microwave-assisted extraction) with extraction yields reaching a plateau after 30 minutes, so this was chosen as the best compromise between yield and analysis time.
Observing the satisfactory results obtained in terms of the extraction yield and purification of the matrix from potential interfering signals, it was deemed unnecessary to introduce further purification steps, in order to maintain high procedure effectiveness and feasibility. Therefore, after centrifugation of the extract, the supernatant was transferred into an autosampler vial and directly injected into the LC-MS/MS system.
Validation of the hemaPEN and LC-MS/MS method
Calibration curves were set up for the four target compounds in samples collected by means of hemaPEN, using blank whole blood fortified with the analytes at seven different concentrations. Good linearity was observed for all analytes: for FLX and NFLX, r2 ≥ 0.9991 was calculated over the 7–750 ng mL−1 concentration range, with LLOQ and LOD values of 7 and 2.5 ng mL−1, respectively. For SRT and NSRT, r2 ≥ 0.9993 was calculated over the 5–500 ng mL−1 range with LLOQ and LOD values of 5 and 1.5 ng mL−1, respectively. The comprehensive results of these assays are reported in Table 2.| Analyte | Linearity range, ng mL−1 | r 2 | LLOQ, ng mL−1 | LOD, ng mL−1 |
|---|---|---|---|---|
| FLX | 7–750 | 0.9996 | 7.0 | 2.5 |
| NFLX | 7–750 | 0.9992 | 7.0 | 2.5 |
| SRT | 5–500 | 0.9997 | 5.0 | 1.5 |
| NSRT | 5–500 | 0.9994 | 5.0 | 1.5 |
The extraction yield and precision assays were carried out at three different concentration levels of FLX, NFLX, SRT and NSRT, (ISs were added at a constant concentration of 50 ng mL−1). The results were satisfactory, with extraction yields always higher than 81% for all analytes and ISs. Precision data were also good, with RSD values lower than 7.3% (<9.4% at the LLOQ level) for intraday data and lower than 9.2% (<12.0% at the LLOQ level) for inter-day data. Complete results are reported in Table 3.
| Analyte | Concentration levela | Extraction yield,b % | Precision, RSD%c | Matrix effect,b % | |
|---|---|---|---|---|---|
| Intra-day | Inter-day | ||||
| a For each analyte, “low” corresponds to the LLOQ, “intermediate”, to an intermediate point and “high” to the upper limit of each linearity curve. b n = 6, mean value. c n = 6. | |||||
| FLX | Low | 87 | 8.7 | 9.5 | 7.4 |
| Intermediate | 90 | 6.5 | 9.1 | 7.2 | |
| High | 92 | 5.4 | 6.3 | 3.6 | |
| NFLX | Low | 86 | 9.0 | 10.2 | 6.3 |
| Intermediate | 88 | 6.8 | 8.4 | 5.8 | |
| High | 90 | 5.5 | 5.9 | 3.3 | |
| SRT | Low | 88 | 8.6 | 9.1 | 6.0 |
| Intermediate | 90 | 6.8 | 7.9 | 4.1 | |
| High | 91 | 5.2 | 6.4 | 1.9 | |
| NSRT | Low | 82 | 9.3 | 11.9 | 7.5 |
| Intermediate | 85 | 7.2 | 9.0 | 6.7 | |
| High | 86 | 5.8 | 6.2 | 4.4 | |
| FLX-D6 | 50 ng mL−1 | 93 | 5.2 | 6.1 | 3.8 |
| NFLX-D6 | 50 ng mL−1 | 92 | 5.3 | 6.0 | 4.0 |
| SRT-D3 | 50 ng mL−1 | 92 | 5.0 | 6.2 | 1.8 |
| NSRT-D4 | 50 ng mL−1 | 89 | 5.5 | 6.1 | 3.4 |
Selectivity and carryover were within the defined limits: maximum interference signal was <20% of the LLOQ area and carryover was <5% of the respective IS area.
The possible matrix effect was also investigated by injecting blank hemaPEN samples from three different subjects, spiked after extraction with the analytes at three concentration levels (ISs at a constant concentration) and comparing the results with those of standard solutions at the same nominal concentrations. Analyte matrix effects were below 8% for all analytes, as shown in Table 3, thus meeting the defined acceptance criterion of 15%.
Evaluation of haematocrit impact
In order to evaluate the possible impact of HCT on the analytical performance of the hemaPEN-generated DBS approach, specific extraction yield and matrix effect assays were carried out, by taking into account three concentration levels for each analyte and three representative HCT values (0.30, 0.50 and 0.70).The extraction yields were demonstrated to be HCT-independent (i.e., always within ±15% of the recovery for hemaPEN-generated DBS from blood with 0.5 HCT), as shown in Fig. 2.
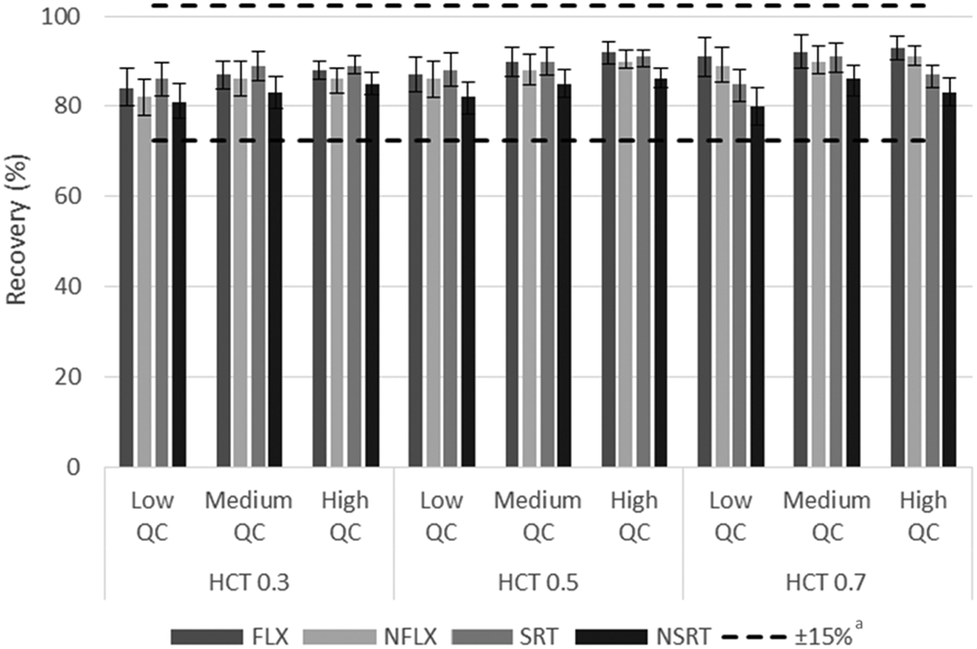 | ||
Fig. 2 Assays results based on the relationship between extraction yield and blood HCT. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Referred to the average values of HCT 0.5. Referred to the average values of HCT 0.5. | ||
The matrix effect was calculated as the ratio of analyte peak areas in the presence of hemaPEN-DBS matrix components to the peak area without the same matrix. The magnitude of the matrix effect was within the 3–9% range for all analytes and all HCT values.
Stability assay results
All the analytes considered in this study were stable in hemaPEN DBS samples for at least 3 months at RT (analyte losses within 6.3% of the nominal concentration) and 5 days at 50 °C (analyte losses <5.2%), thus well within the defined acceptability criterion of 15% analyte loss, when storing the intact hemaPEN devices in their original packages. Complete results of the stability study are reported in Table 4. In particular, it can be seen how stability is satisfactory for all analytes under both considered storage conditions, providing the preconditions for effective storage even at non-controlled temperatures.Evaluation of hemaPEN misuse
Possible undersampling or oversampling of DBS generated by hemaPEN was assessed by carrying out sampling time assays: spiked blank whole blood was subjected to hemaPEN sampling for different times, ranging from 2 to 20 s (the optimal sampling time defined by the manufacturer is 10 s). Then, recoveries were compared and the technical performance (in terms of the number of valid samples generated) was assessed. Complete results are reported in Fig. 3: as can be seen, for valid microsamples no significant undersampling or over-sampling was observed at any of the considered sampling times, as the analytical response ranged from 99.4% to 101.5% of that obtained with the standard 10 s sampling time.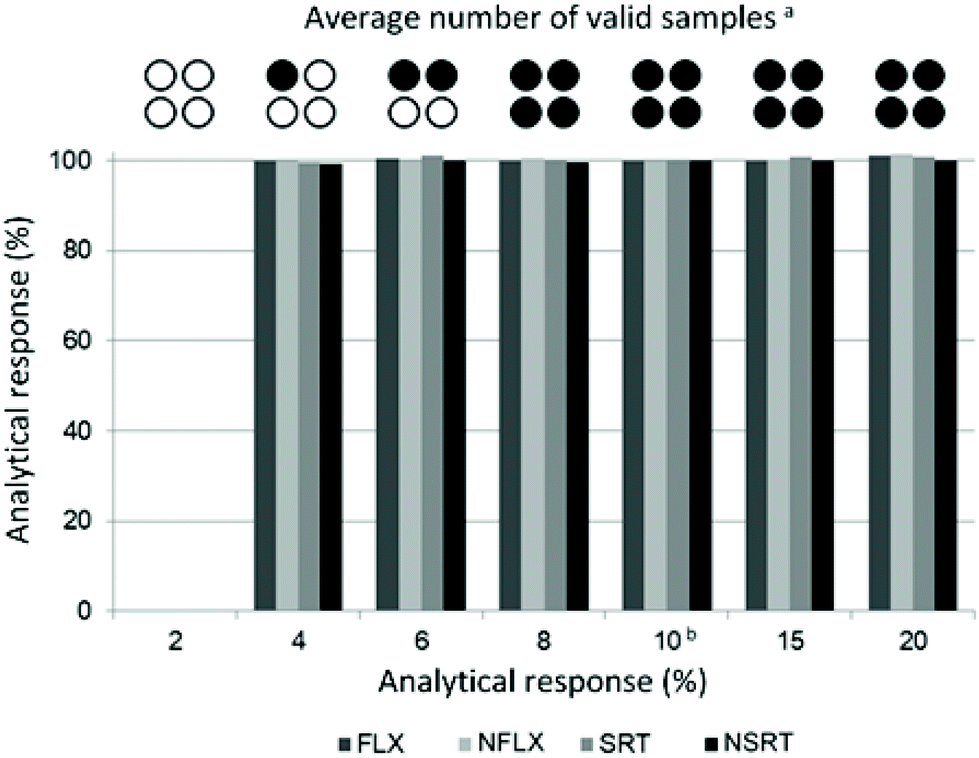 | ||
| Fig. 3 HemaPEN misuse results. an = 6. bSampling time defined by the manufacturer. | ||
As far as technical performance is concerned within the assays carried out in this study, it can be seen that as sampling time decreases, a statistically greater chance of obtaining invalid samples was observed (n = 6). In particular, 8 s of contact time generated 100% valid samples, while 6 s of contact time produced an average of 2 valid samples out of 4. At 4 s, the average was 1 out of 4, while with a contact time of just 2 s it was not possible to generate any DBS sample within the 6 assay repetitions. This is due to the fact that for contact times shorter than the recommended 10 s there is a statistical possibility that not all capillaries are completely filled. Consequently, when the device is closed, the absence of contact between the whole blood matrix and the pre-cut disc prevents the emptying of the capillary, resulting in a null sample. Despite representing a potential sampling issue, there is no risk of oversampling (the capillaries cannot be overfilled) nor undersampling. In fact, instead of having lower-volume samples, a visibly invalid sample is obtained, which would allow the operator to repeat the sampling instead of having analytical underestimates.
Clinical application
After validating the microsampling, pretreatment and analysis workflow of hemaPEN samples, real samples from psychiatric patients undergoing treatment with FLX and SRT were analysed. Whole blood microvolumes were sampled using hemaPEN and the generated DBS samples were pretreated and analysed according to the described procedures. As method application examples, two chromatograms of hemaPEN samples from patients treated with FLX (20 mg day−1) and SRT (50 mg day−1) are shown in Fig. 4a and b, respectively. For these samples, the observed concentrations were: FLX 26.4 ng mL−1 and NFLX 42.7 ng mL−1 (Fig. 4a); and SRT 27.2 ng mL−1 and NSRT 38.1 ng mL−1 (Fig. 4b).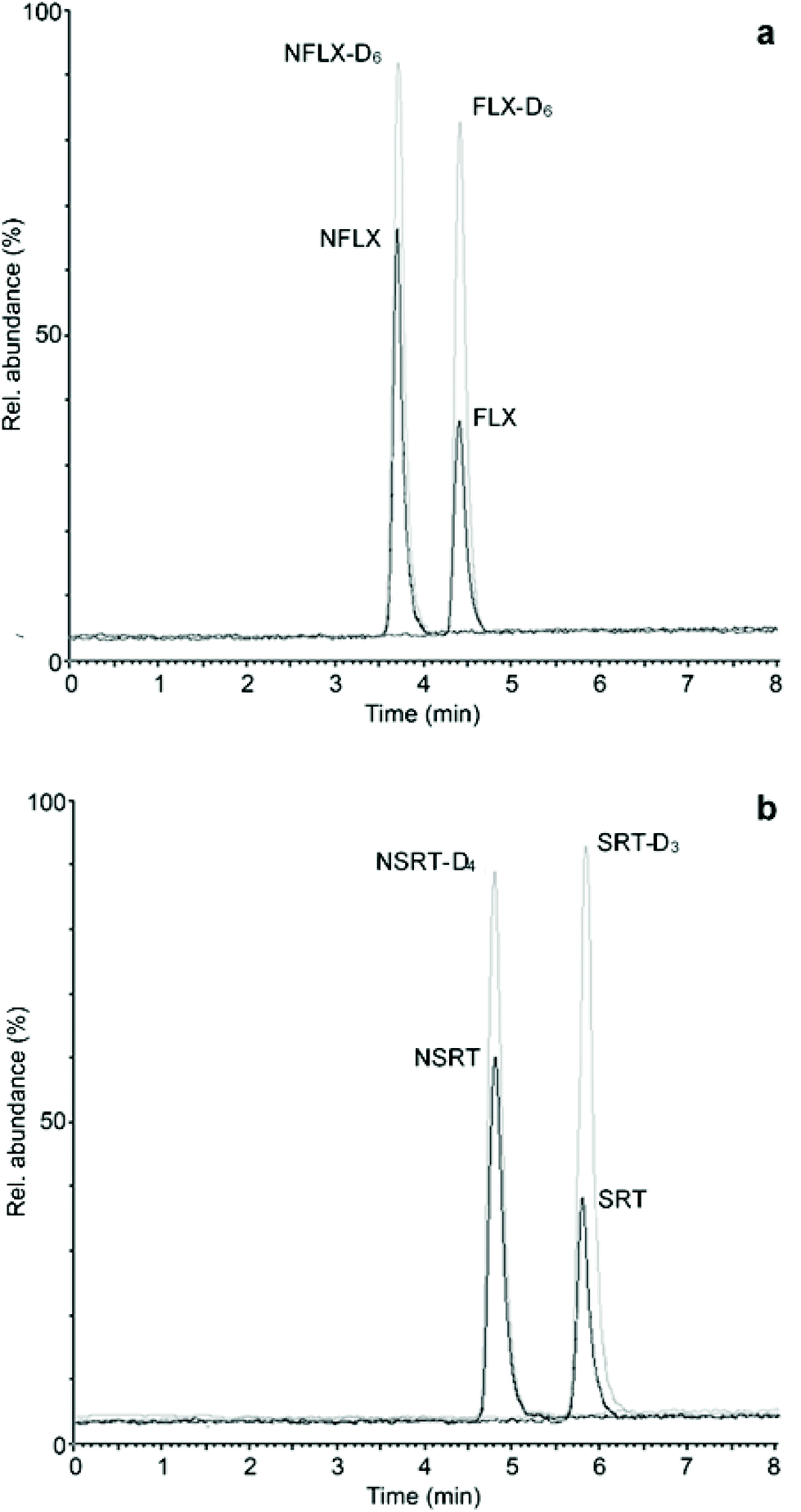 | ||
| Fig. 4 LC-MS/MS chromatograms of hemaPEN samples from patients treated with (a) FLX and (b) SRT. | ||
The same whole blood samples were analysed by means of fully validated procedures based on SPE and plasma analysis and the results obtained from the latter were compared with those from hemaPEN, after converting plasma concentrations to whole blood concentrations by using the respective haematocrit values and mean erythrocyte-to-plasma concentration ratios previously determined for FLX and NFLX (1.08) and for SRT (1.16) and NSRT (1.58).39,40
The results obtained from hemaPEN samples, after conversion factor application, are always in good agreement with those found in the corresponding fluid samples. To effectively compare the two datasets, linear correlation and Bland–Altman plots were generated for hemaPEN DBS/fluid whole blood (Fig. 5). These results, together with correlation analysis, prove the good agreement between hemaPEN and their corresponding fluid matrix (Fig. 5a): correlation analysis produced r2 values ≥0.9896 for the whole dataset (see Table ESI1 of the ESI† for linear regression parameter of each analyte), while the Bland–Altman plot describes a mean bias between hemaPEN and whole blood results of −0.04 ng mL−1 and a 95% limit of agreement (LoA) of between −6.4 ng mL−1 and +6.3 ng mL−1 (Fig. 5b).
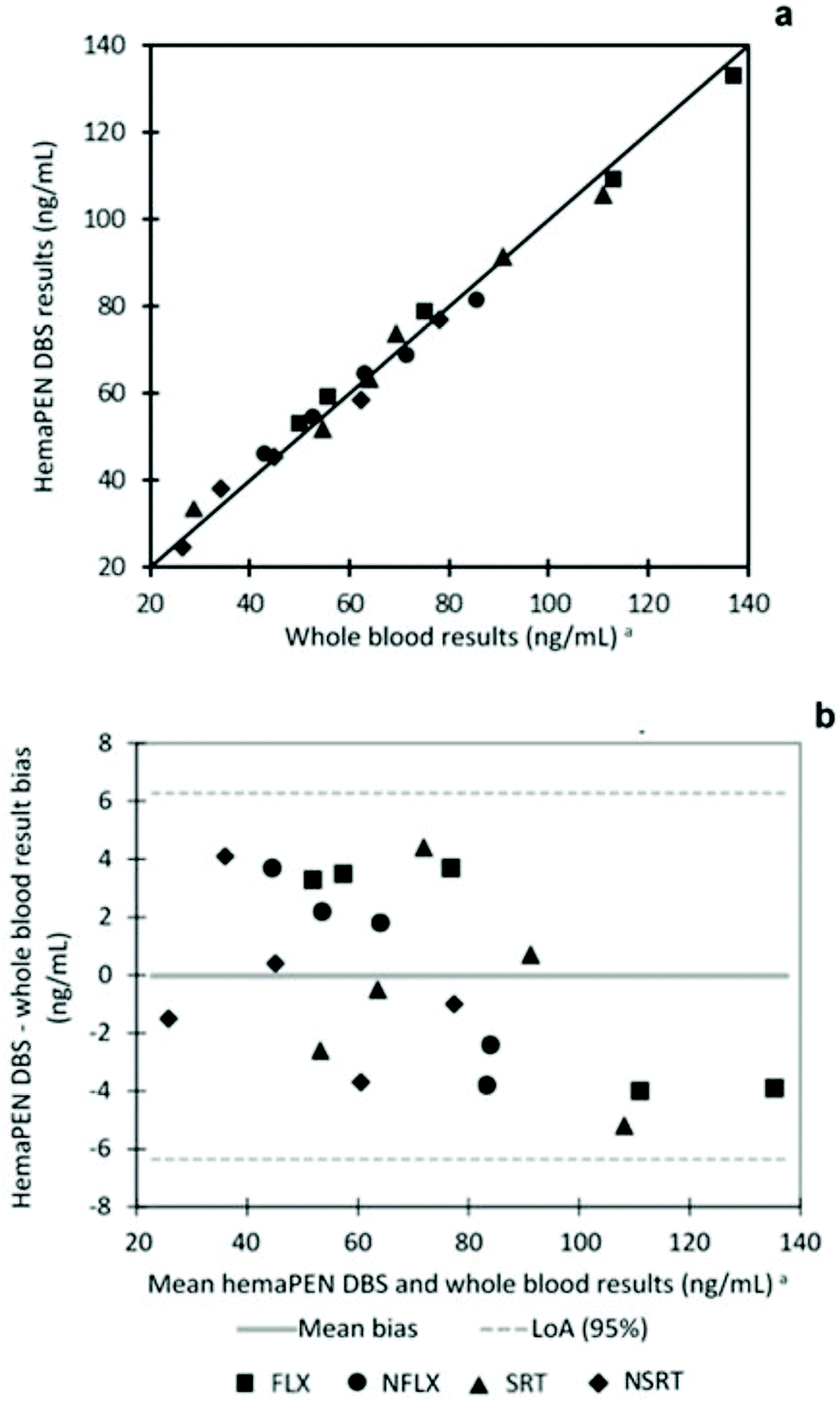 | ||
| Fig. 5 Linearity correlation (a) and Bland Altman (b) plots for the comparison between hemaPEN DBS and fluid whole blood results. aObtained by plasma analysis and converted by means of blood-to-plasma conversion factors. | ||
Finally, accuracy of the developed hemaPEN methodology was assessed by recovery assays at three different concentrations (n = 3 for each concentration level). The results were satisfactory, being always in the 91–106% range.
In conclusion, LC-MS/MS analysis of DBS generated by hemaPEN sampling provide reliable and sound analytical results. This allowed the assessment of the potential applicability of the developed and validated protocols with the prospect of application to capillary blood sampling by fingerpricking and the possible implementation of the method as a routine procedure for the minimally invasive TDM of patients receiving antidepressant therapies.
Conclusions
Accurate TDM practices require reliable, yet feasible analytical approaches in order to promote frequent patient monitoring and therapy personalisation. In this research work, an original analytical workflow has been developed and fully validated, based on miniaturised sampling using the innovative capillary volumetric blood microsampling hemaPEN technology and LC-MS/MS analysis. A straightforward and high-throughput methodology has been obtained to carry out the TDM of patients affected by CNS disorders and those undergoing treatment with SRT and/or FLX, chosen as the target compounds for this study. The developed strategy was found to be valid and reliable for the qualitative and quantitative analysis of the considered drugs and their main active metabolites, providing satisfactory validation results. The accuracy of capillary microsampling volume and the potential for misuse of hemaPEN have been tested in terms of possible undersampling or oversampling, providing solid and promising results. Analyte stability in DBS samples generated by means of hemaPEN and stored without temperature control was remarkable even after 3 months. An in-depth assessment was performed to evaluate the possible influence of HCT on the recovery and matrix effect, which highlighted the outstanding HCT-independence of hemaPEN DBS performances.The innovative hemaPEN technology, able to provide four identical 2.74 μL DBS samples from a single drop of blood, proved to be a promising tool for volumetric sampling of capillary whole blood from patients undergoing pharmacological treatment with CNS drugs. It also has high potential for future implementation in self- and home-sampling procedures. This could pave the road toward feasible, frequent but accurate TDM practices for better clinical decision-making and for therapy personalisation, in the perspective of precision medicine regimens in particular for major depression, but also for CNS symptoms associated with neurodegenerative diseases.
Of course, other microsampling techniques, such as volumetric absorptive microsampling (VAMS), could certainly be envisioned for the same goal. In this case, hemaPEN was deemed more suitable, since it produces DBS, which are a well-known, well-understood and widely used miniaturised matrix. In this way, the possible, future transition to microsampling for routine TDM could be made gentler for personnel and institutions, taking advantage of existing, proven procedures and equipment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Fundamental Oriented Research (RFO) 2019 funds, the Research Projects of National Relevance (PRIN) 2017 funds and by the Italian Institute of Technology of Genoa (Italy). The Authors would like to acknowledge Trajan Scientific and Medical for kindly providing hemaPEN devices and in particular Dr Pawanbir Singh and Dr Florian Lapierre for the valuable discussion.References
- L. Mercolini, M. A. Saracino and M. Protti, Current advances in biosampling for therapeutic drug monitoring of psychiatric CNS drugs, Bioanalysis, 2015, 7, 1925–1942 CrossRef CAS PubMed.
- G. Nys, M. G. M. Kok, A. C. Servais and M. Fillet, Beyond dried blood spot: Current microsampling techniques in the context of biomedical applications, Trends Anal. Chem., 2017, 97, 326–332 CrossRef CAS.
- S. Velghe, S. Capiau and C. P. Stove, Opening the toolbox of alternative sampling strategies in clinical routine: a key-role for (LC-)MS/MS, Trends Anal. Chem., 2016, 84, 61–73 CrossRef CAS.
- V. Ferrone, M. Carlucci, R. Cotellese, P. Raimondi, A. Cichella, L. Di Marco, S. Genovese and G. Carlucci, Development of a dried blood spot HPLC-PDA method for the analysis of linezolid and ciprofloxacin in hospital-acquired pneumonia patients, Drug Test. Anal., 2017, 9, 1611–1619 CrossRef CAS PubMed.
- E. J. Berm, E. Brummel-Mulder, J. Paardekooper, E. Hak, B. Wilffert and J. G. Maring, Determination of venlafaxine and O-desmethylvenlafaxine in dried blood spots for TDM purposes, using LC-MS/MS, Anal. Bioanal. Chem., 2014, 406, 2349–2353 CrossRef CAS PubMed.
- C. Linder, M. Andersson, K. Wide, O. Beck and A. Pohanka, A LC-MS/MS method for therapeutic drug monitoring of carbamazepine, lamotrigine and valproic acid in DBS, Bioanalysis, 2015, 7, 2031–2039 CrossRef CAS PubMed.
- L. Mercolini, R. Mandrioli, M. Protti, A. Conca, L. J. Albers and M. A. Raggi, Dried blood spot testing: a novel approach for the therapeutic drug monitoring of ziprasidone-treated patients, Bioanalysis, 2014, 6, 1487–1495 CrossRef CAS PubMed.
- M. Protti, A. Vignali, T. Sanchez Blanco, J. Rudge, F. Bugamelli, A. Ferranti, R. Mandrioli and L. Mercolini, Enantioseparation and determination of asenapine in biological fluid micromatrices by HPLC with diode array detection, J. Sep. Sci., 2018, 41, 1257–1265 CrossRef CAS PubMed.
- H. Iboshi, F. Yamaguchi, H. Suzuki, M. Kikuchi, M. Tanaka, S. Takasaki, A. Takahashi, M. Maekawa, M. Shimada, Y. Matsuda, Y. Okada and N. Mano, Development of a liquid chromatography-tandem mass spectrometric method for quantification of mycophenolic acid and its glucuronides in dried blood spot samples, Ther. Drug Monit., 2017, 39, 648–653 CrossRef CAS PubMed.
- A. Bhatnagar, M. J. McKay, M. Thaysen-Andersen, M. Arasaratnam, M. Crumbaker, H. Gurney and M. P. Molloy, Bioanalytical evaluation of dried plasma spots for monitoring of abiraterone and Δ(4)-abiraterone from cancer patients, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1126–1127, 121741 CrossRef CAS PubMed.
- B. Osteresch, S. Viegas, B. Cramer and H. U. Humpf, Multi-mycotoxin analysis using dried blood spots and dried serum spots, Anal. Bioanal. Chem., 2017, 409, 3369–3382 CrossRef CAS PubMed.
- R. Mandrioli, L. Mercolini and M. Protti, Blood and plasma volumetric absorptive microsampling (VAMS) coupled to LC-MS/MS for the forensic assessment of cocaine consumption, Molecules, 2020, 25, 1046 CrossRef CAS PubMed.
- M. Protti, R. Mandrioli and L. Mercolini, Tutorial: Volumetric absorptive microsampling (VAMS), Anal. Chim. Acta, 2019, 1046, 32–47 CrossRef CAS PubMed.
- M. G. M. Kok and M. Fillet, Volumetric absorptive microsampling: Current advances and applications, J. Pharm. Biomed. Anal., 2018, 147, 288–296 CrossRef CAS PubMed.
- L. A. Leuthold, O. Heudi, J. Déglon, M. Raccuglia, M. Augsburger, F. Picard, O. Kretz and A. Thomas, New microfluidic-based sampling procedure for overcoming the hematocrit problem associated with dried blood spot analysis, Anal. Chem., 2015, 87, 2068–2071 CrossRef CAS PubMed.
- S. Velghe and C. P. Stove, Evaluation of the Capitainer-B microfluidic device as a new hematocrit-independent alternative for dried blood spot collection, Anal. Chem., 2018, 90, 12893–12899 CrossRef CAS PubMed.
- Trajan scientific and medical. https://www.trajanscimed.com/pages/hemapen. 2020. (Accessed 20 May 2020).
- S. Deprez, L. Paniagua-González, S. Velghe and C. P. Stove, Evaluation of the performance and hematocrit independence of the hemaPEN as a volumetric dried blood spot collection device, Anal. Chem., 2019, 91, 14467–14475 CrossRef CAS PubMed.
- V. Ion, C. Legoff, E. Cavalier, P. Delanaye, A. C. Servais, D. L. Muntean and M. Fillet, Determination of iohexol by capillary blood microsampling and UHPLC-MS/MS, J. Pharm. Anal., 2019, 9, 259–265 CrossRef PubMed.
- S. Capiau, J. W. Alffenaar and C. P. Stove, in Clinical Challenges in Therapeutic Drug Monitoring, ed. W. Clarke and A. Dasgupta, Elsevier, Amsterdam, 2016, Alternative sampling strategies for therapeutic drug monitoring, pp. 279–336 Search PubMed.
- R. Mandrioli, M. Protti and L. Mercolini, Novel Atypical Antipsychotics: metabolism and therapeutic drug monitoring (TDM), Curr. Drug Metab., 2015, 16, 141–151 CrossRef CAS PubMed.
- R. Mandrioli, M. Protti and L. Mercolini, New-generation, non-SSRI antidepressants: therapeutic drug monitoring and pharmacological interactions. Part 1: SNRIs, SMSs, SARIs, Curr. Med. Chem., 2018, 25, 772–792 CrossRef CAS PubMed.
- M. Protti, R. Mandrioli, C. Marasca, A. Cavalli, A. Serretti and L. Mercolini, New-generation, non-SSRI antidepressants: Drug-drug interactions and therapeutic drug monitoring. Part 2: NaSSAs, NRIs, SNDRIs, MASSAs, NDRIs, and others, Med. Res. Rev., 2020, 1–39, DOI:10.1002/med.21671.
- L. Mercolini, R. Mandrioli, G. Finizio, G. Boncompagni and M. A. Raggi, Simultaneous HPLC determination of 14 tricyclic antidepressants and metabolites in human plasma, J. Sep. Sci., 2010, 33, 23–30 CrossRef CAS PubMed.
- C. Saka, An overview of analytical methods for the determination of monoamine oxidase inhibitors in pharmaceutical formulations and biological fluids, Crit. Rev. Anal. Chem., 2017, 47, 1–23 CrossRef CAS PubMed.
- C. Sabbioni, F. Bugamelli, G. Varani, L. Mercolini, A. Musenga, M. A. Saracino, S. Fanali and M. A. Raggi, A rapid HPLC-DAD method for the analysis of fluoxetine and norfluoxetine in plasma from overdose patients, J. Pharm. Biomed. Anal., 2004, 36, 351–356 CrossRef CAS PubMed.
- A. Musenga, E. Kenndler, L. Mercolini, M. Amore, S. Fanali and M. A. Raggi, Determination of sertraline and N-desmethylsertraline in human plasma by CE with LIF detection, Electrophoresis, 2017, 28, 1823–1831 CrossRef PubMed.
- R. Mandrioli, L. Mercolini, M. A. Saracino and M. A. Raggi, Selective serotonin reuptake inhibitors (SSRIs): therapeutic drug monitoring and pharmacological interactions, Curr. Med. Chem., 2012, 19, 1846–1863 CrossRef CAS PubMed.
- C. Hiemke, N. Bergemann, H. W. Clement, A. Conca, J. Deckert, K. Domschke, G. Eckermann, K. Egberts, M. Gerlach, C. Greiner, G. Gründer, E. Haen, U. Havemann-Reinecke, G. Hefner, R. Helmer, G. Janssen, E. Jaquenoud, G. Laux, T. Messer, R. Mössner, M. J. Müller, M. Paulzen, B. Pfuhlmann, P. Riederer, A. Saria, B. Schoppek, G. Schoretsanitis, M. Schwarz, M. S. Gracia, B. Stegmann, W. Steimer, J. C. Stingl, M. Uhr, S. Ulrich, S. Unterecker, R. Waschgler, G. Zernig, G. Zurek and P. Baumann, Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: update 2017, Pharmacopsychiatry, 2018, 51, 9–62 CrossRef CAS PubMed.
- A. C. C. da Silva, J. R. Raasch, T. G. Vargas, G. P. Peteffi, R. Z. Hahn, M. V. Antunes, M. S. Perassolo and R. Linden, Simultaneous determination of fluoxetine and norfluoxetine in dried blood spots using high-performance liquid chromatography-tandem mass spectrometry, Clin. Biochem., 2018, 52, 85–93 CrossRef CAS PubMed.
- J. Déglon, E. Lauer, A. Thomas, P. Mangin and C. Staub, Use of the dried blood spot sampling process coupled with fast gas chromatography and negative-ion chemical ionization tandem mass spectrometry: application to fluoxetine, norfluoxetine, reboxetine, and paroxetine analysis, Anal. Bioanal. Chem., 2010, 396, 2523–2532 CrossRef PubMed.
- Y. Luo, W. Korfmacher, S. Ho, L. Shen, J. Wang, Z. Wu, Y. Guo, G. Snow and T. O'Shea, Evaluation of two blood microsampling approaches for drug discovery PK studies in rats, Bioanalysis, 2015, 7, 2345–2359 CrossRef CAS PubMed.
- L. E. Eibak, A. B. Hegge, K. E. Rasmussen, S. Pedersen-Bjergaard and A. Gjelstad, Alginate and chitosan foam combined with electromembrane extraction for dried blood spot analysis, Anal. Chem., 2012, 84, 8783–8789 CrossRef CAS PubMed.
- M. Moretti, F. Freni, B. Valentini, C. Vignali, A. Groppi, S. D. Visonà, A. M. M. Osculati and L. Morini, Determination of antidepressants and antipsychotics in dried blood spots (DBSs) collected from post-mortem samples and evaluation of the stability over a three-month period, Molecules, 2019, 24, E3636 CrossRef PubMed.
- M. A. Raggi, R. Mandrioli, G. Casamenti, F. Bugamelli and V. Volterra, Determination of fluoxetine and norfluoxetine in human plasma by high-pressure liquid chromatography with fluorescence detection, J. Pharm. Biomed. Anal., 1998, 18, 193–199 CrossRef CAS PubMed.
- R. Mandrioli, M. A. Saracino, S. Ferrari, D. Berardi, E. Kenndler and M. A. Raggi, HPLC analysis of the second-generation antidepressant sertraline and its main metabolite N-desmethylsertraline in human plasma, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 836, 116–119 CrossRef CAS PubMed.
- European Medicines Agency, Guideline on Bioanalytical Method Validation, 2012 Search PubMed.
- U.S. Food and Drug Administration, Bioanalytical Method Validation. Guidance for Industry, 2018 Search PubMed.
- Y. Amitai, E. Kennedy, P. DeSandre, J. Fawcett and H. Frischer, Red cell and plasma concentrations of fluoxetine and norfluoxetine, Vet. Hum. Toxicol., 1993, 35, 134–136 CAS.
- G. T. Vatassery, L. A. Holden, D. K. Hazel and M. W. Dysken, Analysis of sertraline and desmethylsertraline in human plasma and red blood cells, Clin. Biochem., 1997, 30, 565–568 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an01039a |
| ‡ These two authors contributed equally to the study. |
| This journal is © The Royal Society of Chemistry 2020 |