
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntravenous anti-VEGF agents with RGD peptide-targeted core cross-linked star (CCS) polymers modified with indocyanine green for imaging and treatment of laser-induced choroidal neovascularization†
Wenting
Cai‡
a,
Qijing
Chen‡
 b,
Tianyi
Shen
a,
Qian
Yang
ac,
Weinan
Hu
d,
Peng
Zhao
*b and
Jing
Yu
*a
b,
Tianyi
Shen
a,
Qian
Yang
ac,
Weinan
Hu
d,
Peng
Zhao
*b and
Jing
Yu
*a
aDepartment of Ophthalmology, Shanghai Tenth People's Hospital, Tongji University, School of Medicine, Shanghai, 200072, China. E-mail: dryujing@aliyun.com
bInstitute for Translational Medicine, Institute for Biomedical Engineering and Nanoscience, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, 200092, China. E-mail: zp@tongji.edu.cn
cAnhui Medical University, Hefei, 230032, China
dDepartment of Ophthalmology, Anhui University of Science and Technology, Huainan, 232001, China
First published on 20th June 2020
Abstract
Age-related macular degeneration (AMD) is a leading cause of irreversible visual loss among elderly persons, of which wet AMD is characterized by choroidal neovascularization (CNV). We herein developed nanoparticles with good biosafety for effective treatment of choroidal neovascularization (CNV). S-PEG-ICG-RGD-RBZ NPs were synthesized and characterized by ZP, DLS, UV–Vis, TEM and Coomassie Brilliant Blue staining analyses. In our study, the S-PEG-ICG-RGD-RBZ NPs exhibited good biocompatibility in vitro and in vivo. There was no cellular toxicity, dead cells, apoptosis or genotoxicity in the studied concentration range in vitro; meanwhile, intravenous injection of the designed NPs did not cause histological damage or apoptosis in the organs in vivo, including the heart, liver, spleen, lung, kidneys and brain. The designed NPs inhibited VEGF-induced proliferation, cell migration, tube formation and expression of CD31 and VEGF in vitro. Meanwhile, in vivo studies also indicated the inhibition of CNV development by NPs. What's more, the CNV area was imaged after intravenous injection of NPs modified with indocyanine green. The NPs were mainly targeted to CNV areas and did not remain in the other organs. In summary, S-PEG modified with RGD was designed as a powerful carrier to deliver anti-VEGF agents to CNV areas. The smart NPs, which have good cellular compatibility, hold great potential for drug delivery in CNV treatment.
Introduction
Age-related macular degeneration (AMD) is a leading cause of irreversible visual loss among elderly persons.1 There are approximately 21 million AMD patients worldwide.2 The majority of patients with severe visual loss suffer from the exudative form of AMD, which is characterized by choroidal neovascularization (CNV) in the subretinal pigment epithelial space.3 CNV is mainly driven by the vascular endothelial growth factor (VEGF). Repeated intravitreal injections of anti-VEGF agents have been recommended as the standard treatment for CNV in clinical work.4 However, intravitreal injection has the risk of endophthalmitis,5,6 elevated intraocular pressure7,8 and other complications, which can induce severe visual reduction. Thus, it is important to identify noninvasive drug delivery techniques.The application of anti-VEGF agents has been considered the first-line treatment strategy for CNV, including the monoclonal antibody fragment ranibizumab (RBZ), the monoclonal antibody bevacizumab (BVZ) and aflibercept. RBZ is a high-affinity recombinant Fab that neutralizes all subtypes of VEGF-A.9 Recently published studies have reported that RBZ and BVZ can effectively reduce areas of CNV in laser-induced animal models.10,11
In recent years, many kinds of polymers have been widely used as delivery carriers for bioapplications due to their controllable size and multifunctional structures.12,13 Polyethyleneglycol (PEG) is widely used in pharmaceutical and biomedical applications due to its excellent physicochemical and biological properties, including its solubility, nontoxicity, hydrophilicity, ease of chemical modification and lack of immunogenicity.14 PEG with multifunctional modifications has been widely explored as a promising nanocarrier for therapeutic drug delivery,15–17 gene delivery,18–20 and CT/MR imaging.21 Chen et al. reported that PEG loaded with anticancer drugs could improve anticancer efficacy and had pH-sensitive and tumor targeting properties after specific modifications.22 We previously reported that core cross-linked star (CCS) polymers act as intermediates between linear polymers and polymeric nanoparticles.23,24
Integrins are receptors of specific extracellular matrix proteins involved in various diseases associated with angiogenesis such as ocular angiogenesis,25 tumors26 and ischemic stroke.27 Some researchers have reported that integrin receptors, especially αvβ3, are overexpressed in retina or choroid tissues from AMD patients,28,29 which suggests that integrin αvβ3 might act as a drug delivery target for CNV. The arginine–glycine–aspartic acid (RGD) peptide has been reported to specifically bind with integrin αvβ3.30 A previous study reported that intravenous injection of liposomes, surface-modified with the RGD peptide, could deliver photosensitive agents and anti-VEGF agents to CNV lesions and achieve therapeutic effects.31
In this current study, PEG CCS (S-PEG) polymers were synthesized to act as a carrier at first, and then were modified with RGD and indocyanine green (ICG) and loaded with anti-VEGF agents to form S-PEG-ICG-RGD-RBZ NPs for CNV imaging and treatment.32,33
Experimental section
Materials
PPEGMA (Mn = 300), N-hydroxysuccinimide (NHS), a chain transfer agent (CTA), dicyclohexylcarbodiimide (DCC) and triethylamine (TEA) were obtained from Sigma-Aldrich (St Louis, MO). ICG-NH2 was obtained from AAT Bioquest (Sunnyvale, CA). A human retinal pigment epithelium cell line (ARPE-19) was obtained from iCell Bioscience Company. Human umbilical vein endothelial cells (HUVECs), endothelial cell medium (ECM) and endothelial cell growth supplement (ECGS) were purchased from ScienCell (USA). DMEM/F12 cell medium and fetal bovine serum (FBS) were purchased from HyClone (USA). A cell counting kit (CCK)-8 was purchased from Yeasen (Shanghai, China), and an Annexin V-FITC Apoptosis Detection Kit and a Matrigel™ Basement Membrane Matrix were obtained from BD BioScience (San Jose, CA, USA). C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China).Synthesis and characterization of S-PEG-ICG-RGD-RBZ NPs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) of its aqueous solutions.
000) of its aqueous solutions.
Cell viability assay. ARPE-19 cells were seeded in 96-well plates at a density of 5000 cells per well and incubated overnight for adhesion. S-PEG-ICG-RGD-RBZ NPs with increasing concentrations (0–100 μM) were added to each well and incubated for 24 h. Then, 10 μL of the cell counting kit (CCK)-8 (Yeasen, Shanghai, China) reagent was added to each well for 2 h, and cell viability was evaluated at 450 nm. The OD value reflects the proportion of living cells.
EdU (5-ethynyl-2′-deoxyuridine) staining. ARPE-19 cells were seeded on slides and treated with S-PEG-ICG-RGD-RBZ (0, 50, 100 μM) for 24 h. After washing with PBS thrice, the cells were incubated with 2 μM calcein AM and 8 μM PI for 30 min. The solution was removed and washed with PBS, after which the slides were mounted and the images were recorded via fluorescence microscopy.
Flow cytometry assay for evaluating cell apoptosis. ARPE-19 cells and HUVECs were treated with S-PEG-ICG-RGD-RBZ (0, 50, and 100 μM) for 24 h. After 24 h incubation, the cells were washed with PBS, and the Annexin V-FITC Apoptosis Detection Kit was used for staining. The results were recorded via a FASCCanto II flow cytometer (BD).
Comet assay. The cells treated with the NPs were resuspended in 1× PBS and then processed for the comet assay. Firstly, 1% normal melting point agarose was placed on a glass slide. Then, 150 μL of each cell suspension was blended with 200 μL of 1% low-melting point agarose. The mixture was placed on the glass slide and solidification was achieved at 4 °C, followed by lysis in the lysis solution for 6 h at 4 °C. The slides were immersed in neutral unwinding solution to unwind the DNA for 30 min. Next, electrophoresis was performed at 20 V for 30 min in the neutral unwinding solution. The gels were stained with 1× SYBR Green dye for 20 min in the dark after neutralization in cold deionized water. Images were recorded via fluorescence microscopy.
Transwell assay. A total of 5 × 104 cells in 200 μL of FBS-free ECM culture medium were seeded in the upper compartment of a Transwell system (8 mm pore size, Corning, USA); culture medium with 10% FBS was added to the lower compartment. After incubation for 16 h, nonmigratory cells were removed with cotton swabs. After washing with PBS, the migrated cells attached to the bottom membrane were fixed with ethanol for 20 min and stained with 1% crystal violet for 15 min after drying in air. Images in 3 fields were acquired under phase-contrast microscopy.
Wound-healing assay. HUVECs were plated in 6-well plates at a density of 1 × 105 cells per well. Wounds were created with a 200 μL pipette tip in the middle of the monolayer at 80% confluence. Images were recorded at 0 h as an initial point, and the cells were treated as described above. Wound closure was recorded after incubation for 24 and 48 h by measuring the scratch widths, and migration rates were calculated based on the formula (distance/scratch width) × 100%. The experiments were repeated three times.
Tube formation assay. Matrigel was coated in 96-well plates at 37 °C for 30 min for solidification, and 100 μL of HUVECs (5 × 103 per well), which were treated as described above, were seeded into each well. The images were captured with a light microscope after incubation for 6 h.
Immunofluorescence assay. HUVECs were seeded in 35 mm confocal dishes (glass-bottom dishes). After treatment with VEGF and nanoparticles, the cells were fixed with 4% paraformaldehyde for 10 min. The cells were immersed in 0.1% Triton X-100 and 5% BSA dissolved in PBS for 1 h at room temperature to achieve permeabilization and blocking and then were incubated overnight at 4 °C with primary antibodies (all 1
:100 dilution). The cells were incubated with Cy3-conjugated secondary antibodies for 40 min. Next, nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (1:1000 dilution) for 20 min. Images were recorded by confocal microscopy (LSM710; Carl Zeiss, Jena, Germany).
Establishment of laser-induced CNV mouse models. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shanghai Tenth People's Hospital and approved by the Animal Ethics Committee of Shanghai Tenth People's Hospital. C57BL/6 mice (6–8 w, 25–30 g, male) were used to establish laser photocoagulation-induced CNV models as previously described. Intraperitoneal injection of 1% pentobarbital sodium (40 mg kg−1) was used for anesthesia, and topical application of 0.5% tropicamide and 0.5% Alcaine was used to dilate the pupil and achieve ocular surface anesthesia, respectively. Photocoagulation was performed using a 532 nm wavelength laser, and the laser burn was at 150 mW intensity, 0.05 s duration, and 50 μm spot size. In total, 4–6 laser spots were generated between each main retinal vessel at approximately the same distance to the optic nerve head. Bubbles or slight hemorrhage indicated a rupture in the Bruch's membrane and success of the laser photocoagulation-induced CNV mouse models.
Experimental design. The mice were divided into five groups, including the control group (n = 20), PBS group (n = 20), vehicle group (n = 20), NP group (n = 20) and RBZ group (n = 20). The mice in the control groups were normal mice of the same strain. The mice in the PBS, vehicle and NP groups were intravenously injected with PBS, the vehicle and the NPs. The mice in the RBZ group were intravitreally injected with RBZ.
In vivo imaging. In vivo near-infrared reflectance and fluorescence imaging of the ocular fundus of the rodent eye was performed using confocal scanning laser ophthalmoscopy (Spectralis, Heidelberg Engineering Inc., Heidelberg, Germany). The fluorescence images were recorded before NP injection and at predefined time points following NP injection (7, 14, 28, and 90 days). Approximately 0.2 mL of 10% sodium fluorescein (Alcon Japan, Tokyo, Japan) was injected intraperitoneally following NP injection, and the leakage of fluorescein was captured by confocal scanning laser ophthalmoscopy.
Measurement of the CNV area. After anesthetization, 2 mL of PBS containing 50 mg of fluorescein isothiocyanate- (FITC)-dextran was injected into the left ventricle of the heart. The eyes were extracted and fixed in 4% paraformaldehyde for 1 h. The RPE-choroid-sclera flat mounts were prepared after removing the cornea, lens, and vitreous body. Then, the tissues were cut radially into six flaps to be flattened onto a microscope slide, with the RPE section facing up. Next, the tissues were incubated with IB4 (1
:1000) for 30 min and washed with 1× PBS three times. Afterwards, the CNV lesions in the flat mounts were recorded via fluorescence microscopy. The CNV area was measured with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
CNV histology and immunofluorescence. HE assays were performed to stain the CNV area. The CNV length, thickness and area were measured to observe the progression of neovascularization. Platelet-endothelial cell adhesion molecule 1 (CD31) and VEGF were used to observe the CNV area. The paraffin sections were dewaxed, and hydration and antigen retrieval were performed according to the protocol. Then, the slides were blocked with 5% BSA for 1 h at room temperature and incubated with rabbit VEGF antibody (1
:50, Proteintech, Chicago, IL, USA) and mouse CD31 antibody (1:50, Cell Signaling Technology, Danvers, MA, USA) at 4 °C overnight. Next, the slides were stained with Alexa Fluor 488 goat anti-mouse immunoglobulin G (IgG) and Alexa Fluor 546 goat anti-rabbit IgG for 40 min, followed by staining with DAPI (1:1000 dilution) for another 10 min. Photomicrographs were captured using fluorescence microscopy.
Growth curve. The body weights at different time points including 7, 14, 21 and 28 days were recorded in these groups. The changes in body weights were calculated.
Serum assays. Blood samples were collected from the mice after treatment in different groups. Serum was separated from the blood samples by centrifugation at 4600g for 10 min, and was stored at −80 °C until use. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) reflected the liver function, while creatinine (Cr) indicated kidney function. Total cholesterol (T-CHO) and triglyceride (TG) showed blood lipids, and electrolyte assay used the indexes of potassium (K), calcium (Ca), and chlorine (Cl). All these indexes were assayed with commercial kits following the manufacturers’ instructions for biocompatibility detection in vivo.
Hemolysis assay. A hemolysis assay was performed using fresh whole blood. To whole blood (4 mL), 10 mL of NS was added, and the mixture was centrifuged at 3000 rpm for 5 min and then washed with NS until the supernatant was colorless. The erythrocytes were collected to prepare a 2% red cell suspension. The nanoparticles were prepared in NS at different concentrations. NS was used as the negative control with 0% hemolysis, and ddH2O was used as the positive control with 100% hemolysis. Two milliliters of nanoparticles, NS, and ddH2O were incubated for 30 min at 37 °C in an incubator shaker. Then, 2 mL of red cell suspension was added to each group, and mixed as well as incubated for 60 min at 37 °C. The supernatants were collected after centrifugation at 3000 rpm for 5 min. The percentage of hemolysis was measured with a microplate reader at 540 nm absorbance.
HE staining. After injection of NPs for 28 days, the mice were sacrificed, and the organs including the heart, spleen, kidneys, liver, brain and lungs were extracted and fixed in 4% paraformaldehyde. The organs were embedded, sectioned, and stained with H&E according to the protocol. The slides were mounted with neutral gum and imaged with a light microscope.
Terminal dUPT nick-end labeling (TUNEL) assay. The paraffin-embedded organs were sectioned, dewaxed and hydrated as described in a previous study. After incubation with proteinase K, the sections were incubated with a solution mixed with terminal deoxynucleotidyl transferase (TdT) and fluorescein-labeled dUTP according to the manufacturer's protocol. The slides were imaged via a fluorescence microscope.
Metabolism of NPs was observed via the imaging of small animals in vivo. The mice were intravenously injected with NPs and sacrificed at 2 h, 6 h, 16 h and 24 h after injection. The organs including the heart, liver, spleen, lungs, kidneys, brain, muscle and small intestine were extracted, and the images were captured at an absorption wavelength of 780 nm and an emission wavelength of 820 nm.
Statistical analysis. SPSS 19.0 software and Graph Pad Prism version 5 were used for statistical analyses. One-way analysis of variance (ANOVA) was performed among multiple groups. P-Values less than 0.05 were considered to be statistically significant.
Results and discussion
Preparation and characterization of the NPs
L-PEG was synthesized by RAFT polymerization of PEGMA in dioxane. Then, S-PEG was synthesized by RAFT dispersion polymerization in water/ethanol solutions. After synthesis of S-PEG, the fluorophores called ICG were bound with S-PEG to obtain S-PEG-ICG. Finally, RGD and RBZ were reacted with S-PEG under certain reaction conditions. The method used to prepare the S-PEG-ICG-RGD-RBZ NPs is shown in Scheme 1. The synthesized S-PEG-ICG-RGD-RBZ NPs were well characterized via different methods. Due to the formation of particle clusters, the S-PEG-ICG-RGD-RBZ NPs displayed a much larger hydrodynamic size than S-PEG. In the present study, the ZP of S-PEG, S-PEG-ICG and S-PEG-ICG-RGD-RBZ was −4.74 ± 1.56, −3.79 ± 0.47 and −2.42 ± 0.13 mV, respectively. As shown in Fig. 1, the ICG unit was successfully coupled onto the surface of S-PEG based on UV–Vis spectroscopy, and nearly no obvious difference in the surface plasmon resonance peak was observed between S-PEG-ICG and S-PEG-ICG-RGD-RBZ NPs. The TEM micrographs of S-PEG and S-PEG-ICG-RGD-RBZ NPs are shown in Fig. 2. The particle sizes of S-PEG-ICG-RGD-RBZ NPs appeared larger than the sizes of S-PEG due to the formation of particle clusters.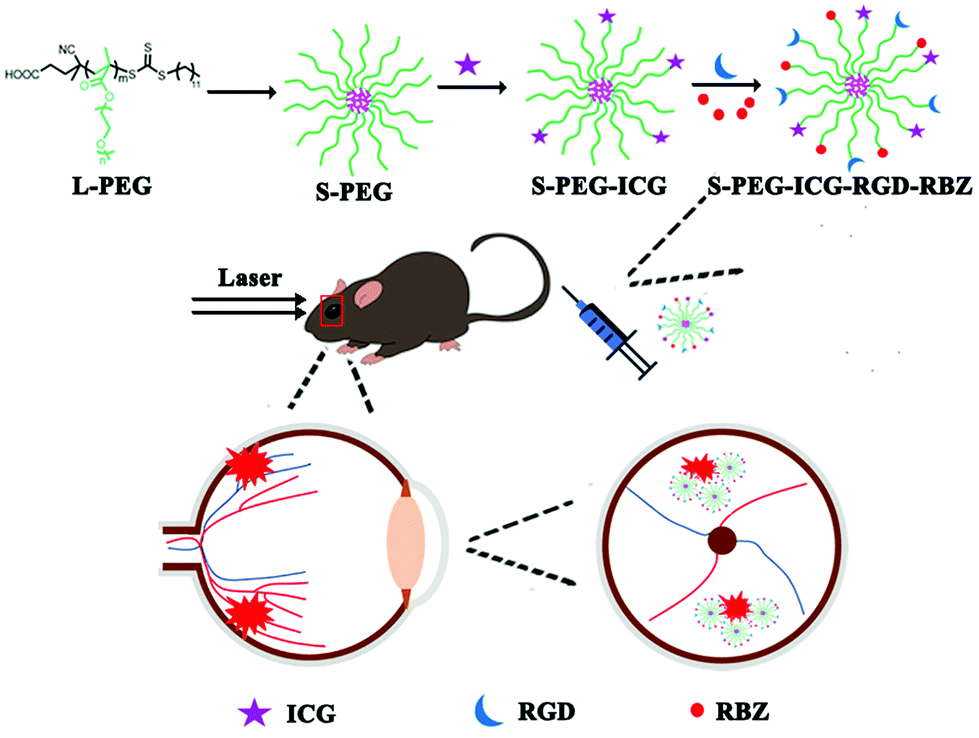 | ||
| Scheme 1 Schematic illustration of the preparation of the S-PEG-ICG-RGD-RBZ NPs. | ||
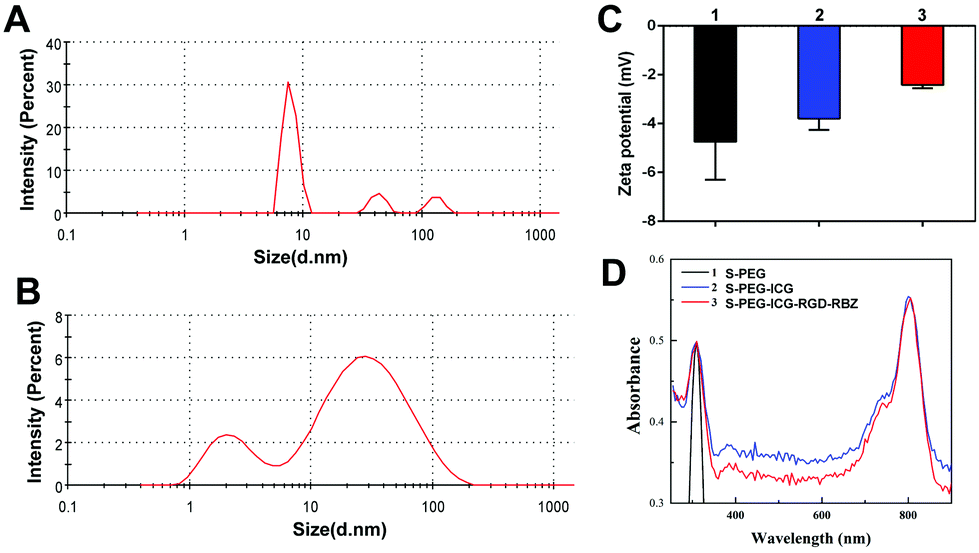 | ||
| Fig. 1 Characterization of NPs was performed by mean particle size, zeta potential and UV–Vis spectrum measurements. (A) Mean particle size of S-PEG. (B) Mean particle size of S-PEG-ICG-RGD-RBZ. (C) Zeta potential of S-PEG (1), S-PEG-ICG (2) and S-PEG-ICG-RGD-RBZ (3), respectively. (D) UV–Vis spectrum of S-PEG (1), S-PEG-ICG (2) and S-PEG-ICG-RGD-RBZ (3), respectively. | ||
 | ||
| Fig. 2 Characterization of NPs was performed by TEM. (A) TEM images of S-PEG and (B) TEM images of S-PEG-ICG-RGD-RBZ NPs. Scale bar: 100 μm. | ||
The conjugation and bioactivity of RBZ on the S-PEG-ICG-RGD-RBZ NPs
As shown in Fig. 3, SDS-PAGE bands were stained with Coomassie Brilliant Blue stain to show the conjugation of RBZ on the S-PEG-ICG-RGD-RBZ NPs. Native RBZ showed a distinctive band at 23 kDa corresponding to the reduction and cleavage of the disulfide bond between the two chains of the Fab fragment.35 This band was also present in the S-PEG-ICG-RGD-RBZ NPs, which indicated the conjugation and bioactivity of RBZ on the S-PEG-ICG-RGD-RBZ NPs. No band was observed in the sample of S-PEG since S-PEG did not conjugate with RBZ.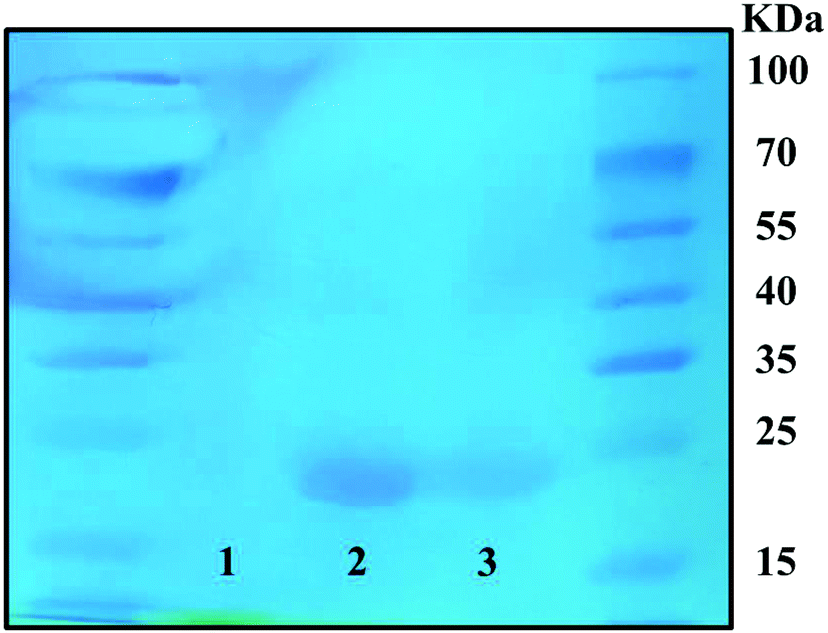 | ||
| Fig. 3 Coomassie Brilliant Blue staining of the SDS-PAGE band was performed to verify the conjugation of RBZ. Group 1: S-PEG, Group 2: RBZ, and Group 3: S-PEG-ICG-RGD-RBZ NPs. | ||
The entrapment efficiency of RBZ on the S-PEG-ICG-RGD-RBZ NPs
As shown in Fig. S1,† the ranibizumab loaded on the S-PEG-ICG-RGD-RBZ NPs was calculated according to the standard curve. The entrapment efficiency of RBZ on S-PEG-ICG-RGD-RBZ NPs was approximately 76.3%.NPs caused no cell viability damage, apoptosis or genotoxicity
Investigation of the potential toxicity of the nanoparticles is a vital key point that needs to be considered prior to their application. To our knowledge, many experiments have been reported to investigate cellular toxicity, including cell viability, morphology, Annexin V-FITC apoptosis assay, comet assay and others.36 To assess the biosafety of the NPs, we performed these experiments to investigate the cell viability, apoptosis and DNA damage, respectively. Since the NPs would be delivered to the CNV lesions, the biosafety of the surrounding RPE cells should be taken into consideration. In the current study, ARPE-19 cells were applied to determine the cytotoxicity of the nanoparticles.The results of the CCK-8 cell viability assay showed that ARPE-19 cells treated with the NPs at a concentration range of 0 to 100 μM had no significant difference in cell viability after 24 h of incubation, as shown in Fig. 4A. According to EdU staining, the green calcein-AM stain could indicate living cells, and the red PI stain could indicate dead cells. The results showed good cell viability after NP treatment (Fig. 4B). In addition, the values of Q2 + Q4 present the cell apoptosis rates from the flow cytometry assay. In Fig. 4C and D, the apoptosis rates were 5.20 ± 0.30%, 5.00 ± 0.75% and 4.97 ± 0.55% in RPE cells with different NP treatments (0, 50, and 100 μM, respectively). Therefore, no significant difference could be observed with or without treatment. It is typically thought that PEG has good biological properties, especially nontoxicity,14 which widens its application in biology.
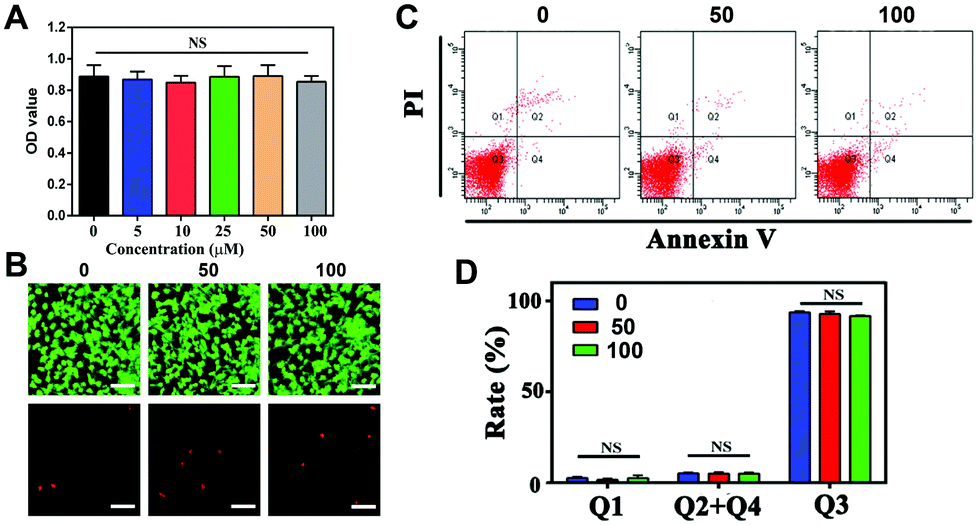 | ||
| Fig. 4 Different methods were used to analyze biosecurity in vitro by CCK-8 assay, EdU staining and Annexin V/PI staining. (A) CCK-8 assay of ARPE-19 cells treated with NPs at different concentrations for 24 h. (B) Fluorescence images of live/dead staining on RPE after incubation with NPs for 24 h. Scale bar: 100 μm. (C and D) Cell apoptosis was measured by Annexin V/PI staining in RPE cells treated with various concentrations of NPs. | ||
The comet assay, also called the single cell gel electrophoresis (SCGE) assay, is a rapid, simple and visual technique for measuring genotoxicity, and the formation of a comet tail indicates DNA breakage. In Fig. S2,† compared with the genetic damage induced by cisplatin as a positive control, there is no comet tail observed in the cells treated with NPs, indicating no genotoxicity in vitro. Some polymers have been reported to interact with negatively charged DNA, inducing genotoxicity.37 Considering this point, a comet assay was necessary in the assessment. Fortunately, compared with the positive control induced by cisplatin, our results provided evidence that no toxicogenomic damage occurred. In summary, the synthesized NPs in our study displayed good cytocompatibility in vitro and were considered safe for the subsequent experiments.
We used ARPE-19 cells to evaluate cell viability, apoptosis, and genotoxicity in vitro. Thus, we found that the S-PEG-ICG-RGD-RBZ NPs had good cytocompatibility in vitro and could be considered safe to complete the subsequent research.
NPs attenuated VEGF-induced HUVEC proliferation, migration and tube formation
As shown in Fig. S3,† cell proliferation was promoted after incubation with VEGF, while the proliferation was inhibited by NP treatment. For the wound-healing test, the width was measured at 0 h, 24 h, and 48 h, as shown in Fig. 5, and the rate of cell migration was calculated via the proportions of (24 h–0 h)/0 h and (48 h–0 h)/0 h. Migration in VEGF-induced HUVECs was greater than that of cells not treated with VEGF at both 24 h and 48 h (**P < 0.01 and ****P < 0.0001, respectively) (Fig. 5A). The migration rates of the four groups were 0.22 ± 0.03, 0.48 ± 0.07, 0.33 ± 0.06, and 0.28 ± 0.02 at 24 h (Fig. 5B). The migration rates decreased from 0.72 ± 0.01 after VEGF stimulation to 0.41 ± 0.06 and 0.34 ± 0.06 after NP treatment at 50 μM and 100 μM, respectively (Fig. 5C). NPs attenuated VEGF-induced cell migration in a concentration-dependent and time-dependent manner. The transwell assay was also performed to evaluate cell migration. The numbers of cells migrating after stimulation with VEGF were increased from 146.33 ± 10.02 to 277.33 ± 18.61 (***P = 0.0004), and NPs reduced these numbers to 157.33 ± 37.17 (**P = 0.0075) and 132.00 ± 29.46 (**P = 0.0019) at concentrations of 50 μM and 100 μM, respectively (Fig. 5D and E).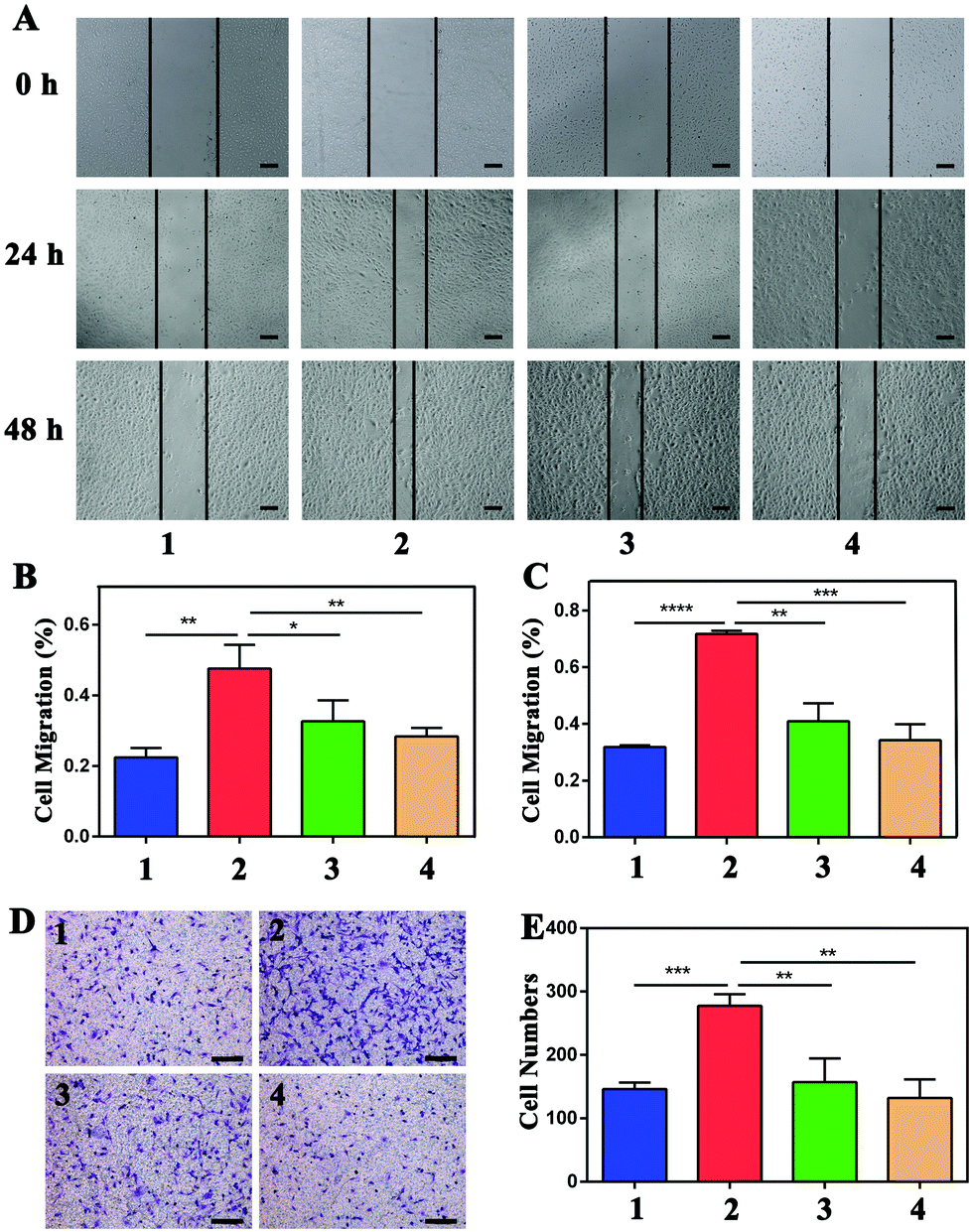 | ||
| Fig. 5 NPs attenuated VEGF-induced HUVEC migration, which was measured by wound-healing assay and transwell assay. (A) After treatment with 20 ng mL−1 of VEGF with or without NPs for 24 h and 48 h, the migration of HUVECs with different treatments was detected via wound-healing assay analysis. Scale bar: 200 μm. Migrated cells at 24 h (B) and 48 h (C) were quantified by counting three random vision fields under a microscope. (D) Transwell assay was also used to detect cell migration in the four groups. Scale bar: 200 μm. (E) The quantified analysis of migrated cells via transwell assay. Group 1: control, Group 2: 20 ng mL−1 VEGF treatment, Group 3: 20 ng mL−1 VEGF with 50 μM NP treatment, and Group 4: 20 ng mL−1 VEGF with 100 μM NPs treatment. | ||
For the tube formation assay, the numbers of nodes, junctions, segments and relative tube lengths were measured to evaluate the NPs in VEGF-induced tube formation. In the four groups including the control, 20 ng mL−1 VEGF treatment, 20 ng mL−1 VEGF with 50 μM NP treatment and 20 ng mL−1 VEGF with 100 μM NP treatment groups, the numbers of nodes were 55.00 ± 16.00, 110.00 ± 13.00, 44.67 ± 14.36 and 25.33 ± 17.01, the numbers of junctions were 16.00 ± 4.58, 31.67 ± 5.51, 12.67 ± 5.03 and 8.00 ± 5.20, and the numbers of segments were 20.67 ± 7.37, 45.33 ± 8.08, 14.33 ± 6.03 and 7.67 ± 6.66. In addition, the relative tube length increased to 1.51 ± 0.08 after VEGF stimulation and was decreased by NP treatment to 0.97 ± 0.18 and 0.75 ± 0.15 at concentrations of 50 μM and 100 μM, respectively (Fig. 6).
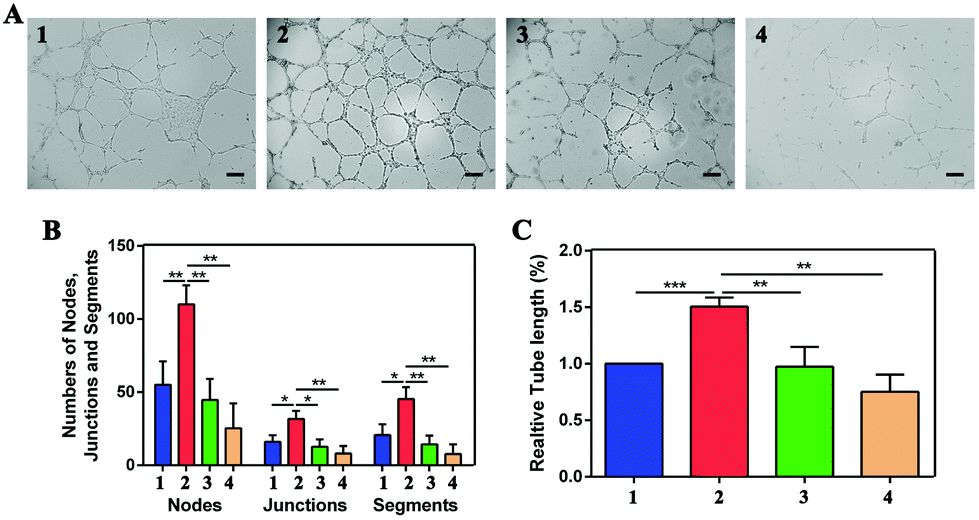 | ||
| Fig. 6 NPs attenuated VEGF-induced HUVEC tube formation. (A) The tube formation was stimulated by VEGF, and inhibited by the incubation of the NPs. Scale bar: 200 μm. (B) The numbers of nodes, junctions and segments, as well as relative tube length (%) (C) were quantified with ImageJ software. Group 1: control, Group 2: 20 ng mL−1 VEGF treatment, Group 3: 20 ng mL−1 VEGF with 50 μM NP treatment, and Group 4: 20 ng mL−1 VEGF with 100 μM NP treatment. | ||
VEGF is one of the strongest inductors of CNV, promoting the process of neovascularization.38 HUVECs stimulated by VEGF have been widely used as a neovascularization cell model.39,40 J. Siedlecki et al. found that HUVECs stimulated by VEGF would promote cell proliferation, migration and tube formation, which is in accordance with the progression of neovascularization.41 K. Hollanders et al. reported that inhibiting endotheliocyte proliferation and VEGF-mediated migration could reduce the formation of new blood vessels, consequently reducing the development of CNV.42 The present study indeed indicates that administration of NPs could effectively attenuate VEGF-induced cell proliferation, migration and tube formation, which provides evidence enabling us to perform our next experiments in vivo.
NPs decreased VEGF-induced expression of CD31 and VEGF
CD31 and VEGF are generally considered angiogenic markers. VEGF could be used as an angiogenesis-stimulating factor to increase the expression of angiogenic markers in HUVECs. Here, we learned from the immunofluorescence assay that the NPs could attenuate the increased expression of CD31 and VEGF after stimulation by the angiogenesis-stimulating factor, as shown in Fig. 7, which indicated that the NPs could effectively inhibit the expression of angiogenesis-stimulating factors.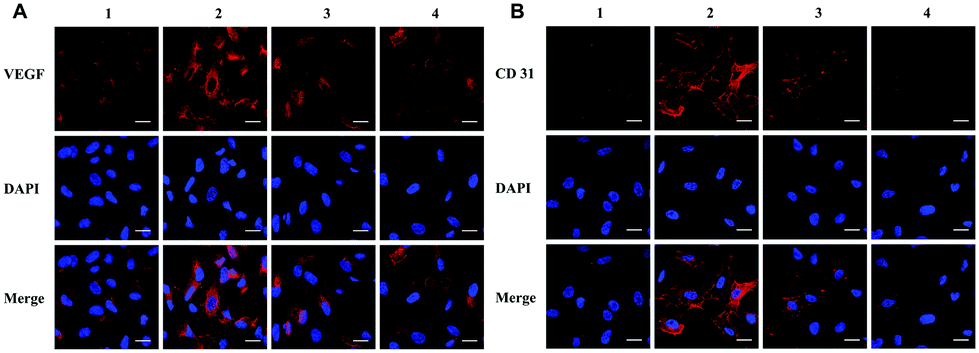 | ||
| Fig. 7 NPs inhibited VEGF-induced increased expression of CD31 and VEGF in HUVECs via immunofluorescence assay. (A) The expression and localization of VEGF in four groups. (B) The expression and localization of CD31 in four groups. Scale bar: 20 μM. Group 1: control, Group 2: 20 ng mL−1 VEGF treatment, Group 3: 20 ng mL−1 VEGF with 50 μM NP treatment, and Group 4: 20 ng mL−1 VEGF with 100 μM NP treatment. | ||
In vivo imaging
In vivo near-infrared reflectance images were captured at different time points. The results showed that NPs could facilitate targeted delivery to CNV lesions, and fluorescence was observed in the CNV areas, as shown in Fig. 8A. With increasing time points, the fluorescence gradually decreased and remained at 3 m. The results showed that the NPs could remain in the CNV areas and work for a long time.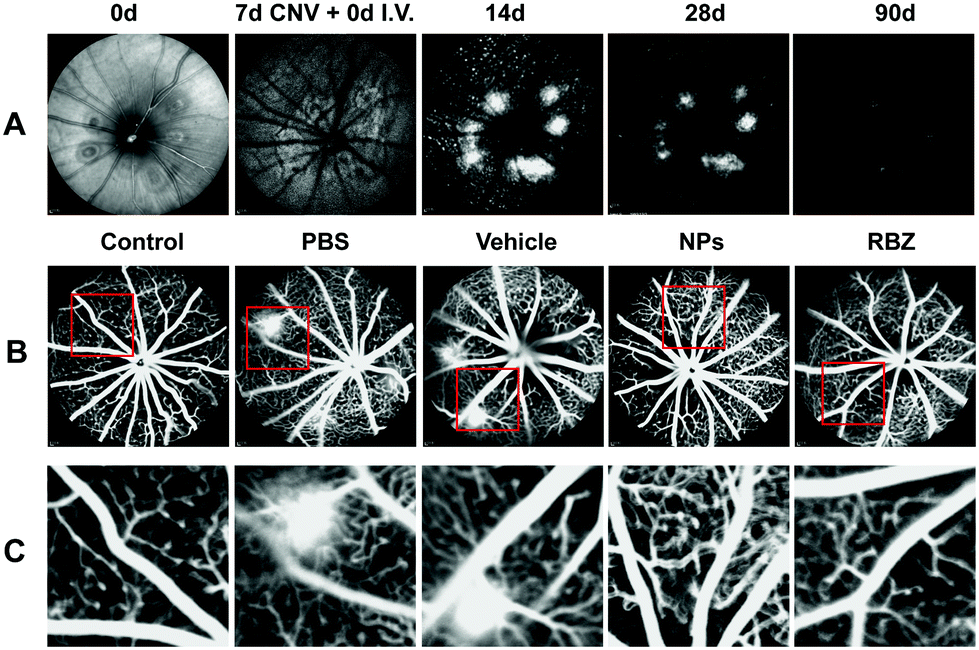 | ||
| Fig. 8 In vivo near-infrared reflectance and fluorescence imaging of the ocular fundus of the rodent eye was performed in laser-induced CNV models. (A) In vivo near-infrared reflectance images were captured at different time points including 0 d and 7 d after laser administration, and 14 d, 28 d, and 90 d after intravenous injection of NPs. (B) FFA was performed to observe the CNV lesion. (C) Amplification of the fluorescein leakage in five groups after intravenous injection of NPs for 28 d. | ||
Intravenous injection of anti-VEGF complexes attenuated neovascularization in mouse models of CNV
The CNV lesion was characterized by fluorescence due to the area of neovascularization. The sizes of the CNV fluorescent halos in the negative group and vehicle-treated group showed similar fluorescein leakage, presenting no apparent differences. In contrast, a reduction in the leakage area of the CNV was observed in the intravenous injection of the NP group and the intravitreal injection of the anti-VEGF group (Fig. 8B and C).To further confirm the efficacious treatment of NPs on the CNV area of the mouse model, we prepared RPE-choroid-sclera flat mounts by perfusion with FITC-dextran and further performed IB4 staining to compare and quantitatively measure the CNV areas in different groups. Based on FITC-dextran and IB4 staining, the mice with CNV with only intravenous injection of PBS showed a lesion area of 12923.18 ± 3325.11 μm2. The vehicle-treated group exhibited neovascularization areas that were similar to those of the PBS-treated group (13106.10 ± 3660.18 μm2). We demonstrated that the areas of the CNV lesions in the NP-treated group (2403.31 ± 320.43 μm2) were significantly decreased compared to those in the negative control and vehicle-treated groups (**p < 0.01). In addition, the mice that received a single intravitreal anti-VEGF injection were set as the positive control group. A reduction to 2529.60 ± 139.08 μm2 was also observed in eyes receiving the intravitreal anti-VEGF injection (**p < 0.01). All these data confirmed the FFA results and demonstrated that intravenous injection of NPs was an efficacious strategy in the regression of CNV lesions (Fig. 9).
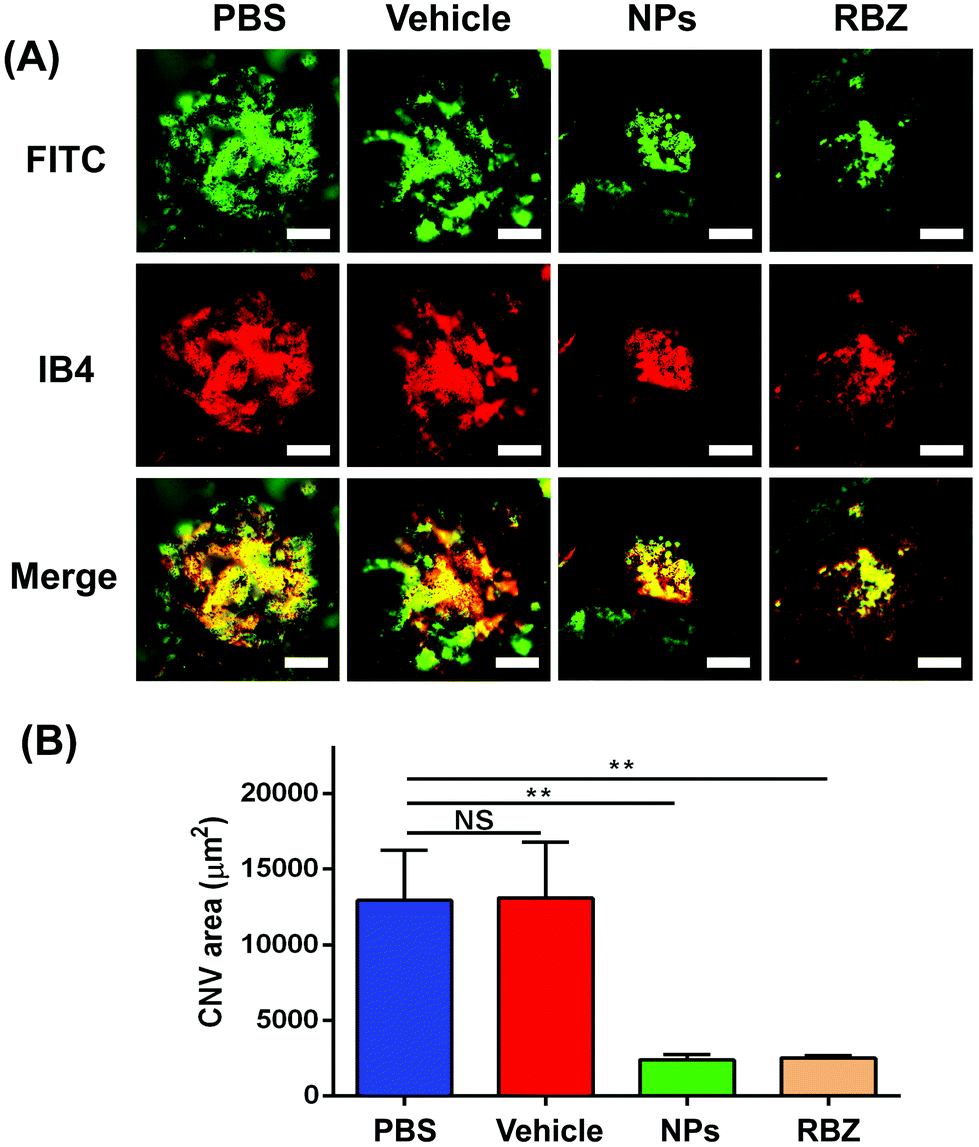 | ||
| Fig. 9 RPE-choroid-sclera flat mounts were prepared by perfusion with FITC-dextran and IB4 staining to measure CNV areas. (A) CNV areas with FITC-dextran and IB4 staining were captured with the fluorescence microscope in four groups. Scale bar: 50 μm. (B) CNV areas were quantified via ImageJ software. **P < 0.01, NS: no significance. | ||
Furthermore, the CNV areas were stained and measured by HE assays. As shown in Fig. 10, CNV disrupted Bruch's membrane and formed in the retinal neuroepithelial layer (RNL), which damaged the normal structure of the retina and induced depigmentation. The CNV areas in the NP-treated group were significantly decreased compared to those in the vehicle-treated group, which indicated the efficacy in arresting CNV formation. Meanwhile, the results of CD31 and VEGF immunofluorescence staining also showed that NPs could inhibit neovascularization via suppressing the expression of CD31 and VEGF (Fig. 11).
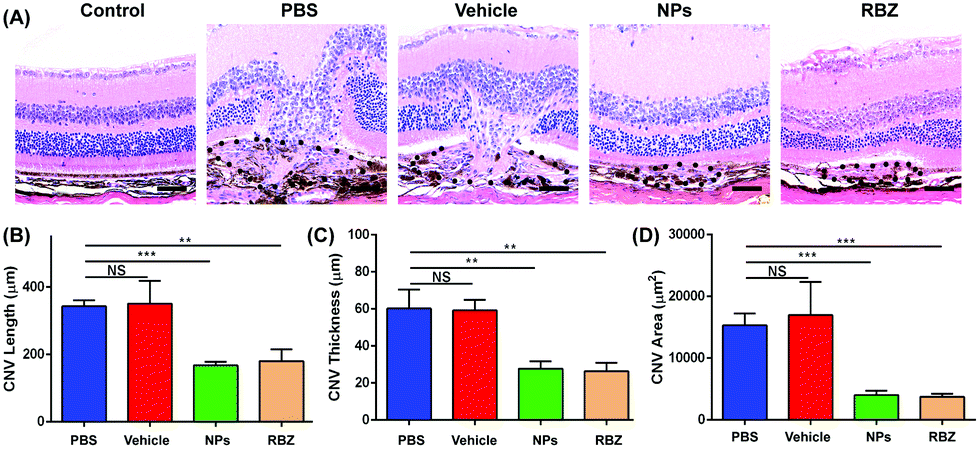 | ||
| Fig. 10 The CNV areas were stained and measured by HE assays. (A) The structures of the retina and choroid were observed via HE staining. The black spot shows the CNV areas. Scale bar: 50 μm. (B,C,D) The CNV length, thickness and areas were quantified in HE staining via ImageJ software. Column 1: intravenous injection of PBS, Column 2: intravenous injection of S-PEG, Column 3: intravenous injection of NPs, and Column 4: intravitreal injection of RBZ. **P < 0.01, ***P < 0.001, NS: no significance. | ||
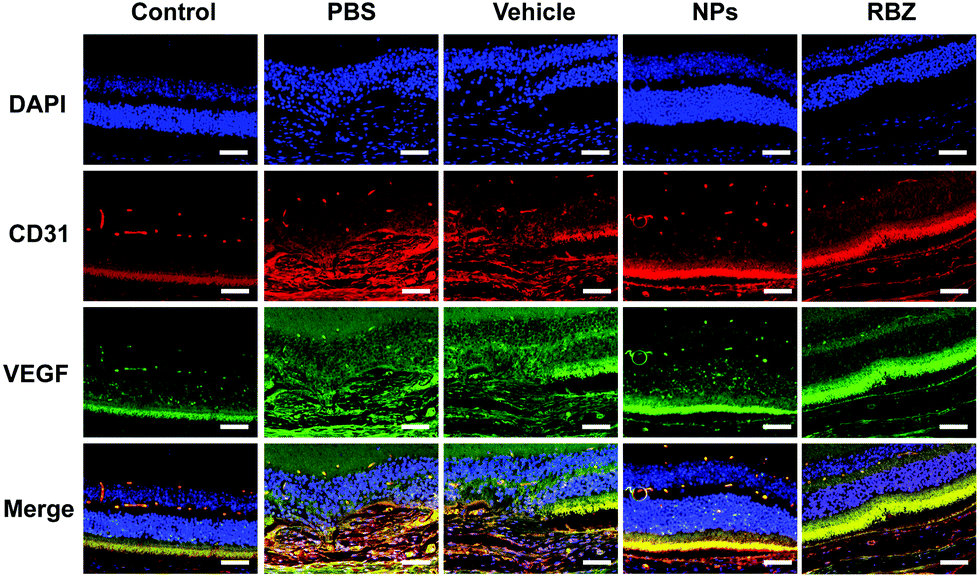 | ||
| Fig. 11 Double immunostaining with endothelial marker CD31 and VEGF in the retina/choroid cryosections was performed in five groups and images were recorded with the fluorescence microscope. Scale bar = 50 μm. | ||
Another pivotal key point is to consider how to deliver NPs to CNV lesions via intravenous administration. M. Friedlander et al. and J. Luna et al. reported that integrin receptors, in particular αvβ3, were overexpressed in the neovascular tissues of AMD patients, which provided evidence that αvβ3 could act as a potential target.28,29
S. R. Singh et al. engineered nanoparticles bound with RGD peptide to perform targeted delivery of the anti-VEGF intraceptor plasmid to CNV lesions. The CNV areas were significantly smaller after treatment with functionalized nanoparticles, which indicated that the RGD peptide could enable targeted delivery.43 Thus, the nanoparticles in our study also contained an engineered RGD peptide to achieve targeted delivery via intravenous administration.
NPs had no adverse effects on the body weight, hemolysis, serological indexes, structures of organs and apoptosis in vivo
The changes in body weights reflected the growth curve of the mice. As shown in Fig. S4,† there were no significant changes in body weights in these five groups at different time points. Since the nanoparticles would circulate around the entire body after intravenous injection, it was necessary to monitor the function of organs. For this purpose, several biochemical indexes including the liver, renal, lipid and electrolyte levels were evaluated. In Fig. 12A–C, the levels of ALT, AST, Cr, T-CHO, TG, K, Ca and Cl showed no apparent differences in these groups. Desirable liver enzyme profiles, renal index, lipid levels and electrolyte levels were observed in our study, and all these indexes are generally applied in the assessment of nanoparticle administration in vivo.44–46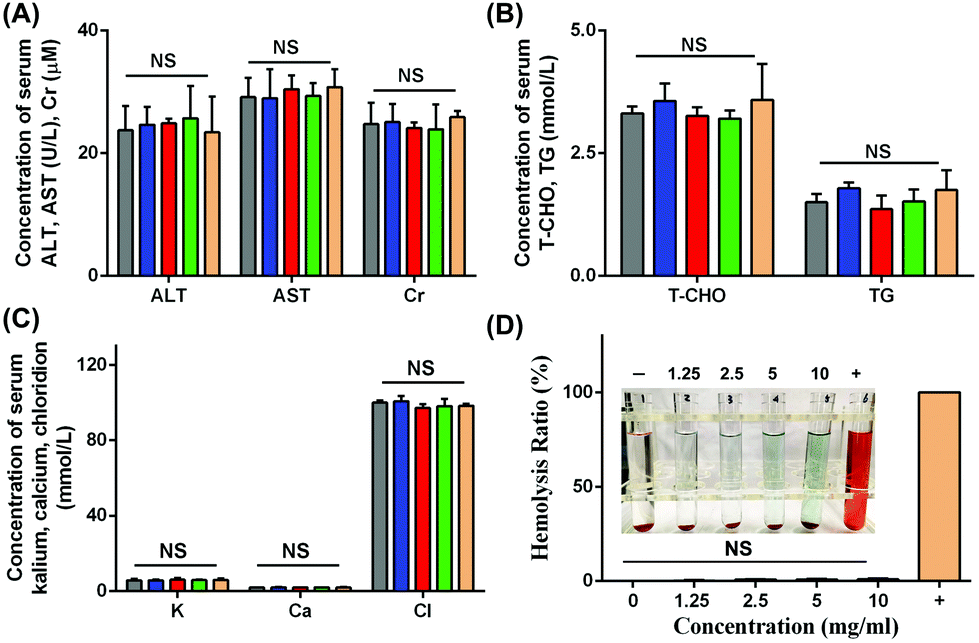 | ||
| Fig. 12 In vivo biosecurity was measured via several serum assays and hemolysis assay. (A) Measurement of liver and renal indexes including ALT, AST and Cr. (B) Measurement of lipid indexes including T-CHO and TG. (C) Measurement of electrolyte levels including K, Ca and Cl. (D) hemolysis rates of different concentrations of NPs were calculated via hemolysis assay. | ||
As shown in Fig. 12D, the hemolysis rates of all concentrations of NPs from 1.25 mg mL−1 to 10 mg mL−1 showed negative values in our study, and the hemolysis rate at 10 mg mL−1 was 0.95 ± 0.52%, which indicated good blood compatibility in intravenous application. In general, the biosafety of NPs should be taken into consideration due to the intravenous injection administration. Blood compatibility plays a crucial role in materials administered by vein application. A hemolysis ratio lower than 10% was regarded as allowable and suitable for intravenous injection.47 In the present study, the hemolysis ratio was low, allowing the intravenous injection.
Since the drug would circulate throughout the whole body blood, the structures and functions of different organs were investigated to evaluate the biosafety in vivo, and altogether, the main organs including the heart, spleen, kidneys, liver, brain and lungs were included. HE staining is a widely used visualization method to assess the structural damage.48 HE staining and TUNEL assay were performed on the main organs of the mice to observe any adverse effects on the organs. As shown in Fig. 13 and Fig. S5,† no histomorphological changes and apoptotic cells were observed in the heart, liver, spleen, lungs, kidneys and brain after intravenous injection of particles in vivo, which indicated biosafety in intravenous application. The results showed a normal cellular and tissue morphology, which confirmed that no damage to the organ structure occurred. The results of TUNEL staining also showed that no apoptosis in organs was found after intravenous injection, indicating the biosafety in systematic applications.
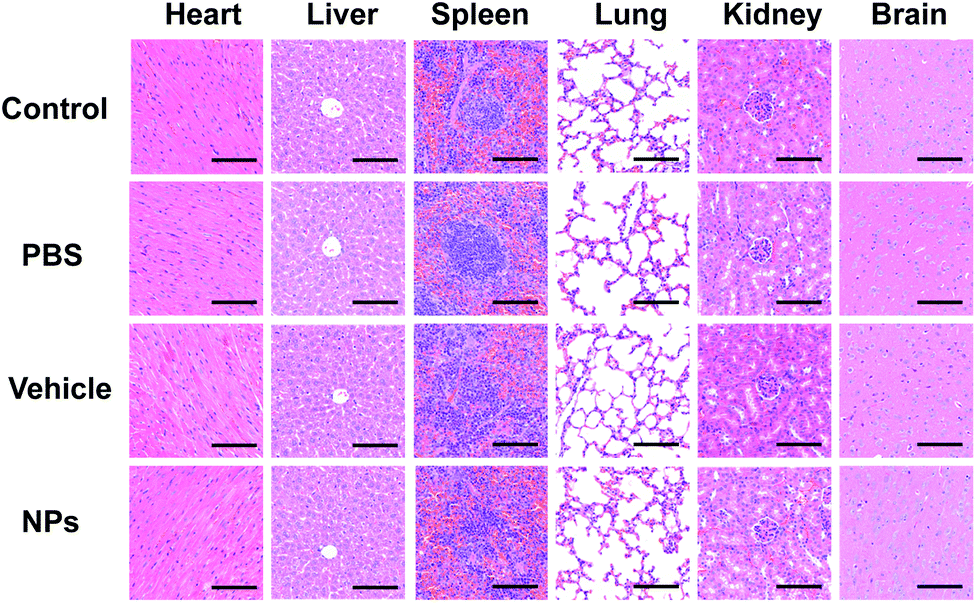 | ||
| Fig. 13 In vivo biosecurity was measured via HE staining of different organs, including the heart, liver, spleen, lungs, kidneys and brain. Scale bar: 100 μm. | ||
NPs did not remain in the main organs in vivo
As shown in Fig. 14, a strong luminous signal was observed in the liver and kidney areas after intravenous injection of NPs in bioluminescence mode. The afterglow signal was gradually reduced when the time increased from 4 h to 24 h. A weak signal was observed in the organs at 24 h, which indicated that the NPs circulated throughout the entire body and did not remain in the main organs for a long time.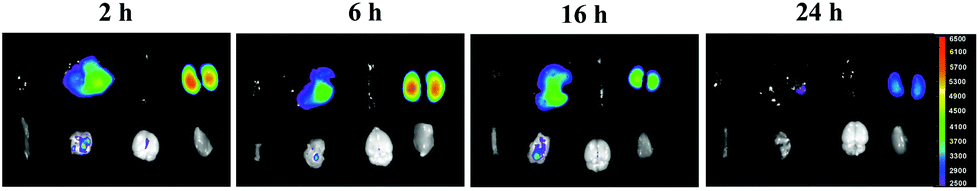 | ||
| Fig. 14 In vivo afterglow imaging for different organs after NP injection was performed via imaging of small animals in vivo at 2 h, 6 h, 16 h and 24 h time points. First line (from left to right): heart, liver, spleen, and kidneys. Second line (from left to right): small intestine, lungs, brain and muscle. | ||
In this study, we used the widely accepted laser-induced CNV mouse model to complete our experiments.49 Laser-induced CNV is a VEGF-dependent process, as demonstrated in previous studies in which the process could be inhibited by anti-VEGF agents. We observed increased expression of VEGF and CD31 in VEGF-induced HUVECs and CNV tissues, which could be inhibited by NP treatment. In addition, in vivo studies demonstrated that the NP group presented a lower CNV area in RPE-choroid-sclera flat mounts and HE staining, as well as less fluorescein leakage in FFA. As anti-VEGF therapy is an important treatment modality in this pathology, our study aimed to improve the anti-VEGF delivery system.
Conclusions
In the present study, S-PEG modified by RGD was designed as a powerful carrier to deliver anti-VEGF agents to CNV areas. The developed NPs had no cellular toxicity and did not induce cell apoptosis or DNA damage in the studied concentration range in vitro. In addition, the NPs could attenuate VEGF-induced HUVEC migration and tube formation in vitro. In an in vivo study, the low hemolysis ratio confirmed the application of intravenous injection, and meanwhile, the relatively unaffected functions of the liver, and kidneys, lipid metabolism and electrolytes as well as the various organ structures ensured the biosafety of NPs for further applications. More importantly, anti-VEGF agents could be delivered to the CNV areas and they remained there for a long time to maintain the efficacy of the anti-VEGF effect. The CNV areas showed a significant decrease as observed by various experiments. Overall, the smart NPs, which have good cellular compatibility, hold great potential for drug delivery in CNV treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored by Shanghai Tenth People's Hospital and Tonji university. W. T. Cai and Q. J. Chen synthesized the nanoparticles and wrote the manuscript. T. Y. Shen and W. N. Hu finished the in vitro experiments. W. T. Cai and Q. Yang performed the in vivo experiments. P. Zhao and J. Yu conducted the experiments. This study was financially supported by the Natural Science Foundation of Shanghai (No. 19ZR1439500), the National Natural Science Foundation of China (No. 21801189), Fundamental Research Funds for the Central Universities (No. 22120180509), Public Welfare Projects of Ningbo (No. 2019C50051) and Medical and Health Research Project of Zhejiang (No. 2019KY643).Notes and references
- G. Adamus, Autoimmun. Rev., 2017, 16, 231–236 CrossRef CAS PubMed.
- G. H. S. Buitendijk, E. Rochtchina, C. Myers, C. M. van Duijn, K. E. Lee, B. E. K. Klein, S. M. Meuer, P. de Jong, E. G. Holliday, A. G. Tan, A. G. Uitterlinden, T. S. Sivakumaran, J. Attia, A. Hofman, P. Mitchell, J. R. Vingerling, S. K. Iyengar, A. Janssens, J. J. Wang, R. Klein and C. C. W. Klaver, Ophthalmology, 2013, 120, 2644–2655 CrossRef PubMed.
- L. S. Lim, P. Mitchell, J. M. Seddon, F. G. Holz and T. Y. Wong, Lancet, 2012, 379, 1728–1738 CrossRef.
- J. Cui, D. Sun, H. Lu, R. Dai, L. Xing, H. Dong, L. Wang, D. Wei, B. Jiang, Y. Jiao, M. M. Jablonski, S. Charles, W. Gu and H. Chen, Eye, 2018, 32, 391–399 CrossRef CAS PubMed.
- M. L. Durand, Clin. Microbiol. Infect., 2013, 19, 227–234 CrossRef CAS PubMed.
- M. M. Sachdeva, A. Moshiri, H. A. Leder and A. W. Scott, J. Ophthalmic Inflammation Infect., 2016, 6, 2 CrossRef PubMed.
- G. Abedi, R. A. Adelman and S. Salim, Semin. Ophthalmol., 2013, 28, 126–130 CrossRef PubMed.
- R. S. Singh and J. E. Kim, Drugs Aging, 2012, 29, 949–956 CrossRef CAS PubMed.
- N. Ferrara, L. Damico, N. Shams, H. Lowman and R. Kim, Retina, 2006, 26, 859–870 CrossRef PubMed.
- Z. Yan, H. Shi, R. Zhu, L. Li, B. Qin, L. Kang, H. Chen and H. Guan, Mol. Vision, 2018, 24, 83–93 CAS.
- F. de Cogan, L. J. Hill, A. Lynch, P. J. Morgan-Warren, J. Lechner, M. R. Berwick, A. F. A. Peacock, M. Chen, R. A. H. Scott, H. Xu and A. Logan, Invest. Ophthalmol. Visual Sci., 2017, 58, 2578–2590 CrossRef CAS PubMed.
- H. Zhang, J. Wang, M. Hu, B. C. Li, H. Li, T. T. Chen, K. F. Ren, J. Ji, Q. M. Jing and G. S. Fu, Biomater. Sci., 2019, 7, 5177–5186 RSC.
- Y. Yang, W. Zhu, L. Cheng, R. Cai, X. Yi, H. He, X. Pan, L. Yang, K. Yang, Z. Liu, W. Tan and M. Chen, Biomaterials, 2020, 246, 119971 CrossRef CAS PubMed.
- C. Sun, K. Du, C. Fang, N. Bhattarai, O. Veiseh, F. Kievit, Z. Stephen, D. Lee, R. G. Ellenbogen, B. Ratner and M. Zhang, ACS Nano, 2010, 4, 2402–2410 CrossRef CAS PubMed.
- X. Huang, W. Liao, G. Zhang, S. Kang and C. Y. Zhang, Int. J. Nanomed., 2017, 12, 2215–2226 CrossRef CAS PubMed.
- Y. Chen, M. Su, Y. Li, J. Gao, C. Zhang, Z. Cao, J. Zhou, J. Liu and Z. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 30519–30535 CrossRef CAS PubMed.
- H. Chen and S. He, Mol. Pharm., 2015, 12, 1885–1892 CrossRef CAS PubMed.
- J. Yang, X. Hao, Q. Li, M. Akpanyung, A. Nejjari, A. L. Neve, X. Ren, J. Guo, Y. Feng, C. Shi and W. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 4485–4497 CrossRef CAS PubMed.
- J. Kim, H. Kim and W. J. Kim, Small, 2016, 12, 1184–1192 CrossRef CAS PubMed.
- Y. Chen, Y. Li, J. Gao, Z. Cao, Q. Jiang, J. Liu and Z. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 490–501 CrossRef CAS PubMed.
- Q. Chen, H. Wang, H. Liu, S. Wen, C. Peng, M. Shen, G. Zhang and X. Shi, Anal. Chem., 2015, 87, 3949–3956 CrossRef CAS PubMed.
- X. Chen, X. Yao, C. Wang, L. Chen and X. Chen, Biomater. Sci., 2015, 3, 870–878 RSC.
- Q. Chen, T. Shi, F. Han, Z. Li, C. Lin and P. Zhao, Sci. Rep., 2017, 7, 8493 CrossRef PubMed.
- Q. Chen, X. Deng and Z. An, Macromol. Rapid Commun., 2014, 35, 1148–1152 CrossRef CAS PubMed.
- W. Q. Wang, F. H. Wang, W. X. Qin, H. Y. Liu, B. Lu, C. Chung, J. Zhu, Q. Gu, W. Shi, C. Wen, F. Wu, K. Zhang and X. D. Sun, Mol. Pharm., 2016, 13, 2881–2890 CrossRef CAS PubMed.
- L. W. Contois, A. Akalu, J. M. Caron, E. Tweedie, A. Cretu, T. Henderson, L. Liaw, R. Friesel, C. Vary and P. C. Brooks, Angiogenesis, 2015, 18, 31–46 CrossRef CAS PubMed.
- J. J. Bi and L. Yi, J. Huazhong Univ. Sci. Technol., Med. Sci., 2014, 34, 299–305 CrossRef CAS PubMed.
- M. Friedlander, C. L. Theesfeld, M. Sugita, M. Fruttiger, M. A. Thomas, S. Chang and D. A. Cheresh, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9764–9769 CrossRef CAS PubMed.
- J. Luna, T. Tobe, S. A. Mousa, T. M. Reilly and P. A. Campochiaro, Lab. Invest., 1996, 75, 563–573 CAS.
- A. J. Schraa, R. J. Kok, H. E. Moorlag, E. J. Bos, J. H. Proost, D. K. Meijer, L. F. de Leij and G. Molema, Int. J. Cancer, 2002, 102, 469–475 CrossRef CAS PubMed.
- J. L. Wang, Y. Xi, Y. L. Liu, Z. H. Wang and Q. Zhang, Invest. Ophthalmol. Visual Sci., 2013, 54, 7983–7989 CrossRef CAS PubMed.
- R. B. Rush and S. W. Rush, J. Ophthalmol., 2015, 2015, 642624 Search PubMed.
- J. Meyer, A. Cunea, D. Sonntag-Bensch, P. Welker, K. Licha, F. G. Holz and S. Schmitz-Valckenberg, Invest. Ophthalmol. Visual Sci., 2014, 55, 6204–6212 CrossRef CAS PubMed.
- N. Elsaid, T. L. Jackson, Z. Elsaid, A. Alqathama and S. Somavarapu, Mol. Pharm., 2016, 13, 2923–2940 CrossRef CAS PubMed.
- S. Grisanti and A. Tura, Ophthalmologe, 2010, 107, 1123–1132 CrossRef CAS PubMed.
- J. Matuszak, J. Baumgartner, J. Zaloga, M. Juenet, A. E. da Silva, D. Franke, G. Almer, I. Texier, D. Faivre, J. M. Metselaar, F. P. Navarro, C. Chauvierre, R. Prassl, L. Dezsi, R. Urbanics, C. Alexiou, H. Mangge, J. Szebeni, D. Letourneur and I. Cicha, Nanomedicine, 2016, 11, 597–616 CrossRef CAS PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS.
- M. Kliffen, H. S. Sharma, C. M. Mooy, S. Kerkvliet and P. T. de Jong, Br. J. Ophthalmol., 1997, 81, 154–162 CrossRef CAS PubMed.
- A. B. Cerezo, R. Hornedo-Ortega, M. A. Alvarez-Fernandez, A. M. Troncoso and M. C. Garcia-Parrilla, Nutrients, 2017, 9, 249 CrossRef PubMed.
- S. L. Park, S. Y. Won, J. H. Song, S. Y. Lee, W. J. Kim and S. K. Moon, Am. J. Chin. Med., 2016, 44, 61–76 CrossRef CAS PubMed.
- J. Siedlecki, C. Wertheimer, A. Wolf, R. Liegl, C. Priglinger, S. Priglinger and K. Eibl-Lindner, Graefes Arch. Clin. Exp. Ophthalmol., 2017, 255, 963–972 CrossRef CAS PubMed.
- K. Hollanders, T. Van Bergen, N. Kindt, K. Castermans, D. Leysen, E. Vandewalle, L. Moons and I. Stalmans, Invest. Ophthalmol. Visual Sci., 2015, 56, 1335–1348 CrossRef CAS PubMed.
- S. R. Singh, H. E. Grossniklaus, S. J. Kang, H. F. Edelhauser, B. K. Ambati and U. B. Kompella, Gene Ther., 2009, 16, 645–659 CrossRef CAS PubMed.
- O. Oyeyemi, O. Morenkeji, F. Afolayan, K. Dauda, Z. Busari, J. Meena and A. Panda, Front. Pharmacol., 2018, 9, 562 CrossRef.
- M. A. K. Abdelhalim, H. A. Qaid, Y. Al-Mohy and M. S. Al-Ayed, Int. J. Nanomed., 2018, 13, 7765–7770 CrossRef CAS PubMed.
- L. Zhang, J. Chang, Y. Zhao, H. Xu, T. Wang, Q. Li, L. Xing, J. Huang, Y. Wang and Q. Liang, Int. J. Nanomed., 2018, 13, 2051–2064 CrossRef CAS PubMed.
- H. Xu, D. Yang, C. Cai, J. Gou, Y. Zhang, L. Wang, H. Zhong and X. Tang, Acta Biomater., 2015, 16, 156–168 CrossRef CAS PubMed.
- V. P. Nguyen, Y. Li, W. Qian, B. Liu, C. Tian, W. Zhang, Z. Huang, A. Ponduri, M. Tarnowski, X. Wang and Y. M. Paulus, Sci. Rep., 2019, 9, 5945 CrossRef PubMed.
- J. A. Lavine, M. Farnoodian, S. Wang, S. R. Darjatmoko, L. S. Wright, D. M. Gamm, M. S. Ip, C. M. Sorenson and N. Sheibani, Invest. Ophthalmol. Visual Sci., 2017, 58, 299–308 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm02086a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |