
Ligand targeting and peptide functionalized polymers as non-viral carriers for gene therapy
Khan
Muhammad†
a,
Jing
Zhao†
a,
Ihsan
Ullah
a,
Jintang
Guo
 ab,
Xiang-kui
Ren
abc and
Yakai
Feng
*abc
ab,
Xiang-kui
Ren
abc and
Yakai
Feng
*abc
aSchool of Chemical Engineering and Technology, Tianjin University, Yaguan Road 135, Tianjin 300350, P. R. China
bCollaborative Innovation Center of Chemical Science and Chemical Engineering (Tianjin), Tianjin 300350, P. R. China
cKey Laboratory of Systems Bioengineering (Ministry of Education), Tianjin University, Tianjin 300072, P. R. China. E-mail: yakaifeng@tju.edu.cn
First published on 10th October 2019
Abstract
Polymeric gene carriers have been developed to deliver therapeutic genes for treating various genetic diseases. They may diminish the problems related with viral vectors in terms of safety, immunogenicity and mutagenesis. But inefficient endosomal release, cytoplasmic transport and nuclear entry are the main limiting issues in the usage of polymeric carriers. Different strategies have been proposed to functionalize gene carriers for individually overcoming these barriers. Towards this aim, various polymeric carriers have been developed and further modified with certain ligands and peptides to achieve properties like good cargo DNA protection, excellent colloidal stability, high cellular uptake efficiency, efficient endo/lysosome escape, efficient import into the nucleus and specificity to target cells. These multifunctional polymeric carriers are beneficial for efficient gene delivery. In this review, we provide a comprehensive summary of the interaction between gene and polymeric carriers and the mechanism and challenges of gene carriers. We also highlight and discuss the recent developments and advancements in the synthesis of ligand targeting and peptide functionalized gene carriers.
1. Introduction
Gene therapy has been proved to possess important therapeutic abilities and it has significant prospect for the treatment of inherited and life threatening diseases caused by genetic deficiencies and abnormalities, such as cancers, acquired immune deficiency syndrome, cardiovascular diseases, and certain autoimmune disorders.1–4 Gene therapy can be used in the most common sense as a technique to deliver genes into chromosomes of target tissues or cells to regulate or replace abnormal genes.5 This therapy can overcome the limitations related with the direct administration of therapeutic proteins, including systemic toxicity, low bioavailability, in vivo instability, and high hepatic and renal clearance rates.6Theoretically, gene therapy is a simple therapeutic method but in practice it is a very complicated process due to several hurdles that need to be overcome by the transgene to reach the targeted cell-nucleus, where it expresses correctly.7 After strong efforts in this field for the last 20 years, gene therapy is now considered as the most modern and beneficial approach for the treatment of various genetic diseases, such as mitochondrial related diseases,8 blindness,9 muscular dystrophy,10 cystic fibrosis11 and certain cancers.12 The first successful study was performed in the 1990s to treat severe combined immunodeficiencies.13 2597 clinical trials of gene therapy had been reported by the end of 2017 in different countries,14i.e., USA (63.3%), UK (8.5%), Germany (3.5%), China (3.2%), France (2.3%), Switzerland (1.9%), Japan (1.7%), Netherlands (1.4%), Australia (1.2%), Spain (1.2%) and others (11.8%).
There are two major gene carriers, i.e., viral vectors and non-viral carriers. Despite their efficient delivery capability, severe limitations of viral vectors such as low DNA/RNA loading capacity, immunogenicity and toxicity have stimulated new approaches towards the development of non-viral carriers. Non-viral carriers compromise highly efficient gene delivery systems due to their facile chemistry, flexible manufacturing and safe profiles. Several kinds of nanoscale DNA/drug carriers have been developed, such as polymers,15 dendrimers,16 responsive materials,17,18 organic–inorganic hybrids,19,20 two dimensional nanomaterials21 and lipid-based materials. However, only a few types are considered for clinical trials such as 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP)-cholesterol, polyethyleneimine (PEI), poly(ethylene glycol)-polyethyleneimine-cholesterol (PEG-PEI-cholesterol), PEI-mannose-dextrose and so on.22 Polymeric carriers can condense DNA/RNA molecules into nanometer- or micrometer-sized complexes via electrostatic interaction between opposite charges, defend the cargo from enzymatic/non-enzymatic degradation, and improve cellular communication via electrostatic interaction.23
Non-viral carriers are still facing complications from administration to localization in the nucleus. Common barriers include instability of the complexes in extracellular spaces, poor cellular uptake, low endosomal escape and their translocation into the nucleus for transcription. Besides, biological barriers such as immunological responses have also been described.24 The chemical and physical barriers are the most unfavorable factors for efficient gene delivery compared with biological barriers.25 A broad knowledge of the barriers and mechanism is necessary for the development of a stable and efficient gene delivery system. Before discussion on efficient DNA carriers, we review the mechanism and challenges involved in the DNA delivery process.
2. Mechanism of the gene delivery process
Gene delivery is a multistep process that involves the condensation of the gene, cellular attachment, cellular uptake and intracellular trafficking. Its mechanism usually depends on the cell type (Fig. 1).26 | ||
| Fig. 1 Schematic presentation of the gene delivery process. Reproduced with permission from ref. 26, copyright [2014], [Ivyspring International Publisher]. | ||
2.1. Condensation of the gene
DNA having negative charge needs to be loaded on cationic carriers and condensed into compact nanoparticles (polyplexes) of a size less than 200 nm to facilitate efficient delivery.22,27 DNA condensation is typically due to the electrostatic interaction between the positive charge of the carriers and the negative charge of the phosphate groups of DNA. Thus, the construction of synthetic non-viral carriers has been constrained by the compromise between transfection efficiency and cytotoxicity, which is controlled by carrier surface charge. A higher surface charge means stronger DNA binding ability, which is beneficial for efficient cellular uptake and gene transfection. But it is also associated with higher cytotoxicity. Excess of positive charge can lead to the interaction of DNA carriers with polyanionic biomolecules in the bloodstream, which may cause the inhibition of many cellular processes.28 Thus, strong DNA binding ability and minimum cytotoxicity are two key parameters for an efficient carrier.2.2 Cellular attachment
There are two ways for gene carriers to reach the cell. One way is non-specific cellular attachment. This process is due to the electrostatic interaction between the opposite charges of polyplexes and the cell surface. Even though the adhesion of anionic carriers has been reported, positive nanoparticles are beneficial for strong cellular attachment and high cellular uptake. They have a greater chance to succeed in the transfection process.29 For non-specific targeting carriers, the attachment has been suggested to be motivated by glycosaminoglycans (GAGs).30 GAGs have a dense glycocalyx of large O-linked oligosaccharides with negatively charged repeating disaccharide units.31 They may act as “receptors” and assist the entry of nanoparticles. However, in numerous cases, GAGs cannot induce internalization on their own and it has been proposed that they can stimulate other receptors which may lead to signaling processes to start the uptake.31The absence of specific targeting moieties may lead to internalization in other types of cells rather than in the desired one. In this regard, the gene carriers should have special chemical structures or peptide sequences that assist their interaction with the target cell membrane. For this purpose, several ligands and peptide sequences such as folate (FA),32 transferrin,33 monoclonal antibodies (mAb), and Arg-Gly-Asp (RGD), Arg-Glu-Asp-Val (REDV) and Cys-Ala-Gly (CAG) peptides have been used to modify gene carriers.34 They can induce the carriers to target cells and overexpress receptors. Many research studies have demonstrated that targeting delivery can enhance cellular attachment and uptake, especially in cancer cells and endothelial cells.35–38
Although active targeting of gene carriers is supposed to be a good approach, it has to be considered that the attachment and binding of the carriers efficiently to the cell membrane do not guarantee the completion of the delivery process. It needs further processes for ultimate success.
2.3 Cellular uptake
Polyplexes are considered as a good cargo for efficient cellular uptake. To reach the nuclei of target cells, the therapeutic DNA must pass through certain biological barriers via energy dependent pathways.39 Cellular uptake is classified as either endocytic or nonendocytic pathways. The endocytic pathways include phagocytosis, clathrin-mediated endocytosis (CME), caveolae-mediated endocytosis (CvME) and macropinocytosis (Fig. 2).39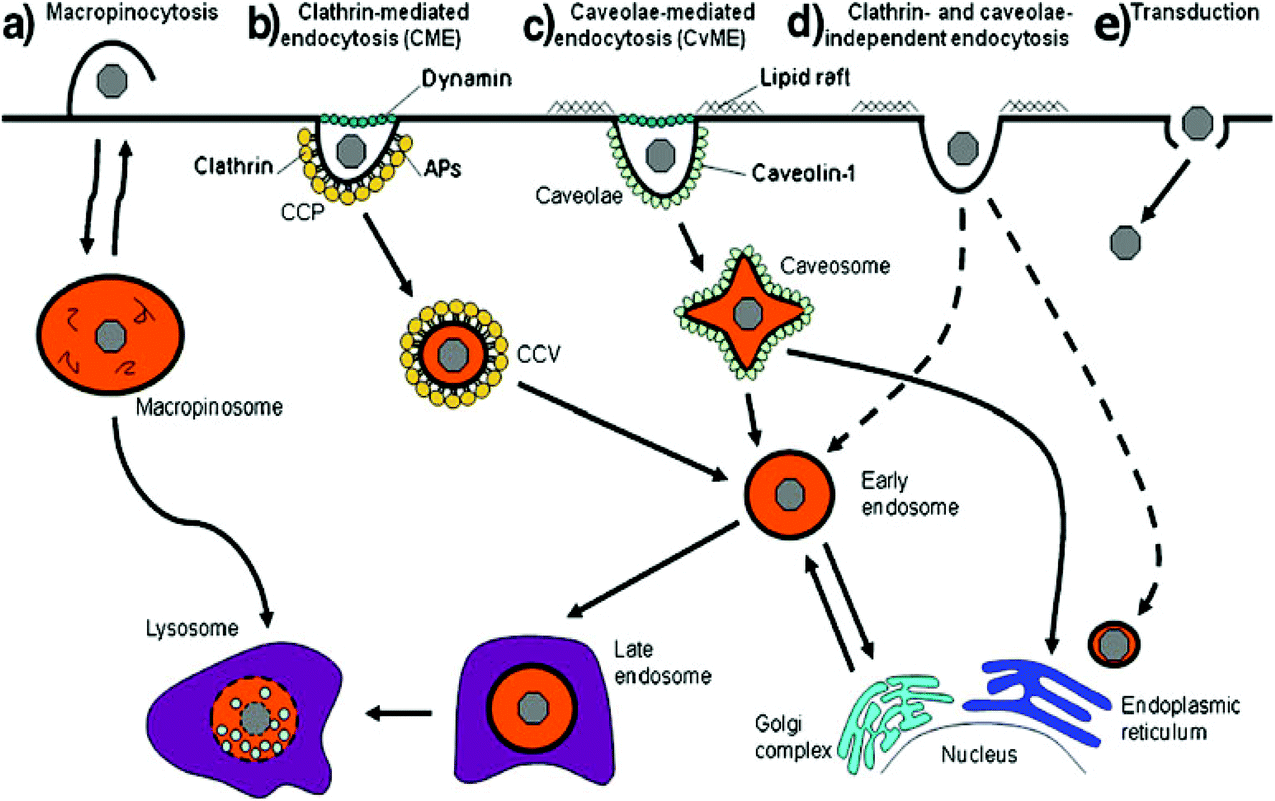 | ||
| Fig. 2 Different endocytic pathways: (a) macropinocytosis, (b) clathrin-mediated endocytosis, (c) caveolae-mediated endocytosis, (d) clathrin- and caveolae-independent endocytosis and (e) transduction. Dashed lines represent alternative pathways. Reproduced with permission from ref. 39, copyright [2012], [Dove Medical Press Ltd]. | ||
Phagocytosis mostly occurs in professional phagocytes such as macrophages, monocytes, neutrophils and dendritic cells. Nonprofessional phagocytosis has also occurred in fibroblasts, epithelial cells and endothelial cells.40 Large polyplexes and lipoplexes are recommended to be taken up by phagocytosis. Opsonization is an important step in the phagocytosis pathway. Particles or complexes are first opsonized by opsonins in the bloodstream, followed by binding to phagocytes and then ingestion.40,41 Following ingestion, phagosomes attach to lysosomes to produce phagolysosomes where they undergo acidic and enzymatic degradation42 (Fig. 3). That is why there is a need to design gene carriers that protect genes from enzymatic degradation.
 | ||
| Fig. 3 Different stages of phagocytosis. (1) Recognition in the bloodstream through opsonisation. (2) The opsonized complex attached to the cell. (3) Complexes are ingested into a phagosome. (4) Formation of a phagolysosome. Reproduced with permission from ref. 40, copyright [2010], [Elsevier]. | ||
Clathrin-mediated endocytosis depends on receptors. Transferrin has been utilized as a ligand of gene carriers to enhance the endocytosis of complexes.43 After binding to the cell surface receptor, clathrin assembles together right on the cytosolic surface of the membrane, which initiates the deformation of the membrane into coated pits (100–150 nm in size).44 This process continues until vesicle splitting takes place, which needs dynamin, a kind of GTPase, and cholesterol. Subsequent to endocytosis, endosomes mature into late endosomes via acidification by proton pumps of the endosomal membrane and are lastly attached to lysosomes.43 Some gene carriers such as PEI can discharge genes by the proton sponge effect in the acidic environment of endosomes. The nitrogen atoms of PEI are protonated to consume endosomal protons, which causes an increase in the chloride anion concentration, and as a result the endosomal membrane breaks due to osmotic swelling, finally releasing the complexes.45 The payload is released by a flip-flop mechanism in the case of lipoplexes, where positively charged lipids interact with negatively charged lipids from the cytoplasm-facing monolayer of the endosomes. This leads to the ejection of the payload into the cytoplasm.46
CvME starts in a special flask-shaped structure on the cell membrane called the caveola, which is a type of cholesterol and sphingolipid enriched smooth invagination. CvME usually takes place in the vessel wall lining monolayer of endothelial cells.39 The diameter of the caveola ranges from 50 to 100 nm and is usually between 50 and 80 nm with a neck of 10–50 nm.47 It is also a type of cholesterol- and dynamin-dependent and receptor-mediated pathway. The splitting of the caveola from the membrane is mediated by the GTPase dynamin, which is present in the neck of the caveola, and leads to the production of the cytosolic caveolar vesicle.41 Some receptors present in the caveola, such as insulin receptors48 and epidermal growth factor receptor (a type of receptor in ovarian cancer),49 can mediate CvME. The budding from the caveola is a type of caveolin-1-having caveosome.50 The intracellular fate of the caveosome varies from that of CME. CvME is usually considered as an alternative pathway which can bring the carriers into the Golgi apparatus and/or endoplasmic reticulum in order to circumvent normal lysosomal degradation.
Macropinocytosis is a type of distinct pathway that non-specifically takes up a huge amount of fluid-phase contents by fluid-phase endocytosis (FPE).51 Usually it is induced by growth factors such as macrophage colony stimulating factor-1 (CSF-1), epidermal growth factor (EGF) and tumour promoting factor. It is also dependent on Rac-1 (an intracellular signalling protein). Macropinocytosis takes place through the formation of actin-driven membrane protrusions, which is similar to phagocytosis. However, in this case, the protrusions do not zipper up the ligand-coated particle; instead, they collapse onto and fuse with the plasma membrane.41 The macropinosomes have no seeming coat structure and are varied in size, but are generally larger than 0.2 μm in diameter. During this course, the small GTPases, Ras-related in brain (Rab) proteins, are vital for vesicle fission from the cell membrane.52
Endocytosis of the genetic materials causes poor intracellular availability due to the acidic and enzymatic environment of lysosomes. So non-endocytic pathways are favourable for high intracellular availability of genetic materials. Some technologies are intended to mediate the non-endocytic pathways. One is microinjection, by which each cell is inserted with genes by using glass capillary pipettes. The second one is permeabilization by applying pore-forming reagents such as streptolysin O or anionic peptides such as the HA2 subunit of influenza virus hemagglutinin. The third one is electroporation, in which an electric field produces pores in the cell. The abovementioned technologies are extremely worrisome processes and not suitable for in vivo gene delivery.
Other non-endocytic pathways are related to the formation of a hole in the cell membrane. Protein transduction domains (PTDs) such as HIV-1 transactivator of transduction sequence (TAT) have the ability for this process. These peptides can directly penetrate the cell membrane. Poly(amidoamine) (PAMAM) was also studied for the non-endocytic pathway mechanism. The cationic PAMAM can diffuse into the cells through a small hole in the membrane. The mechanism is considered to be a non-specific pathway which is not receptor mediated and lacks selective uptake.43
Fusion with the cell membrane is another non-endocytic pathway, which is special for lipoplexes, as they can directly release DNA into the cytoplasm before entering the endocytic pathways. However, many pieces of evidence suggest that fusion contributes slightly to the overall uptake of lipoplexes, while there is a very good contribution of CME in the uptake of lipoplexes.53
2.4 Intracellular trafficking
Endo/lysosomal escape is one of the most critical issues for gene delivery. Non-viral delivery systems may be locked in and degraded in the lysosomes if their cellular uptake pathways include endo/lysosomes. Some of the uptake pathways involve endo/lysosomes, such as CME and phagocytosis, while CvME delivers vectors into the Golgi apparatus or endoplasmic reticulum instead of lysosomes, thus avoiding normal lysosomal degradation. Similarly, macropinocytosis does not have any links with endo/lysosomes, but some studies have proposed that it involves lysosomes.54Numerous methods have been tried to promote early endosomal escape of gene delivery vehicles and many hypotheses have been proposed to explain these processes. The proton sponge hypothesis has been proposed for cationic polymers such as PEI and PAMAM dendrimers.55 For cationic-lipid based delivery systems, the flip-flop mechanism was suggested to be favourable for their endosomal escape mechanism.46 Cell-penetrating peptides (CPPs) signify another class of promising candidates as non-viral nucleic acid carriers,56 for example, TAT, pep analogues, GALA, MPG, CADY, LAH4 derivatives, etc. Their mechanisms of promoting endosomal release are still challenging. It has been suggested that the opening of transient pores in the lipid bilayer of endosomes is involved; alternatively CPPs may undergo conformational changes in response to the acidification inside the endosomes, leading to destabilization of the endosomal membrane bilayer. Photochemical internalization is a novel light-based strategy that aims to trigger endosomal escape.57 A photosensitizer is confined in the endosomal membrane and causes the destabilization of membrane on illumination and prompting the release of endosomal contents into the cytosol.58
Although there are different types of possible mechanisms and strategies, most of the non-viral carriers remain in the endo/lysosomes, irrespective of their transfection efficiency.30 Furthermore, autophagy may be induced by nanoparticles to form autophagosomes, followed by eventual digestion after grouping with lysosomes into phagolysosomes.59 This procedure, in addition to the probability of exocytosis events,60 includes a main hurdle for efficient transfection that may be overcome by designing suitable delivery systems.
After endosomal escape, DNA should be free or unpacked from the original carriers, while it needs to overcome the most difficult barrier, i.e., the nuclear envelope, to enter into the nucleus. DNA enters the nucleus during the process of mitosis when the nuclear envelope is broken down.61 The plasmids can also enter the nucleus in the absence of cell division but this process is slow and very inefficient, resulting in very low nuclear entry because the structure is too preventive and only molecules below 50 kDa are allowed to pass via the diffusion process. Fortunately, the nuclear import of plasmid DNA can be enhanced by specific DNA sequences. Plasmids containing the SV40 enhancer can target the nucleus of non-dividing cells within a few hours. This SV40 sequence, named DNA nuclear targeting sequence, is beneficial for nuclear import.
3. Ligand targeting and peptide functionalized polymers
Despite the large number of ongoing clinical studies, many challenges remain in realizing the complete benefits of gene therapy. Many polymers such as dextran,62–64 cationic polyrotaxane,65 chitosan,66 pullan67 and cyclodextran68 have been explored for gene delivery. The ideal gene delivery carriers should possess many properties like DNA protection, excellent colloidal stability, high cellular uptake efficiency, efficient endo/lysosomal escape, efficient nuclear import and DNA unpacking. Some of the most popular cationic polymers like PEI, poly(L-lysine) (PLL) and PAMAM dendrimers cannot fulfil these requirements. It is necessary to develop carriers with targeting ligands and functionalized with certain peptides to overcome the challenges and achieve clinically acceptable properties.PEI and its derivatives are widely used as cationic polymeric carriers.69–73 PEI is available in a linear structure (l-PEI) and a branched structure (b-PEI). b-PEI has 25% primary amines, 50% secondary amines and 25% tertiary amines. b-PEI can successfully condense plasmids into colloidal particles that efficiently transfect DNA into various cells both in vitro and in vivo. These particles possess a spherical shape and have a narrow particle size distribution, which probably lead to high cellular uptake and good transfection efficiency.74 The gene delivery efficiency depends on the degree of polymerization (molecular weight), branching (topology) and buffering capacity.
PEI has high charge density, because every third atom on the PEI backbone is a nitrogen atom. In the case of l-PEI, all of the nitrogen atoms are protonatable, whereas in b-PEI, only two-thirds of them can be protonated.75 The protonation level of PEI increases from 20% to 40% when the pH decreases from 7 to 5. During the maturation of the endosome, the ATPase proton pump shifts protons from the cytosol into the endosome. At this stage, PEI polyplexes are protonated and the proton concentration increases within the endosome, therefore the osmotic pressure of the endosome increases, which finally causes the endosomal rupture.76 Therefore, PEI may boost intracellular trafficking by buffering the endosomal compartments, thus protecting the DNA from lysosomal degradation and helping DNA release through lysosomal disruption.
The homogeneous and small sized PEI/DNA complexes are beneficial for high gene expression in mature mouse brain. The in vitro transfection efficiency of PEI increases with the molecular weight. However, the in vivo efficiency drops with Mw. Some contradictory results have been reported on the influence of PEI branching density. The transfection efficiency of PEI is relatively higher than most non-viral carriers, but its non-biodegradability and intolerable toxicity produce hurdles for clinical applications.77–79
The transfection efficiency can be improved by increasing cellular uptake via joining target-specific ligands. Folate (FA), hyaluronic acid (HA), peptides and antibodies are commonly used ligands.80–83 These ligands are conjugated onto the backbone of carriers either directly or by spacers where the ligands can be disconnected from the backbone in a controlled mode.84
3.1. Folate based targeting polymers
Folate receptors are cysteine-rich cell-surface glycoproteins that integrate folate with strong affinity to mediate cellular uptake of folate. Folate receptors (FRα) are expressed in different cancer cells to meet the requirement of folate for fast division of cells under low folate conditions. The folate dependency of various tumour cells gives a direction to synthesize folate conjugated polymers for targeting delivery.85 FA based targeting polymers showed higher transfection efficiency in folate receptor overexpressing cells.86Folate was linked to PLL with a PEG spacer to obtain FA-PEG-PLL.87 This polymer coated onto the complexes of PEI/DNA for receptor mediated gene transfer. They showed considerably greater transfection efficiency in KB cells than in folate receptor deficient A549 cells. Interestingly, these formulated complexes also displayed much higher transfection efficiency than PEI/DNA or lipofectamine/DNA complexes in the presence of 10% serum. Folate can increase cellular uptake by receptor-mediated endocytosis, and the PEG segment well stabilized the complexes by enhancing their solubility and decreasing the non-specific adsorption of serum proteins. These complexes also showed much lower cytotoxicity than PEI/DNA complexes. Besides, FA and PEG were also used to modify PEI for minicircle DNA (mcDNA) delivery. The targeting delivery of mcDNA to tumour cells gave a good result in gene expression as compared with the conventional plasmid delivery system. In this delivery system, PEG conjugation decreases cytotoxicity through shielding of the positive charge of PEI.88 Reducing the positive charge of cationic carriers without disturbing their transfection efficiency is somewhat difficult. To achieve this goal, pH responsive charge-convertible ternary complexes were developed.89 These ternary complexes possessed negative charge at pH 7.4 and possessed positive charge at pH 6.5 with a particle size of 150 nm. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay demonstrated that they had low cytotoxicity, especially lower at pH 7.4 than at pH 6.5.89 Folate conjugated PEI-g-PCL-b-PEG was prepared and used for targeted gene delivery. In this synthesis, the folate moiety was conjugated at the distal PEG by click chemistry and then PEI was grafted by the Michael addition reaction. Click chemistry and Michael addition reactions are a useful method to retain the biological activity of folate and to avoid difficult cleaning procedures. This approach linked the folate ligand to a flexible PEG spacer and probably increased their target binding efficacy. This copolymer possessed low cytotoxicity and could condense DNA at a N/P ratio >2. Furthermore, the cellular uptake and transfection efficiency of PEI-g-PCL-b-PEG-Fol/pDNA complexes were examined in vitro in FR-positive KB cells. They showed good cellular uptake and 14-fold higher transfection efficiency than the control group.90 A monovalent/divalent FA modified polyamidoamine–polyethylenimine (PME) through a PEG spacer was developed and the effect of FA density was examined. The introduction of different FA densities at the end of PEG had no negative effects on the condensation ability, cytotoxicity and hemolysis of their DNA complexes. The cellular uptake and transfection efficiency were considerably increased with the increase of the FA density on PEGylated PME.91
The folate-chitosan-graft-polyethyleneimine (FA-CS-g-PEI) copolymer was synthesized and used as a good cancer cell targeting gene carrier. This carrier exhibited high ability to form complexes with shRNA. These complexes possessed suitable physiochemical properties, high transfection efficiency and cancer cell specificity.92 In particular, they had low cytotoxicity due to the presence of biocompatible chitosan, the low molecular weight of PEI (1.8 kDa), and the shielding of the primary amines of PEI by chitosan. Besides chitosan, cyclodextrins as biocompatible and solubility-enhancing moieties are usually used to modify gene carriers. DNA carriers were developed by grafting 0.6 kDa PEI onto β-cyclodextrin and attaching FA as a targeting ligand. The resultant carriers possessed low cytotoxicity and high specific delivery ability to tumour cells.93
Hydroxyl terminated PAMAM dendrimers were attached to S-methyl-L-cysteine via an acid-labile ester bond, named β-thiopropionate bond, and then FA was attached through a PEG linker to produce an acid-labile delivery carrier.94 This carrier possessed strong gene condensation ability and low cytotoxicity to KB and HepG2 cells. It exhibited higher transfection efficiency than PAMAM/DNA. Importantly, this nanocarrier displayed long circulation time along with excellently targeted accumulation in the tumour site in vivo.94 Besides, a multifunctional gene delivery system was developed as follows: pDNA and high mobility group box 1 (HMGB1) were condensed with PAMAM dendrimers to produce a binary complex (PAMAM/HMGB1/DNA, denoted as PHDs) and then coated with folate-PEG-carboxylated chitosan (FA-PEG-CCTs) to obtain the ternary complexes (FA-PEG-CCTs/PAMAM/HMGB1/DNA, denoted as FPCPHDs).95 HMGB1 is a protein with the amino acid sequence known as nuclear localization signals, which can improve DNA nuclear import. FPCPHDs showed long circulation time in vivo to target the tumor tissue and are beneficial for high cellular uptake by cancer cells through FR-mediated endocytosis. They efficiently and rapidly discharged from endosomes and accumulated in the nucleus. Thus they can transfect target cells with high gene transfection and expression efficiency.95
A supramolecular approach was applied to construct a single gene carrier system, i.e., PCD-SS-PDMAEMA/Ad-PEG-FA (Fig. 4). This delivery system consisted of host and guest modules. PCD-SS-PDMAEMA acted as the host module and consisted of a poly(β-cyclodextrin) backbone and disulfide-linked PDMAEMA arms, while Ad-PEG-FA acted as the guest module and was composed of adamantyl and folate terminated PEG. This resultant gene carrier could efficiently condense DNA into stable nanosized polyplexes, release DNA in a reducing environment, significantly enhance hemocompatibility and precisely transfer both DNA and siRNA into folate-receptor positive cells by folate-mediated targeting.96 It possessed simultaneously many functions required to overcome delivery barriers.
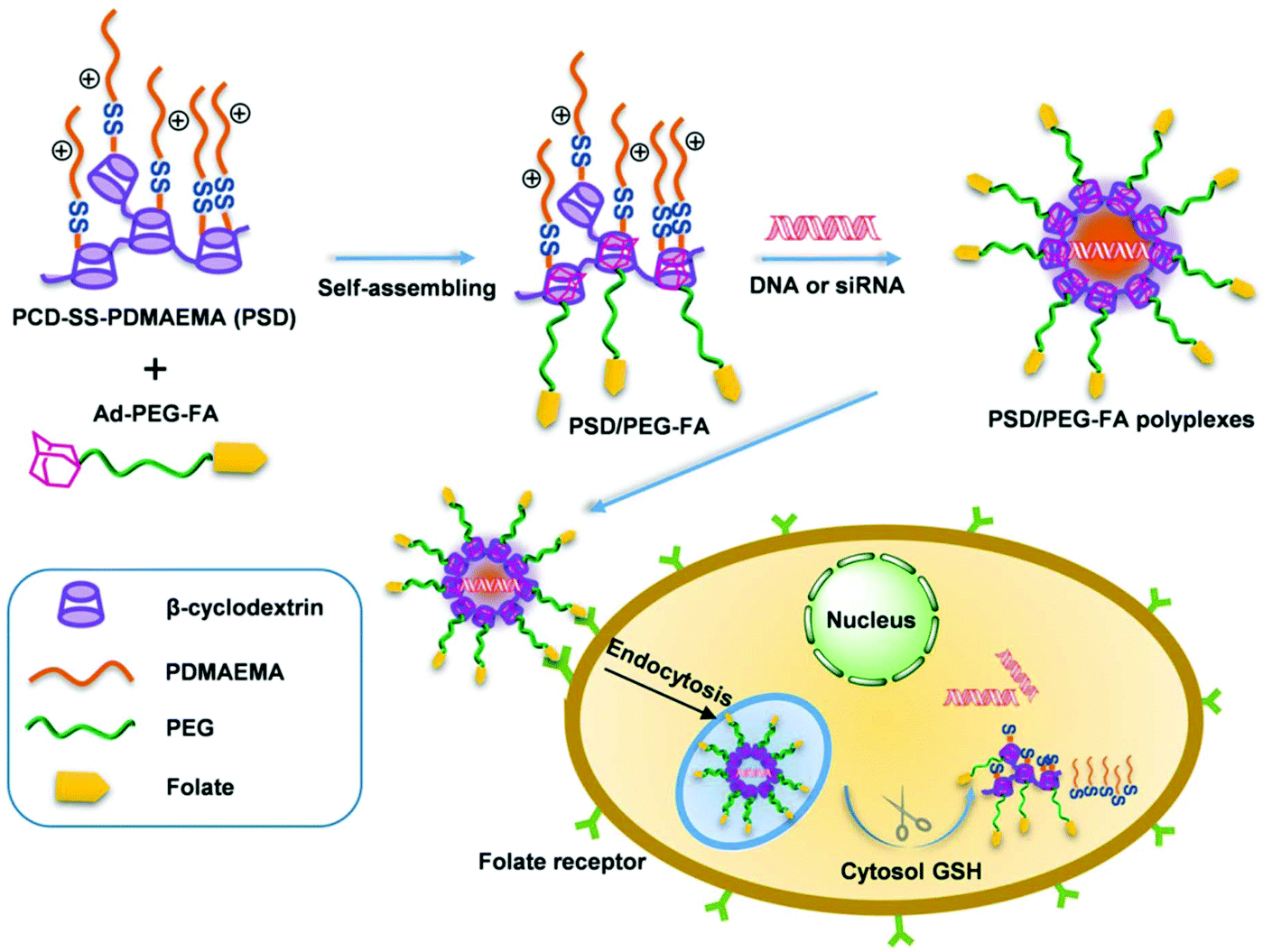 | ||
| Fig. 4 Schematic illustration of the host–guest self-assembly process, receptor-mediated specific cellular uptake, and glutathione-triggered intracellular gene release from the PSD/PEG-FA supramolecular gene delivery system. Reproduced with permission from ref. 96, copyright [2014], [American Chemical Society]. | ||
A multifunctional, dual targeted, pH-responsive siRNA nano-carrier served as smart polymeric nanoparticles (SPNs) to simultaneously target two hallmarks of the tumour microenvironment. It was designed in such a way to develop their selectivity compared to the conventional targeting approaches by energetically delivering siRNA payloads only to the positions where both of these two hallmarks exist. In this approach, the mixed micelle was prepared from two polymers having the same pH-responsive, endosomolytic core forming block but different hydrophilic corona-forming blocks. One polymer consisted of 2 kDa PEG attached to the FA group while the second polymer was composed of y-shaped 20 kDa PEG attached to the core block through proximity-activated targeting (PAT) MMP7-cleavable peptides. In the mixed micelle, SPNs were prepared from the FA-based polymer (FA-SPN) and PAT-based polymer (PAT-SPN) (Fig. 5). It has been observed that cellular uptake and siRNA knockdown of a 50% FA/50% PAT formulation depend upon both proteolytic activation and FA receptor engagement. Furthermore, MMP activation and the delivery of siRNA to breast cancer cells expressing the FA receptor achieved greater than 50% protein-level knockdown of a model gene with very low cytotoxicity.97
 | ||
| Fig. 5 Formation of a mixed micelle from FA-SPN and PAT-SPN. Reproduced with permission from ref. 97, copyright [2014], [American Chemical Society]. | ||
A tumour targeting gene delivery system (Dex-SSBAP30-FA) was synthesized in three steps. In the first step, the bioreducible cationic polyamide (pSSBAP) with a primary amine ending group was prepared by a stepwise polycondensation reaction of bis(p-nitrophenyl)-3,3′-dithiodipropanoate and 1,4-bis(3-aminopropyl)piperazine (BAP). In the second step, two cationic dextran conjugates, i.e., Dex-SSBAP6 and Dex-SSBAP30, were prepared. Finally, folate was attached to Dex-SSBAP30 by carbodiimide chemistry. pSSBAP was highly effective for in vitro gene delivery against MCF-7 and SKOV-3 cell lines. It was explained that both two conjugates possess the capability to bind DNA to form polyplexes with better colloidal stability under physiological conditions. In vitro transfection experiments showed that the transfection efficiency of polyplexes of Dex-SSBAP30 is comparable to that of linear PEI or Lipofectamine2000. Furthermore, Dex-SSBAP30-FA was effective for targeted gene delivery to the SKOV-3 tumour xenografted in a nude mouse model by intravenous injection, prompting an advanced level of gene expression in the tumour cells as compared to Dex-SSBAP30.98
A star shaped bioreducible cationic polymer (γ-CD-OEI-SS-FA) was synthesized from FA, γ-cyclodextrin (γ-CD) and oligoethylenimine (OEI) for targeted gene delivery (Fig. 6). This cationic polymer was composed of a γ-CD core and multiple OEI arms with FA attached through a bioreducible disulfide bond. This polymer was examined for its gene delivery properties in FR-positive KB cells and FR-negative A549 cells in contrast with cationic polymers such as PEI (25 kDa), γ-CD-OEI and γ-CD-OEI-FA polymers. The γ-CD-OEI-SS-FA polymer showed similar cytotoxicity to γ-CD-OEI and γ-CD-OEI-FA polymers, and exhibited very low cytotoxicity as compared to PEI (25 kDa). Furthermore, the γ-CD-OEI-SS-FA polymer exhibited good DNA condensation ability and formed complexes with the particle size varying from 100 to 150 nm and the positive zeta potential ranging from 20 to 40 mV at N/P ratios of 10 to 60.99
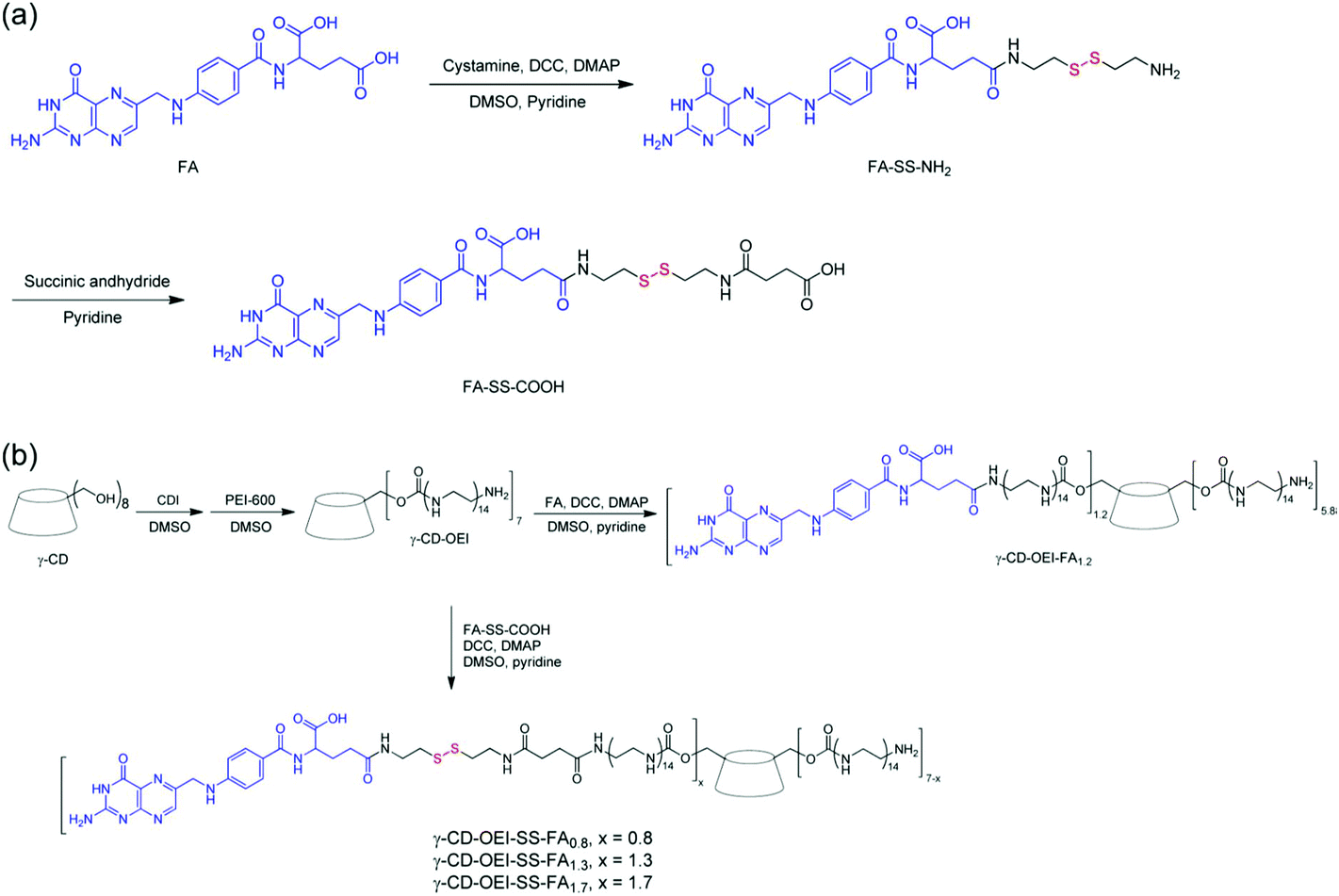 | ||
| Fig. 6 (a) Synthesis of FA with the disulfide linker FA-SS-COOH and (b) γ-CD-OEI-FA and γ-CD-OEI-SS-FA. Reproduced with permission from ref. 99, copyright [2013], [American Chemical Society]. | ||
A multifunctional bioreducible targeted supramolecular co-delivery system was developed from a star-shaped cationic polymer consisting of γ-CD and OEI arms with FA conjugated via a disulphide linkage. This supramolecular complex system can deliver the anticancer drug paclitaxel (PTX) and DNA. The FA-targeted function enhanced gene transfection in the FR-positive KB cells. The redox sensitive disulphide linker helped to detach the FA group from the carriers after the FR-mediated endocytosis. PTX was delivered to the cell with DNA, which further boosted the transfection efficiency even at a low N/P ratio in the FR-positive KB cells.100
A folated PEG-oligoaminoamide copolymer was composed of eight units of succinoyl-tetraethylene pentaamine (Stp), two cysteines, PEG and a folate group.101 The Stp units provided the positive charge for siRNA binding, while cysteine was beneficial for disulfide-based polyplex stabilization. This copolymer complexed with lipo-oligomers for siRNA delivery to treat leukemia. These delivery systems exhibited strong cellular internalization in FR-rich cells.101
3.2. HA based targeting polymers
CD44 is a cell adhesion molecule. CD44 is overexpressed in numerous cancer cells and impaired endothelium in atherosclerotic lesions.102 HA is a basic ligand of CD44. HA is a naturally occurring mucopolysaccharide, consisting of a tandem disaccharide repeat of β-1,4-D-glucuronic acid-β-1,3-D-N-acetylglucosamine. HA exhibits biodegradability, nontoxicity, non-immunogenicity and non-inflammation.103–105 The electrostatic interaction is a simple and straightforward strategy to introduce targeting ligands into delivery systems. HA has dual benefits in gene delivery, namely, it neutralizes the cationic charge density of polyplexes and also increases specificity toward the wanted receptor and cell type.106HA is usually used as coating material for gene complexes in order to enhance their serum stability by reducing the surface positive charge, and also to enable cancer cell targeting through CD44 receptors.107 For example, HA was coated on the pH-responsive, reversibly crosslinked PEI-based polyplexes (K-PEI/DNA) for efficient and targeted gene delivery.108 The result indicated that K-PEI/DNA polyplexes quickly degraded into low molecular weight segments in endosomes which was beneficial for them to release DNA efficiently. HA coating modification improved the polyplexes’ stability under physiological conditions and also reduced the cytotoxicity.108 Aldehyde group-bearing HA was applied to shield PEI/DNA complexes through electrostatic and chemical crosslinking.109 This dual crosslinking method condensed PEI/DNA into complexes with a size of 190 nm and a zeta potential of 15 mV. Cell and animal experiments demonstrated that these complexes exhibited good stability in the serum and long circulation time in vivo. These dual-crosslinked nanoparticles had good accumulation ability in tumours.109
In order to develop the reducible shielding materials, HA sequentially reacted with cystamine, dithiothreitol and 3-mercaptopropionic acid to obtain the disulfide bond modified HA (HA-SS-COOH).110 A reduction-sensitive disulfide bond was introduced between the carboxyl group and HA backbone. This reducible shielding material coated the PEI/pDNA complexes to develop the more effective gene delivery system, i.e., PEI/pDNA/HA-SS-COOH ternary complexes.110 These ternary complexes possessed low cytotoxicity, and they could greatly enhance cellular uptake and transfection efficiency. More interestingly, HA-SS-COOH shielding was superior to that of HA due to the extra responsive de-shielding function.110 Another modified HA was prepared by conjugating ethylamine-bridged epigallocatechin gallate (EGCG) dimers to HA using a carbodiimide-mediated coupling reaction.111 The conjugates were attached to the binary system of PEI/pDNA to obtain the ternary gene delivery system of PEI/pDNA/HA-EGCG (Fig. 7). These conjugates could stabilize the complexes via the strong DNA-binding affinity of green tea catechin, because EGCG can interact with DNA via hydrogen bonding, π–π stacking and hydrophobic interactions. More importantly, they facilitated the transport into CD44-overexpressing cells via receptor-mediated endocytosis. Thus this system boosted nuclear entry of pDNA compared to the binary system. The HA-green tea catechin conjugates showed promising applications for CD44-targeting delivery of DNA-based therapeutics.111
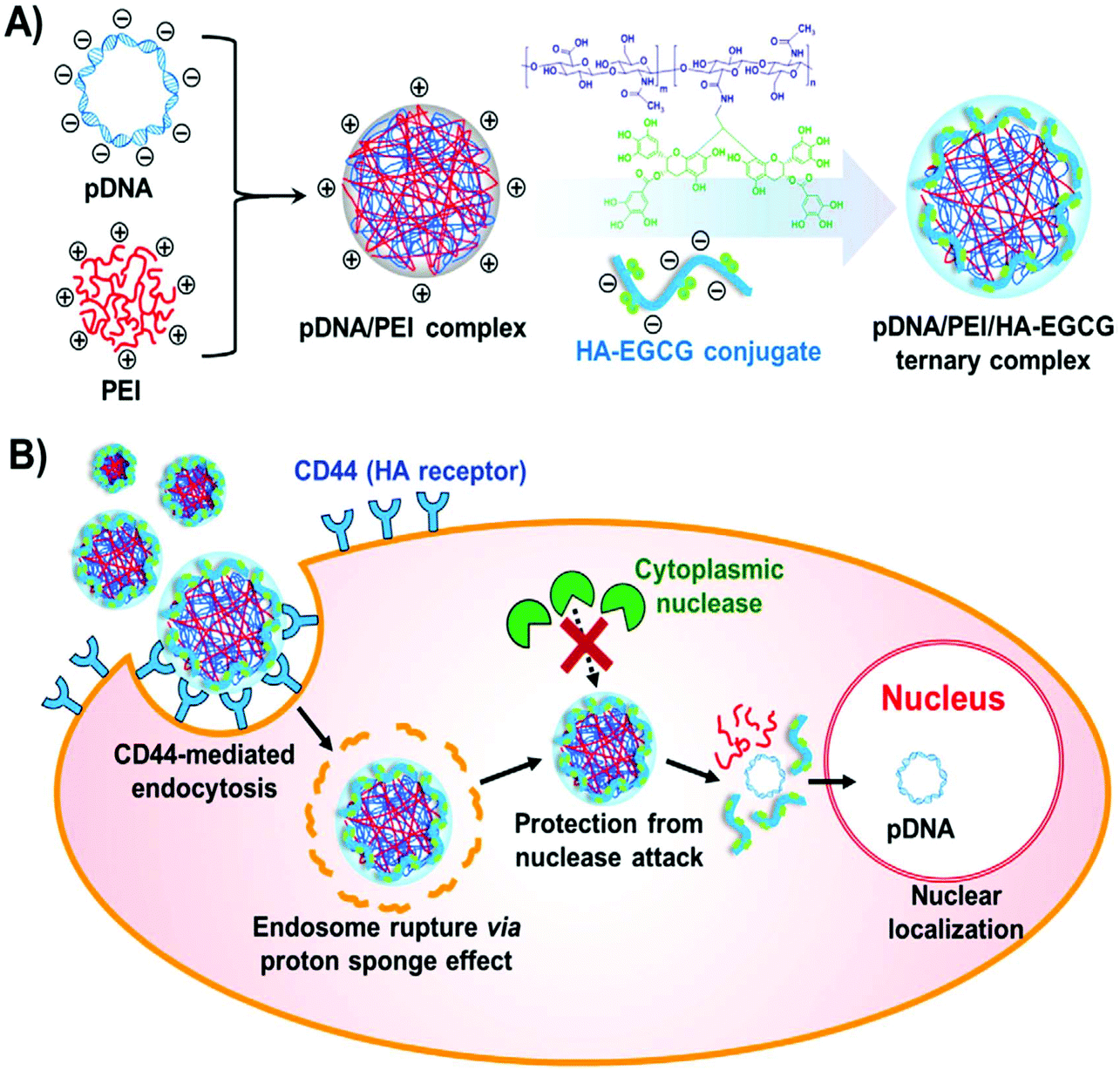 | ||
| Fig. 7 Formation of a ternary complex. (A) Synthetic route of PEI/pDNA/HA-EGCG ternary complex formation. (B) Diagrammatic illustration of receptor mediated cellular uptake, endosomal escape and nuclear localization. Reproduced with permission from ref. 111, copyright [2016], [Elsevier]. | ||
HA was also used to modify gene complexes via host–guest interactions. HA was conjugated with aminomethyladamantane to produce a pendant polymer (HA-Ad).112 The hydrophobic adamantane acted as a guest ligand to attach to the hydrophobic cavity of PEI modified β-CD (CD-PEI+) to produce a strong guest–host relationship. The formed complexes (HA-Ad:CD-PEI+:pDNA) were used for targeting delivery through CD44 receptors (Fig. 8). These HA-based complexes exhibited better target-specific cellular localization in CD44+ HeLa cells. Good transfection efficiencies were found in CD44+ HeLa cells compared with the commercial standard Lipofectamine2000, but low transfection efficiencies in CD44− HeLa and NIH 3T3 fibroblasts. Furthermore, these complexes were attached to the cell surface within 2–4 h. Internalization in acidic endosome/lysosome compartments occurred between 4–9 h, and pDNA cargoes were released within 24 h.112
 | ||
| Fig. 8 Synthetic route of the HA-Ad:CD-PEI+:pDNA complex. Reproduced with permission from ref. 112, copyright [2016], [American Chemical Society]. | ||
Owing to the polyanionic property, HA is usually used to shield the positive charge of gene carriers. HA can decrease the toxicity and enhance the blood stability of the complexes, especially beneficial for receptor-mediated endocytosis. For example, chitosan oligosaccharide (CSO)-grafted PEI (CSO-PEI) was synthesized by an imine reaction between the amine group of PEI and the aldehyde group of periodate-oxidized CSO, and then coated with HA.113 Their siRNA nanoparticles effectively suppressed endometriotic lesion formation with atrophy and degeneration of the ectopic endometrium. The rat models of endometriosis showed that the epithelial cells of the ectopic endometrium had lower CD44 expression than the control.113 HA was coated on the surface of binary gene complexes to produce ternary complexes. These ternary complexes can effectively protect the condensed pDNA and siRNA from enzymatic degradation by DNase I. They can improve the stability in sodium heparin solution or serum in vitro.114
HA nanoshells with a high molecular weight (>260 kDa) on nanoparticles could significantly promote specific CD44 targeting. But HA coated nanoparticles had a large particle size distribution, especially, when using high molecular weight HA. In order to overcome this problem, the high molecular weight HA (HHA) was conjugated with the poly(D,L-lactide-co-glycolide)-poly(ethylene glycol) (PLGA-PEG) copolymer to obtain HHA-PEG-PLGA.115 HHA-PEG-PLGA could encapsulate the DOTAP/pDNA (D/P) lipoplex with high encapsulation efficiency. The micelles had a narrow size distribution with a low polydispersity index (<0.3) and negative zeta potential. PEG and HA were beneficial for high stability of the micelles owing to the shielding effect of hydrophilic PEG and ionic HA surrounding the outer shell of the micelles. Interestingly, these HHA-conjugated micelles not only showed high selectivity and high transfection efficiency to cancer cells, but also demonstrated low cytotoxicity to normal cells.115
Recently, targeting peptides were used to modify HA to obtain multi-target materials.116,117 For example, retro-inverso D-peptide (RIF7) exhibits great binding ability within the Annexin 1 receptor, which is expressed in tumour vasculature cells and certain tumour cells like B16F10 and U87MG cells.118 The RIF7 modified HA (RIF7-HA) was coated onto the gene complexes via the layer-by-layer method to produce the core–shell ternary complexes for the dual targeting delivery of short hairpin RNA-encoding plasmids. These complexes considerably improved the survival time of tumour-bearing mice and significantly reduced tumour growth because they possessed extremely specific tumour-targeting ability. HA and RIF7 effectively increased the therapeutic effect for CD44-positive tumours.118
3.3. Peptide functionalized targeting polymers
Peptide-based ligands are very specific and capable molecules for targeting delivery of complexes in vitro and in vivo. The combination of specific peptides into gene carriers allows the translocation of nucleic acid therapeutics to the chosen location in living organisms.119 RGD is one of the most commonly used small peptides for targeting delivery. RGD grafted cationic polymers show ∼100-fold effective gene delivery in integrin overexpressing fibroblasts. RGD grafted PEI was prepared for the delivery of sFlt-1 specific DNA into primary endothelial cells. sFlt-1 is a natural inhibitor of the VEGF receptor. The fruitful inhibition of angiogenic tissue growth was recorded during in vitro studies. But their poor selectivity and non-specific interactions with smooth muscle cells restricted their applications in vivo.120 When the integrin targeting linear and cyclic RGD (cRGD) peptides were grafted onto the bPEI-PEG copolymer, the resulted polymers were suitable for enhanced targeting function and transfection.121When the RGD peptide and HIV-1 TAT peptide were coated onto PEI/DNA complexes, their cellular uptake was expressively improved and the transfection efficiency in neuronal cells was high.122 A multifunctional DNA delivery carrier had a cRGD moiety and a hydrophobic cholesteryl moiety on the α- and ω-ends of PEG-PAs(DET) as a targeting ligand for enhancing the affinity to the tumour. This nanocarrier showed significant tumour growth suppression as a result of its antiangiogenic effect.123 A well-tailored and multifunctional core–shell ternary gene delivery system (RRPHC) was developed to treat aggressive melanoma (Fig. 9).124 This system had multiple tumour targeting, nucleus targeting and depth penetration abilities for in vivo gene delivery. RRPHC was composed of a core and a capsid-like shell. The core was heptafluorobutyric acid modified PEI (1.8 kDa). This fluorinated PEI could effectively condense pDNA, helped in endosomal escape and possessed nucleus targeting ability. The capsid-like shell was composed of negatively charged multifunctional RGD-R8-PEG-HA (RRPH). RGD-R8 possessed specific targeting and high penetrating abilities due to the specific RGD and cell penetrating peptide R8. This system showed good cellular uptake via dual receptor mediated endocytosis and high gene transfection efficiency in B16F10 cells, which was better than the commercial transfection agents such as PEI (25 kDa), Lipofectamine2000 and Lipofectamine3000.124
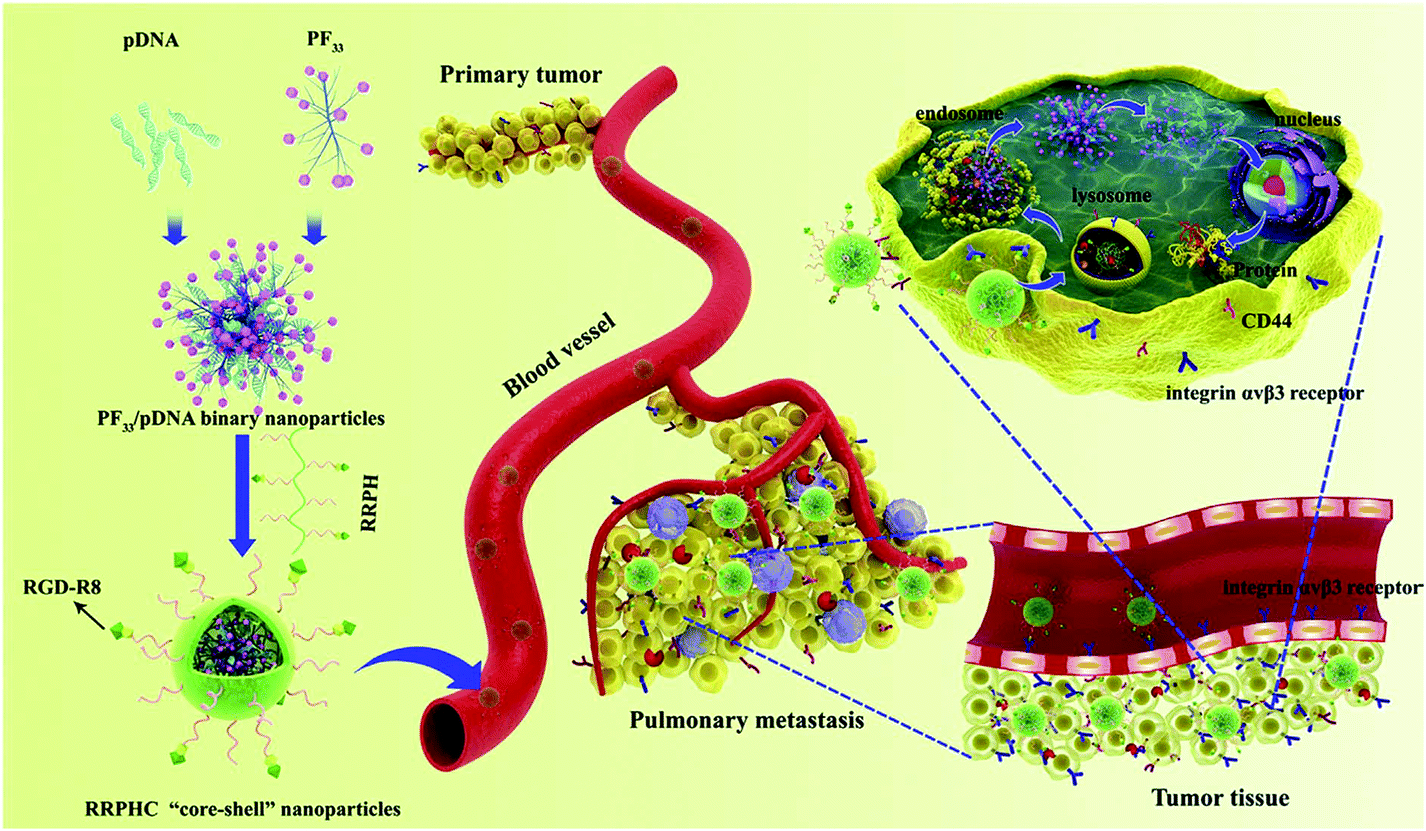 | ||
| Fig. 9 Schematic illustration of the multi-stage and multiple tumour-targeting RRPHC ternary gene delivery system. Reproduced with permission from ref. 124, copyright [2016], [Elsevier]. | ||
Recently, CAG peptide-modified materials showed specific adhesion for human umbilical vein endothelial cells (HUVECs) and served as effective gene carriers for targeting delivery of the pZNF580 plasmid to HUVECs.125 The high CAG content of the complexes was beneficial for high transfection efficiency of HUVECs in vitro and in vivo. CAGW-modified polymeric micelles with different hydrophobic cores (PLGA and PLMD) also exhibited low cytotoxicity, strong cellular uptake ability and high transfection efficiency.126 Another strategy for targeting HUVEC transfection involved the REDV peptide. PEI (1.8 kDa and 10 kDa) was conjugated to a biodegradable diblock copolymer of poly(ethylene glycol) monomethyl ether-block-poly(L-lactide-co-glycolide) (mPEG-b-PLGA) to develop a gene carrier, i.e., mPEG-b-PLGA-g-PEI. Furthermore, the Cys-Arg-Glu-Asp-Val-Trp (CREDVW) peptide was attached to their micelle surface to enable them to have special recognition to ECs. This special structure of PEG, REDV, low molecular weight PEI and biodegradable PLGA was beneficial for low cytotoxicity and high transfection efficiency.127 In order to increase the CREDVW content in the carriers, the comb-like copolymer having many side functional groups was used to link CREDVW. The resulted polymers could condense pDNA with a suitable size and showed high internalization efficiency owing to the high content of CREDVW.128 The REDV modified copolymers with different molar ratios of REDV to PEI can easily control the properties of the resultant complexes.129 The star-shaped copolymer of poly(lactide-co-3(S)-methyl-morpholine-2,5-dione)6 (star-(PLMD)6) was prepared by ring-opening polymerization and then grafted by PEI, PEG and REDV. The star-(PLMD)6 copolymer can easily form nanoparticles owing to the hydrogen bond in PLMD blocks. These nanoparticles have a stable core of PLMD and a hydrophilic shell of PEI and PEG. They condensed pEGFP-ZNF580 with a suitable size through electrostatic interaction with PEI. These complexes showed high selectivity for ECs, high transfection efficiency and migration of ECs.130
The TAT peptide has been proven to promote cellular uptake and gene delivery owing to its high cell membrane penetration. For example, the TAT peptide was grafted onto chitosan (CS) to obtain TAT-g-CS to deliver functional siRNA to tumour cells. The results showed that TAT-g-CS/siRNA nanoparticles inhibited the proliferation of 4T1-Luc tumour cells in vitro. It also showed a strong inhibitory effect on the growth and metathesis of a malignant breast tumour in vivo.131 Recently, polyhedral oligomeric silsesquixane (POSS) has attracted much interest in gene delivery because POSS is biocompatible and has multifunctional groups for linking targeting molecules. The integrated multifunctional REDV-G-TAT-G-NLS-C sequence was conjugated to POSS by hetero-bifunctional poly(ethylene glycol). This polymeric carrier could condense the pZNF580 plasmid into a suitable size. In order to enhance endosomal escape, the histidine enriched peptides of REDV-TAT-NLS-Hn (oligohistidine sequences with n = 4, 8 and 12) were incorporated on the surface of complexes to obtain the ternary gene delivery system. REDV-TAT-NLS-Hn peptides have TAT (typical cell penetrating peptide), NLS (nuclear localization signal), and Hn (n = 4, 8 and 12). This ternary delivery system showed high internalization ability, good cellular uptake, low cytotoxicity, rapid endosomal escape and good translocation properties.132 It has been indicated that Hn is beneficial for high gene transfection.133 Besides, biocompatible star/comb-shaped POSS copolymer hybrids were also prepared from functional peptides. They were composed of a hydrophobic POSS core, eight cationic poly(2-dimethylaminoethyl methacrylate) middle blocks, a hydrophilic comb-shaped PEGMA block and a multifunctional functional CAG-TAT-NLS peptide. These gene carriers with a multifunctional peptide showed high cellular uptake, good nuclear internalization and high transfection efficiency compared to PEI (25 kDa).134 POSS-(C-G-NLS-G-TAT) was prepared by applying “thiol–ene” click chemistry between TAT-G-NLS-G-C and POSS (Fig. 10).135 This cationic POSS-(C-G-NLS-G-TAT) condensed pZNF580 to produce binary complexes (BCPs). They were further modified by the anionic polymer of PLL-g-CAGW21%-g-Acon (n = 47%, 57% and 64%) by electrostatic interaction to prepare ternary gene complexes (TCPs). TCPs were stable in the blood owing to their neutral charge; meanwhile, they could preferentially transfect ECs due to the presence of CAGW. cis-Aconitic amide bonds were cleaved in the acidic environment of endosomes and as a result PLL-g-CAGW21% and BCPs were regenerated. After endosomal escape, the BCPs entered into the cytoplasm and moved toward the nucleus with the help of the nucleus targeting peptide. It was noted that TCPs possessed good hemocompatibility and cytocompatibility and expressively enhanced gene delivery efficiency. Furthermore, the transfected HUVECs achieved high proliferation, high migration and particularly high neovascularization in vitro and in vivo.135
 | ||
| Fig. 10 Schematic illustration showing the design of TCPs and multitargeting gene delivery. (A) The chemical structures of POSS-(C-G-NLS-TAT)16, PLL-g-CAGW21%-g-Acon and PLL-g-CAGW21%. (B) The formation of TCPs, the multitargeting gene delivery process and the angiogenesis of HUVECs in vivo. Reproduced with permission from ref. 135, copyright [2019], [Royal Society of Chemistry]. | ||
A brain targeted gene delivery system (DGL-PEG-Leptin30) was developed from dendrigraft poly(L-lysine) (DGL), polyethylene glycol (PEG) and a 30-amino-acid peptide (leptin30). Leptin30 is a brain targeted ligand which is derived from the endogenic hormone leptin. DGL-PEG-Leptin30 nanoparticles accumulated in the brain after intravenous injection, and showed high transfection efficiency and low toxicity.136 The nucleolar translocation signal (NoLS) sequence of the LIM Kinase 2 (LIMK2) protein (LIMK2 NoLS peptide, LNP) could not only transport genes to the nucleus but also achieve the characteristic cell-penetrating function.137 LNP is a type of efficient CPP for crossing the blood–brain barrier. LNP was used to synthesize DGL-PEG-LNP for glioma gene therapy. The LNP-modified nanoparticles maintained the cell penetrating characteristics of LNP. The conjugation of LNP effectively increased the blood–brain barrier-crossing efficiency, cellular uptake and gene expression within the tumour cells.137
The Ser-Arg-Leu (SRL) peptide was also used to develop high efficiency carriers for brain gene delivery. The SRL peptide was linked to PAMAM dendrimers using bifunctional PEG. This nanocarrier was internalized by brain capillary ECs through clathrin and caveolae mediated endocytosis. This gene delivery system showed good transfection efficiency and low cytotoxicity.138 Importantly, the SRL peptide functionalized carrier was efficient for brain targeting gene delivery both in vitro and in vivo. Angiopep was demonstrated to exhibit high transcytosis ability and parenchymal accumulation owing to its targeting capacity to low-density lipoprotein receptor-related protein-1 (LRP-1). PAMAM dendrimers were modified with angiopep through bifunctional PEG, and complexed with DNA to form PAMAM-PEG-angiopep/DNA nanoparticles. They were internalized by brain capillary ECs through energy dependent endocytosis, i.e., clathrin and caveolae mediated endocytosis. Besides, they were also partly internalized by macropinocytosis. Angiopep-modified nanoparticles showed high efficiency in crossing the blood–brain barrier and accumulated in the brain. Thus these angiopep-modified nanoparticles with high gene expression in the brain showed great potential to be applied in brain-targeting therapy.139 MC11 is a peptide with the sequence of MQLPLATGGGC. MC11 is capable of selectively binding fibroblast growth factor receptor (FGFR), and can act to target FGFR cells via the receptor mediated gene delivery pathway. MC11 was conjugated to a star-shaped cationic copolymer constructed from eight-armed PEG and LMw-PEI.140 The resultant carrier showed high ability to condense DNA with a suitable particle size for cellular uptake and possessed low cytotoxicity in HepG2 and PC3 cell lines.140 More importantly, this carrier could deliver a reporter gene in a targeted manner into tumor tissues by systemic injection. The in vivo results showed that the complexes were much more effective in inhibiting tumor growth.140 The host–guest supramolecular complexation was also used to introduce PEG into MC11 functionalized gene carriers. The advanced multifunctional delivery system (γ-CD-PEI-MC11/Ad-SS-PEG) was synthesized from adamantyl-SS-PEG (Ad-SS-PEG) and γ-CD-PEI-MC11 (Fig. 11). This smart delivery system showed low cytotoxicity and high efficiency for targeted gene delivery for cancer therapy.141
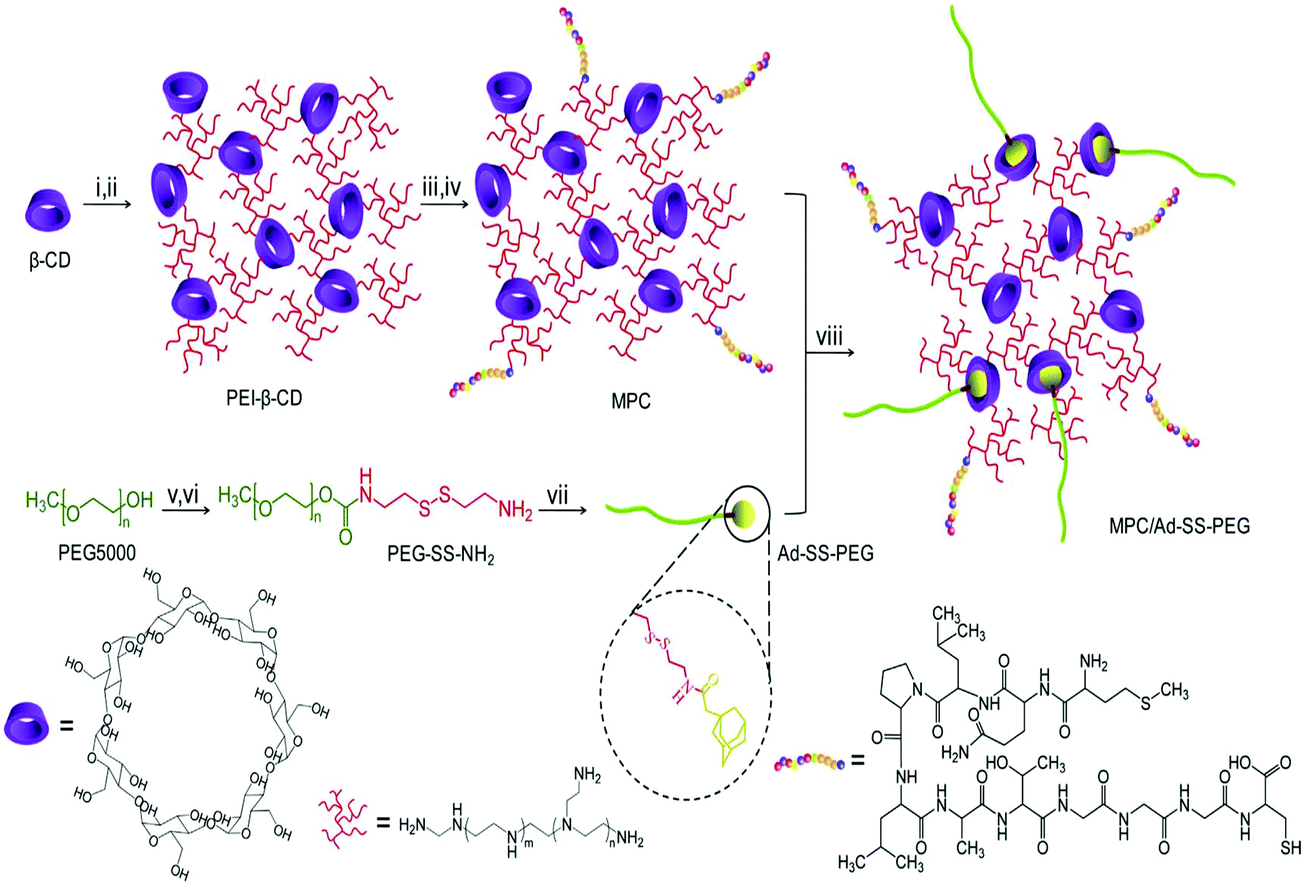 | ||
| Fig. 11 Synthetic route of MPC/ad-SS-PEG, (i) CDI, DMF, 4 h, (ii) PEI (600), DMSO/PBS, 12 h, (iii) SPDP, DMSO, 16 h, (iv) MC11, DMSO/PBS, 12 h, (v) CDI, DMF, overnight, (vi) cystamine dihydrochloride, DMSO, 24 h, (vii) adamantine acetic acid, DCM, 24 h, and (viii) r.t., 12 h. Reproduced with permission from ref. 141, copyright [2013], [Elsevier]. | ||
A triblock copolymer of poly(ethylene glycol)-b-poly(L-lysine)-b-poly(L-leucine) was applied for the systematic co-delivery of docetaxel (DOC) and siRNA-Bcl-2 toward MCF7 breast cancer xenografts in murine models.142 The hydrophobic poly(L-leucine) core entrapped DOC, while the PLL cationic backbone was used for electrostatic binding with siRNA-Bcl-2. This delivery system exhibited strong application in synergistic delivery of siRNA and chemotherapeutic drugs. The multi-component PLL-based gene delivery system was composed of a membrane disrupting peptide, a nucleic acid binding component, a protective coat layer and outer targeting ligands; therefore, it was very useful for gene and drug delivery. Furthermore, it showed high tolerance ability in metabolic activity assay.143
RNA interference technology is an important tool for Alzheimer's disease. In order to treat this disease, a rabies virus glycoprotein (RVG)-modified poly(mannitol-co-PEI) gene transporter (PMT) was used to cross the blood–brain barrier and target neuronal cells.144 The osmotically active PMT increased receptor mediated endocytosis by stimulating caveolar endocytosis, thus avoiding lysosomal degradation. High transfection efficiency and successful brain-target beta-secretase 1 suppression were observed.144 Besides, a well-developed multifunctional carrier was synthesized by the combination of dendrigraft PLL, PEG and brain targeting ligand (RVG29) for the double therapy treatment of Alzheimer's disease. RVG29 is a 29-amino-acid peptide derived from a rabies virus glycoprotein. DNA and peptide drugs could be delivered into the brain simultaneously by this targeting system. Neurofibrillary tangles and memory loss were decreased in mice with Alzheimer's disease.145 Artery wall binding peptide (AWBP) is a specific targeting peptide with a Cys-Gly-Arg-Ala-Leu-Val-Asp-Thr-Leu-Lys-Phe-Val-Thr-Gln-Ala-Glu-Gly-Ala-Lys sequence. AWBP was attached to PLL by a PEG linkage to obtain a AWBP-PEG-g-PLL copolymer. This copolymer combined with DNA to form complexes with a size of 100 nm and showed high transfection efficiency (above 180-fold) as compared to PLL and poly(ethylene glycol)-g-poly(L-lysine) (PEG-g-PLL) in bovine aorta ECs and smooth muscle cells. The presence of free AWBP in the transfection medium minimized the transfection efficiency of AWBP-PEG-g-PLL, indicating that AWBP-PEG-g-PLL could be applied as a tissue-selective gene carrier.146 Besides, dual targeting gene carriers can be developed by linking two kinds of functional peptides. For example, the peptide YC25 and the peptide CP9 can recognize fibroblast growth factor receptors and integrins on the cell surface, respectively.147 These two peptides were used to functionalize PEI to obtain the dual-receptor targeted carrier. This carrier exhibited a higher transgene efficiency in cell lines with positive expression of the fibroblast growth factor receptor and integrin than single peptide modified PEI or unmodified PEI. The in vivo experiment results also demonstrated high transfection efficiency.147
3.4. Antibody based targeting polymers
Antibodies and immunoglobulins are big glycoproteins. Immunoglobulins are involved in some basic processes within the immune systems such as opsonisation, phagocytosis, complement dependent cytotoxicity (CDC) and antibody-dependent cytotoxicity (ADCC). Five types of immunoglobulins have been discovered, namely IgA, IgD, IgE, IgG and IgM, and each type is characterized by special structural characteristics.148Tumour specific antigens such as human epidermal growth factor receptor-2 (HER2) and prostate specific antigens serve as efficient and specific targeting moieties for tumor therapies. For example, the antibodies for a prostate specific membrane antigen were attached to PEI/DNA complexes for the treatment of prostate cancer cells. The in vivo results showed that the antibody attached complexes showed 20-fold improvement over PEI/DNA complexes.149 However, there are some problems related with the use of antibody ligands, such as poor stability, immunogenicity and rapid elimination. Fortunately, the use of antibody fragments can diminish both essential and practical problems encountered during the use of whole antibodies. The small size of antibody fragments is beneficial for low immunogenicity. They have high loading capabilities, good orientation of targeting ligands, and high efficacy.150
An approach was developed for the effective treatment of colitis through the delivery of TNF-α siRNA condensed within F4/80 Fab’ targeted PEG-PLA polymersomes. Fab’ targeted TNF-α siRNA can reduce colonic inflammation.151 A tumor-targeting Fab’ fragment of mAB 323/A3 was combined onto the periphery of a lipid-coated PDMAEMA polyplex. The cellular uptake and transfection efficiency were particularly enhanced as compared to the unconjugated analogues in human ovarian carcinoma (OVCAR-3) cell lines.152
A monoclonal antibody against the leukemia-specific JL-1 antigen (anti-JL-1-Ab) was conjugated with PLL by iodate-mediated oxidation of a carbohydrate moiety in the Fc domain of the antibody, followed by reaction with PLL.153 The anti-JL-1-Ab-PLL conjugate could specially interact with the target antigen (JL-1) of leukemia cells (Molt 4) and was fruitfully internalized into the cells. The anti-JL-1-Ab-PLL conjugate showed higher transfection efficiency than PLL and lipofectin.153
3.5. Others
For targeting delivery, phenylboronic acid (PBA) has been used as a target ligand owing to its specific binding ability to sialylated epitopes which are overexpressed on the surface of various types of tumours. Compared to other targeting moieties, PBA exhibits low immunogenicity and can be easily used to modify many carriers. For example, PBA was used to modify cationic PEI.154 The PBA modified carriers showed high cellular uptake toward tumor cells because the PBA moieties could strongly bind to sialylated glycoproteins. Interestingly, the chemical bonding of PBA with the diol in the sugar enabled the carriers with dual stimuli-responsive properties (acidic endosomal pH and intracellular ATP inside the cells). These dual stimuli-responsive carriers can deliver DNA for high transfection efficiency with low cytotoxicity.Star-shaped cyclodextrin-PLL derivatives (CD-PLLD) were prepared by ‘click’ coupling of per-6-azido-β-cyclodextrin with propargyl focal point PLL for the co-delivery of the chemotherapy drug DOC and MMP-9 siRNA (pMR3) for cancer therapy.155 The CD-PLLD/DOC/pMR3 complexes prompted more significant apoptosis and reduced intrusiveness in nasopharyngeal epithelial carcinoma in contrast to DOC or pMR3 used alone. These star-shaped copolymers have been proved to possess good blood compatibility and minor cytotoxicity as compared to hyperbranched PEI (25 kDa). The mannosylated chitosan-graft-polyethylenimine (Man-CS-g-PEI) copolymer was used as a targeting carrier for the antigen presenting cells having a mannose receptor. This copolymer possessed superior ability for forming complexes with suitable physiochemical properties for the delivery system.156 A galactosylated poly(ethylene glycol)-chitosan-graft-spermine copolymer was developed as a hepatocellular-targeting carrier with strong gene condensation ability. It also exhibited a high level of gene expression efficiency in vivo and in vitro with low cytotoxcity.157 The conjugation of targeting ligands to PEGylated chitosan is considered as one of the famous techniques for increasing specificity. Hepatocyte targeting gene carriers such as galactosylated poly(ethylene glycol)-chitosan-graft-polyethylenimine showed strong condensation ability for DNA, low cytotoxicity and high hepatocyte specificity.158 A pullulan-based conjugated system was developed for liver-targeting gene delivery. Pullulan is a linear carbohydrate with intrinsic liver targeting properties, which interacts with the asialoglycoprotein receptor.67,159
Biotinylated transferrin was also used to modify disulphide containing PEI (PEI-SS) through the biotin–aridin bridge in order to obtain a tumour targeted gene carrier, i.e., biotinylated transferrin/aridin/biotinylated disulphide containing PEI bioconjugates (TABP-SS). TABP-SS complexed with the tumour compressor gene p53 for targeting therapeutic gene delivery. They showed significantly lower cytotoxicity and much higher transfection efficiency in HepG2 and HeLa cells due to the specific interaction between transferrin ligands and their receptor in tumour cells.160 The cationic polyurethane having a disulphide bond and a 1,4-bis(3-aminopropyl)piperazine residue was used as biodegradable multifunctional gene carrier for targeting delivery to ovarian SKOV-3 cells with high gene expression.161
The intelligent nanocarrier systems can be used for combining therapies, i.e., chemotherapy and gene therapy, against human cancer. For this purpose, an amphiphilic copolymer (PEI-SS-PCL-SS-PEI) was prepared through the conjugation of low molecular weight PEI (LMw-PEI) and poly(ε-caprolactone) (PCL) via a cleavable disulphide linkage (Fig. 12). This copolymer tended to form positively charged nanoparticles with a hydrophobic core and a hydrophilic shell, thus enhancing the encapsulation ability of anticancer drugs and genes. It can simultaneously deliver drugs and genes into target cells. In vitro cytotoxicity tests and LDH indicated that it had very low cytotoxicity compared with PEI (25 kDa).162
 | ||
| Fig. 12 Synthetic route of PEI-SS-PCL-SS-PEI with a disulphide linker. (a) Activation of the PCL-diol group, (b) synthesis of PCL-cystamine, (c) conversion of the cystamine group into carboxylic acid, and (d) synthesis of PEI-SS-PCL-SS-PEI. (i) CDI, anhydrous DCM, r.t., 24 h, (ii) cystamine dihydrochloride, DMSO, pyridine, TEA, r.t., 48 h, (iii) succinic anhydride, anhydrous DCM. TEA, r.t., 24 h, (iv) DCC, NHS, r.t., 48 h, and (v) LMw-PEI, DCM, r.t., 48 h. Reproduced with permission from ref. 162, copyright [2016], [Elsevier]. | ||
Silica nanoparticles are an attractive candidate for the development of safe and efficient non-viral carriers.163 Large pore cubic mesoporous silica nanoparticles can be functionalized with PLL for gene delivery.164 The mesoporous silica nanoparticles consisted of multiple cavities (28 nm in size). More interestingly, low molecular weight PLL (2.9 kDa) was covalently immobilized on the outer surface and inner cavities of the nanoparticles. They showed high gene condensation ability and superior transfection of HeLa cells with comparatively low cytotoxicity at a concentration up to 100 μg mL−1. The PLL conjugates established colloidally stable nanoplexes with pEGFP-C2 plasmid DNA, and showed high transfection efficiency in MCF-7 human breast cancer cells.164
Glycol chitosan-based polymers also showed high transfection efficiency and low cytotoxicity in human epithelial ovary carcinoma (HeLa) cells, human embryonic kidney (HEK293) cells and human hepatocellular liver carcinoma (HepG2) cells.165 Multifunctional and well-developed mitochondria-targeted nano-carriers were synthesized for co-delivery of chemotherapeutic drugs and siRNA. For this purpose, the therapeutic drug lonidamine (LND) and the mitochondria targeting ligand (TPP) were attached to the biocompatible chitosan-g-polyethylenimine (CP) copolymer through an amide bond to form the TPP-CP-LND (denoted as TCPL) copolymer and then the stealthy, pH-responsive, detached and tumor targeted copolymer poly(acrylic acid)-polyethylene glycol-folic acid (PPF) coated to the rationally prepared hierarchical mitochondria targeted co-delivery system (TCPL/SiBcl-2/PPFNPs) for cancer therapy (Fig. 13). The unique structure of these carriers endowed them with the capacity of prolonged blood circulation. They simultaneously released siBcl-2 into the cytoplasm and delivered LND to the mitochondria. The synergistic effects sufficiently improved the anticancer efficacy.166
 | ||
| Fig. 13 (A) Scheme of formation of the hierarchical targeted delivery system for co-delivery of siRNA and LND. (B) Schematic illustration of the proposed mechanism of the mitochondria apoptosis pathway synergistically motivated by siBcl-2 and LND. Reproduced with permission from ref. 166, copyright [2015], [Elsevier]. | ||
4. Conclusion and future perspective
Many efforts have been made to develop polymeric carriers to treat various types of genetic diseases, but the gene delivery systems have not met the standard requirement to achieve clinical importance. Gene delivery involves several steps and each step has its own barriers. Therefore, the delivery needs various requirements to overcome these barriers. Viruses have been developed to competently address the requirements to avoid the barriers by hiding from the body defense system and applying intracellular machineries of the host cells for transferring their genetic information. Knowledge has been obtained from viruses, and polymeric cargoes should be wisely designed at the molecular level to mimic the important characteristics of the viruses and compete with viral vectors. The polymeric cargoes are designed to have three functional parts, i.e., cationic polymer part, hydrophilic polymer part and cell or tissue specific ligand moiety. The first part is used to condense DNA and produce complexes, the second part can increase solubility, stability and biocompatibility of the complexes in biological fluids, while the third part mainly enhances selectivity toward the target cells or tissues. The plasmid DNA is composed of two functional elements: (1) promoter for the tissue specific or the stimuli responsive expression of the therapeutic protein and (2) cis-element that interacts with karyophilic proteins for the improvement in cellular trafficking and translocation of the plasmid. The ideal polymeric carriers can load DNA and form stable complexes, which possess high stability in biological solution, hide from the host immune system and transfer DNA to the desired target. After localization within the cell, the polymeric gene carriers should have suitable functionalities, such as escape from the endocytic pathways, supply complexes to the vicinity of the nucleus and unpack in response to pH or a redox environment. Although it is still challenging to incorporate all functionalities to diminish the barriers, smart molecular engineering of functional polymers provides an emerging strategy to mimic most of the viral infection machineries. Multidisciplinary approaches will be required to succeed in the rational design of virus mimicking polymeric carriers in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Key R&D Program of China (Grant No. 2016YFC1100300), the National Natural Science Foundation of China (Grant No. 51673145, 51873149, 21875157 and 31370969), and the International Science & Technology Cooperation Program of China (Grant No. 2013DFG52040).References
- N. Hosseinahli, M. Aghapour, P. H. G. Duijf and B. Baradaran, J. Cell. Physiol., 2018, 233, 5574–5588 CrossRef CAS.
- Y. Xiao, K. Shi, Y. Qu, B. Y. Chu and Z. Y. Qian, Mol. Ther.–Methods Clin. Dev., 2019, 12, 1–18 CrossRef CAS PubMed.
- Y. S. Siu, L. Li, M. F. Leung, K. L. D. Lee and P. Li, Biointerphases, 2012, 7, 16 CrossRef CAS PubMed.
- J.-J. Nie, B. Qiao, S. Duan, C. Xu, B. Chen, W. Hao, B. Yu, Y. Li, J. Du and F.-J. Xu, Adv. Mater., 2018, 30, 1801570 CrossRef PubMed.
- B. Shi, M. Zheng, W. Tao, R. Chung, D. Jin, D. Ghaffari and O. C. Farokhzad, Biomacromolecules, 2017, 18, 2231–2246 CrossRef CAS.
- L. Hamuro, G. Kijanka, F. Kinderman, H. Kropshofer, D. Bu, M. Zepeda and V. Jawa, J. Pharm. Sci., 2017, 106, 2946–2954 CrossRef CAS.
- M. Foldvari, D. W. Chen, N. Nafissi, D. Calderon, L. Narsineni and A. Rafiee, J. Controlled Release, 2016, 240, 165–190 CrossRef CAS.
- M. Tachibana, P. Amato, M. Sparman, J. Woodward, D. M. Sanchis, H. Ma, N. M. Gutierrez, R. Tippner-Hedges, E. Kang, H.-S. Lee, C. Ramsey, K. Masterson, D. Battaglia, D. Lee, D. Wu, J. Jensen, P. Patton, S. Gokhale, R. Stouffer and S. Mitalipov, Nature, 2013, 493, 627–631 CrossRef CAS.
- K. Mancuso, W. W. Hauswirth, Q. Li, T. B. Connor, J. A. Kuchenbecker, M. C. Mauck, J. Neitz and M. Neitz, Nature, 2009, 461, 784–787 CrossRef CAS.
- J. R. Mendell, K. Campbell, L. Rodino-Klapac, Z. Sahenk, C. Shilling, S. Lewis, D. Bowles, S. Gray, C. Li, G. Galloway, V. Malik, B. Coley, K. R. Clark, J. Li, X. Xiao, J. Samulski, S. W. McPhee, R. J. Samulski and C. M. Walker, N. Engl. J. Med., 2010, 363, 1429–1437 CrossRef CAS.
- T. J. Burney and J. C. Davies, Appl. Clin. Genet., 2012, 5, 29–36 CAS.
- T. Harris, Nat. Rev. Drug Discovery, 2010, 9, 660–660 CrossRef PubMed.
- N. Nayerossadat, T. Maedeh and P. A. Ali, Adv. Biomed. Res., 2012, 1, 27 CrossRef PubMed.
- S. L. Ginn, A. K. Amaya, I. E. Alexander, M. Edelstein and M. R. Abedi, J. Gene Med., 2018, 20, e3015 CrossRef.
- J. Chang, X. H. Xu, H. P. Li, Y. T. Jian, G. Wang, B. He and Z. W. Gu, Adv. Funct. Mater., 2013, 23, 2691–2699 CrossRef CAS.
- Y. Cao, X. X. Liu and L. Peng, Front. Chem. Sci. Eng., 2017, 11, 663–675 CrossRef CAS.
- L. Xu, M. Y. Zhao, Y. D. Yang, Y. Liang, C. Z. Sun, W. X. Gao, S. Li, B. He and Y. J. Pu, J. Mater. Chem. B, 2017, 5, 9157–9164 RSC.
- L. Xu, Y. D. Yang, M. Y. Zhao, W. X. Gao, H. Zhang, S. Li, B. He and Y. J. Pu, J. Mater. Chem. B, 2018, 6, 1076–1084 RSC.
- P. Yan, N. Zhao, H. Hu, X. Lin, F. Liu and F.-J. Xu, Acta Biomater., 2014, 10, 3786–3794 CrossRef CAS.
- K. L. Guo, X. Y. Zhao, X. G. Dai, N. N. Zhao and F. J. Xu, J. Gene Med., 2019, 21, e3084 CrossRef.
- F. Yin, B. B. Gu, Y. N. Lin, N. Panwar, S. C. Tjin, J. L. Qu, S. P. Lau and K.-T. Yong, Coord. Chem. Rev., 2017, 347, 77–97 CrossRef CAS.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS.
- C. V. Bruggen, J. K. Hexum, Z. Tan, R. J. Dalal and T. M. Reineke, Acc. Chem. Res., 2019, 525, 1347–1358 Search PubMed.
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671–681 CrossRef CAS.
- C. H. Jones, C.-K. Chen, A. Ravikrishnan, S. Rane and B. A. Pfeifer, Mol. Pharm., 2013, 10, 4082–4098 CrossRef CAS.
- L. Jin, X. Zeng, M. Liu, Y. Deng and N. He, Theranostics, 2014, 4, 240–255 CrossRef.
- U. Lächelt and E. Wagner, Chem. Rev., 2015, 115, 11043–11078 CrossRef.
- J. Zhou, J. Liu, C. J. Cheng, T. R. Patel, C. E. Weller, J. M. Piepmeier, Z. Jiang and W. M. Saltzman, Nat. Mater., 2011, 11, 82–90 CrossRef.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef.
- D. Vercauteren, J. Rejman, T. F. Martens, J. Demeester, S. C. De Smedt and K. Braeckmans, J. Controlled Release, 2012, 161, 566–581 CrossRef CAS.
- M. E. Favretto, R. Wallbrecher, S. Schmidt, R. van de Putte and R. Brock, J. Controlled Release, 2014, 180, 81–90 CrossRef CAS PubMed.
- B. Bahrami, M. Mohammadnia-Afrouzi, P. Bakhshaei, Y. Yazdani, G. Ghalamfarsa, M. Yousefi, S. Sadreddini, F. Jadidi-Niaragh and M. Hojjat-Farsangi, Tumor Biol., 2015, 36, 5727–5742 CrossRef CAS.
- C. Tros de Ilarduya and N. Düzgüneş, Expert Opin. Drug Delivery, 2013, 10, 1583–1591 CrossRef CAS.
- X. K. Ren, Y. K. Feng, J. T. Guo, H. X. Wang, Q. Li, J. Yang, X. F. Hao, J. Lv, N. Ma and W. Z. Li, Chem. Soc. Rev., 2015, 44, 5680–5742 RSC.
- T. Yang, B. Li, S. Qi, Y. Liu, Y. Gai, P. Ye, G. Yang, W. Zhang, P. Zhang, X. He, W. Li, Z. Zhang, G. Xiang and C. Xu, Theranostics, 2014, 4, 1096–1111 CrossRef CAS.
- J. Park, S. S. Park, N. J. Jo and C. S. Ha, J. Nanosci. Nanotechnol., 2019, 19, 6217–6224 CrossRef.
- S. Lakkadwala, B. dos Santos Rodrigues, C. W. Sun and J. Singh, J. Controlled Release, 2019, 307, 247–260 CrossRef CAS PubMed.
- J. Varshosaz and M. Farzan, World J. Gastroenterol., 2015, 21, 12022 CrossRef CAS.
- F. C. Pérez-Martínez, J. Guerra, I. Posadas and V. Ceña, Pharm. Res., 2011, 28, 1843–1858 CrossRef.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS.
- M. Aouadi, G. J. Tesz, S. M. Nicoloro, M. Wang, M. Chouinard, E. Soto, G. R. Ostroff and M. P. Czech, Nature, 2009, 458, 1180–1184 CrossRef CAS PubMed.
- S. Xiang, H. Tong, Q. Shi, J. C. Fernandes, T. Jin, K. Dai and X. Zhang, J. Controlled Release, 2012, 158, 371–378 CrossRef CAS PubMed.
- M. Kaksonen and A. Roux, Nat. Rev. Mol. Cell Biol., 2018, 19, 313–326 CrossRef CAS.
- H. Hatakeyama, E. Ito, H. Akita, M. Oishi, Y. Nagasaki, S. Futaki and H. Harashima, J. Controlled Release, 2009, 139, 127–132 CrossRef CAS.
- C. J. Barile, E. C. M. Tse, Y. Li, J. P. Gewargis, N. A. Kirchschlager, S. C. Zimmerman and A. A. Gewirth, Biophys. J., 2016, 110, 2451–2462 CrossRef CAS.
- Z. Wang, C. Tiruppathi, R. D. Minshall and A. B. Malik, ACS Nano, 2009, 3, 4110–4116 CrossRef CAS PubMed.
- S. Fagerholm, U. Ortegren, M. Karlsson, I. Ruishalme and P. Strålfors, PLoS One, 2009, 4, e5985 CrossRef PubMed.
- Y. Ning, T. Buranda and L. G. Hudson, J. Biol. Chem., 2007, 282, 6380–6387 CrossRef CAS.
- R. Gupta, C. Toufaily and B. Annabi, Biochimie, 2014, 107, 188–202 CrossRef CAS.
- A. L. Silva, L. I. F. Moura, B. Carreira, J. Conniot, A. I. Matos, C. Peres, V. Sainz, L. C. Silva, R. S. Gaspar and H. F. Florindo, Biomed. Appl. Funct. Nanomater., 2018, 399–448 Search PubMed.
- L. Kou, J. Sun, Y. Zhai and Z. He, Asian J. Pharm. Sci., 2013, 8, 1–10 CrossRef CAS.
- I. S. Zuhorn, R. Kalicharan and D. Hoekstra, J. Biol. Chem., 2002, 277, 18021–18028 CrossRef CAS.
- X.-X. Zhang, P. G. Allen and M. Grinstaff, Mol. Pharm., 2011, 8, 758–766 CrossRef CAS.
- H. K. Shete, R. H. Prabhu and V. B. Patravale, J. Nanosci. Nanotechnol., 2014, 14, 460–474 CrossRef CAS.
- A. Ahmad, S. Ranjan, W. Zhang, J. Zou, I. Pyykkö and P. K. J. Kinnunen, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 544–553 CrossRef CAS.
- E. Yaghini, R. Dondi, K. J. Edler, M. Loizidou, A. J. MacRobert and I. M. Eggleston, Nanoscale, 2018, 10, 20366–20376 RSC.
- T. Endoh and T. Ohtsuki, Adv. Drug Delivery Rev., 2009, 61, 704–709 CrossRef CAS PubMed.
- J. J. Li, D. Hartono, C.-N. Ong, B.-H. Bay and L.-Y. L. Yung, Biomaterials, 2010, 31, 5996–6003 CrossRef CAS.
- M. Sramkova, L. Parente, T. Wigand, M.-P. Aye, A. Shitara and R. Weigert, Front. Cell Dev. Biol., 2015, 2, 74 Search PubMed.
- X. Zhou, X. Liu, B. Zhao, X. Liu, D. Zhu, N. Qiu, Q. Zhou, Y. Piao, Z. Zhou, J. Tang and Y. Shen, J. Controlled Release, 2016, 234, 90–97 CrossRef CAS.
- Y. Hu, H. F. Wang, H. Q. Song, M. Young, Y. Q. Fan, F. J. Xu, X. J. Qu, X. Lei, Y. Liu and G. Cheng, Biomater. Sci., 2019, 7, 1543–1553 RSC.
- Z. H. Wang, W. B. Li, J. Ma, G. P. Tang, W. T. Yang and F. J. Xu, Macromolecules, 2011, 44, 230–239 CrossRef CAS.
- Z.-H. Wang, Y. Zhu, M.-Y. Chai, W.-T. Yang and F.-J. Xu, Biomaterials, 2012, 33, 1873–1883 CrossRef CAS.
- H.-Q. Song, Y. Qi, R.-Q. Li, G. Cheng, N. Zhao and F.-J. Xu, Polym. Chem., 2018, 9, 2281–2289 RSC.
- H.-Q. Song, X.-B. Dou, R.-Q. Li, B.-R. Yu, N.-N. Zhao and F.-J. Xu, Acta Biomater., 2015, 12, 156–165 CrossRef CAS.
- M. R. Rekha and C. P. Sharma, Acta Biomater., 2011, 7, 370–379 CrossRef CAS.
- H. Hu, H.-Q. Song, B.-R. Yu, Q. Cai, Y. Zhu and F.-J. Xu, Polym. Chem., 2015, 6, 2466–2477 RSC.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS.
- A. Zintchenko, A. Philipp, A. Dehshahri and E. Wagner, Bioconjugate Chem., 2008, 19, 1448–1455 CrossRef CAS.
- Y. X. Zhang, J. Zhou, S. N. Ma, Y. Y. He, J. Yang and Z. W. Gu, Biomacromolecules, 2019, 20, 1899–1913 CrossRef CAS.
- Q. Li, X. F. Hao, J. T. Guo, X.-K. Ren, S. H. Xia, W. C. Zhang and Y. K. Feng, Macromol. Rapid Commun., 2019, 40, 1800916 CrossRef PubMed.
- X. F. Hao, Q. Li, H. N. Wang, K. Muhammad, J. T. Guo, X. K. Ren, C. C. Shi, S. H. Xia, W. C. Zhang and Y. K. Feng, J. Mater. Chem. B, 2018, 6, 5975–5985 RSC.
- M. Neu, D. Fischer and T. Kissel, J. Gene Med., 2005, 7, 992–1009 CrossRef CAS.
- A. P. Pandey and K. K. Sawant, Mater. Sci. Eng., C, 2016, 68, 904–918 CrossRef CAS.
- L. M. P. Vermeulen, S. C. De Smedt, K. Remaut and K. Braeckmans, Eur. J. Pharm. Biopharm., 2018, 129, 184–190 CrossRef CAS.
- D. O. Forcato, A. E. Fili, F. E. Alustiza, J. L. Martínez, S. B. Abel, M. O. Nicotra, A. P. Alessio, N. Rodríguez, C. Barbero and P. Bosch, Cytotechnology, 2017, 69, 655–665 CrossRef CAS.
- M. D. Giron-Gonzalez, R. Salto-Gonzalez, F. J. Lopez-Jaramillo, A. Salinas-Castillo, A. B. Jodar-Reyes, M. Ortega-Munoz, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Bioconjugate Chem., 2016, 27, 549–561 CrossRef CAS.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS.
- Y. X. Liu, Y. H. Zong, Z. X. Yang, M. Luo, G. L. Li, W. Yingsa, Y. L. Cao, M. Xiao, T. J. Kong, J. He, X. Y. Liu and J. D. Lei, ACS Sustainable Chem. Eng., 2019, 7, 3614–3623 CrossRef CAS.
- S. N. Huang, S. F. Duan, J. Wang, S. J. Bo, X. J. Qiu, C. M. Li, Y. Liu, L. J. Yan, Z. Z. Zhang and Y. R. Hu, Adv. Funct. Mater., 2016, 26, 2532–2544 CrossRef CAS.
- O. Koenig, B. Neumann, C. Schensak, H. P. Wendel and A. Notel, PLoS One, 2019, 14, e0212584 CrossRef CAS.
- H. Tian, L. Lin, J. Chen, X. Chen, T. G. Park and A. Maruyama, J. Controlled Release, 2011, 155, 47–53 CrossRef CAS.
- K. Kunath, T. Merdan, O. Hegener, H. Häberlein and T. Kissel, J. Gene Med., 2003, 5, 588–599 CrossRef CAS.
- C. Chen, J. Ke, X. E. Zhou, W. Yi, J. S. Brunzelle, J. Li, E.-L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486–489 CrossRef CAS.
- T. T. Le and D. Kim, Mater. Sci. Eng., C, 2019, 101, 464–471 CrossRef CAS.
- K. C. Cho, S. H. Kim, J. H. Jeong and T. G. Park, Macromol. Biosci., 2005, 5, 512–519 CrossRef CAS.
- C. Zhang, S. Gao, W. Jiang, S. Lin, F. Du, Z. Li and W. Huang, Biomaterials, 2010, 31, 6075–6086 CrossRef CAS.
- A. Guo, Y. Wang, S. Xu, X. Zhang, M. Li, Q. Liu, Y. Shen, D. Cui and S. Guo, Colloids Surf., B, 2017, 152, 58–67 CrossRef CAS.
- L. Liu, M. Zheng, T. Renette and T. Kissel, Bioconjugate Chem., 2012, 23, 1211–1220 CrossRef CAS.
- D. Cao, S. Tian, H. Huang, J. Chen and S. Pan, Mol. Pharm., 2015, 12, 240–252 CrossRef CAS.
- H.-L. Jiang, C.-X. Xu, Y.-K. Kim, R. Arote, D. Jere, H.-T. Lim, M.-H. Cho and C.-S. Cho, Biomaterials, 2009, 30, 5844–5852 CrossRef CAS.
- H. Yao, S. S. Ng, W. O. Tucker, Y.-K.-T. Tsang, K. Man, X. Wang, B. K. C. Chow, H.-F. Kung, G.-P. Tang and M. C. Lin, Biomaterials, 2009, 30, 5793–5803 CrossRef CAS.
- K. Chen, Q. Chen, K. Wang, J. Zhu, W. Li, W. Li, L. Qiu, G. Guan, M. Qiao, X. Zhao, H. Hu and D. Chen, Int. J. Pharm., 2016, 509, 314–327 CrossRef CAS.
- M. Wang, H. Hu, Y. Sun, L. Qiu, J. Zhang, G. Guan, X. Zhao, M. Qiao, L. Cheng, L. Cheng and D. Chen, Biomaterials, 2013, 34, 10120–10132 CrossRef CAS.
- J. Liu, L. Xu, Y. Jin, C. Qi, Q. Li, Y. Zhang, X. Jiang, G. Wang, Z. Wang and L. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 14200–14210 CrossRef CAS PubMed.
- H. M. Li, M. Miteva, K. C. Kirkbride, M. J. Cheng, C. E. Nelson, E. M. Simpson, M. K. Gupta, C. L. Duvall and T. D. Giorgio, Biomacromolecules, 2015, 16, 192–201 CrossRef CAS.
- Y. Song, B. Lou, P. Zhao and C. Lin, Mol. Pharm., 2014, 11, 2250–2261 CrossRef CAS PubMed.
- F. Zhao, H. Yin, Z. X. Zhang and J. Li, Biomacromolecules, 2013, 14, 476–484 CrossRef CAS.
- F. Zhao, H. Yin and J. Li, Biomaterials, 2014, 35, 1050–1062 CrossRef CAS.
- D.-J. Lee, E. Kessel, T. Lehto, X. Liu, N. Yoshinaga, K. Padari, Y.-C. Chen, S. Kempter, S. Uchida, J. O. Rädler, M. Pooga, M.-T. Sheu, K. Kataoka and E. Wagner, Bioconjugate Chem., 2017, 28, 2393–2409 CrossRef CAS.
- L. Zhao, J. A. Hall, N. Levenkova, E. Lee, M. K. Middleton, A. M. Zukas, D. J. Rader, J. J. Rux and E. Puré, Arterioscler. Thromb. Vasc. Biol., 2007, 27, 886–892 CrossRef CAS.
- L. Ding, Y. Jiang, J. Zhang, H.-A. Klok and Z. Zhong, Biomacromolecules, 2018, 19, 555–562 CrossRef CAS.
- P. Kesharwani, S. Banerjee, S. Padhye, F. H. Sarkar and A. K. Iyer, Biomacromolecules, 2015, 16, 3042–3053 CrossRef CAS PubMed.
- Z. X. Cai, H. B. Zhang, Y. Wei, Y. Wei and F. S. Gong, Biomacromolecules, 2017, 18, 1677–1696 CrossRef CAS.
- K. C. R. Bahadur, B. Thapa and P. Xu, Macromol. Biosci., 2012, 12, 637–646 CrossRef.
- B. Jiang, H. He, L. Yao, T. Zhang, J. Huo, W. Sun and L. Yin, Biomacromolecules, 2017, 18, 877–885 CrossRef CAS.
- H. He, Y. Bai, J. Wang, Q. Deng, L. Zhu, F. Meng, Z. Zhong and L. Yin, Biomacromolecules, 2015, 16, 1390–1400 CrossRef CAS.
- Y. Zhang, L. Liu, L. Lin, J. Chen, H. Tian, X. Chen and A. Maruyama, Acta Biomater., 2018, 65, 349–362 CrossRef CAS.
- Y. He, G. Cheng, L. Xie, Y. Nie, B. He and Z. Gu, Biomaterials, 2013, 34, 1235–1245 CrossRef CAS.
- K. Liang, K. H. Bae, F. Lee, K. Xu, J. E. Chung, S. J. Gao and M. Kurisawa, J. Controlled Release, 2016, 226, 205–216 CrossRef CAS PubMed.
- V. Badwaik, L. J. Liu, D. Gunasekera, A. Kulkarni and D. H. Thompson, Mol. Pharmaceutics, 2016, 13, 1176–1184 CrossRef CAS.
- M.-D. Zhao, J.-L. Cheng, J.-J. Yan, F.-Y. Chen, J.-Z. Sheng, D.-L. Sun, J. Chen, J. Miao, R.-J. Zhang, C.-H. Zheng and H.-F. Huang, Int. J. Nanomed., 2016, 11, 1323–1336 CrossRef CAS.
- J. Gu, X. Chen, X. Ren, X. Zhang, X. Fang and X. Sha, Bioconjugate Chem., 2016, 27, 1723–1736 CrossRef CAS.
- W. J. Lin, W.-C. Lee and M.-J. Shieh, Carbohydr. Polym., 2017, 155, 101–108 CrossRef CAS.
- R. R. Fan, D. Chuan and H. Hou, Nanoscale, 2019, 11, 11285–11304 RSC.
- K. Huang, Y. H. He and Z. H. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 22175–22180 Search PubMed.
- J. Gu, X. Chen, X. Fang and X. Sha, Acta Biomater., 2017, 57, 156–169 CrossRef CAS.
- Y. Sun, Z. Yang, C. Wang, T. Yang, C. Cai, X. Zhao, L. Yang and P. Ding, Acta Biomater., 2017, 60, 23–37 CrossRef CAS.
- M. Ahmed, Biomater. Sci., 2017, 5, 2188–2211 RSC.
- D. Pezzoli, P. Tarsini, L. Melone and G. Candiani, J. Drug Delivery Sci. Technol., 2017, 37, 115–122 CrossRef CAS.
- J. S. Suk, J. Suh, K. Choy, S. K. Lai, J. Fu and J. Hanes, Biomaterials, 2006, 27, 5143–5150 CrossRef CAS.
- Z. Ge, Q. Chen, K. Osada, X. Liu, T. A. Tockary, S. Uchida, A. Dirisala, T. Ishii, T. Nomoto, K. Toh, Y. Matsumoto, M. Oba, M. R. Kano, K. Itaka and K. Kataoka, Biomaterials, 2014, 35, 3416–3426 CrossRef CAS.
- L. Li, L. J. Song, X. Yang, X. Li, Y. Z. Wu, T. He, N. Wang, S. L. X. Yang, Y. Zeng, L. Yang, Q. J. Wu, Y. Q. Wei and C. Y. Gong, Biomaterials, 2016, 111, 124–137 CrossRef CAS.
- J. Yang, X. Hao, Q. Li, M. Akpanyung, A. Nejjari, A. L. Neve, X. Ren, J. Guo, Y. Feng, C. Shi and W. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 4485–4497 CrossRef CAS.
- X. F. Hao, Q. Li, H. N. Wang, K. Muhammad, J. T. Guo, X. K. Ren, C. C. Shi, S. S. Xia, W. C. Zhang and Y. K. Feng, ACS Biomater. Sci. Eng., 2018, 4, 2870–2878 CrossRef CAS.
- X. F. Hao, Q. Li, J. Lv, L. Yu, X. K. Ren, L. Zhang, Y. K. Feng and W. C. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 12128–12140 CrossRef CAS.
- Q. Li, X. F. Hao, J. Lv, X. K. Ren, K. Y. Zhang, I. Ullah, Y. K. Feng, C. C. Shi and W. C. Zhang, J. Mater. Chem. B, 2017, 5, 1673–1687 RSC.
- J. Yang, W. Liu, J. Lv, Y. K. Feng, X. K. Ren and W. C. Zhang, J. Mater. Chem. B, 2016, 4, 3365–3376 RSC.
- J. Lv, X. F. Hao, Q. Li, M. Akpanyung, A. Nejjari, A. L. Neve, X. K. Ren, Y. K. Feng, C. C. Shi and W. C. Zhang, Biomater. Sci., 2017, 5, 511–522 RSC.
- F. Yang, W. Huang, Y. Li, S. Liu, M. Jin, Y. Wang, L. Jia and Z. Gao, Biomaterials, 2013, 34, 5689–5699 CrossRef CAS.
- X. F. Hao, Q. Li, H. Ali, S. S. A. Zaidi, J. T. Guo, X. K. Ren, C. C. Shi, S. H. Xia, W. C. Zhang and Y. K. Feng, J. Mater. Chem. B, 2018, 6, 4251–4263 RSC.
- Q. Li, X. F. Hao, S. S. A. Zaidi, J. T. Guo, X. K. Ren, C. C. Shi, W. C. Zhang and Y. K. Feng, J. Nanobiotechnol., 2018, 16, 29 CrossRef.
- J. Wang, S. S. A. Zaidi, A. Hasnain, J. T. Guo, X. K. Ren, S. H. Xia, W. C. Zhang and Y. K. Feng, ACS Biomater. Sci. Eng., 2018, 4, 2155–2168 CrossRef CAS.
- B. Gao, Q. P. Zhang, K. Muhammad, X. K. Ren, J. T. Guo, S. H. Xia, W. C. Zhang and Y. K. Feng, Biomater. Sci., 2019, 7, 2061–2075 RSC.
- Y. Liu, J. Li, K. Shao, R. Huang, L. Ye, J. Lou and C. Jiang, Biomaterials, 2010, 31, 5246–5257 CrossRef CAS PubMed.
- H. Yao, K. Wang, Y. Wang, S. Wang, J. Li, J. Lou, L. Ye, X. Yan, W. Lu and R. Huang, Biomaterials, 2015, 37, 345–352 CrossRef CAS.
- A. Zarebkohan, F. Najafi, H. R. Moghimi, M. Hemmati, M. R. Deevband and B. Kazemi, Eur. J. Pharm. Sci., 2015, 78, 19–30 CrossRef CAS.
- W. Ke, K. Shao, R. Huang, L. Han, Y. Liu, J. Li, Y. Kuang, L. Ye, J. Lou and C. Jiang, Biomaterials, 2009, 30, 6976–6985 CrossRef CAS.
- D. Li, Y. Ping, F. Xu, H. Yu, H. Pan, H. Huang, Q. Wang, G. Tang and J. Li, Biomacromolecules, 2010, 11, 2221–2229 CrossRef CAS.
- Y. Ping, Q. Hu, G. Tang and J. Li, Biomaterials, 2013, 34, 6482–6494 CrossRef CAS.
- C. Zheng, M. Zheng, P. Gong, J. Deng, H. Yi, P. Zhang, Y. Zhang, P. Liu, Y. Ma and L. Cai, Biomaterials, 2013, 34, 3431–3438 CrossRef CAS.
- M. Soliman, R. Nasanit, S. R. Abulateefeh, S. Allen, M. C. Davies, S. S. Briggs, L. W. Seymour, J. A. Preece, A. M. Grabowska, S. A. Watson and C. Alexander, Mol. Pharm., 2012, 9, 1–13 CrossRef CAS.
- T.-E. Park, B. Singh, H. Li, J.-Y. Lee, S.-K. Kang, Y.-J. Choi and C.-S. Cho, Biomaterials, 2015, 38, 61–71 CrossRef CAS PubMed.
- Y. Liu, S. An, J. Li, Y. Kuang, X. He, Y. Guo, H. Ma, Y. Zhang, B. Ji and C. Jiang, Biomaterials, 2016, 80, 33–45 CrossRef CAS PubMed.
- J.-W. Nah, L. Yu, S. Han, C.-H. Ahn and S. W. Kim, J. Controlled Release, 2002, 78, 273–284 CrossRef CAS.
- D. Li, G. P. Tang, J. Z. Li, Y. Kong, H. L. Huang, L. J. Min, J. Zhou, F. P. Shen, Q. Q. Wang and H. Yu, J. Biomater. Sci., Polym. Ed., 2007, 18, 545–560 CrossRef CAS.
- D. A. Richards, A. Maruani and V. Chudasama, Chem. Sci., 2017, 8, 63–77 RSC.
- V. Shargh, H. Hondermarck and M. Liang, Nanomedicine, 2016, 11, 63–79 CrossRef CAS PubMed.
- A. Srivastava, I. B. O'Connor, A. Pandit and J. G. Wall, Prog. Polym. Sci., 2014, 39, 308–329 CrossRef CAS.
- H. Laroui, E. Viennois, B. Xiao, B. S. B. Canup, D. Geem, T. L. Denning and D. Merlin, J. Controlled Release, 2014, 186, 41–53 CrossRef CAS.
- E. Mastrobattista, R. H. Kapel, M. H. Eggenhuisen, P. J. Roholl, D. J. Crommelin, W. E. Hennink and G. Storm, Cancer Gene Ther., 2001, 8, 405–413 CrossRef CAS.
- W. Suh, J.-K. Chung, S.-H. Park and S. W. Kim, J. Controlled Release, 2001, 72, 171–178 CrossRef CAS.
- J. Kim, M. Lee, H. Kim, D. Park, J. Kim and J. Kim, Biomaterials, 2016, 75, 102–111 CrossRef CAS.
- K. S. Siu, D. Chen, X. Zheng, X. Zhang, N. Johnston, Y. Liu, K. Yuan, J. Koropatnick, E. R. Gillies and W.-P. Min, Biomaterials, 2014, 35, 3435–3442 CrossRef CAS.
- H.-L. Jiang, Y.-K. Kim, R. Arote, D. Jere, J.-S. Quan, J.-H. Yu, Y.-J. Choi, J.-W. Nah, M.-H. Cho and C.-S. Cho, Int. J. Pharm., 2009, 375, 133–139 CrossRef CAS.
- J.-H. Kim, A. Minai-Tehrani, Y.-K. Kim, J.-Y. Shin, S.-H. Hong, H.-J. Kim, H.-D. Lee, S.-H. Chang, K.-N. Yu, Y.-B. Bang, C.-S. Cho, T.-J. Yoon, D.-Y. Yu, H.-L. Jiang and M.-H. Cho, Biomaterials, 2012, 33, 1894–1902 CrossRef CAS PubMed.
- H.-L. Jiang, J.-T. Kwon, E.-M. Kim, Y.-K. Kim, R. Arote, D. Jere, H.-J. Jeong, M.-K. Jang, J.-W. Nah, C.-X. Xu, I.-K. Park, M.-H. Cho and C.-S. Cho, J. Controlled Release, 2008, 131, 150–157 CrossRef CAS.
- X.-C. Yang, Y.-L. Niu, N.-N. Zhao, C. Mao and F.-J. Xu, Biomaterials, 2014, 35, 3873–3884 CrossRef CAS.
- X. Zeng, Y.-X. Sun, W. Qu, X.-Z. Zhang and R.-X. Zhuo, Biomaterials, 2010, 31, 4771–4780 CrossRef CAS.
- J. Cheng, X. Tang, J. Zhao, T. Shi, P. Zhao and C. Lin, Acta Biomater., 2016, 30, 155–167 CrossRef CAS.
- P. Davoodi, M. P. Srinivasan and C.-H. Wang, Acta Biomater., 2016, 39, 79–93 CrossRef CAS PubMed.
- X. Lin, N. Zhao, P. Yan, H. Hu and F.-J. Xu, Acta Biomater., 2015, 11, 381–392 CrossRef CAS.
- S. B. Hartono, W. Gu, F. Kleitz, J. Liu, L. He, A. P. J. Middelberg, C. Yu, G. Q. Lu and S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS PubMed.
- Y. H. Lee, H. I. Park and J. S. Choi, Carbohydr. Polym., 2016, 137, 669–677 CrossRef CAS.
- B.-F. Zhang, L. Xing, P.-F. Cui, F.-Z. Wang, R.-L. Xie, J.-L. Zhang, M. Zhang, Y.-J. He, J.-Y. Lyu, J.-B. Qiao, B.-A. Chen and H.-L. Jiang, Biomaterials, 2015, 61, 178–189 CrossRef CAS PubMed.
Footnote |
| † Khan Muhammad and Jing Zhao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |