
Integration of metabolomics and proteomics to reveal the metabolic characteristics of high-intensity interval training†
Jingjing
Zhao
,
Yang
Wang
,
Dan
Zhao
,
Lizhen
Zhang
,
Peijie
Chen
* and
Xin
Xu
 *
*
Shanghai anti-doping laboratory, Shanghai University of Sport, Changhai Road 399, Shanghai, 200438, China. E-mail: chenpeijie@sus.edu.cn; xxu2000@outlook.com; Fax: +86-021-65506702; Tel: +86-021-65506702
First published on 24th July 2020
Abstract
High-intensity interval training (HIIT) can elicit a greater training stimulus to improve maximal aerobic capacity and is frequently applied in professional sports training. Although select metabolic pathways have been examined, the omics-scale molecular response to the HIIT has not been fully characterized. The longitudinal multi-omic profiling, including metabolome and proteome was performed on urine samples from 23 healthy young soccer players before and after the HIIT exercise. Metabolomics revealed the metabolomic changes during the HIIT, including steroid hormone metabolites, amino acid biosynthesis and relevant metabolites. Furthermore, changes in protein expression in metabolic pathways involved in energy metabolism, oxidative stress as well as immune pathways were found by proteomics, which provide the foundation for understanding metabolomic changes during the HIIT. There was a significant association between the HIIT and the urinary omic profiles, with the alteration in metabolic pathways associated with long-term adaptation to training. Future studies should focus on the validation of these findings and the development of metabolic models to monitor the training intensity and the adaptation in athletes.
1. Introduction
In the last twenty years, the emergence and rapid development of ‘-omic’ techniques, such as metabolomics or proteomics, has greatly accelerated the process of physiological or pathophysiological research. The intrinsic properties of urine make it an ideal source for metabolomic and proteomic studies. Urine can provide information from proximal tissues and blood perfusing distant organs.1 Most importantly, changes in the blood may even be magnified in the urine, as urine does not have the homeostatic mechanism of blood.2 Moreover, urine samples are easy to obtain and are non-invasive. Therefore, urinary metabolome and proteome have been widely used for the disease marker screening, disease diagnosis and disease process monitoring.3–5 In recent years, urine proteome and metabolome have also been used to study the effects of physical activity and exercise.6–8High-intensity interval training (HIIT), as a modern exercise program for physical activity, generally consists of alternating periods of intensive aerobic exercise with periods of passive or active moderate/mild intensity recovery.9 Since the HIIT can offer the possibility to maintain high-intensity exercise for longer periods than continuous exercise, it has been believed to elicit a greater training stimulus to improve maximal aerobic capacity, and is frequently applied in professional sports training as in soccer7 and exercise rehabilitation in coronary artery disease.10,11 Currently, the HIIT is widely applied in sports and exercise, and the multi-omics of urine may shed light on the new understanding of human metabolomics during exercise. Once the training intensity exceeds the adaptation in athletes, the overtraining might cause a decrease in sports performance and further damage the athlete's health. Several studies have shown that physical activity has a distinct effect on the human urinary metabolome and contains significant amount of metabolites from the purine synthesis pathway, glycolysis, the Krebs cycle, glucocorticoid metabolism, androgen metabolism, amino acid and fatty acid oxidation and the gastrointestinal microbiome.12–15 Given the profound effects of exercise, it is essential to carry out multi-omics-based studies on the impact of HIIT exercise.
Metabolomics can offer quantitative information on the metabolic profiles related to exercise to identify pivotal biomarkers associated with their performance and potentially sports-related disorders.3,16,17 Non-targeted metabolomics allows the detection of changes in response to various physiological states, as well as identifies metabolic signatures with potential translational impact.18,19 Previous studies have indicated that plasma lactate20,21 and adenine breakdown products22 were upregulated in the metabolomics profiling of athletes who underwent intensive exercise, indicating the occurrence of anaerobic metabolism and ATP cycling, respectively. Intensive exercise was also shown to trigger changes in amino acids, including a moderate upregulation of glutamate in skeletal muscle leading to the release of alanine to promote ammonia metabolism and the concentrations of these metabolites correspond to changes in the plasma.21,23 Most metabolic profiling studies are currently performed using quadrupole-time-of-flight (QTOF)-MS due to the high-resolution, sensitivity, rapid data acquisition, and high mass accuracy. It is undoubtedly a suitable system for untargeted metabolomics analysis in our study.
Proteomic techniques have been used to gain insight into changes in proteins in different physiological conditions associated with exercise. Quantitative proteomic techniques were performed to characterize the exercise-induced secretion of extracellular vehicles (EVs)-containing proteins.24 A recent report indicated that biological pathways involved in cardiopulmonary exercise response were discovered by multi-omics techniques including metabolome, lipidome, immunome, proteome and transcriptome.8 The molecular response to exercise was assessed based on omic techniques not only in healthy individuals but also in individuals at risk for cardiometabolic disease.25 Although omic studies related to exercise have been developed in serum,26 plasma8,14,18 and skeletal muscle,15,27 the metabolomic and proteomic response to HIIT has not been fully characterized, which is vital for maintaining the best training condition of athletes.
In this study, metabolomics and proteomics were integrated to analyse urine specimens collected before and after HIIT exercise to investigate the metabolic and protein response to HIIT in-depth. Differential metabolites were determined by the combination of VIP values and p values. Metabolic pathways associated with the biosynthesis and metabolism of amino acids, steroid hormone metabolism and others were analysed. Metabolic characteristics were further investigated by the proteomic analysis to reveal the differential protein expression during HIIT exercise. Our study demonstrated the prospectives and feasibility of multi-omics for exercise, and the assessment of changes in metabolites and proteins could provide valuable characteristics of different exercises for future study.
2. Experimental
For the metabolomic and proteomic characterization of HIIT exercise, twenty-three healthy players from the Youth Team of a professional soccer club volunteered to take part in this study. The details of the descriptive characteristics of the participants, the design of the HIIT exercise, as well as chemicals and reagents used in this study are described in the ESI.† The protocol for this study was approved by the ethics committee of the Shanghai University of Sport, China (no. 102772019RT050). Urine samples were collected pre-exercise in the morning (first urine, labelled as Pre), 30 min post-exercise (labelled as Post) and 18 hours after recovery (labelled as Rec). A total of 69 urine samples were collected, aliquoted and stored at −80 °C until analysis.2.1 Metabolite extraction
For untargeted metabolomic analysis, urine samples stored in the refrigerator were thawed at 4 °C. For the analysis, 100 μL of the urine sample was thoroughly mixed with 200 μL of ACN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (1:1, v/v), followed by centrifugation at 12000 rpm for 15 min; 200 μL of supernatant was then transferred to an LC auto-sampler vial (Thermo Fisher, UK). Blanks were prepared by replacing urine with Milli Q water. A quality control (QC) sample was prepared by mixing 10 μL of each sample and treating it as above, which was used to ensure the stability of the analytical conditions.
:MeOH (1:1, v/v), followed by centrifugation at 12000 rpm for 15 min; 200 μL of supernatant was then transferred to an LC auto-sampler vial (Thermo Fisher, UK). Blanks were prepared by replacing urine with Milli Q water. A quality control (QC) sample was prepared by mixing 10 μL of each sample and treating it as above, which was used to ensure the stability of the analytical conditions.
2.2 Protein extraction, digestion
Urine samples of ten subjects were randomly selected to execute the proteomic analysis. The protease inhibitor cocktail was added to each sample to avoid proteolysis immediately after the urine was thawed. The urine was centrifuged with 4000g at 4 °C for 20 min to remove cells and debris. The sample was subsequently passed through a 0.22 μm filter membrane with ultra-low protein-binding capacity and the flow-through was saved. Acetone (five times the sample volumes, pre-chilled at −20 °C) was added to precipitate the proteins at −20 °C for 4 h. The precipitates were resuspended in a lysis solution containing 1% SDS and 8 M urea in water, followed by vortexing. The protein concentration was determined by the enhanced bicinchoninic acid (BCA) protein assay kit (Beyotime Biotechnology, Shanghai, China), and then 200 μg of proteins were transferred into a new Eppendorf tube. The proteins were reduced with hydrochloride (TCEP) at the final concentration of 10 mM at 37 °C for 1 h and then alkylated with iodoacetamide (IAM) at the final concentration of 20 mM in the dark at room temperature for 40 min. Acetone (five times the sample volume, pre-chilled at −20 °C) was added to precipitate the proteins at −20 °C for 4 h. The precipitates were rinsed with 90% acetone wash solution and then centrifuged at 12000g at 4 °C for 20 min. The supernatant was removed and the samples were air-dried thoroughly at room temperature. Proteins were then re-dissolved in 100 mM triethylammonium bicarbonate (TEAB) and the enzymatic digestion was performed with 4 μg sequence-grade modified trypsin (Promega, Madison, WI) at 37 °C overnight.
2.3 LC-MS/MS analysis
For untargeted metabolomic analysis, UPLC-QTOF-MS analysis was performed on a Triple TOF 6600 mass spectrometer (SCIEX, Boston, USA) coupled to a LC-30AD series Ultra Performance Liquid Chromatography system (Shimadzu, Kyoto, Japan) in both positive and negative ion modes. The mass scanning range was m/z 100–1200 in full data storage mode. The drying gas temperature was set at 550 °C in positive mode and 450 °C in negative mode. The ion spray voltage was 5500 V in positive mode and −4500 V in negative mode. Atomization gas pressure, auxiliary heating gas pressure and curtain gas pressure in both ionization modes were set at 55, 55 and 40 psi, respectively. The separation was performed using an Acquity UPLC BEH Amide column (100 × 2.1 mm, 1.7 μm, Waters, Milford, MA, USA) in binary gradient mode. The mobile phases in different ionization modes were as follows: (A) 10 mM ammonium formate and 0.1% (v/v) formic acid in water and (B) pure ACN in positive mode; (A) 25 mM ammonium formate and 4 mM ammonium hydroxide in water and (B) pure ACN in negative mode. The flow rate was 300 μL min−1. The gradient was programmed as follows: initial conditions of 95% of mobile phase B followed by a linear gradient from 95% to 65% of mobile phase B in 14 min and then from 65% to 40% in 2 min; the 40% elution of mobile phase B was held for 2 min and then a gradient of 0.1 min was used to return to the initial conditions, which were held for an additional 4.9 min. Autosampler and column temperatures were set to 4 °C and 45 °C, respectively and the injection volume used was 2 μL. Samples were run in a stratified method with between-subject samples placed in randomised order. One QC sample and one blank vial were run after each of the 10 urine samples.For proteomic analysis, the peptide mixture of thirty samples was fractionated by high pH separation using the Ultimate 3000 system (ThermoFisher Scientific, MA, USA) connected to a reverse-phase XBridge C18 column (4.6 × 250 mm, 5 μm, Waters, Milford, MA, USA) to build the spectral library by data-dependent acquisition (DDA) mode. The experimental details are given in the ESI.†
The samples were re-dissolved in 30 μL solvent A (A: 0.1% formic acid in water) and analysed by on-line nanospray LC-MS/MS on an Orbitrap Lumos coupled to an EASY-nLC 1200 system (Thermo Fisher Scientific, MA, USA). Then, 1.5 μL peptide sample was loaded onto the analytical column (Acclaim PepMap C18, 75 μm × 25 cm) with 60 min gradient, from 8% to 36% B (B: 0.1% formic acid in 80% ACN). The column flow rate was maintained at 600 nL min−1. The electrospray voltage of 2 kV versus the inlet of the mass spectrometer was used. The mass spectrometer was run under data-independent acquisition (DIA) mode, and automatically switched between MS and MS/MS mode. The parameters were as follows: (1) MS: scan range (m/z) = 350–1350; resolution = 120000; AGC target = 4 × 105; maximum injection time = 50 ms; (2) HCD-MS/MS: resolution = 30000; AGC target = 3 × 105; collision energy = 32; stepped CE = 5%; (3) DIA was performed with variable isolation window, and each window overlapped 1 m/z, and the window number was 60.
2.4 Data analysis
For untargeted metabolomic analysis, the instrument software XCMS plus (AB Sciex, USA) was used to process all data files obtained by UPLC-QTOF-MS. Raw data were treated by performing peak extraction, peak matching, peak alignment and normalization preprocessing. The main parameters were set as follows: the retention time range was 1–23 min; the retention time and m/z tolerance were 0.1 min and 10 ppm respectively; the response threshold was 100 counts; the isotope peaks were removed. The obtained data set matrix was then imported into the data analysis software XCMS plus.To setup the spectral library, raw data of DDA were processed and analysed by Spectronaut 13 (Biognosys AG, Switzerland) with default settings to generate an initial target list. Spectronaut was set up to search the database of Homo_sapiens_2017 (20414 entries). Q-Value (FDR) cut-offs on precursor and protein levels were applied at 1%.
For proteomic analysis, the raw data of DIA were processed and analysed by Spectronaut 13 with default settings, retention time prediction type set to dynamic iRT. Data extraction was determined by Spectronaut 13 based on the extensive mass calibration. Spectronaut Pulsar X determined the ideal extraction window dynamically depending on iRT calibration and gradient stability. Q-value (FDR) cut-off on precursor and protein levels was applied at 1%. Decoy generation was set to mutated, which is similar to scrambled but will only apply a random number of AA position swamps (min = 2, max = length/2). All selected precursors passing the filters are used for quantification. The average top 3 filtered peptides that passed the 1% FDR cut off were used to calculate the major group quantities.
2.5 Identification of metabolites and proteins
Multivariable statistical analysis of metabolome and proteome data was processed using Metaboanalyst 4.0 (McGill University, Quebec, Canada).28 The different metabolites and proteins were determined by the combination of the VIP (variable importance in the projection) value >1 in partial least squares regression-discriminant analysis (PLS-DA) models and the p values (<0.05) from two-tailed t-tests on the normalized peak intensities. Fold change was calculated as the average normalized peak intensity ratio between two groups.The structural identification of differential metabolites was performed by matching the mass spectra with the METDNA library, which is available online (http://metdna.zhulab.cn/), including accuracy mass, MS/MS spectra and online databases: METLIN (http://www.metlin.scripps.edu). Functional annotation of proteins was carried out based on the euKaryotic orthologous groups of proteins (KOG) database and Gene Ontology (GO) annotations (https://www.ebi.ac.uk/QuickGO/).
2.6 Pathway enrichment analysis
Pathway analysis was implemented on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, which is available online (https://www.kegg.jp/). The pathway analysis module in the tool explores the results interactively and gives detailed instructions and outcomes to obtain a better understanding of the pathways involved in the conditions under study. By uploading the differential metabolites, the built-in pathway library (human) and hypergeometric test for over-representation analysis were employed. The network of activated proteins was generated with the help of the STRING database.293 Results and discussion
In this study, the longitudinal multi-omic profiling of urine was performed before and after HIIT in participants. As shown in the workflow (Scheme 1), 23 participants underwent HIIT exercise, and urine samples of pre-exercise, post-exercise and the recovery were collected (n = 69). In-depth multi-omic profiling was conducted on each sample containing untargeted metabolomics and proteomics. The longitudinal multi-omic dataset was used to characterize the dynamic molecular response to HIIT as well as the influence of HIIT on athlete performance. | ||
| Scheme 1 . Workflow for the mass spectrometry (MS)-based multi-omics profile analysis on urine specimens to research the influence of high intensity interval training (HIIT). | ||
3.1 UPLC-QTOF-MS analysis for metabolic profiling
The Yo–Yo intermittent recovery (YYIR) test has been used frequently in a variety of sports. The YYIR test consists of repeated exercise bouts performed at progressively increasing speeds, interspersed with 10 s active rest periods and performed until the subject is exhausted. Therefore, the YYIR test was chosen as the HIIT model in this study. To investigate the impact of HIIT on the metabolic characteristics in human urine, the samples of pre-exercise, post-exercise and the recovery were collected and a metabolic profiling analysis was performed on three groups samples. The UPLC-QTOF-MS system was able to obtain metabolic profiling for the aforementioned samples in the positive ion mode and the negative ion mode. TICs of QC samples in both positive and negative ion modes are shown in Fig. 1a and Fig. S1 (ESI†), respectively. As shown in Fig. 1a and Fig. S1,† TICs of QC samples overlapped well, indicating that the analytical system had good stability and repeatability. Creatinine is considered to be an ideal endogenous standard in urine QC samples. In the positive ion mode, there was a good overlap of creatinine for chromatograms (Fig. 1b). Furthermore, the relative standard deviation (RSD) values of retention time, peak height and peak area of endogenous creatinine were calculated, which were lower than 2%. In the negative ion mode, the RSD values of retention time and the peak area of creatinine were calculated, and a similar result to the positive mode (Fig. S2†) was obtained. The aforementioned results indicated that the datasets can be used to establish the mathematical model.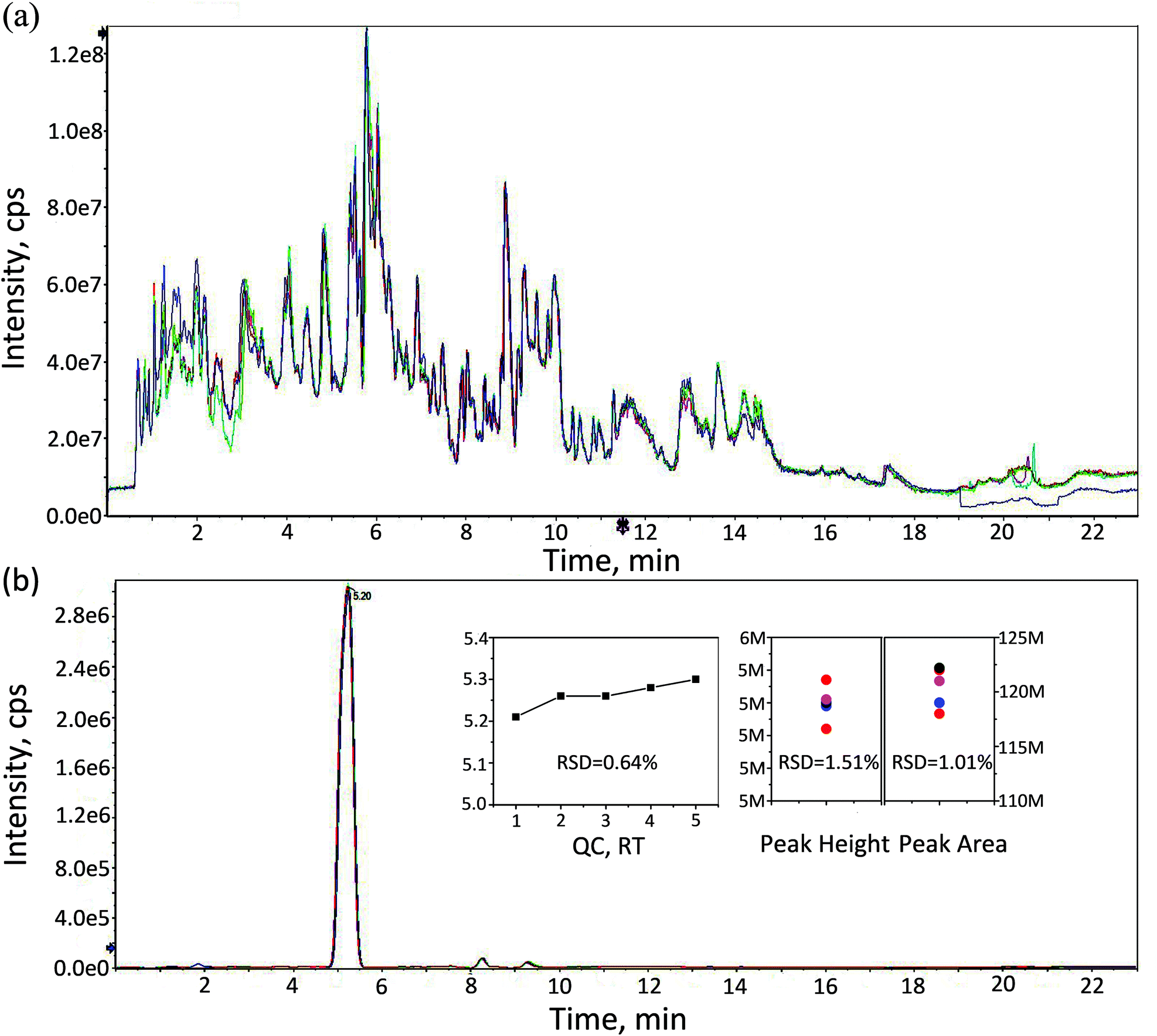 | ||
| Fig. 1 (a) LC-MS total ion chromatography (TIC) of QC data in the positive ion mode. (b) Extracted ion chromatogram (EIC) of creatinine and relative standard deviation (RSD) of retention time, peak height and peak area. RSDRT = 0.64%, RSDHeight = 1.51%, RSDArea = 1.01%. | ||
Data were treated by performing peak extraction, peak matching, peak alignment and normalization pre-processing in the XCMS plus software. Metabolites were identified by matching the mass spectra with open source databases (METDNA,30 METLIN31) including accuracy mass and MS/MS spectra in positive and negative ion modes respectively. A total of 1568 metabolites was identified after removing substances co-identified by both ion models. Those identified metabolites were associated with glucose, lipid, amino acid, and energy metabolism, such as 6-acetyl-glucose, adenosine, deoxya-denosine, hypoxanthine, 3-ketolactose, tyrosine, lysine, thymine, glutathione (GHS), cysteine, γ-gultamyl-cysteine, testosterone, cortisol, creatine, creatine phosphate and others. Adenosine, deoxyadenosine, hypoxanthine and xanthosine are involved in adenosine triphosphate (ATP) synthesis and degradation. Previous studies have indicated that the active γ-glutamyl cycle plays an important role in the GHS-mediated radical detoxification during oxidative stress.6 Testosterone and cortisol are related to endurance; it has been shown that exercise can cause changes in sex steroid hormone concentrations in the serum of non-athletes as well as athletes. Creatine and creatine phosphate play essential roles in the storage and transmission of phosphate-bound energy.
3.2 Metabolomic changes in response to HIIT
Compared to conventional statistical analysis, multivariate statistical analysis can effectively reduce data dimensions. PLS-DA, as one of the supervised multivariate analysis in omics techniques, has been used to identify biomarkers and provides vital information for investigating the pathogenesis of the disease and the mechanism of drug toxicity.32,33 In untargeted metabolomics, PLS-DA was useful for identifying the main factors affecting the dependent variable.34,35 Herein, PLS-DA was performed on the identified metabolites of pre-exercise, post-exercise and recovery under both positive and negative ion modes to figure out the significant variables among the different samples (Fig. 2). As shown in Fig. 2a and b, datasets from pre-exercise, immediately post-exercise and recovery are represented by red, green and blue, respectively. After executing PLS-DA on the obtained data, there was a separation of clusters in PLS-DA plots for different group samples, especially in negative ion mode. The separation of clusters indicated that significant differences existed among the analysed samples for both positive and negative ion modes.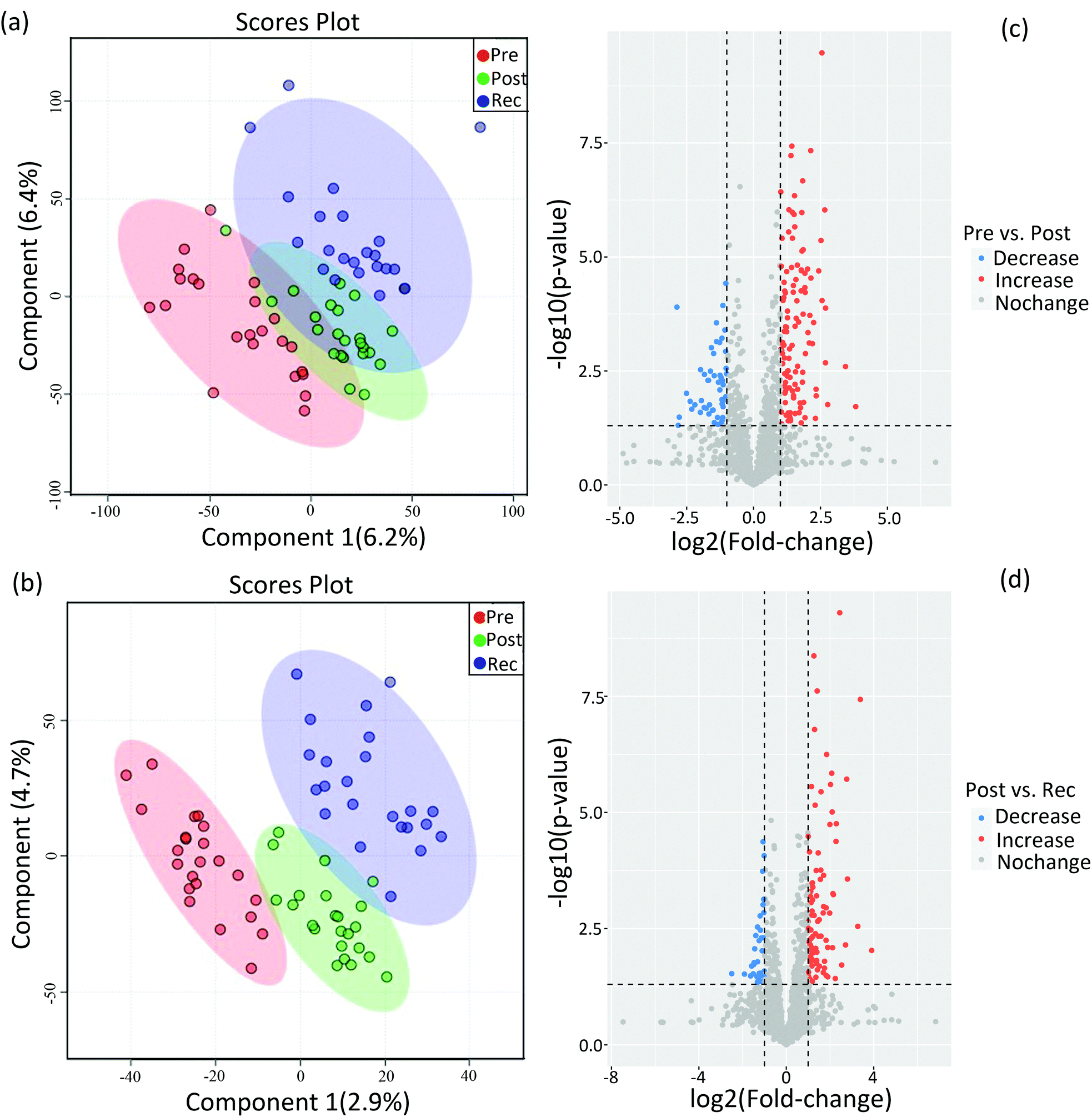 | ||
| Fig. 2 PLS-DA plot for metabolomic datasets of urine samples collected pre-exercise (Pre), post-exercise (Post) and after recovery (Rec) in the positive ion mode (a) and the negative ion mode (b). (c) Volcano plots of metabolites with log2(FC) as the horizontal axis and −log10(p value) as the vertical axis. FC, fold change, of post-exercise to pre-exercise. (d) Volcano plots of metabolites with log2(FC) as the horizontal axis and −log10(p value) as the vertical axis. FC, fold change, of the recovery to post-exercise. | ||
Differential metabolites were determined by the combination of VIP value >1 and p values < 0.05. Volcano plots in Fig. 2c show changes in metabolites on comparing post-exercise to pre-exercise. Red represents the increase in the concentrations of metabolites and blue represents the decrease in the concentrations. These metabolites contained 3-sulfino-alanine, 3-beta-galactosyl-sn-glycerol, glutamate, N-acetylornithine, sorbitol, hypoxanthine and so on. The changes in metabolites in the recovery as compared with post-exercise are shown in Fig. 2d. Volcano plots clearly show that many metabolites, such as adenine, androstenedione, testosterone, histidinol, tryptophan and sphingosine 1-phosphate, were up- or downregulated in the recovery as compared to post-exercise. In this study, 59 differential metabolites were identified by simultaneously comparing the datasets of urine metabolites from pre-exercise, post-exercise and recovery, which are listed in sheet 1 of the Supplemental Dataset.
In order to analyse the roles of differential metabolites in the HIIT process, differential metabolites were uploaded to the KEGG pathway database. In the pathway enrichment of the differential metabolites, the built-in pathway library (human) and hypergeometric test for over-representation analysis were employed. Heat maps of the pathway enrichment analysis are displayed in Fig. S3† (comparing post-exercise to pre-exercise) and Fig. S4† (comparing the recovery to post-exercise). As shown in the figures, the main pathways affected by HIIT exercise are the amino acid metabolism pathways (such as tyrosine, tryptophan, lysine, cysteine, methionine), purine metabolism, CoA biosynthesis and glycolysis, most of which are associated with energy metabolism. Differential pathway enrichment analysis of 59 differential metabolites identified from urine samples collected pre-exercise, post-exercise and during the recovery is shown in Fig. 3, which largely involved the biosynthesis and metabolism of amino acids and steroid hormone biosynthesis. Relationships among most of the differential metabolites in the biosynthesis and metabolism of amino acids and steroid hormone biosynthesis are shown in Fig. 4a and c. Significant changes in the differential metabolites in pre-exercise, post-exercise and recovery are represented by the histogram in Fig. 4.
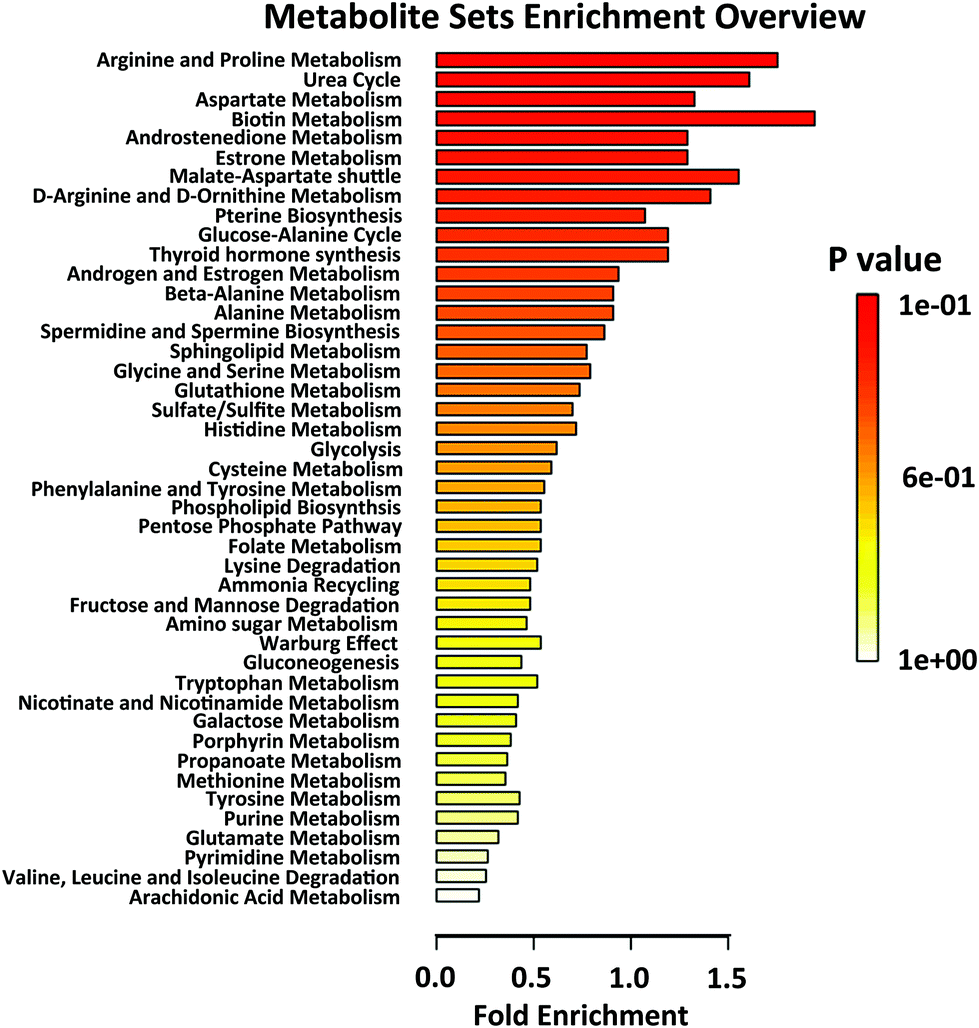 | ||
| Fig. 3 Differential pathway enrichment analysis of all differential metabolites identified from urine samples collected pre-exercise (Pre), post-exercise (Post) and at recovery (Rec). | ||
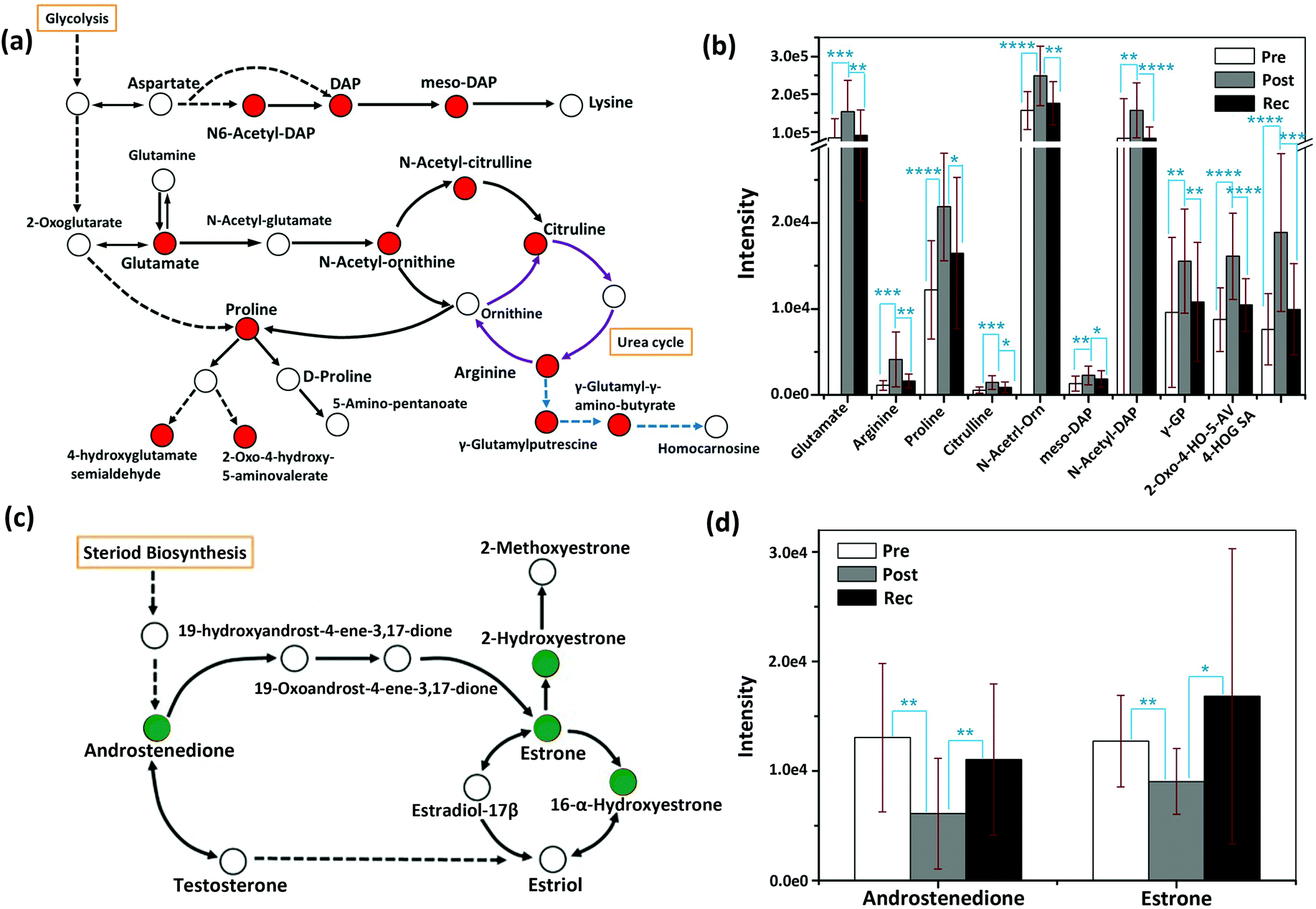 | ||
| Fig. 4 Schematic overview of the metabolic changes in the biosynthesis and metabolism of amino acids (a) and steroid hormone biosynthesis (c); the relative abundance of important metabolites in the pathway of biosynthesis and metabolism of amino acids (b), as well as steroid hormone biosynthesis pathway (d), measured from the samples of pre-exercise, post-exercise and recovery. DAP, orn, GP, 2-Oxo-4-HO-5-AV and 4-HOG SA represent diaminoheptanedioate, ornithine, glutamyl-putrescine, 2-oxo-4-hydroxy-5-aminovalerate and 4-hydroxyglutamate semialdehyde, respectively. Red in Fig. 4a indicates that the identified differential metabolites were upregulated after exercise and downregulated in the recovery. The purple arrows represent the urea cycle and the blue arrows are the parts of arginine metabolism. Green in Fig. 4c indicates that the identified differential metabolites were downregulated after exercise and upregulated in the recovery. Error bars represent the standard deviation. “*”, “**”, “***” and “****” indicate p values smaller than 0.05, 0.01, 0.001 and 0.0001, respectively. | ||
Arginine, proline and their metabolites, which are mostly associated with carbohydrate metabolism,8 increased post-exercise. In the recovery sample, they tended to decrease to pre-exercise levels. As an important small molecule, glutamate plays a key role during HIIT exercise. Glutamate is the feedstock for the citric acid cycle of classical energy metabolism, the raw material for the synthesis of GSH in oxidative stress and the immune system, as well as a well-known excitatory neurotransmitter, which participates in regulating neuronal excitation in the central nervous system (CNS).36
The upregulation of glutamate after HIIT exercise is consistent with the literature and may be the result of the response of oxidative stress and the improvement of the immune system.37 The circulation between arginine and citrulline (purple cycle in Fig. 4a) is part of the urea cycle. Exhaustive exercise may lead to a disorder of the urea cycle.38 In the current study, arginine and citrulline significantly increased after HIIT exercise (P < 0.001), suggesting that the urea cycle might be elevated with HIIT exercise, in line with a reported rise in blood serum ornithine after heavy exercise.38 Steroid hormone metabolism is mainly related to a signalling system that mediates changes in the metabolic and cellular processes of skeletal muscle and neural and connective tissue as a function of exercise.39 Testosterone induces anabolic and anticatabolic mechanisms involved in muscle tissue growth, recovery and remodelling, and performance enhancement.40 Previous studies have reported that testosterone is the major promoter of muscle growth and the subsequent increase in muscle strength in response to resistance training in men.41–43 Total testosterone and free testosterone are elevated directly following heavy resistance exercise.40 The changes in steroid hormones are also reflected in our metabolic study. In steroid hormone biosynthesis, androstenedione, as a precursor of testosterone, was decreased post-exercise and elevated in the recovery.
3.3 Proteomics changes in the response to HIIT
Metabolism can best reflect the phenotypic characteristics and immediate stress response of organisms. Metabolic changes are influenced by the changes in enzymes. Therefore, to further the research on metabolomic changes in response to HIIT, proteomic analysis was performed on the urine samples collected pre-exercise, post-exercise and at recovery. A total of 1146 proteins were identified on combining the pre-exercise, post-exercise and recovery groups. PLS-DA and volcano plot analysis were performed on the proteome data of pre-exercise, post-exercise and recovery, shown in Fig. S5 and S6.† Results showed that proteins identified from pre-exercise and post-exercise were clustered into two discriminative groups in score plots of PLS-DA, indicating that proteins in the urine were significantly influenced by HIIT (Fig. S5†). Volcano plots showed that many proteins were up- or downregulated (Fig. S6†) in post-exercise as compared to pre-exercise. The comparison of proteins identified in the recovery and post-exercise likewise showed obvious clustering and variation as seen in Fig. S5 and S6.† The changes in the differential proteins between pre-exercise and post-exercise are visually displayed in the heatmap (Fig. S7†). We observed 51 proteins with significant upregulation and 51 with significant downregulation (fold change (FC) >2 or <0.5 and p-value < 0.05) post-exercise as compared to pre-exercise. There are 134 significantly differential proteins between the recovery and post-exercise groups, including 74 proteins with significant downregulation, as shown in the heatmap of Fig. S8.† In order to explore the function of differential proteins, the significant proteins were uploaded to the KOG database.KOG functional annotation for the proteins with significant variations is displayed in Fig. 5. Comparing post-exercise to pre-exercise, 102 differential proteins were distributed in 17 classes of functions with red columns indicating upregulation and black columns indicating downregulation; 134 differential proteins identified from the post-exercise and the recovery groups were distributed in 20 classes of functions. The annotation results revealed that energy-production-associated proteins [C] were upregulated after exercise, and were naturally downregulated in the recovery. This is due to the energy consumption promoting the upregulation of energy-related proteins in the process of exercise. Similarly, proteins associated with defense mechanisms [V], inorganic ion transport and metabolism [P], secondary metabolites biosynthesis, transport and catabolism [Q] were upregulated after exercise and downregulated in the recovery. Many proteins with different sorts of functions were also significantly perturbed during exercise and recovery, including carbohydrate transport and metabolism [G], post-translational modification [O], signal transduction [T], transport and metabolism of amino acid [E], [I] lipid transport and metabolism [I], etc.
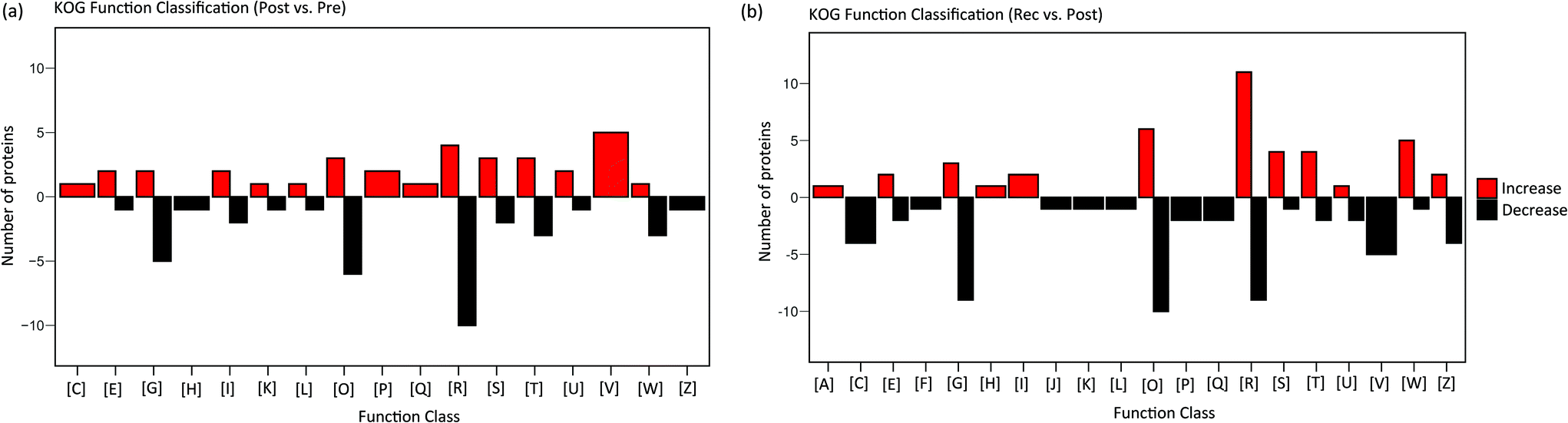 | ||
| Fig. 5 KOG functional annotation for the differential proteins: (a) 102 proteins with significant variations in post-exercise as compared to pre-exercise, which were distributed in 17 classes of functions. (b) 134 proteins with significant variations in the recovery compared to post-exercise, which were distributed in 20 classes of functions. Red columns indicate upregulation and black columns indicate downregulation. Note: [A] RNA processing and modification; [C] energy production and conversion; [E] amino acid transport and metabolism; [F] nucleotide transport and metabolism; [G] carbohydrate transport and metabolism; [H] coenzyme transport and metabolism; [I] lipid transport and metabolism; [J] Translation, ribosomal structure and biogenesis; [K] Transcription; [L] Replication, recombination and repair; [O] posttranslational modification, protein turnover, chaperones; [P] inorganic ion transport and metabolism; [Q] secondary metabolites biosynthesis, transport and catabolism; [R] general function prediction only; [S] function unknown; [T] signal transduction mechanisms; [U] intracellular trafficking, secretion, and vesicular transport; [V] defense mechanisms; [W] extracellular structures; [Z] cytoskeleton. | ||
The KEGG pathway plot in Fig. S9† further elucidated the significant pathways that the differential proteins were involved in. Most of the differential proteins were related to metabolism, including carbohydrate metabolism, lipid metabolism, amino acid metabolism, etc. Furthermore, there are some proteins that are relevant to transport and catabolism in cellular processes, signal transduction, the immune system as well as the endocrine system. From the untargeted proteome data, KOG functional annotation and KEGG pathway analysis, we found some proteins crucially involved in the processes of exercise and recovery with significant upregulation or downregulation (p-value < 0.05) (Fig. 6). These proteins play vital roles in promoting human health.
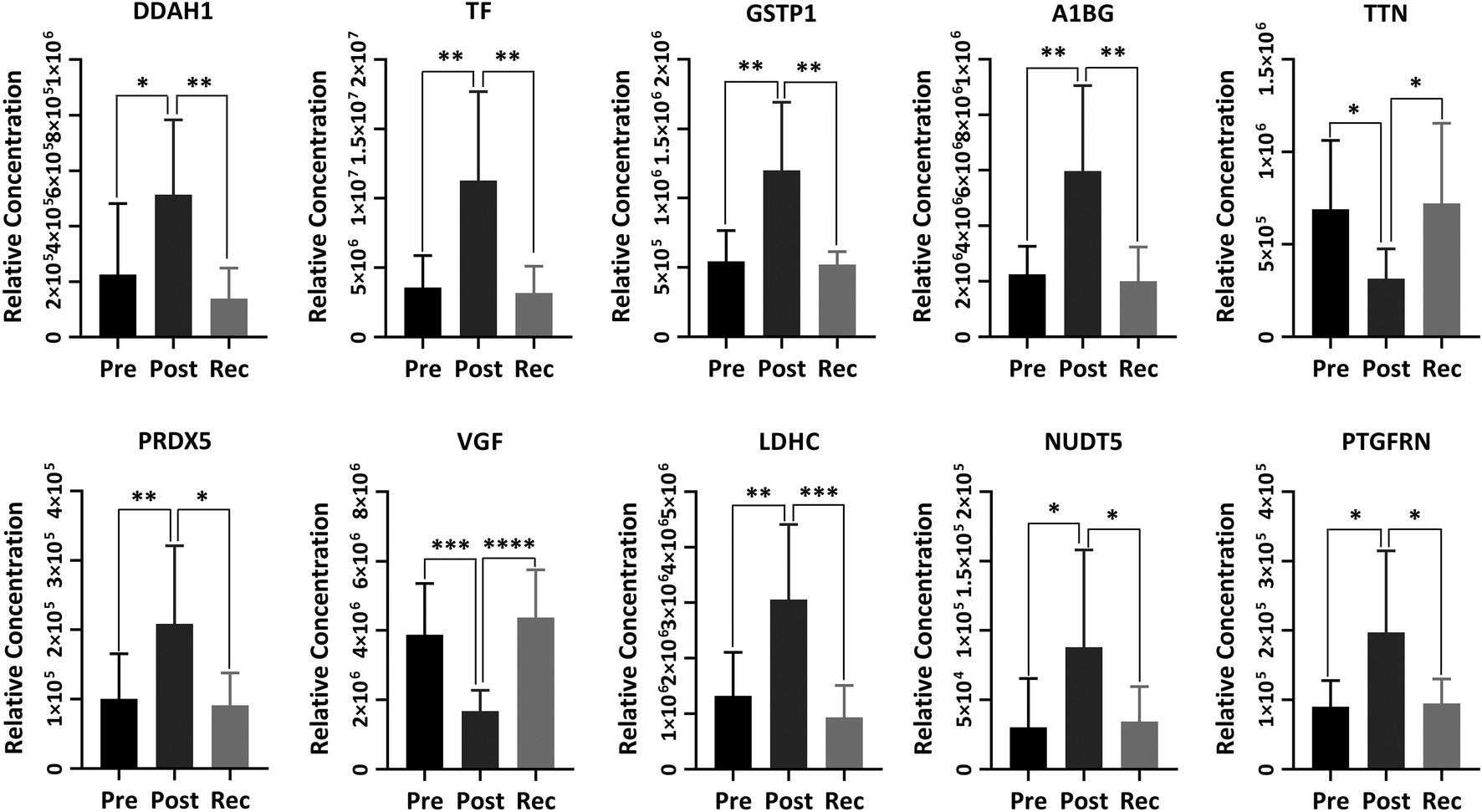 | ||
| Fig. 6 The relative abundance of important proteins identified from the pre-exercise, post-exercise and recovery samples. Error bars represent the standard deviation. DDAH1, TF, GSTP1, A1BG, TTN, PRDX5, VGF, LDHC, NUDT5 and PTGFRN represent N(G),N(G)-dimethylarginine dimethyl-aminohydrolase 1, serotransferrin, glutathione S-transferase P, α-1B-glycoprotein, titin, peroxiredoxin-5, neurosecretory protein VGF peptides, L-lactate dehydrogenase C chain, ADP-sugar pyrophosphatase and prostaglandin F2 receptor negative regulator, respectively. “*”, “**”, “***” and “****” indicate p values smaller than 0.05, 0.01, 0.001 and 0.0001, respectively. | ||
N(G),N(G)-Dimethylarginine dimethyl-aminohydrolase 1 (DDAH1) is a crucial protein for eliminating asymmetric dimethylarginine (ADMA) and monomethyl arginine (L-NMMA), which are strongly predictive of premature cardiovascular disease and death.44,45 Thus, the dysfunction of DDAH1 may lead to an increased cardiovascular risk. It has been well proved that DDAH1 plays a crucial role in promoting cardiac angiogenesis. Our recent study proved that moderate exercise elevated DDAH1 protein expression in mice.45 In the current study, DDAH1 protein expression was upregulated after HIIT exercise and downregulated in recovery, the variational trend of which was also found in serotransferrin (TF), glutathione S-transferase P (GSTP1) and α-1B-glycoprotein (A1BG). TF is relevant to mineral absorption and hypoxia-inducible factor 1 (HIF-1) signalling pathway.46 GSTP1 is a member of the glutathione S-transferases (GSTs), a family of enzymes playing an important role in detoxification and in the antioxidant defense system. There is some evidence indicating that GSTP1 may be beneficial for exercise performance.47 A1BG has been proved to be associated with glycemic control.48 Titin (TTN), one of the biomarkers of muscle damage, was not increased after HIIT exercise, indicating that this exercise program protected the muscle to some extent instead of damaging the muscle.49 Peroxiredoxin-5 (PRDX5) with a high expression in human tissues is a member of the family of mammalian proteins that neutralize reactive oxygen species and are sensitive to oxidative stress.50 We found that PRDX5 was upregulated after HIIT exercise from Fig. 6. Some research indicated that the high basal expression of PRDX5 is coordinated with the expression of nuclear genes encoding mitochondrial proteins and that the PRDX5 protein might play a major role in the permanent defense against reactive oxygen species produced by mitochondria.51 Neurosecretory protein VGF peptides have been proved to play a critical role in the control of energy homeostasis, as well as important neuroendocrine functions.52 Thus, VGF showed a significant decrease after HIIT exercise to maintain energy homeostasis. Proteins associated with energy production, such as the L-lactate dehydrogenase C chain (LDHC) and ADP-sugar pyrophosphatase (NUDT5), were increased after HIIT exercise and returned to the baseline after recovery. The research has shown that subjects in a fracture group showed increased LDHC levels as compared to non-fracture subjects.53 Prostaglandin F2 receptor negative regulator (PTGFRN), with upregulation after exercise, is an inhibitor of prostaglandin signalling and follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion, and thus can regulate sex steroid hormones.54 Changes in other differential proteins between post-exercise and recovery are listed in sheet 2 and sheet 3 of the Supplemental Data.
Protein–protein interaction (PPI) network analysis of all differential proteins was applied using the STRING database, which can provide known and predicted PPI, to summarize the influence of HIIT exercise on proteomics as a whole. As shown in Fig. S10,† comparing post-exercise to pre-exercise, most of the differential proteins have known or predicted interactions and there is one closely associated cluster (blue cycle) involved in immunological function. The cluster with the blue cycle in Fig. S11† was also associated with immunologic function and the proteins in the red cluster that participated in glycolysis were relevant to the energy metabolism. In short, the proteomic response to HIIT exercise is conspicuous and manifests as changes in protein expression associated with energy production, immune function, and metabolism.
4. Conclusions
For the first time, the integration of metabolomics and proteomics has been applied to reveal the metabolic characteristics of HIIT exercise in youth soccer players. Differential metabolites associated with amino acid metabolism were significantly upregulated after exercise, which may result from oxidative stress induced by HIIT exercise as found in proteomic analysis. The changes in steroid hormone metabolism were downregulated after exercise and upregulated during the recovery, which may indicate the increase in muscle growth following HIIT exercise. The expression of proteins in urine was also regulated by HIIT exercise and the energy consumption promoted the upregulation of energy-related proteins in the process of exercise. Most of the differential proteins were related to carbohydrate metabolism, immune system and cardiovascular signalling transduction, which extended our metabolomic findings. Metabolomics and proteomics reflected the influence of HIIT on urine metabolic characteristics from the level of metabolisms and proteins, respectively. Assessment of these changes could provide a valuable measure of the current physical status of the athletes, which may be beneficial for arranging future training programs and preventing possible disorders associated with overtraining.Conflicts of interest
The authors declare that they have no conflicts of interest in relation to the work described here.Acknowledgements
This work was supported by the postdoctoral science foundation of China (Grant No. 2018M642057).Notes and references
- J. Wu and Y. Gao, Expert Rev. Proteomics, 2015, 12, 623–636 CrossRef CAS.
- M. Li, Adv. Exp. Med. Biol., 2015, 845, 13–19 CrossRef.
- S. Wagner, K. Scholz, M. Sieber, M. Kellert and W. Voelkel, Anal. Chem., 2007, 79, 2918–2926 CrossRef CAS.
- E. A. Palmer, H. J. Cooper and W. B. Dunn, Anal. Chem., 2019, 91, 14306–14313 CrossRef CAS.
- N. Bergman and J. Bergquist, Analyst, 2014, 139, 3836–3851 RSC.
- L. M. Heaney, K. Deighton and T. Suzuki, J. Sports Sci., 2019, 37, 959–967 CrossRef.
- G. Quintas, X. Reche, J. D. Sanjuan-Herraez, H. Martinez, M. Herrero, X. Valle, M. Masa and G. Rodas, Metabolomics, 2020, 16, 45 CrossRef CAS.
- K. Contrepois, S. Wu, K. J. Moneghetti, D. Hornburg, S. Ahadi, M. S. Tsai, A. A. Metwally, E. Wei, B. Lee-McMullen, J. V. Quijada, S. Chen, J. W. Christle, M. Ellenberger, B. Balliu, S. Taylor, M. G. Durrant, D. A. Knowles, H. Choudhry, M. Ashland, A. Bahmani, B. Enslen, M. Amsallem, Y. Kobayashi, M. Avina, D. Perelman, S. M. Schüssler-Fiorenza Rose, W. Zhou, E. A. Ashley, S. B. Montgomery, H. Chaib, F. Haddad and M. P. Snyder, Cell, 2020, 181, 1112–1130 CrossRef CAS.
- Z. Kong, X. Fan, S. Sun, L. Song, Q. Shi and J. Nie, PLoS One, 2016, 11, e0158589 CrossRef.
- I. B. Garcia, J. A. R. Arias, D. J. R. Campo, I. M. Gonzalez-Moro and M. C. Poyatos, Rev. Esp. Cardiol., 2019, 72, 233–243 CrossRef.
- J. C. Quindry, B. A. Franklin, M. Chapman, R. Humphrey and S. Mathis, Am. J. Cardiol., 2019, 123, 1370–1377 CrossRef.
- C. Enea, F. Seguin, J. Petitpas-Mulliez, N. Boildieu, N. Boisseau, N. Delpech, V. Diaz, M. Eugene and B. Dugue, Anal. Bioanal. Chem., 2010, 396, 1167–1176 CrossRef CAS.
- A. Pechlivanis, S. Kostidis, P. Saraslanidis, A. Petridou, G. Tsalis, V. Mougios, H. G. Gika, E. Mikros and G. A. Theodoridis, J. Proteome Res., 2010, 9, 6405–6416 CrossRef CAS.
- I. Cervenka, L. Z. Agudelo and J. L. Ruas, Science, 2017, 357, eaaf9794 CrossRef.
- J. W. Starnes, T. L. Parry, S. K. O’Neal, J. R. Bain, M. J. Muehlbauer, A. Honcoop, A. Ilaiwy, P. M. Christopher, C. Patterson and M. S. Willis, Metabolites, 2017, 7, 40 CrossRef.
- F. Al-Khelaifi, I. Diboun, F. Donati, F. Botre, M. Alsayrafi, C. Georgakopoulos, K. Suhre, N. A. Yousri and M. A. Elrayess, Sports Med. Open, 2018, 4, 2 CrossRef.
- B. Yan, J. A. G. Wang, H. Lu, X. Huang, Y. Liu, W. Zha, H. Hao, Y. Zhang, L. Liu, S. Gu, Q. Huang, Y. Zheng and J. Sun, J. Appl. Physiol., 2009, 106, 531–538 CrossRef CAS.
- G. D. Lewis, L. Farrell, M. J. Wood, M. Martinovic, Z. Arany, G. C. Rowe, A. Souza, S. Cheng, E. L. McCabe, E. Yang, X. Shi, R. Deo, F. P. Roth, A. Asnani, E. P. Rhee, D. M. Systrom, M. J. Semigran, R. S. Vasan, S. A. Carr, T. J. Wang, M. S. Sabatine, C. B. Clish and R. E. Gerszten, Sci. Transl. Med., 2010, 2, 33ra37 Search PubMed.
- K. C. Bedi, N. W. Snyder, J. Brandimarto, M. Aziz, C. Mesaros, A. J. Worth, L. L. Wang, A. Javaheri, I. A. Blair, K. B. Margulies and J. E. Rame, Circulation, 2016, 133, 706–716 CrossRef CAS.
- M. L. Goodwin, J. E. Harris, A. Hernandez and L. B. Gladden, J. Diabetes Sci. Technol., 2007, 1, 558–569 CrossRef.
- R. Berton, M. S. Conceicao, C. A. Libardi, R. R. Canevarolo, A. F. Gaspari, M. P. Chacon-Mikahil, A. C. Zeri and C. R. Cavaglieri, J. Sports Sci., 2017, 35, 1211–1218 CrossRef.
- W. Dudzinska, A. Lubkowska, B. Dolegowska, K. Safranow and K. Jakubowska, Eur. J. Appl. Physiol., 2010, 110, 1155–1162 CrossRef CAS.
- A. Leibowitz, Y. Klin, B. F. Gruenbaum, S. E. Gruenbaum, R. Kuts, M. Dubilet, S. Ohayon, M. Boyko, E. Sheiner, Y. Shapira and A. Zlotnik, Acta Neurobiol. Exp., 2012, 72, 385–396 Search PubMed.
- M. Whitham, B. L. Parker, M. Friedrichsen, J. R. Hingst, M. Hjorth, W. E. Hughes, C. L. Egan, L. Cron, K. I. Watt, R. P. Kuchel, N. Jayasooriah, E. Estevez, T. Petzold, C. M. Suter, P. Gregorevic, B. Kiens, E. A. Richter, D. E. James, J. F. P. Wojtaszewski and M. A. Febbraio, Cell Metab., 2018, 27, 237–251 CrossRef CAS.
- S. M. Schüssler-Fiorenza Rose, K. Contrepois, K. J. Moneghetti, W. Zhou, T. Mishra, S. Mataraso, O. Dagan-Rosenfeld, A. B. Ganz, J. Dunn, D. Hornburg, S. Rego, D. Perelman, S. Ahadi, M. R. Sailani, Y. Zhou, S. R. Leopold, J. Chen, M. Ashland, J. W. Christle, M. Avina, P. Limcaoco, C. Ruiz, M. Tan, A. J. Butte, G. M. Weinstock, G. M. Slavich, E. Sodergren, T. L. McLaughlin, F. Haddad and M. P. Snyder, Nat. Med., 2019, 25, 792–804 CrossRef.
- D. C. Nieman, R. A. Shanely, N. D. Gillitt, K. L. Pappan and M. A. Lila, J. Proteome Res., 2013, 12, 4577–4584 CrossRef CAS.
- D. J. Klein, K. H. McKeever, E. T. Mirek and T. G. Anthony, Front. Physiol., 2020, 11, 110 CrossRef.
- J. Chong, O. Soufan, C. Li, I. Caraus, S. Z. Li, G. Bourque, D. S. Wishart and J. G. Xia, Nucleic Acids Res., 2018, 46, W486–W494 CrossRef CAS.
- D. Szklarczyk, J. H. Morris, H. Cook, M. Kuhn, S. Wyder, M. Simonovic, A. Santos, N. T. Doncheva, A. Roth, P. Bork, L. J. Jensen and C. von Mering, Nucleic Acids Res., 2017, 45, D362–D368 CrossRef CAS.
- X. Shen, R. Wang, X. Xiong, Y. Yin, Y. Cai, Z. Ma, N. Liu and Z. J. Zhu, Nat. Commun., 2019, 10, 1516 CrossRef.
- C. A. Smith, G. O'Maille, E. J. Want, C. Qin, S. A. Trauger, T. R. Brandon, D. E. Custodio, R. Abagyan and G. Siuzdak, Ther. Drug Monit., 2005, 27, 747–751 CrossRef CAS.
- S. Benito, A. Sanchez-Ortega, N. Unceta, F. Andrade, L. Aldamiz-Echevarria, M. A. Goicolea and R. J. Barrio, Analyst, 2018, 143, 4448–4458 RSC.
- A. Zhang, H. Sun, Y. Han, Y. Yuan, P. Wang, G. Song, X. Yuan, M. Zhang, N. Xie and X. Wang, Analyst, 2012, 137, 4200–4208 RSC.
- K. Yang, M. L. Duley and J. Zhu, J. Agric. Food Chem., 2018, 66, 1386–1393 CrossRef CAS.
- L. P. Paris, C. H. Johnson, E. Aguilar, Y. Usui, K. Cho, L. T. Hoang, D. Feitelberg, H. P. Benton, P. D. Westenskow, T. Kurihara, J. Trombley, K. Tsubota, S. Ueda, Y. Wakabayashi, G. J. Patti, J. Ivanisevic, G. Siuzdak and M. Friedlander, Metabolomics, 2016, 12, 15 CrossRef.
- Y. Zhang, X. J. Yu, W. S. Chen, H. L. Gao, K. L. Liu, X. L. Shi, X. Y. Fan, L. L. Jia, W. Cui, G. Q. Zhu, J. J. Liu and Y. M. Kang, Sci. Rep., 2016, 6, 37467 CrossRef CAS.
- F. Navarro, A. V. N. Bacurau, G. B. Pereira, R. C. Araujo, S. S. Almeida, M. R. Moraes, M. C. Uchida, L. F. B. P. Costa Rosa, J. Navalta, J. Prestes and R. F. Bacurau, Eur. J. Appl. Physiol., 2013, 113, 1343–1352 CrossRef CAS.
- W. Zhou, G. Zeng, C. Lyu, F. Kou, S. Zhang and H. Wei, J. Sports Sci. Med., 2019, 18, 253–263 Search PubMed.
- W. J. Kraemer, N. A. Ratamess and B. C. Nindl, J. Appl. Physiol., 2017, 122, 549–558 CrossRef CAS.
- J. L. Vingren, W. J. Kraemer, N. A. Ratamess, J. M. Anderson, J. S. Volek and C. M. Maresh, Sports Med., 2010, 40, 1037–1053 CrossRef.
- M. S. Tremblay, J. L. Copeland and W. Van Helder, J. Appl. Physiol., 2004, 96, 531–539 CrossRef CAS.
- C. P. Sharp and D. R. Pearson, J. Strength Cond. Res., 2010, 24, 1125–1130 CrossRef.
- A. J. Peckett, D. C. Wright and M. C. Riddell, Metabolism, 2011, 60, 1500–1510 CrossRef CAS.
- J. Leiper, M. Nandi, B. Torondel, J. Murray-Rust, M. Malaki, B. O'Hara, S. Rossiter, S. Anthony, M. Madhani, D. Selwood, C. Smith, B. Wojciak-Stothard, A. Rudiger, R. Stidwill, N. Q. McDonald and P. Vallance, Nat. Med., 2007, 13, 198–203 CrossRef CAS.
- X. Shi, X. Luo and X. Xu, BioSci. Trends, 2020, 14, 115–122 CrossRef CAS.
- R. Beard, D. C. A. Gaboriau, A. D. Gee and E. W. Tate, Chem. Sci., 2019, 10, 10772–10778 RSC.
- A. Zarebska, Z. Jastrzebski, M. Kaczmarczyk, K. Ficek, A. Maciejewska-Karlowska, M. Sawczuk, A. Leonska-Duniec, P. Krol, P. Cieszczyk, P. Zmijewski and N. Eynon, Biol. Sport, 2014, 31, 261–266 CrossRef CAS.
- N. Piyaphanee, Q. Ma, O. Kremen, K. Czech, K. Greis, M. Mitsnefes, P. Devarajan and M. R. Bennett, Proteomics: Clin. Appl., 2011, 5, 334–342 CAS.
- S. Li, M. Liang, D. Gao, Q. Su and I. Laher, J. Cardiovasc Transl. Res., 2019, 12, 404–414 CrossRef.
- C. Brinkmann, N. Chung, U. Schmidt, T. Kreutz, E. Lenzen, T. Schiffer, S. Geisler, C. Graf, G. Montiel-Garcia, R. Renner, W. Bloch and K. Brixius, Scand. J. Med. Sci. Sports, 2012, 22, 462–470 CrossRef CAS.
- A. Kropotov, N. Usmanova, V. Serikov, B. Zhivotovsky and N. Tomilin, FEBS J., 2007, 274, 5804–5814 CrossRef CAS.
- S. R. Salton, G. L. Ferri, S. Hahm, S. E. Snyder, A. J. Wilson, R. Possenti and A. Levi, Front. Neuroendocrinol., 2000, 21, 199–219 CrossRef CAS.
- T. Miyamoto, Y. Oguma, Y. Sato, T. Kobayashi, E. Ito, M. Tani, K. Miyamoto, Y. Nishiwaki, H. Ishida, T. Otani, H. Matsumoto, M. Matsumoto and M. Nakamura, Sci. Rep., 2018, 8, 18019 CrossRef CAS.
- K. Ahmed, M. P. LaPierre, E. Gasser, R. Denzler, Y. Yang, T. Rulicke, J. Kero, M. Latreille and M. Stoffel, J. Clin. Invest., 2017, 127, 1061–1074 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an01287d |
| This journal is © The Royal Society of Chemistry 2020 |