
Tumor regression and potentiation of polymeric vascular disrupting therapy through reprogramming of a hypoxia microenvironment with temsirolimus
Haiyang
Yu
abc,
Na
Shen
*bc,
Yanli
Bao
a,
Li
Chen
 *a and
Zhaohui
Tang
bc
*a and
Zhaohui
Tang
bc
aDepartment of Chemistry, Northeast Normal University, 5268 Renmin Street, Changchun 130024, People's Republic of China. E-mail: chenl686@nenu.edu.cn
bKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun 130022, People's Republic of China. E-mail: nshen@ciac.ac.cn
cJilin Biomedical Polymers Engineering Laboratory, Changchun, 130022, P. R. China
First published on 22nd October 2019
Abstract
Although the polymeric vascular disrupting agent (poly(L-glutamic acid)-graft-methoxy poly(ethylene glycol)/combretastatin A4) nanoparticles (CA4-NPs) has great potential to inhibit cancer growth, it is still a challenge to avert tumor recurrence and metastasis after treatment. It is mainly tightly associated with hypoxia induced by CA4-NPs, which can activate many downstream genes regulating tumor growth and metastasis. Herein, to relieve a tumor hypoxia microenvironment, the mTOR inhibitor temsirolimus was employed to modulate the tumor microenvironment when treated with CA4-NPs. In vitro MTT experiments strongly verified that the combination of temsirolimus with polymeric CA4-NPs exhibited an additive toxicity to 4T1 cells. An in vivo study with the 4T1 mammary adenocarcinoma model revealed that consistent with the proposed scenario, combination therapy with CA4-NPs plus temsirolimus suppressed tumor growth significantly more strongly compared to either CA4-NPs or temsirolimus monotherapy, and the inhibition rate to 4T1 tumor with a volume of 300 mm3 was 71%. The mechanism underling combination treatment was confirmed by western blotting and immunofluorescence staining, and the results demonstrated that temsirolimus could inhibit HIF1α expression. Thus, this work provides a mechanistic rationale for the use of VDAs in combination with the mTOR inhibitor to enhance anticancer efficacy, delaying tumor recurrence and inhibiting tumor metastasis.
1. Introduction
Hypoxia is increasingly recognized as a key microenvironmental factor in the regulation of tumor growth and metastasis and is associated with hypoxia-inducible transcription factor (HIF) activation. Hundreds of genes are activated in response to HIF-1 and HIF-2, such as VEGF, matrix metalloproteinases, and endothelins, as well as secretion of chemokines and cytokines, including CCL5, C–X–C motif chemokine 12 (CXCL12 or SDF-1),1 and C–X–C motif receptor 4 (CXCR4) expression,2 which are critical elements in tumor growth and metastasis. Clinically, hypoxia also influences the resistance and metastasis to radiotherapy and chemotherapy, as well as poor patient survival, indicating that hypoxia may contribute to tumor resistance and progression to therapy.3–5 In addition, hypoxia occurs after treatment, such as surgery,6,7 photodynamic therapy,8,9 hepatic artery embolization,10 angiogenesis inhibitor,11 and vascular disrupting agents (VDAs).12,13VDAs have great therapeutic potential in cancer therapy. Several small molecule VDAs are under investigation in clinical trials.14,15 VDAs selectively destroy the vasculature of solid tumors, resulting in decreased oxygen and nutrition supplement and extensive tumor necrosis. However, tumor regrowth after VDA treatment is frequent,16–18 which is associated with a hypoxia microenvironment,13,19,20 compromising the efficiency of VDA therapy.16,17,21 Polymeric vascular disrupting agent (poly(L-glutamic acid)-graft-methoxy poly(ethylene glycol)/combretastatin A4) nanoparticles (CA4-NPs) showed high accumulation inside treated tumors and mainly distributed around the tumor vessels, resulting in effective vascular disruption.22 Extensive studies have demonstrated that the administration of VDAs significantly increases the level of hypoxia in solid tumors,13,23 which contributes to tumor recurrences and metastasis after treatment.
Since hypoxia, especially HIF1α, is an upstream regulator of many factors related to recurrence and metastasis, to choose a drug which inhibits hypoxia is of great significance. The mammalian target of the rapamycin (mTOR) inhibitor is increasingly recognized as a master regulator of fundamental cellular functions, whose deregulation may underlie neoplastic transformation and progression. The mTOR inhibitor, temsirolimus, is reported to have antiangiogenic potential through the inhibition of HIF1α and vascular endothelial growth factor (VEGF),24 linking to the targeting of mTOR/HIF1 α/VEGF signaling.25 Thus, we hypothesize that temsirolimus should have a synergistic anti-cancer effect with VDAs.
2. Experimental
2.1 Materials
Combretastatin A4 (CA4) was purchased from Taizhou Gaogang District Jinyan Biotechnology Co., Ltd, China. 2,4,6-Trichlorobenzoyl chloride was purchased from Tianjin Heowns Biochemical Technology Co., Ltd, China. 4-Dimethylaminopyridine (DMAP) was supplied by Aladdin Reagent Co., Ltd, China. Temsirolimus was bought from Shanghai Biopharmaleader Co., Ltd, China. All solvents were purchased from Sinopharm Chemical Reagent Co., Ltd, China.2.2 Cell culture
4T1 mouse triple negative breast cancer cell lines were purchased from KeyGen Biotechnology Co. Ltd. 4T1 cells were cultured in complete Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin in a humidified incubator with 5% CO2 at 37 °C (approximately 20% O2). Cells were cultured at a density of 0.2–0.3 × 106 cells per mL overnight to reach 70% confluency before drug treatment.2.3 Hypoxia exposure
Hypoxic conditions were established as previously described.26 Cells were cultured under a continuous flow of a humidified mixture of 95% N2 and 5% CO2 gas certified to <10 ppm O2 at 37 °C.2.4 Synthesis of CA4-NPs
CA4-NPs were prepared using the Yamaguchi esterification reaction, as previously described with slight modifications.27,28 Briefly, PLG-g-mPEG (mPEG5k/PLG = 2/1, wt/wt) (4.00 g), CA4 (1.00 g, 3.16 mmol), 2,4,6-trichlorobenzoyl chloride (1.56 g, 6.40 mmol), DMAP (0.46 g, 3.77 mmol), triethylamine (TEA, 0.9 mL, 6.46 mmol) in DMF were reacted at 60 °C for 6 h. The reaction mixtures were then precipitated into excess diethyl ether, filtered and dried. The obtained solid was salted by sodium hydrocarbonate solution, and dialyzed against distilled water (MWCO 3500). CA4-NPs were obtained after lyophilization.The hydrodynamic radius of CA4-NPs dispersed in PBS (pH 7.4) was measured by dynamic laser scattering (DLS), using a Brookhaven ZetaPlus Analyzer (Brookhaven Instrument, USA) with a 90° scatter angle. 1H NMR (300 M, NaOD/D2O) of CA4-NPs: δ 6.55–6.70 ppm (phenyl group), 6.39 ppm (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH–), 4.12 and 4.36 ppm (t, –CH<), 3.72 ppm (s, –OCH3, belong to CA4), 3.68 ppm (s, –CH2CH2O–), 3.36 ppm (s, –OCH3), 2.06–1.84 ppm (m, –CH2COO–, m, >CHCH2–). The drug loading content (DLC %) was calculated using the following equation:
CH–), 4.12 and 4.36 ppm (t, –CH<), 3.72 ppm (s, –OCH3, belong to CA4), 3.68 ppm (s, –CH2CH2O–), 3.36 ppm (s, –OCH3), 2.06–1.84 ppm (m, –CH2COO–, m, >CHCH2–). The drug loading content (DLC %) was calculated using the following equation:
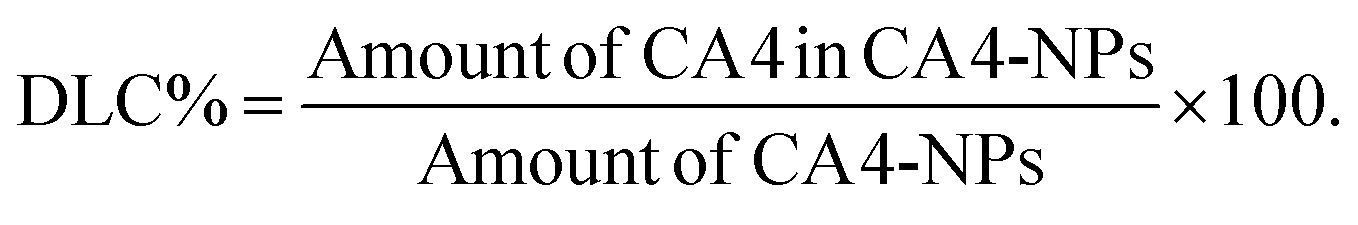
The CA4 content was evaluated by high-performance liquid chromatography (HPLC, mobile phase: acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water = 80:20, v/v, flow rate: 1.0 mL min−1, reverse phase C18 and UV-Vis detector at 305 nm). The test method of DLC was as follows: firstly, the CA4-NPs was hydrolyzed completely using sodium hydroxide, then the solution was acidified using phosphoric acid. The amount of CA4 in the final solution and free CA4 standard liquid was determined by HPLC.
:water = 80:20, v/v, flow rate: 1.0 mL min−1, reverse phase C18 and UV-Vis detector at 305 nm). The test method of DLC was as follows: firstly, the CA4-NPs was hydrolyzed completely using sodium hydroxide, then the solution was acidified using phosphoric acid. The amount of CA4 in the final solution and free CA4 standard liquid was determined by HPLC.
2.5 In vitro cytotoxicity assay
The cytotoxicities of CA4-NPs or/and temsirolimus were studied using an MTT assay. 4T1 cells were seeded in 96-well plates with a density of 8000 cells per well in DMEM containing 10% FBS overnight. The culture medium was replaced by fresh medium containing various concentrations of drugs for another 24 or 48 h. Then, the medium in each well was replaced with 200 μL media containing 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) reagent (0.5 mg mL−1 in PBS), and incubated with cells for another 2–4 h at 37 °C. The supernatant in each well was removed and 150 μL dimethyl sulfoxide (DMSO) was added to dissolve formazan crystals. The absorbance at 490 nm was measured using a Biotek microplate reader. The relative cell viabilities were calculated using the following equation: (Asample/Acontrol) × 100%, where Asample and Acontrol represent the absorbances of the sample wells and control wells, respectively. The data were analyzed with Origin software and presented as means ± SD (n = 3). Points were connected with B-Spline, and the half-maximal inhibitory concentrations (IC50) were calculated according to the log (inhibition)–response curve.2.6 Western blotting
Western blotting was performed with whole cell lysates as reported previously.29 4T1 cells were treated with 10, 20 and 40 μg mL−1 temsirolimus for 48 h or 20 μg mL−1 temsirolimus under normoxia or hypoxia conditions for 48 h. Whole cell proteins were separated by lysing the cells with 1× cell lysis buffer [2 mM Tris-HCl (pH 7.5), 15 mM NaCl, 0.1% Triton] containing proteinase inhibitors. After protein lysates were quantified with a BCA protein assay kit (Thermo), 40 μg of protein was loaded into 10% SDS-PAGE gels and transferred to PVDF membranes (GE Healthcare). β-Actin was used as an internal reference. HIF1α antibody used was purchased from Sartorius, and β-actin antibody was obtained from Sigma. The proteins detected were visualized using an enhanced chemiluminescence western blot detection system (AI600, GE Healthcare Co. Ltd).2.7 Hematoxylin and eosin (H&E) and immunofluorescence (IF) staining
Tumors were collected from our animal studies, fixed with 4% paraformaldehyde, embedded with paraffin, sectioned at 5 μm thickness. The slides were heated in a dry oven at 60 °C for 30 min, deparaffinized in xylene for 10 min, rehydrated through graded ethanol and rinsed in deionized water. Then, the tissues were stained with hematoxylin and eosin. Necrotic areas with some ‘ghost’ cells and nuclear debris were circled in the microscopy images and quantified with ImageJ. For immunofluorescence staining, after dehydration, the slides were then incubated in boiled 10 mM citric acid buffer (pH 6.0) for 10 min to unmask antigens. Endogenous peroxidase was quenched using 3% hydrogen peroxide for 20 min. Nonspecific binding was blocked with 5% BSA for 1 h. Primary antibodies against HIF1α (Sartorius) or Ki-67 (Cell Signaling Technology) were incubated at 4 °C overnight. Then, FITC-labelled anti-mouse secondary antibody was incubated at room temperature for 1 h. The nuclei were stained with DAPI. The fluorescence intensities of HIF1α or Ki-67 were quantified with ImageJ. Apoptosis of tumor tissues was detected by TUNEL. A TUNEL staining kit was obtained from KeyGen and processed according to the manufacturer's instructions. CLSM images were taken using a laser scanning confocal microscope (Zeiss). TUNEL signals were quantified with ImageJ.2.8 Animal studies
Six to eight-week-old female BALB/c mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. 4T1 cells (2 × 106 per mouse) were injected subcutaneously into the right flank of BALB/c mice to generate the metastatic 4T1 mammary adenocarcinoma model. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Jilin University and approved by the Animal Ethics Committee of Jilin University.2.9 Antitumor evaluation in vivo
Approximately 2 × 106 4T1 cells were injected subcutaneously into the right flank of each mouse for tumor generation. When the tumor volumes were approximately 180–200 mm3, mice were divided into four groups (n = 6 mice per group), treated with PBS, CA4-NPs (30 mg kg−1 on CA4 basis, intravenous on days 1 and 8), temsirolimus (20 mg kg−1, intraperitoneal on days 1, 2, 3, 4 and 8, 9, 10, 11), and CA4-NPs plus temsirolimus (30 mg kg−1 on CA4 basis, intravenous on days 1 and 8 and 20 mg kg−1, intraperitoneal on days 1, 2, 3, 4 and 8, 9, 10, 11). Tumor sizes and mouse weights were monitored every two days post-treatment. The tumor volume was calculated using the following formula 1/2 × a × b2, where a and b represent the length and width of the tumor, respectively. The tumor growth inhibition rates (IR) were calculated using the formula IR (%) = (1 − TVt/TVc) × 100, where TVt and TVc are the mean tumor volume of the treated and control groups, respectively. To identify potential lung metastases, lungs were stained intratracheally with 15% India Black Ink solution at the end of the experiment. All the mice were sacrificed, and India ink (15%) were subsequently perfused into the lungs through the trachea.30 Then, lungs were collected and soaked in Fekete's solution (70% ethanol:formalin:glacial acetic acid = 20:2:1, v/v/v) at room temperature for 2–3 days, and imaged. Tumor metastasis sites presented as white spots on black-stained lungs. As tumor metastasis nodules were too large, we quantified the area of metastatic foci instead of numbers. The ImageJ software was employed. The metastatic rate (%) in each whole lung was calculated and compared using the following equation.where Area (black) represents the area of the non-metastatic site, which was stained black and Area (whole lung) represents the area of the whole lung.
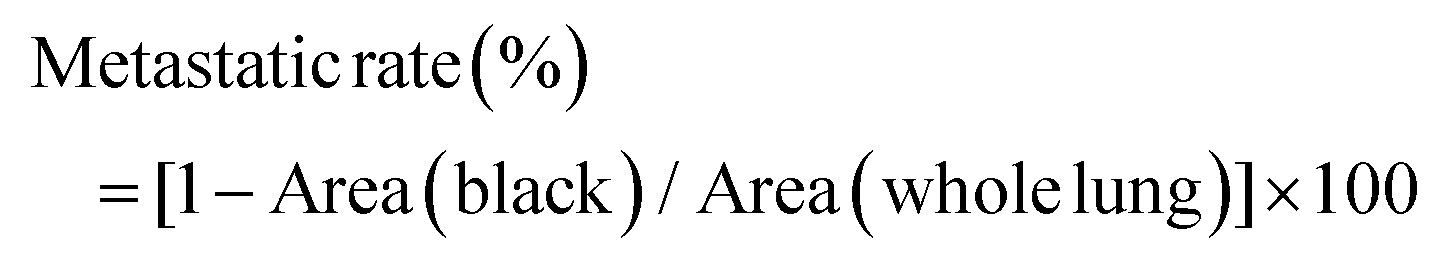
The front or back of the lung was processed and analyzed using the same method.
2.10 Statistical analysis
Statistical differences for multiple groups were obtained using a one-way ANOVA with the GraphPad Prism 6.0 Software. *p < 0.05 was considered to be statistically significant, whereas **p < 0.01 and ***p < 0.001 were considered to be highly and extremely significant.3. Results and discussion
3.1 Synthesis and characterization of CA4-NPs
The preparation strategy of CA4-NPs is shown in Scheme 1. Firstly, poly(L-glutamic acid)-graft-methoxy poly(ethylene glycol)/combretastatin A4 (PLG-CA4) was synthesized through the Yamaguchi reaction, then CA4-NPs were obtained by dialysing in sodium hydrocarbonate solution. Typical peaks in the 1H NMR spectrum of CA4-NPs at δ 6.35–6.70 ppm was the peak of the protons of CA4 (Fig. 1A). The CA4 loading content of CA4-NPs and the free CA4 content were determined by HPLC (Fig. 1B). The DLC % of CA4-NPs was 19.6 wt%. The size of CA4-NPs was measured by DLS, and the hydrodynamic radius (Rh) of CA4-NPs was 45.6 ± 8.4 nm (Fig. 1C). Compare with PLG-CA4, the CA4-NPs had obviously higher water solubility.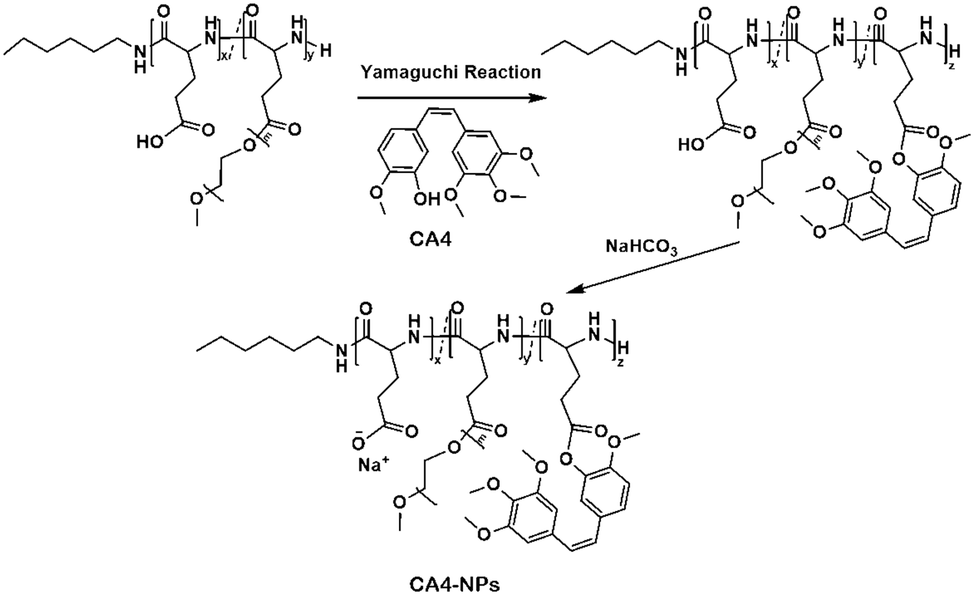 | ||
| Scheme 1 Synthesis of CA4-NPs. | ||
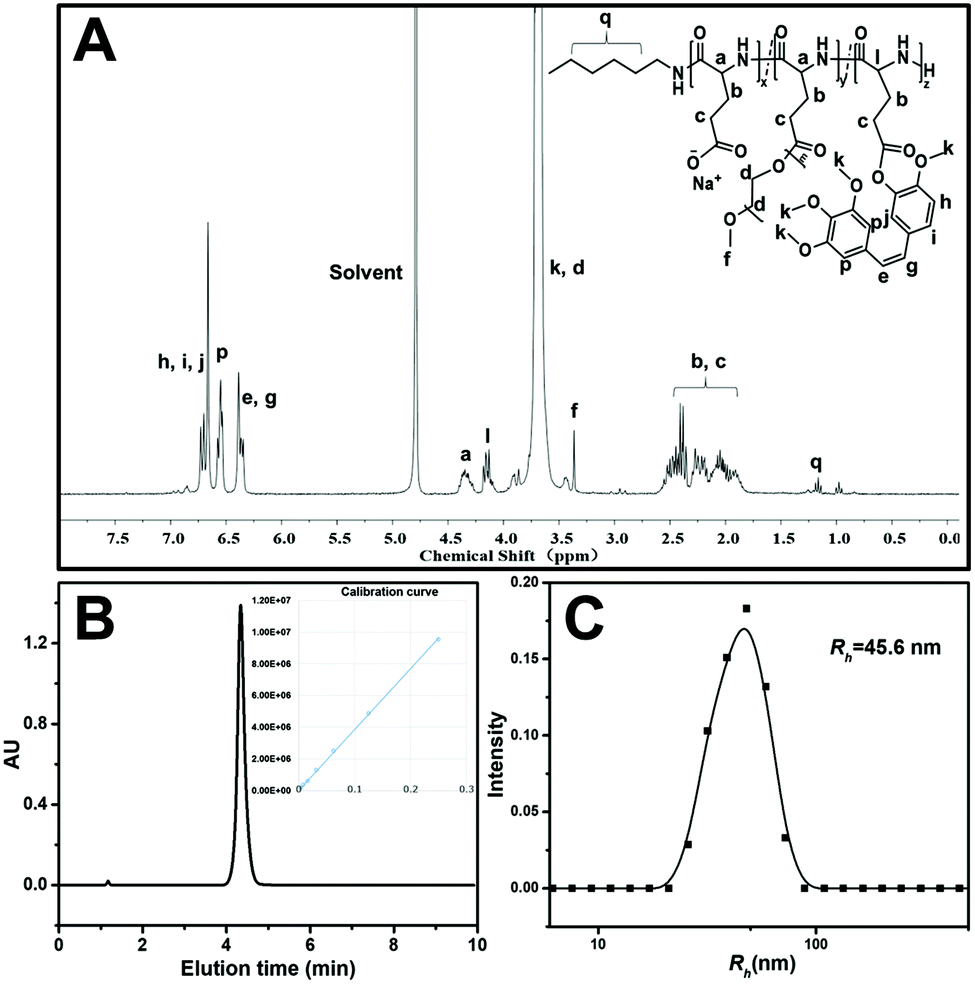 | ||
| Fig. 1 Characterization of CA4-NPs: (A) 1H NMR spectra of CA4-NPs in D2O; (B) HPLC curves of free CA4; the inset is the calibration curve of the CA4 peak area vs. concentration. (C) Size distribution plots of CA4-NPs. | ||
3.2 CA4-NPs and temsirolimus inhibit 4T1 cell growth
The cytotoxic effects of CA4-NPs alone and in combination with temsirolimus on the 4T1 tumor cell line were evaluated. 4T1 cells were treated with different concentrations of CA4-NPs or/and temsirolimus for 24 and 48 h (the concentration ratio of CA4 to temsirolimus was 3:8 wt/wt with 1:5 dilution), and MTT assay was performed (Fig. 2), and the viabilities of the 4T1 cells treated with CA4-NPs was around 70% at the maximum concentrations (CA4-NPs, 75 μg mL−1 eq. to CA4) at 24 h, indicating that the toxicity of CA4-NPs was low. However, the toxicity of CA4-NPs to 4T1 cells increased at 48 h, as the viabilities of the 4T1 cells treated with CA4-NPs were around 40% at the maximum concentrations (CA4-NPs, 75 μg mL−1 eq. to CA4). The toxicity of temsirolimus with low concentration (<10 μg mL−1) was low, while the toxicity of temsirolimus with high concentration (>10 μg mL−1) was strong at both 24 and 48 h. As to the combined CA4-NPs and temsirolimus, the viability of 4T1 cells treated with CA4-NPs and temsirolimus only had significant differences from temsirolimus at low concentration of temsirolimus (<10 μg mL−1), or had significant differences from CA4-NPs at high concentration of temsirolimus (>10 μg mL−1). This indicated that CA4-NPs and temsirolimus did not have a synergistic effect to inhibit 4T1 tumor cell growth. CA4-NPs targeted tubulin of endothelium cells at neo blood vessels, further disrupting the skeleton of endothelium cells and destroying the tumor blood cell. However, temsirolimus targeted the tumor cells, which could block the activity of mTOR and its downstream signals. Thus, this study took advantage of the different roles of the two drugs, even though they did not have synergistic toxicity to tumor cells.
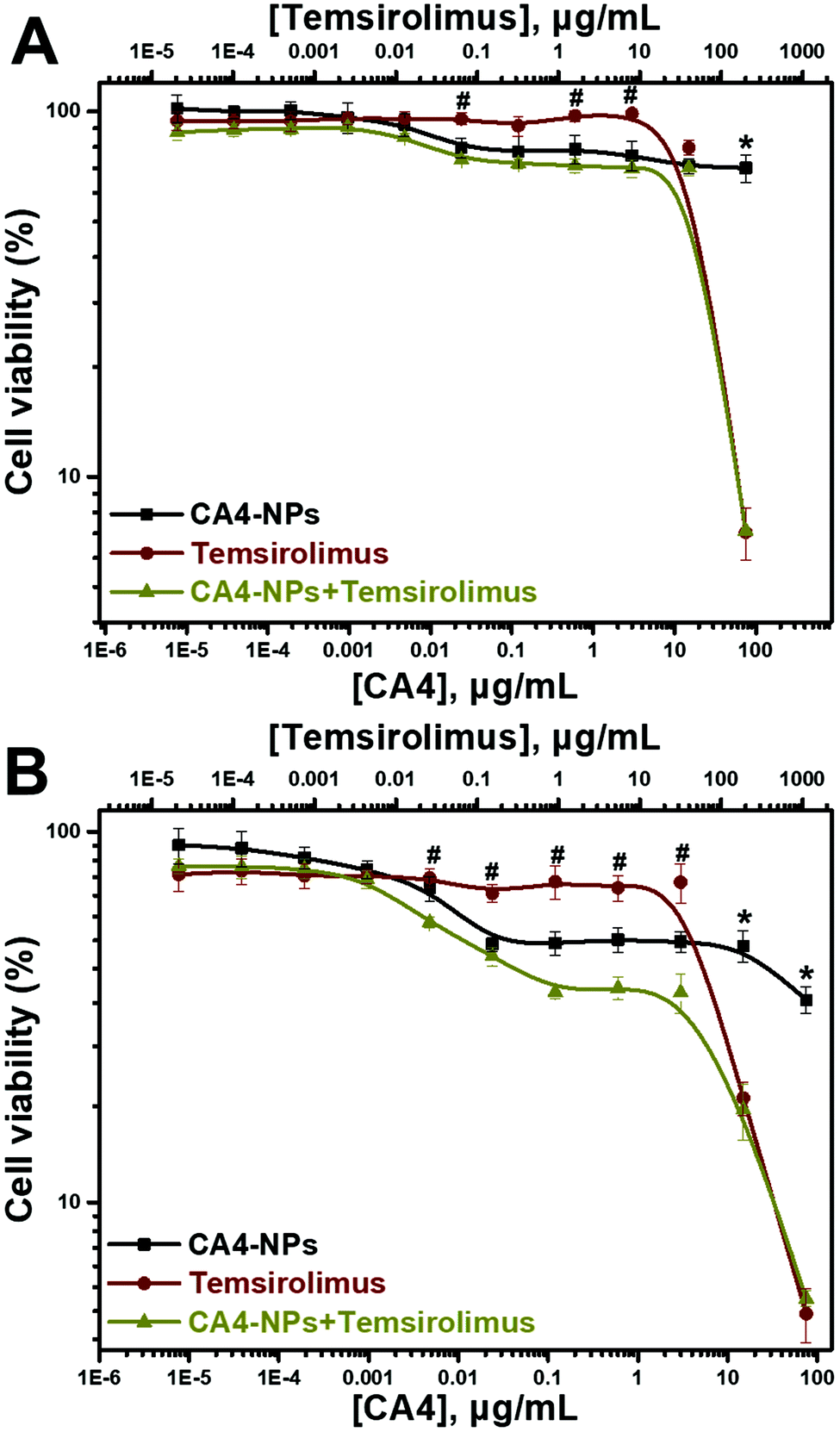 | ||
| Fig. 2 In vitro cytotoxicities of CA4-NPs, temsirolimus and CA4-NPs + temsirolimus to 4T1 cells for 24 h (A) or 48 h (B) (n = 3, *p < 0.05, the CA4-NPs group compared with the CA4-NPs + temsirolimus group at the corresponding concentrations, #p < 0.05, the temsirolimus group compared with the CA4-NPs + temsirolimus group at the corresponding concentrations). | ||
3.3 Temsirolimus could downregulate HIF1α expression under normoxia or hypoxia conditions in vitro
Next, we investigated the microenvironment modulation capacity of temsirolimus, especially, the downregulation of HIF1α expression. 4T1 cells were treated with different concentrations of temsirolimus. Western blot assay was carried out, and it showed that HIF1α expression significantly decreased to 75%, 65% and 11% after 10, 20 and 40 μg mL−1 temsirolimus treatment, respectively (Fig. 3A). Thus, temsirolimus could induce HIF1α downregulation in a dose-dependent manner. Then, 4T1 cells were subjected to temsirolimus under normoxia or hypoxia conditions. The cell lysates were used in western blotting, and the results indicated that HIF1α expressed more under hypoxia conditions than under normoxia conditions, as its expression under hypoxia conditions is significantly 0.45 fold higher than that under normoxia conditions. Critically, temsirolimus could reduce HIF1α expression under both normoxia and hypoxia conditions, because 20 μg mL−1 temsirolimus made HIF1α expression reduced to 74% under normoxia conditions, whilst it made HIF1α expression reduced to 40% under hypoxia conditions (Fig. 3B). Given the hypoxic status of the tumor microenvironment of VDA treatment31 and the inhibition of HIF1α (the critical factor in hypoxia) induced under hypoxia conditions by temsirolimus, CA4-NPs has the potential to exhibit the synergistic anti-cancer effect combined with temsirolimus. | ||
| Fig. 3 (A). 4T1 cells were treated with different concentrations of temsirolimus for 48 h. Then, the cell lysates were subjected to western blotting for HIF1α expression analysis. (B) 4T1 cells were treated with 20 μg mL−1 temsirolimus under normoxia or hypoxia conditions for 48 h. Western blotting was used to evaluate the HIF1α expression. In (A) and (B), the representative image is shown in the left panel, and the quantification result is shown in the right panel. One-way ANOVA was used for statistical comparisons, ***p < 0.001, **p < 0.01 or *p < 0.05. | ||
3.4 Temsirolimus and CA4-NPs exhibit the synergistic anti-tumor effect in vivo
Next, the anticancer effect of CA4-NPs plus temsirolimus was investigated. The in vivo anti-tumor effects of combined temsirolimus and CA4-NPs were then evaluated in 4T1 tumor-bearing female BALB/c mice. When the tumors reached 300 mm3, mice were administered PBS, 30 mg kg−1 CA4-NPs (eq. to CA4, i.v., once a week, twice), 20 mg kg−1 temsirolimus (i.p., initial four consecutive days a week, two weeks) or 30 mg kg−1 CA4-NPs (eq. to CA4, i.v., once a week, twice) + 20 mg kg−1 temsirolimus (i.p., initial four consecutive days a week, two weeks) (Fig. 4A). On day 15, the tumor inhibition rates reached 71% in mice receiving the combination treatment, which were significantly higher than all other groups. The tumor inhibition rates in the CA4-NPs group and the temsirolimus group were 48.9% and 25.8%, respectively (Fig. 4B). These data confirmed the favorable anti-tumor efficacy of combined CA4-NPs and temsirolimus. As to drug toxicity, CA4-NPs doses of 30 mg kg−1 (eq. to CA4) resulted in a greater body weight loss of 18%, compared with temsirolimus 20 mg kg−1 (6%) and the combination of CA4-NPs 30 mg kg−1 (eq. to CA4) + temsirolimus 20 mg kg−1 (10%). Moreover, body weight loss of the combined group was less than the CA4-NPs group, while much more than the temsirolimus group during the treatment period (Fig. 4C). The data shows that CA4-NPs could inhibit tumor growth to some extent, but the system toxicity was the strongest among different administrations. The metastasis may be considered to explain the above phenomenon. Thus, the metastasis of lung was investigated using India ink assays.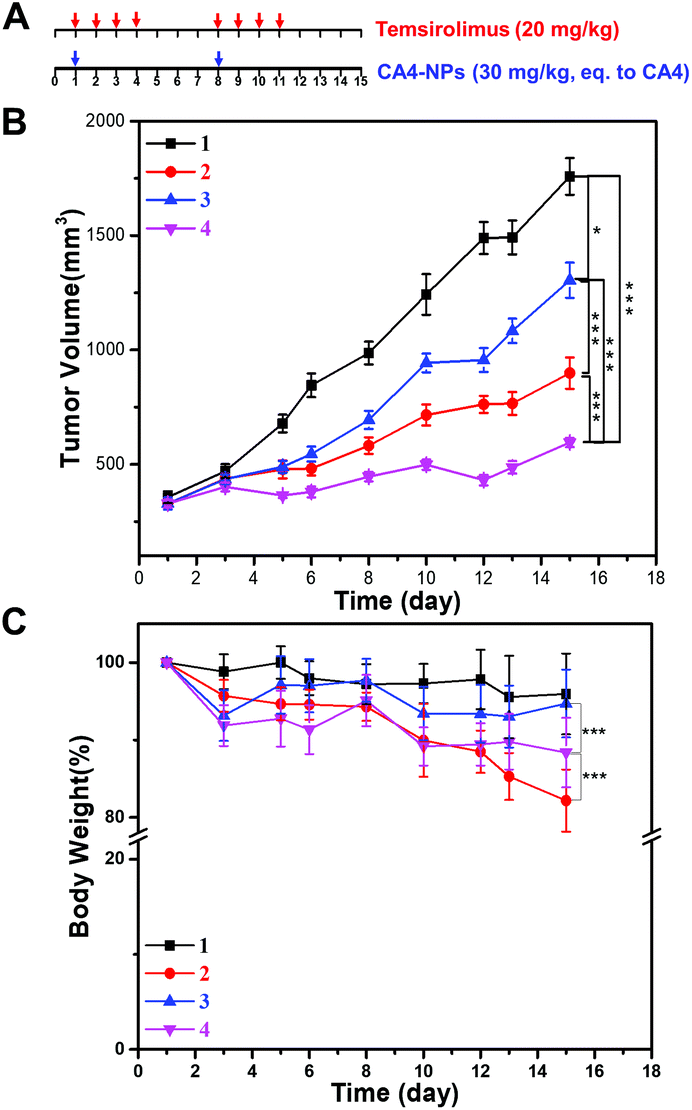 | ||
| Fig. 4 Therapeutic efficacy of the cooperative drug delivery platform on 4T1 tumor-bearing BALB/c mice. (A) Schematic image for drug treatment. (B) Tumor growth curve. (C) Body weight changes during the entire observation period. (1) Saline; (2) 30 mg kg−1 CA4-NPs (eq. to CA4); (3) 20.0 mg kg−1 temsirolimus; (4) 30 mg kg−1 CA4-NPs (eq. to CA4) + 20.0 mg kg−1 temsirolimus. CA4-NPs was administered on the 1st and 8th day, temsirolimus was administered on the 1st, 2nd, 3rd, 4th, 8th, 9th, 10th and 11th day (n = 6). One-way ANOVA was used for statistical comparisons, ***p < 0.001, **p < 0.01 or *p < 0.05. | ||
To observe pulmonary metastatic nodules, mice were euthanized and lungs were perfused with India ink through the trachea and destained in Fekete's solution. The white pulmonary metastasis nodules were photographed (Fig. 5A). The metastatic rate was calculated and quantified by the percent of the metastatic area. The metastatic rate of the combination of CA4-NPs 30 mg kg−1 (eq. to CA4) + temsirolimus 20 mg kg−1 was 18%, while the metastatic rate of CA4-NPs 30 mg kg−1 and temsirolimus 20 mg kg−1 group was 46% and 35%, respectively (Fig. 5B). The data indicated that lung metastasis was significantly inhibited by combination treatment (Fig. 5). This further accounted partly for the reduced body weight in the CA4-NPs group, as which was influenced by metastasis and drug toxicity. Ultimately, 4T1-bearing mice in the combination treatment groups lived more healthily than those in each single treatment group.
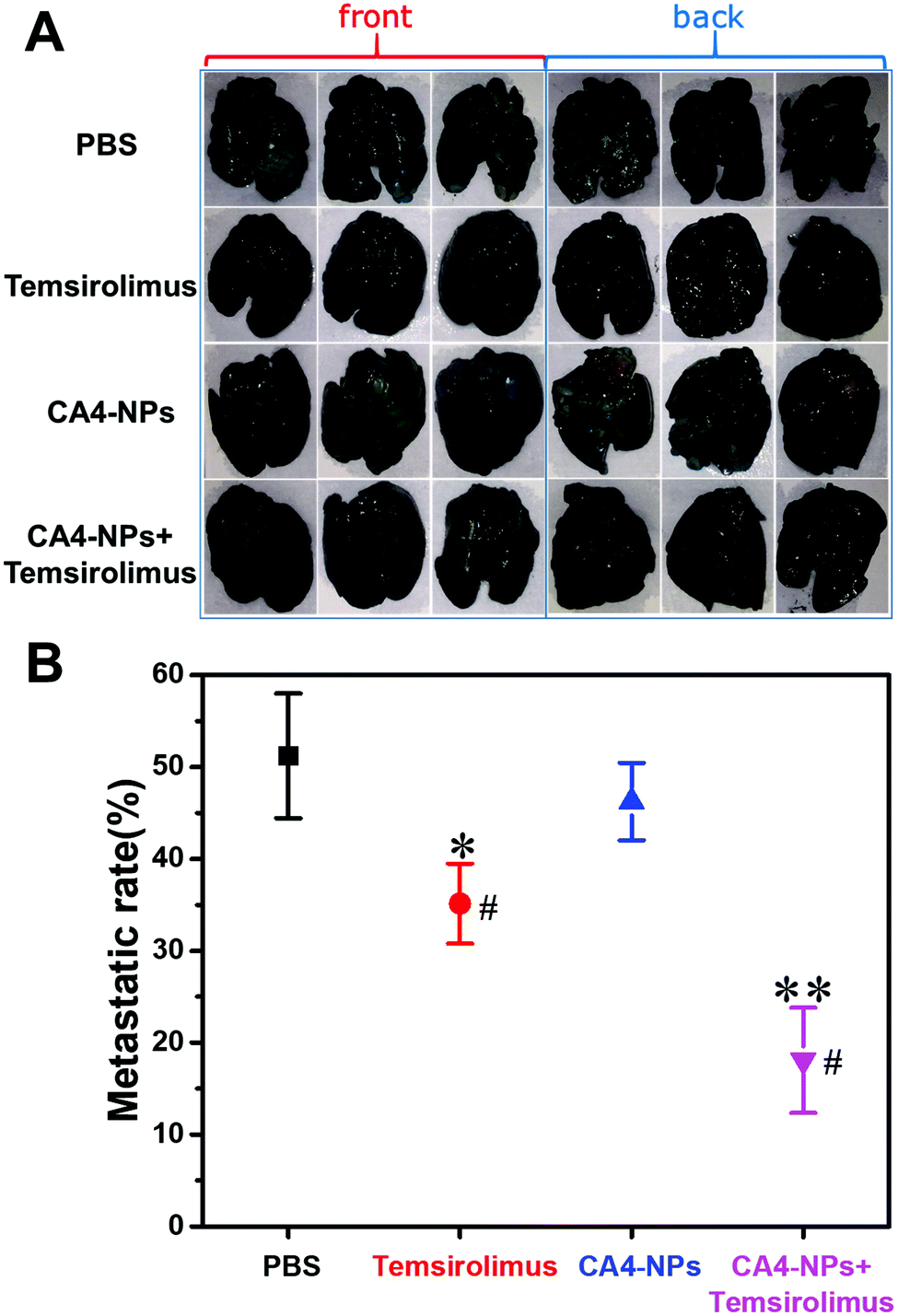 | ||
| Fig. 5 Images and quantification of white lung metastasis nodules with positive India ink staining (A) images of lung metastasis nodules after treatment. (B) Quantification of lung metastasis nodules (n = 3, *p < 0.05, **p < 0.01, compared with the PBS group, #p < 0.05, compared with the CA4-NPs group). | ||
3.5 Mechanism confirmation of the combination treatment in vivo
To further evaluate the therapeutic effects, the excised tumors were processed for histology staining. H&E staining results showed a diffuse tumor cell growth with rare signs of necrosis or apoptosis in untreated tumors (PBS group), and most confluent areas of dead cells with the typical loss of nuclei could be found in the CA4-NPs + temsirolimus group (Fig. 6A). The tumor necrosis areas were calculated thrice in different observation fields. CA4-NPs induced more necrotic areas compared to PBS, and the necrotic area in the CA4-NPs + temsirolimus group was significantly more than that in CA4-NPs (Fig. 6B). Since HIF1α is the critical factor in regulating tumor regrowth and metastasis, the IF staining was applied to analyse tumor HIF1α expression. It showed that CA4-NPs increased HIF1α expression, while temsirolimus reduced HIF1α expression. Thus, the CA4-NPs + temsirolimus group showed significantly less HIF1α expression compared to the CA4-NPs group (Fig. 6A and C).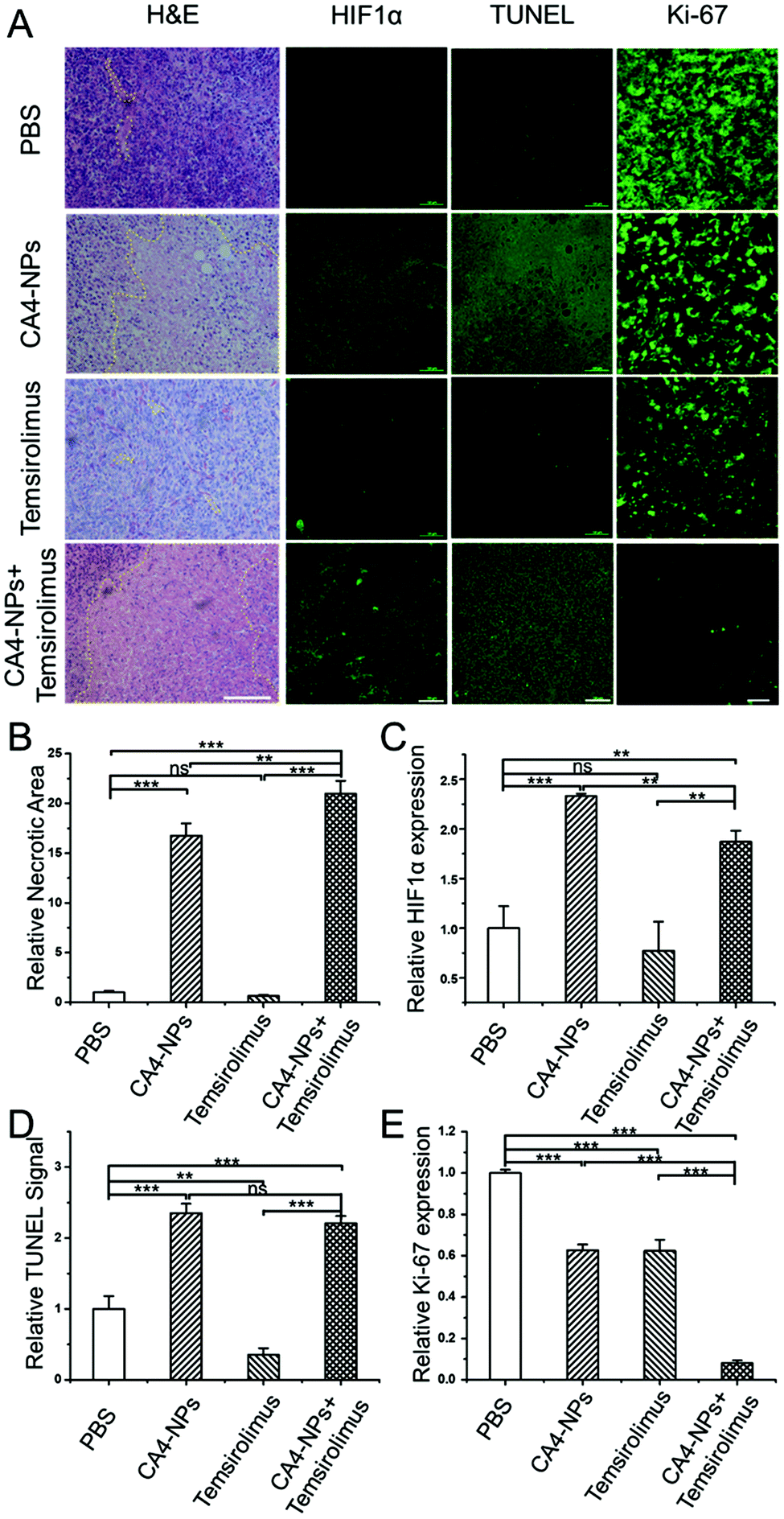 | ||
| Fig. 6 (A) Representative images of IF staining for indicated antibody and H&E staining in tumor. For H&E, HIF1α and TUNEL, scale bar was 100 μm; for Ki-67, scale bar was 20 μm. Necrotic areas were circled with yellow dot lines in H&E staining images. (B) Quantification of tumor necrotic areas in response to the H&E image (n = 3). (C) Quantitative analysis of the expression of HIF1α in tumor tissues measured by immunofluorescence staining assay (n = 3). (D) Relative intensities of tumor sections from TUNEL staining (n = 3). (E) Quantitative analysis of the expression of ki-67 in tumor tissues measured by immunofluorescence staining assay (n = 3). For (B)–(E), one-way ANOVA was used for statistical comparisons, ***p < 0.001, **p < 0.01 or *p < 0.05. Note: “ns” represented not significant. | ||
Finally, the apoptosis promotion and proliferation inhibition effect were detected, respectively. In terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) analyses,32 which allowed for the visualization and quantification of apoptotic cells,33 the CA4-NPs + temsirolimus group exhibited significantly the strongest TUNEL signal, which indicated that CA4-NPs + temsirolimus induced more apoptosis (Fig. 6A and D). Ki-67 is a cellular marker for proliferation.34 In the fluorescence staining assay, the CA4-NPs + temsirolimus group has the weakest Ki-67 signal, reflecting that CA4-NPs + temsirolimus have the strongest anti-tumor effect (Fig. 6A and E). Ample evidence indicates that the combinational strategy of CA4-NPs + temsirolimus can greatly cause tumor cell necrosis and apoptosis to inhibit tumor growth.
4. Conclusions
Collectively, we first developed a combinational strategy with CA4-NPs and temsirolimus for the treatment of breast cancer with rapid proliferation and potential metastasis. CA4-NPs selectively destroyed blood vessels, generating profound hypoxia and the high level of HIF1α inside tumors, which was not adverse to therapy. This study proves temsirolimus to downregulate HIF1α expression, potentiating the tumor growth and metastasis inhibition of CA4-NPs. It blocked the upstream signaling pathway from the microenvironment response of CA4-NPs treatment, delaying tumor recurrence and inhibiting tumor metastasis. In practice, combination therapy with CA4-NPs plus temsirolimus suppressed tumor growth significantly more strongly compared to either CA4-NPs or temsirolimus monotherapy, the inhibition rate to 4T1 tumor with a volume of 300 mm3 was 71% or the metastatic rate was reduced to 18%. The observations additionally provide a mechanistic rationale for the use of VDAs in combination with the mTOR inhibitor to enhance anticancer efficacy.Conflicts of interest
There are no conflicts of declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Projects 51973025, 51503202, 51673189, 51873206, 51829302, 51833010, and 51520105004), the Jilin Province (20190103033JH), and the Ministry of Science and Technology of China (Project 2018ZX09711003-012).Notes and references
- A. Palazon, A. W. Goldrath, V. Nizet and R. S. Johnson, Immunity, 2014, 41, 518–528 CrossRef CAS.
- S. Vanharanta, W. P. Shu, F. Brenet, A. A. Hakimi, A. Heguy, A. Viale, V. E. Reuter, J. J. D. Hsieh, J. M. Scandura and J. Massague, Nat. Med., 2013, 19, 50–56 CrossRef CAS.
- E. B. Rankin and A. J. Giaccia, Cell Death Differ., 2008, 15, 678–685 CrossRef CAS.
- M. Schindl, S. F. Schoppmann, H. Samonigg, H. Hausmaninger, W. Kwasny, M. Gnant, R. Jakesz, E. Kubista, P. Birner, G. Oberhuber and S. Austrian Breast Colorectal Canc, Clin. Cancer Res., 2002, 8, 1831–1837 CAS.
- Y. Yamamoto, M. Ibusuki, Y. Okumura, T. Kawasoe, K. Kai, K. Iyama and H. Iwase, Breast Cancer Res. Treat., 2008, 110, 465–475 CrossRef CAS.
- L. Strachan and D. W. Noble, J. R. Coll. Surg. Edinb., 2001, 46, 297–302 CAS.
- J. M. Ehrenfeld, L. M. Funk, J. Van Schalkwyk, A. F. Merry, W. S. Sandberg and A. Gawande, Can. J. Anaesth., 2010, 57, 888–897 CrossRef.
- D. Cui, J. G. Huang, X. Zhen, J. C. Li, Y. Y. Jiang and K. Y. Pu, Angew. Chem., 2019, 58, 5920–5924 CrossRef CAS.
- D. D. Wang, H. H. Wu, W. Q. Lim, S. Z. F. Phua, P. P. Xu, Q. W. Chen, Z. Guo and Y. L. Zhao, Adv. Mater., 2019, 31, 1901893 CrossRef.
- G. Dong, X. H. Lin, H. H. Liu, D. M. Gao, J. F. Cui, Z. G. Ren and R. X. Chen, Oncol. Lett., 2019, 18, 1831–1839 Search PubMed.
- B. Sennino and D. M. McDonald, Nat. Rev. Cancer, 2012, 12, 699–709 CrossRef CAS.
- M. F. Chen, X. P. Lei, C. Z. Shi, M. H. Huang, X. B. Li, B. J. Wu, Z. Q. Li, W. L. Han, B. Du, J. Y. Hu, Q. L. Nie, W. Q. Mai, N. Ma, N. H. Xu, X. Y. Zhang, C. L. Fan, A. H. Hong, M. H. Xia, L. P. Luo, A. D. Ma, H. S. Li, Q. Yu, H. R. Chen, D. M. Zhang and W. C. Ye, J. Clin. Invest., 2017, 127, 3689–3701 CrossRef.
- A. F. Welford, D. Biziato, S. B. Coffelt, S. Nucera, M. Fisher, F. Pucci, C. Di Serio, L. Naldini, M. De Palma, G. M. Tozer and C. E. Lewis, J. Clin. Invest., 2011, 121, 1969–1973 CrossRef CAS.
- D.-Y. Oh, T.-M. Kim, S.-W. Han, D.-Y. Shin, Y. G. Lee, K.-W. Lee, J. H. Kim, T.-Y. Kim, I.-J. Jang, J.-S. Lee and Y.-J. Bang, Cancer Res. Treat., 2016, 48, 28–36 CrossRef CAS.
- B. J. Monk, M. W. Sill, J. L. Walker, C. J. Darus, G. Sutton, K. S. Tewari, L. P. Martin, J. M. Schilder, R. L. Coleman, J. Balkissoon and C. Aghajanian, J. Clin. Oncol., 2016, 34, 2279–2286 CrossRef CAS.
- M. R. Horsman and D. W. Siemann, Cancer Res., 2006, 66, 11520–11539 CrossRef CAS.
- G. M. Tozer, C. Kanthou and B. C. Baguley, Nat. Rev. Cancer, 2005, 5, 423–435 CrossRef CAS.
- D. W. Siemann, D. J. Chaplin and M. R. Horsman, Cancer, 2004, 100, 2491–2499 CrossRef CAS.
- S. M. Galbraith, R. J. Maxwell, M. A. Lodge, G. M. Tozer, J. Wilson, N. J. Taylor, J. J. Stirling, L. Sena, A. R. Padhani and G. J. Rustin, J. Clin. Oncol., 2003, 21, 2831–2842 CrossRef CAS.
- Y. Shaked, T. Tang, J. Woloszynek, L. G. Daenen, S. Man, P. Xu, S.-R. Cai, J. M. Arbeit, E. E. Voest, D. J. Chaplin, J. Smythe, A. Harris, P. Nathan, I. Judson, G. Rustin, F. Bertolini, D. C. Link and R. S. Kerbel, Cancer Res., 2009, 69, 7524–7528 CrossRef CAS.
- J. J. Kim and I. F. Tannock, Nat. Rev. Cancer, 2005, 5, 516–525 CrossRef CAS.
- T. Liu, D. Zhang, W. Song, Z. Tang, J. Zhu, Z. Ma, X. Wang, X. Chen and T. Tong, Acta Biomater., 2017, 53, 179–189 CrossRef CAS.
- A. B. Iversen, M. Busk and M. R. Horsman, Acta Oncol., 2013, 52, 1320–1326 CrossRef CAS.
- D. Del Bufalo, L. Ciuffreda, D. Triscinoglio, M. Desideri, F. Cognetti, G. Zupi and M. Milella, Cancer Res., 2006, 66, 5549–5554 CrossRef CAS.
- X. Wan, N. Shen, A. Mendoza, C. Khanna and L. J. Helman, Neoplasia, 2006, 8, 394–401 CrossRef CAS.
- T. Y. Reynolds, S. Rockwell and P. M. Glazer, Cancer Res., 1996, 56, 5754–5757 CAS.
- T. Liu, D. Zhang, W. Song, Z. Tang, J. Zhu, Z. Ma, X. Wang, X. Chen and T. Tong, Acta Biomater., 2017, 53, 179–189 CrossRef CAS PubMed.
- Y. Wang, H. Yu, D. Zhang, G. Wang, W. Song, Y. Liu, S. Ma, Z. Tang, Z. Liu, K. Sakurai and X. Chen, Acta Biomater., 2019, 92, 229–240 CrossRef CAS.
- N. Shen, F. Yan, J. Pang, N. Zhao, N. Gangat, L. Wu, A. M. Bode, A. Al-Kali, M. R. Litzow and S. Liu, Clin. Cancer Res., 2017, 23, 6254–6266 CrossRef CAS PubMed.
- J. Yang, H. Su, W. Sun, J. Cai, S. Liu, Y. Chai and C. Zhang, Theranostics, 2018, 8, 1966–1984 CrossRef CAS.
- S. Yang, Z. Tang, C. Hu, D. Zhang, N. Shen, H. Yu and X. Chen, Adv. Mater., 2019, 31, 1805955 CrossRef.
- H.-Y. Gong, Y.-G. Chen, X.-S. Yu, H. Xiao, J.-P. Xiao, Y. Wang and X.-T. Shuai, Chin. J. Polym. Sci., 2019 DOI:10.1007/s10118-019-2272-6.
- W. Gorczyca, F. Traganos, H. Jesionowska and Z. Darzynkiewicz, Exp. Cell Res., 1993, 207, 202–205 CrossRef CAS.
- T. Scholzen and J. Gerdes, J. Cell. Physiol., 2000, 182, 311–322 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |