
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycosylated cyclophellitol-derived activity-based probes and inhibitors for cellulases†
Casper
de Boer‡
 a,
Nicholas G. S.
McGregor‡
b,
Evert
Peterse
a,
Sybrin P.
Schröder
a,
Bogdan I.
Florea
a,
Jianbing
Jiang
a,
Jos
Reijngoud
c,
Arthur F. J.
Ram
c,
Gilles P.
van Wezel
c,
Gijsbert A.
van der Marel
a,
Jeroen D. C.
Codée
a,
Herman S.
Overkleeft
*a and
Gideon J.
Davies
*b
a,
Nicholas G. S.
McGregor‡
b,
Evert
Peterse
a,
Sybrin P.
Schröder
a,
Bogdan I.
Florea
a,
Jianbing
Jiang
a,
Jos
Reijngoud
c,
Arthur F. J.
Ram
c,
Gilles P.
van Wezel
c,
Gijsbert A.
van der Marel
a,
Jeroen D. C.
Codée
a,
Herman S.
Overkleeft
*a and
Gideon J.
Davies
*b
aLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2300 RA Leiden, The Netherlands. E-mail: h.s.overkleeft@chem.leidenuniv.nl
bYork Structural Biology Laboratory, Department of Chemistry, The University of York, Heslington, York YO10 5DD, UK. E-mail: Gideon.davies@york.ac.uk
cInstitute of Biology Leiden, Leiden University, Sylviusweg 72, 2333 BE Leiden, The Netherlands
First published on 28th July 2020
Abstract
Cellulases and related β-1,4-glucanases are essential components of lignocellulose-degrading enzyme mixtures. The detection of β-1,4-glucanase activity typically relies on monitoring the breakdown of purified lignocellulose-derived substrates or synthetic chromogenic substrates, limiting the activities which can be detected and complicating the tracing of activity back to specific components within complex enzyme mixtures. As a tool for the rapid detection and identification of β-1,4-glucanases, a series of glycosylated cyclophellitol inhibitors mimicking β-1,4-glucan oligosaccharides have been synthesised. These compounds are highly efficient inhibitors of HiCel7B, a well-known GH7 endo-β-1,4-glucanase. An elaborated activity-based probe facilitated the direct detection and identification of β-1,4-glucanases within a complex fungal secretome without any detectable cross-reactivity with β-D-glucosidases. These probes and inhibitors add valuable new capacity to the growing toolbox of cyclophellitol-derived probes for the activity-based profiling of biomass-degrading enzymes.
Introduction
Activity-based protein profiling (ABPP) is a powerful technique used to detect and identify active enzymes using modified covalent inhibitors. Following early successes in the detection and identification of proteases,1 this technique has been extended to glycoside hydrolases using a variety of strategies,2,3 including labelled cyclophellitol derivatives.4Though often considered synonymous with cellulases, β-1,4-glucanases are enzymes which recognize β-1,4-linked glucan chains which are characteristic of both the cellulosic and hemicellulosic (i.e. mixed-linkage glucans, xyloglucans, and glucomannans) fractions of plant biomass.5 The catalytic actions of a variety of retaining β-1,4-glucanases contribute to the breakdown of lignocellulosic polysaccharides.6,7 The efficient and specific profiling of β-1,4-glucanases is thus a valuable tool in the study of biomass-degrading organisms.
Efforts have been made to profile β-1,4-glycanases using different “warhead” chemistries. Activity-based probes (ABPs) based on a 2,4-dinitrophenyl 2-deoxy-2-fluoro-β-xylobioside/cellobioside modified with an affinity tag at the 4’ position proved effective probes for retaining β-D-glucanases and β-xylanases with particularly good specificity.2,8,9 However, the slow hydrolysis of the enzyme-probe complex and the weak initial binding of these probes necessitated the use of high probe concentration (∼0.5–1 mM) and time-limited labelling for ABPP. Later experiments using difluoromethylphenyl glycosides and N-haloacetyl glycosylamines demonstrated a unique capacity to label inverting glycoside hydrolases, but suffered from significant non-specific labelling.3
Cyclophellitol is an inhibitor of β-D-glucosidases originally isolated from the Phellinus mushroom.10 This cyclitol is an isostere of a glucoside where the acetal group is replaced by an epoxide. Taking advantage of the catalytic machinery of a retaining glycoside hydrolase, this epoxide undergoes an acid-catalysed ring opening addition to form a non-hydrolysable ester in place of the normal glycosyl-enzyme intermediate, irreversibly inactivating the enzyme.11
ABPs built around synthetic cyclitols, having configurations which target α- and β-D-glucosidases,12–14 β-D-glucuronidases,15 β-D-xylosidases,16 α- and β-D-galactosidases,17,18 and α-L-arabinofuranosidases,19 among others, have consistently been shown to covalently modify the catalytic nucleophiles of cognate retaining glycosidases. These cyclophellitol-derived ABPs generally bind with good specificity, high affinity, and complete irreversibility.
Recent work has shown that cyclophellitol derivatives can be glycosylated, enabling the development of inhibitors and probes which react specifically with endo-glycanases.16 This was first demonstrated with the development of an inhibitor and a collection of ABPs for β-1,4-xylanases. Being built around a xylobiose core with an alkylated aziridine warhead, these probes proved to be potent covalent inhibitors of GH10 β-1,4-xylanases, but showed cross-reactivity with β-D-xylosidases when applied to the direct detection of β-xylanases within fungal secretomes. This cross-reactivity was traced back to the internal hydrolysis of the probe by the action of β-D-xylosidases.
Building on this understanding, here we report the development of cyclophellitol-derived ABPs designed to target β-1,4-glucanases, some of the most abundant glycoside hydrolases in nature. To detect and profile these enzymes, a collection of 4-O substituted (carbohydrate numbering) cyclophellitols have been synthesised and tested for their ability to covalently modify HiCel7B, a well-known endo-β-1,4-glucanase. Through biochemical, structural, and mass spectrometric analyses, we have identified a potent substrate-mimicking probe architecture which shows resistance to hydrolysis by exo-glucosidase and endo-glucanase activities within a fungal secretome.
Experimental
Synthesis of β-D-glucanase inhibitors and probe
Chemical synthesis details and compound analysis is reported in the ESI.†Testing β-D-glucanase inhibitors and probes
HiCel7B was a kind gift from Martin Schülein (sadly now deceased) at Novozymes A/S (Lyngby, Denmark). The pH-activity profile of HiCel7B acting on 4-methylumbelliferyl β-D-cellobioside (4MU-GG) was measured by combining 5 μL of 1 μg mL−1 HiCel7B in 200 mM 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:7 succinate–phosphate–glycine (SPG) buffer prepared at various pH values with 45 μL of 0.1 mM substrate in quadruplicate. The reactions were incubated at 25 °C for 30 minutes prior to the addition of 5 μL of 1 M Na2CO3, transfer of 50 μL to a black 384-well plate and fluorescence measurement (λex = 360 nm, λem = 450 nm). Rates were determined using a 4-methylumbelliferone calibration series prepared in 0.1 M Na2CO3.
:7:7 succinate–phosphate–glycine (SPG) buffer prepared at various pH values with 45 μL of 0.1 mM substrate in quadruplicate. The reactions were incubated at 25 °C for 30 minutes prior to the addition of 5 μL of 1 M Na2CO3, transfer of 50 μL to a black 384-well plate and fluorescence measurement (λex = 360 nm, λem = 450 nm). Rates were determined using a 4-methylumbelliferone calibration series prepared in 0.1 M Na2CO3.
Intact MS of HiCel7B bound to different inhibitors was performed according to McGregor et al.19 Briefly, enzyme was diluted to 0.1 mg mL−1 (∼2.2 μM) in 20 mM sodium phosphate buffer pH 7. Compounds 1, 5, or 13 were added to a final concentration of 5 μM and incubated at 25 °C. Samples taken at 1 hour were diluted with 4 volumes of 1% formic acid, 10% acetonitrile and analysed. Additional experiments with 5 were performed using different concentrations of inhibitor and enzyme as indicated in the text.
Inhibition kinetics for compounds 1, 5, and 13 acting on HiCel7B were measured at 25 °C using 4MU-GG following a method described previously.19 Briefly, enzyme was diluted in 50 mM sodium phosphate buffer pH 7. Substrate was dissolved in DMSO to give a 10 mM stock which was diluted with ultrapure water. Inhibitors were dissolved in and diluted with ultrapure water with the exception of 13 which was dissolved in DMSO to give a 5 mM stock which was diluted with ultrapure water. The enzyme and substrate concentrations used in the continuous inhibition assays were 10 ng mL−1 (∼220 pM) and 50 μM, respectively. The KM value for the interaction of HiCel7B with 4MU-GG under the assay conditions (corrected for inner filter effect) was measured to be 76 μM (Fig. S1, ESI†) and this was used as a correction factor to determine the KI values in Table 1 from the apparent KI determined from fitting of kappvs. [inhibitor] curves.
| Compound | HiCel7B | ||
|---|---|---|---|
| K I (μM) | k inact (min−1) | k inact/KI (s−1 M−1) | |
| 1 | 31 ± 4 | 0.84 ± 0.08 | 450 |
| 5 | 1.9 ± 0.3 | 0.35 ± 0.03 | 3100 |
| 13 | 3.9 ± 0.3 | 0.50 ± 0.03 | 2100 |
Enzyme crystallisation, diffraction, and structure solution
Deglycosylated HiCel7B was desalted into 20 mM pH 8 Tris–HCl buffer and concentrated to 12 mg mL−1 using a 30 kDa MWCO centricon. Building on previous reports,20,21 crystallisation conditions were re-screened using the PACT Suite and AmSO4 Suite (Qiagen) crystallisation screens. High quality tetragonal bipyramidal crystals grew consistently at 20 °C from a 2:1 mixture of protein solution:well solution, where well solution was 0.15 M sodium citrate, 0.8 M ammonium sulfate, 1 M lithium sulfate (Fig. S2, ESI†). Crystal soaks were performed in a solution composed of 0.1 mM ligand in mother liquor for 5 hours at 20 °C prior to transfer into mother liquor supplemented with 20% glycerol and cryo-cooling in LN2.
Crystals were diffracted at Diamond Light Source (Harwell, UK) on beamline I03 at a wavelength of 0.9762 Å and automatically processed using the Xia222 pipeline with 3dii. Computation was carried out using programs from the CCP4 suite23 unless otherwise stated. All crystal structure figures were generated using Pymol (Schrodinger). Data collection and processing statistics for all structures are given in Table S1 (ESI†).
Data for HiCel7B bound to compound 1 were collected to 1.88 Å. Data were also collected out to 1.2 Å in a higher space group (P42212) following a soak with 13, though the structure was found to be unliganded. The structure of 1-bound HiCel7B was solved in the P4122 space group by molecular replacement using Phaser24 with the known structure (PDBID: 2A39) as the search model. Ligand 1 was built using the existing restrains for β-D-glucose (BGC) and cyclophellitol (YLL) with Coot,25 and structures were refined by alternating rounds of manual model building and density refinement using Coot and REFMAC526 respectively.
In-gel fluorescence
The pH-labelling profile of HiCel7B reacting with the probe was measured by combining 10 μL of 1 μg mL−1 HiCel7B in 200 mM SPG buffer prepared at various pH values with 10 μL of 10 μM 14 in quadruplicate. The reactions were incubated at 25 °C for 10 minutes prior to the addition of 8 μL of 4× SDS-PAGE loading dye and heating to 95 °C for 2 minutes. 10 μL of each reaction was separated on a 10% SDS-PAGE gel prior imaging using the Cy5 laser/filter settings on a Typhoon 5 scanner (GE Healthcare). Bands were integrated using ImageQuant (GE Healthcare).Secretome staining was performed using two aliquots of 20 μL xylan-grown Aspergillus niger secretome (day 4 samples prepared as described previously16). To each was added 5 μL of 0.5 M pH 5 McIlvane buffer. To one was then added 5 μL of 60 μM 14 and to the other was added 5 μL of 60 μM 19 (10 μM final ABP concentration). These were incubated for 30 minutes at 37 °C. The reactions were then split in two and one half (15 μL) was diluted with 5 μL of water. The other half (15 μL) of the reaction with 19 was then supplemented with 5 μL of 40 μM 14 and the other half of the reaction with 14 was supplemented with 5 μL of 40 μM 19 (10 μM final ABP concentration). The reactions were incubated for a further 30 minutes at 37 °C before being diluted with 8 μL of 4× SDS-PAGE loading dye, separated on a 4–20% SDS-PAGE gel (Bio-Rad) and imaged for fluorescence using the Cy2 and Cy5 laser/filter settings on a Typhoon 5 scanner (GE Healthcare).
Biotin–avidin enrichment proteomic analysis
5 aliquots of 1 mL of xylan-grown Aspergillus niger secretome were thawed from −80 °C, centrifuged at 12000 × g for 15 minutes to remove particulate and combined. 0.5 mL of this was then subsampled into 9 separate Lo-Bind 2.0 mL tubes. To three tubes was added 55 μL of 1 mM compound 1 in ultrapure water, and to the rest 55 μL of ultrapure water. All samples were incubated for 1 hour at 37 °C prior to the addition of 60 μL of compound 15 in 10% DMSO to the samples treated with 1 and three of the samples not treated with 1. 10% DMSO was added to the remaining three samples. All of the samples were incubated for a further 2 hours at 37 °C. Proteins were denatured by heating to 95 °C for 5 minutes following the addition of 70 μL of 10× denaturing buffer (500 mM Na-HEPES, pH 7.5, 50 mM DTT, 5% SDS). Once cooled to RT, thiols were alkylated by the addition of 70 μL of 0.25 M iodoacetamide and incubation in the dark for 30 minutes. Samples were transferred to 5 mL Eppendorf tubes and proteins were precipitated by the addition of 3.2 mL of chilled acetone followed by incubation at −20 °C for 1 hour. Proteins were collected by centrifugation (14000 × g for 1 minute) and the supernatant was discarded. The pellet was washed with 3 mL of cold acetone and air-dried. The pellet was then resuspended in 50 μL of 8 M urea, 10 mM HEPES, pH 7.2 and diluted with 150 μL of 0.05% SDS in phosphate-buffered saline (PBS). This was shaken overnight at 20 °C to dissolve. The samples were then diluted with a further 200 μL of 0.05% SDS in PBS and centrifuged to collect any insoluble residue. The supernatant was transferred to a 2 mL Eppendorf tube and mixed with 25 μL of Pierce Avidin Agarose beads (Thermo Fisher Scientific) which had been washed twice with PBS. Following 3 hours of mixing by inversion, beads were collected by centrifugation for 2 minutes at 2500 × g. The supernatant was removed and the beads were washed with 500 μL of 0.5% SDS in PBS once, 500 μL of 2% SDS at 65 °C for 10 minutes once, 500 μL of 0.5% SDS in PBS again, then 500 μL of 2 M urea followed by two washes with 500 μL of ultrapure water. The beads were finally resuspended in 20 μL of on-bead digestion buffer and trypsinisation, StageTip desalting, LC-MS/MS data acquisition, and data processing were performed as described previously.27 Peptides were identified by searching against a database of A. niger NRRL3 proteins28 supplemented with streptavidin, avidin, yeast enolase and trypsin. The combined search results were filtered for a minimum of two unique peptides with a false-discovery rate of 4%. Label-free quantification was performed using Progenesis QI (Waters). Following chromatographic alignment, peaks were integrated and assigned. Protein abundance was estimated using the integrated intensity of non-conflicting peptides. Results of this analysis for all identified proteins can be found in Table S2 (ESI†).
Results and discussion
Synthesis of potential cellulase inhibitors and probes
Cellobiose configured cyclophellitol (1) was synthesized via previously reported procedures.16 Glycosylation of cyclophellitol acceptor 2 with cellobiose-derived N-phenyl trifluoroacetimidate 3 afforded pseudo-trisaccharide 4 (Scheme 1). The deprotection sequence: benzoyl removal by NaOMe in MeOH, followed by short hydrogenation over a high loading of Pearlman's catalyst,29 yielded inhibitor 5.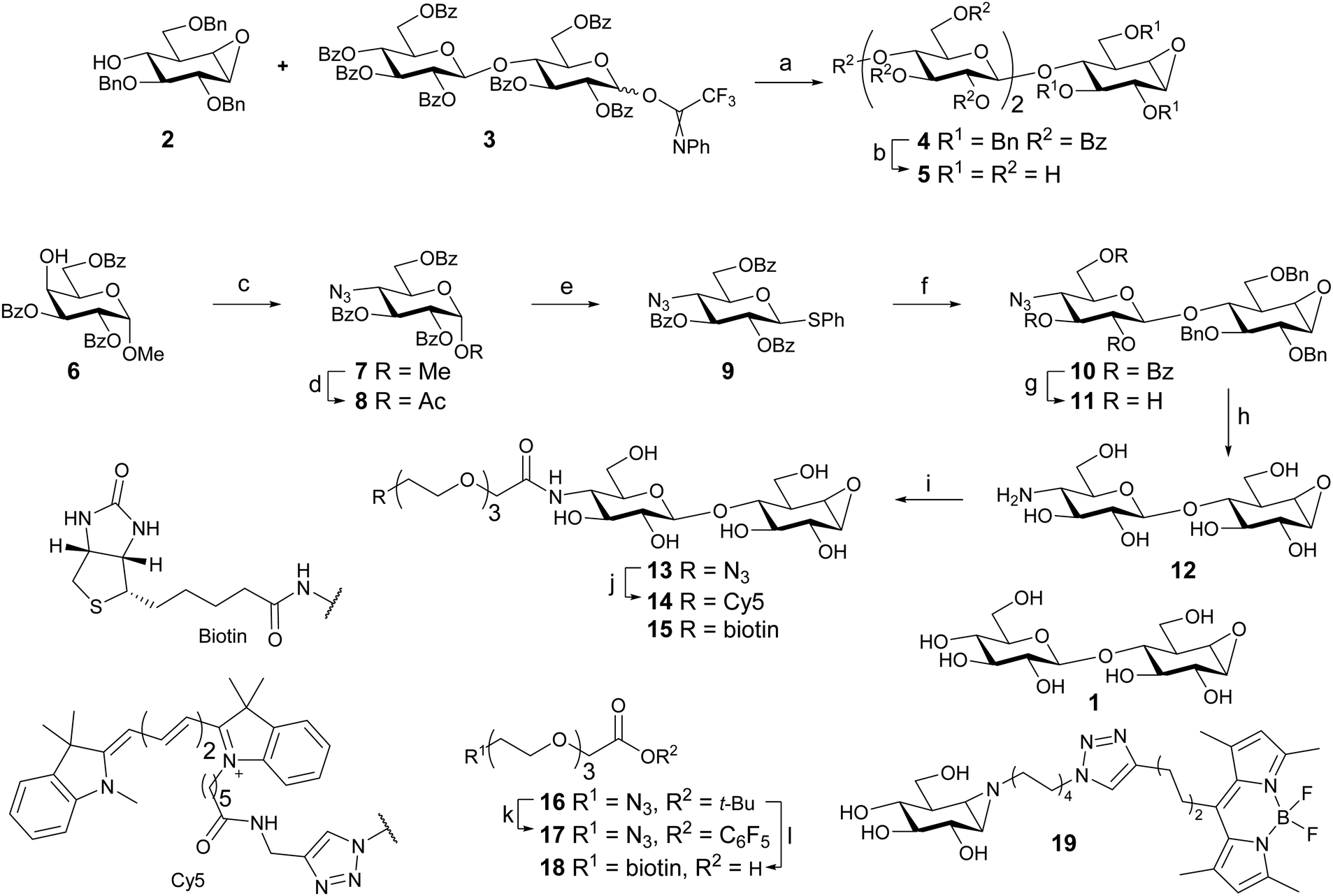 | ||
| Scheme 1 Reagent and conditions: (a) TSMOTf, DCM, −15 °C to 0 °C, 45%. (b) (i) NaOMe, MeOH. (ii) H2, Pd(OH)2/C, H2O, MeOH, dioxane, 40%. (c) (i) Tf2O, pyr, DCM, −55 °C to rt. (ii) NaN3, DMF, 80 °C, 90%. (d) Ac2O, AcOH, H2SO4. (e) HSPh, BF3·Et2O, DCM, 46% over 2 steps. (f) 2, Ph2SO, Tf2O, TTBP, DCM, −70 °C to rt, 64%. (g) NaOMe, MeOH, DCM, 60%. (h) Na (s), t-BuOH, NH3, −60 °C, 53%. (i) 17, DIPEA, DMF, 27%; or 18, PFPOH, DIC, Et3N, DMF, 44%. (j) Cy5 alkyne, THPTA, CuI, DIPEA, DMSO, 46%. (k) (i) TFA, DCM; (ii) PFPOH, DIC, DMAP, DCM, 67%. (l) (i) PPh3, H2O, THF, 88%; (ii) BiotinNHS, DIPEA, DMF, 83%; (iii) TFA, DCM, quant. | ||
To gain access to cellobiose configured ABPs 4-deoxy-4-azido-thioglucoside donor 9 was synthesized. The methods are similar to a published synthesis of 4-deoxy-4-fluoro-thioglucoside donors.30 The axial 4-OH of partially protected methyl α-D-galactopyranoside 6 was activated as a triflate and substituted by sodium azide leading to 7. Acid-catalyzed displacement of the anomeric methoxy group afforded anomeric acetate 8. Introduction of the anomeric thiophenol yielded donor 9.
The glycosylation reaction was improved, compared to that employed in the inhibitor synthesis. Application of a pre-activation protocol (Tf2O/Ph2SO) circumvents the use of relatively high temperatures and long reaction times required to activate this type of donor using NIS/TfOH. It also allows the activation of the donor to take place without the presence of the acid-labile epoxide. Disaccharide 10 was obtained in 64% yield without the use of a large excess of donor. Unreacted acceptor (2) was also recovered indicating the stability of the epoxide functionality under these conditions. Increasing the amount of donor (9) led to diminished yield and complex mixtures. This was presumably due to the reaction of the epoxide in the product with the excess activated donor.31
Following the synthesis of disaccharide 10, the benzoyl esters were removed with NaOMe affording 11. Staudinger reduction of the azide followed by benzyl removal under Birch conditions afforded fully deprotected 12.
Azide-terminated triethylene glycol t-butyl ester 1632 was deprotected using trifluoroactic acid and DIC/DMAP mediated esterification with pentafluorophenol afforded activated ester 17. The amine in 12 was selectively acylated with 17, yielding probe 13 following semi-preparative HPLC purification. Cy5-labeled probe 14 was obtained after copper catalyzed click reaction of 13 with Cy5 alkyne. Biotin-labeled probe 15 was synthesized in one step from 12 by amide bond formation with biotin-terminated spacer 18, obtained from 16 in 3 steps.
BODIPY green-labeled β-glucosidase probe 19 was obtained by methods developed for the previously reported BODIPY red variant using BODIPY green alkyne.33,34
Testing potential cellulase inhibitors and probes with HiCel7B
Humicola insolens Cel7B (HiCel7B) was chosen as a model β-D-glucanase since it is well-characterized, has good hydrolytic performance with chromogenic substrates, can be readily crystallised, and has been studied in our lab previously.20,21,35Compound 1 proved to be an efficient covalent inhibitor of HiCel7B, with a ki/KI of 450 M−1 s−1 (Fig. 1 and Table 1). Intact MS confirmed complete, single labelling after 60 minutes at 25 °C (Fig. 2A). These kinetics compare favourably with the reported requirement to incubate F. oxysporum EG I with 8.25 mmol of 3,4-epoxybutyl β-D-cellobioside for 3 hours at 40 °C to achieve complete inhibition.36
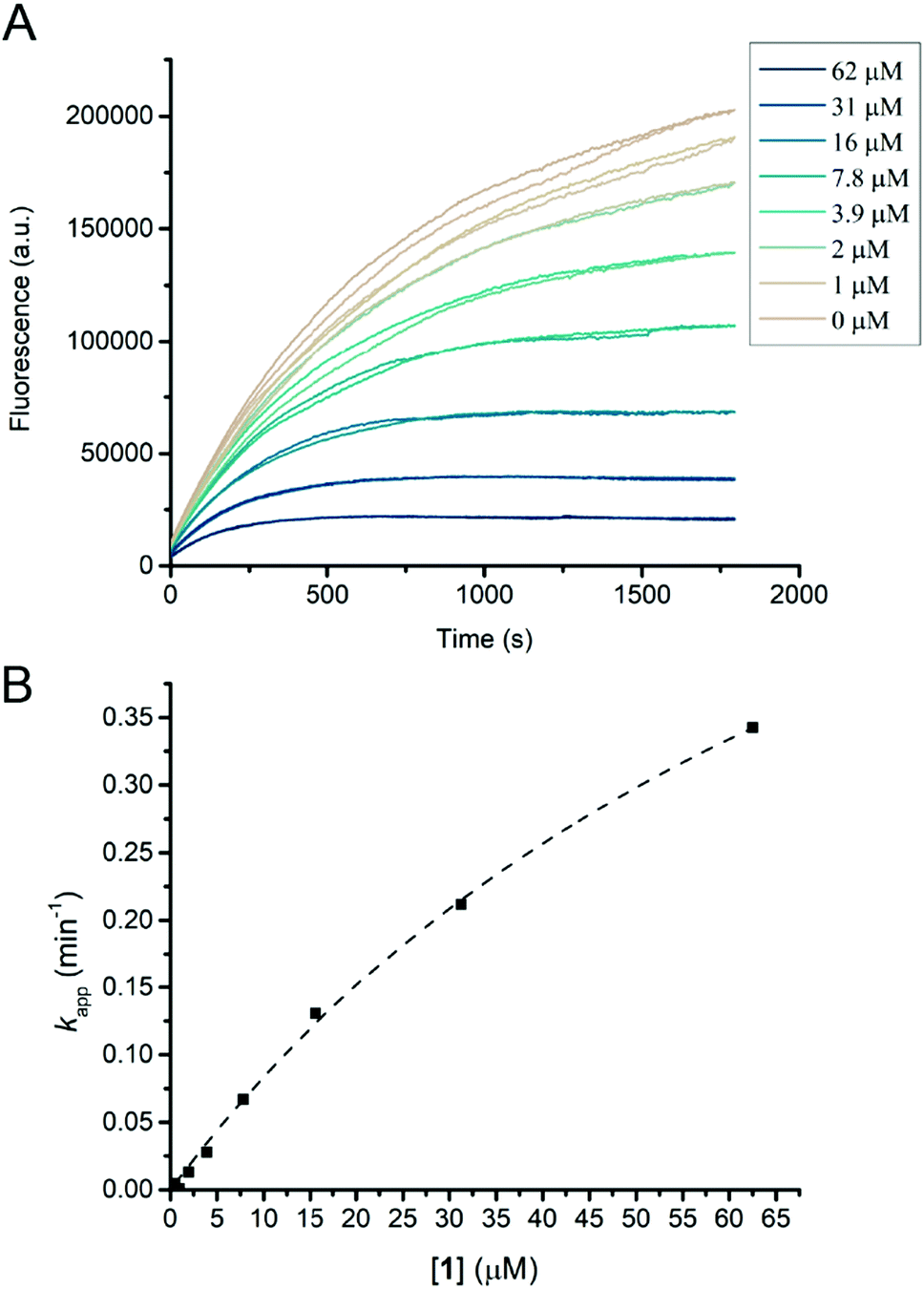 | ||
| Fig. 1 HiCel7B inhibition kinetics with inhibitor 1. (A) Plot of fluorescence vs. time for HiCel7B in the presence of different concentrations of inhibitor 1. Each line represents a single measurement. (B) Plot of apparent decay constant (kapp) extracted from an exponential decay fit of the curves shown in panel A vs. inhibitor concentration with the hyperbolic fit shown as a dotted line. Each point is the average of two measurements. | ||
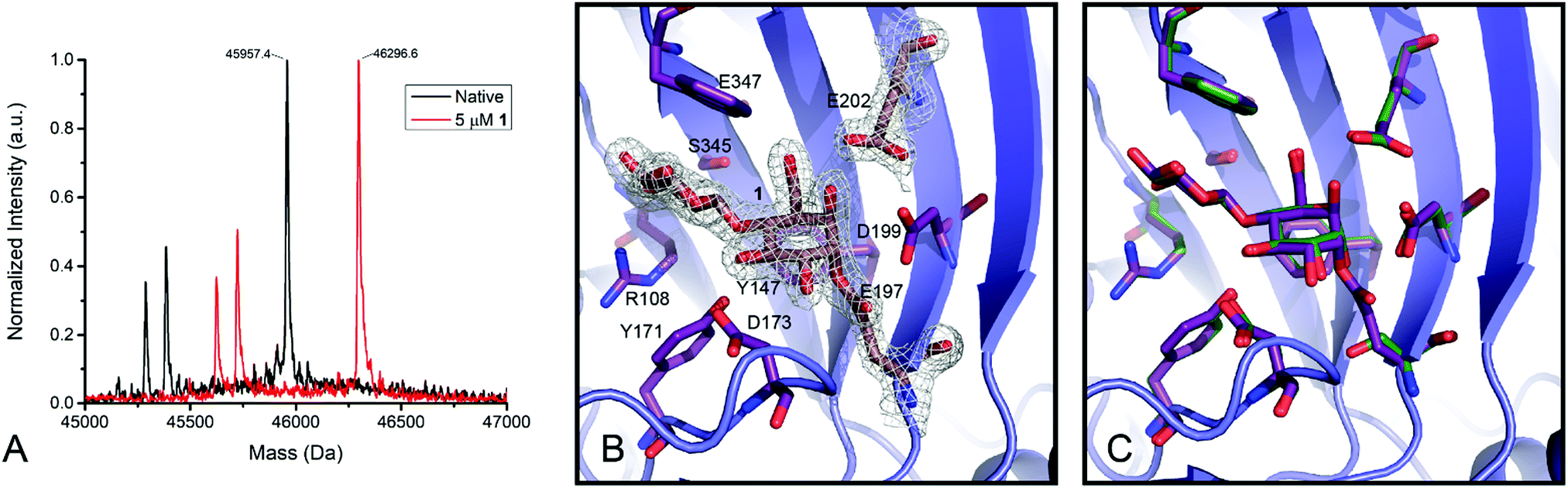 | ||
| Fig. 2 Labelling of HiCel7B with 1. (A) Deconvoluted intact mass spectra of HiCel7B with (red) and without (black) incubation with 1. Δm (expected) = 338, Δm (observed) = 339. (B) Structure of the HiCel7B:1 complex showing 2Fo–Fc electron density at 2σ. Amino acids having polar or hydrophobic contacts with 1 are shown as purple sticks, 1, E197, and E202 are shown as tan sticks. (C) Overlay of 1 complex with Cellobiose:HiCel7B(E197S) complex (PDB ID: 1OJK). Active site residues and cellobiose from PDB ID 1OJK are shown in green, active site residues and 1 from PDB ID 6YOZ (this work) are shown in purple. | ||
The addition of another β-1,4-linked glucose residue to the non-reducing terminus to give 5 improved the performance of the inhibitor roughly 7-fold (Table 1 and Fig. S3, ESI†), however intact MS with 2.2 μM enzyme and 5 μM inhibitor revealed minimal labelling (Fig. S4, ESI†). Treatment with 5 gave small peaks with mass differences of both ∼338 Da (equivalent to addition of 1) and ∼500 Da (expected). Soaking HiCel7B crystals with 0.1 mM 5 also gave an unliganded enzyme structure. Repeating the intact MS experiment with a higher inhibitor concentration (50 μM) resulted in more overall labelling, but still a dominant mass difference of ∼338 Da. Lowering the inhibitor (5 μM) and enzyme concentrations (0.5 μM) gave overall weaker signal, showing incomplete labelling, with a mass difference attributable to 5 as the dominant modification (Fig. S4, ESI†). We interpret these results as indicative of an internal hydrolysis of 5 to give primarily a mixture of cellobiose and cyclophellitol, which is unreactive, and secondarily a mixture of 1 and glucose, which gives rise to the smaller observed mass difference. The observed concentration-dependence suggests that both hydrolytic processes have a higher KM than the KI of the interaction between HiCel7B and 5. Thus, the course of inhibition of HiCel7B with 5 is enzyme- and inhibitor concentration-dependent, being affected by the KM of the two possible hydrolytic pathways and the KI values for 5 and 1. To hopefully avoid the complication of internal hydrolysis, 13 was built on a β-1,4-glucosyl cyclophellitol inhibitor core.
Probe 13 turned out to be a strong inhibitor of HiCel7B, reacting with a ki/KI of 2100 M−1 s−1, comparable to that of the 5. Intact MS confirmed complete single labelling at a 5:2.2 probe:enzyme stoichiometric ratio (Fig. S4, ESI†), confirming efficient labelling without hydrolysis. Modifying the azide handle of 13 with Cy5 gave compound 14, which is an effective probe for in-gel fluorescence-based detection of HiCel7B. A serial dilution of HiCel7B with 14 gave significant signal for the HiCel7B band from as little as 1.6 pg of enzyme per well (Fig. S5, ESI†). Probe 14 also facilitated measurement of the pH-labelling profile for HiCel7B (Fig. S6A, ESI†). Comparison to the pH-activity profile for the hydrolysis of 4MU-GG shows significant similarity between the pH-labelling profile and pH-activity profile, particularly above pH 5 (Fig. S6B, ESI†).
The structure of the HiCel7B complex with inhibitor 1
Soaking HiCel7B crystals with 1 yielded the complex shown in Fig. 2B. This confirmed that 1 binds in the expected manner, mimicking the 4C1 conformation of two glucose units of cellobiose previously observed in the −1 and −2 subsites (Fig. 2C).37 Binding of the inhibitor had no significant impact on the structure of the active site, inducing no conformational change following addition to the catalytic nucleophile. This is in spite of the epoxide oxygen forming an extremely close (2.3 Å) contact with the general acid/base. Extending beyond the −2 subsite, in which essential hydrophobic stacking with W347 and hydrogen bonding interactions with R108, Y147, and S345 are formed, the active site broadens significantly, suggesting a lack of a specific −3 subsite, possibly accounting for the weak selectivity between 5 and 13.Enzyme detection and identification by in-gel fluorescence and biotin–avidin enrichment
To test the ability of the probe to stain β-D-glucanases in fungal secretomes without staining β-D-glucosidases, an A. niger xylan-grown secretome was stained with 14 or 19, followed by 19 or 14, respectively (Fig. 3A). 19-stained bands were present at molecular weights of ∼45 kDa, ∼60 kDa, ∼100 kDa, and ∼130 kDa. 14-stained bands were present at ∼35 kDa, ∼40 kDa (faint), ∼60, and ∼80 kDa. Notably, the gel shows minimal overlap between the staining of the two probes and no apparent preclusion of staining of one probe by the other. This suggests that among the retaining glycoside hydrolases secreted by A. niger, there is no hydrolysis of 14 and no cross-reactivity between 14 and 19. Thus, 4-O substitution appears to reduce cross-reactivity with exo-glycosidases compared to the previously reported xylanase probes.16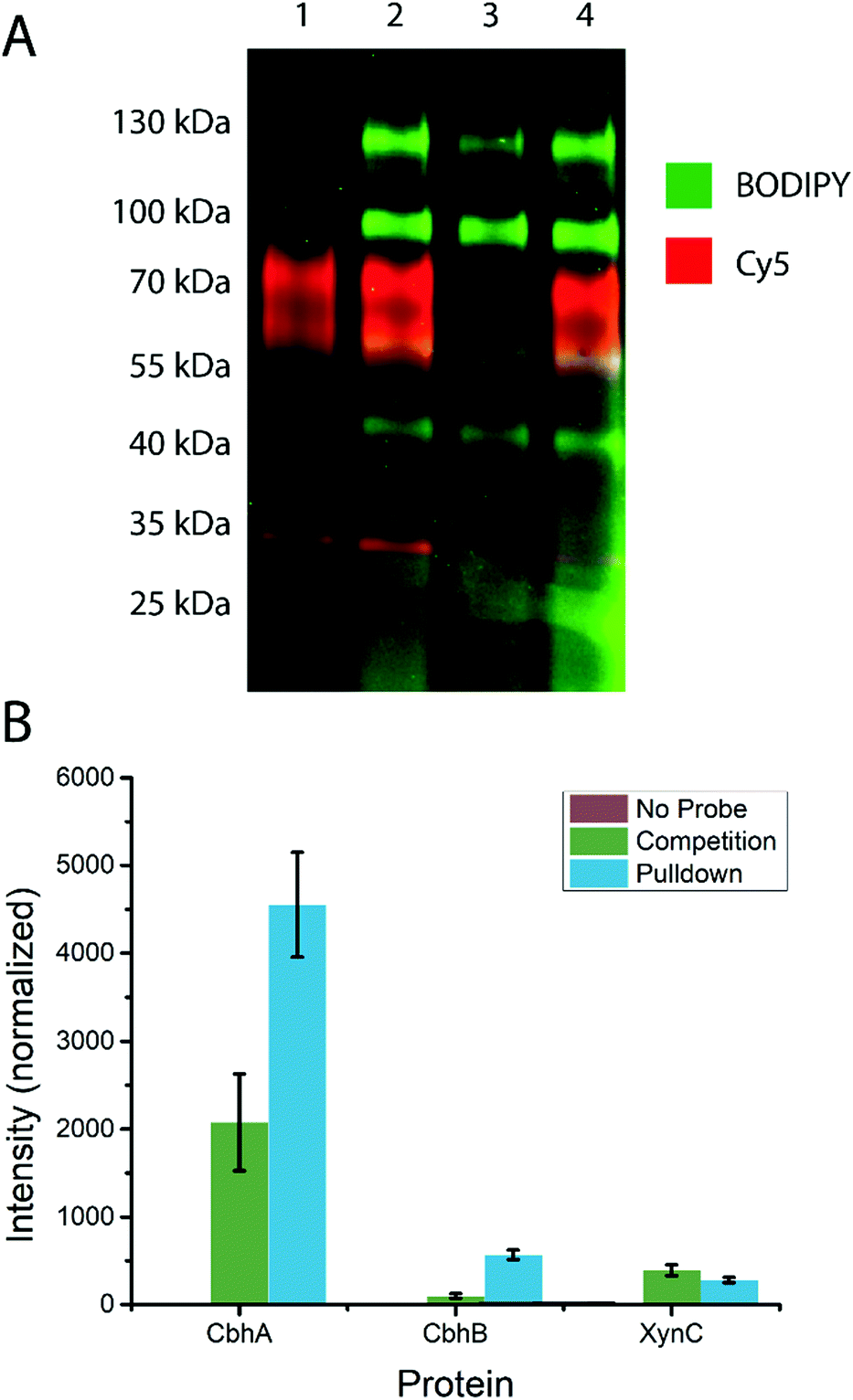 | ||
| Fig. 3 (A) Staining of the A. niger xylan-grown secretome with 14 and 19. Dual channel fluorescence scan of a 4–20% SDS-PAGE is shown. Lane 1 contains 14-stained secretome, lane 2 contains 14-stained secretome subsequently stained with 19, lane 3 contained 19-stained secretome, and lane 4 contains 19-stained secretome subsequently stained with 14. (B) Label-free quantification of proteins identified from the A. niger secretome following treatment with probe 15 and biotin–avidin enrichment. The “No Probe” sample is the negative control, the “Competition” sample was pre-treated with inhibitor 1, and the “Pulldown” sample was only treated with probe 15. The observed signal intensity was normalised to a 10 fmol μl−1 trysinised yeast enolase internal standard added to each sample prior to analysis. | ||
Based on the known content of this secretome, we tentatively assigned the 60 and 80 kDa 14-stained bands as CbhA and CbhB respectively, two GH7 cellulases.39 We also assigned the ∼40 kDa band as EglB,40 a GH5 endo-β-D-glucanase, and the ∼35 kDa band as XynC, an abundant GH10 xylanase likely stained due to a loose enzyme-substrate specificity comparable to other fungal GH10 xylanases.41,42 We tentatively assign the 100 and 130 kDa 19-stained bands as GH3 enzymes, possibly BglA, BglM, and XlnD,38 which have been detected in this secretome previously.16
To test the specificity of our cellulase probe architecture, we used the biotinylated derivative (probe 15) and performed a biotin–avidin pulldown enrichment prior to on-bead digestion, peptide identification, and label-free quantification. Three samples were prepared: a negative control, a probe 15-treated sample, and a sample treated with probe 15 after treatment with inhibitor 1. The only proteins from A. niger detected at elevated levels in the probe 15-treated samples relative to the negative control were CbhA, CbhB, and XynC, confirming our assignment of the major bands observed by in-gel fluorescence. Label-free quantification showed a significant drop in CbhA and CbhB signal following treatment with inhibitor 1, but revealed no significant drop in XynC signal, suggesting that XynC was minimally inhibited (Fig. 3B). Thus, the probe architecture presented here shows specificity towards known GH7 cellulases within the context of a complex fungal secretome.
Conclusions
β-1,4-Glucanases form the foundations of lignocellulose-degrading systems. Being produced by a wide variety of saprotrophic, symbiotic, and pathogenic microorganisms, the ability to detect small quantities of these enzymes directly offers a variety of opportunities for activity-based protein profiling. We have shown here that a cellobiose-mimicking cyclophellitol derivative is a potent inhibitor of β-1,4-glucanases. We have also shown that extension of this inhibitor from the 4′ position with glucose enhances inhibitor binding, but facilitates inhibitor hydrolysis. Fortuitously, extension with a PEG linker enhances binding without facilitating inhibitor hydrolysis. Furthermore, the addition of a detection tag to the linker gives a potent and selective activity-based probe which can be applied to the direct detection of β-1,4-glucanases within a fungal secretome.Data deposition
Coordinates and structure factors have been deposited with the PDB, with accession codes 6OYZ (HiCel7B soaked with 1) and 6YP1 (HiCel7B Soaked with 13). Results from the label-free quantitation proteomic experiment have been deposited in the PRIDE database with the accession code PXD019930.Author contributions
GJD and HO conceived the study. CB, EP, SPS, JJ, G van der M, and JDCC performed or supervised organic synthesis. BIF performed proteomics. JR, AFJR and GPvanW prepared Aspergillus cultures and secretomes. NGSM performed structural biology, intact mass spectrometry, kinetic measurements, and in-gel fluorescence. Manuscript preparation was led by NGSM with help from other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (Post-Doctoral Fellowship to NGSM), the Royal Society (Ken Murray Research Professorship to GJD), the Biotechnology and Biological Sciences Research Council (BBSRC) (grant BB/R001162/1 to GJD), the Netherlands Organization for Scientific Research (NWO TOP grant 2018-714.018.002 to HSO), the European Research Council (ERC-2011-AdG-290836 “Chembiosphing” to HSO), Diamond Light Source (proposal 18598), and the staff of beamline I03 for assistance with crystal testing and data collection.Notes and references
- L. I. Willems, H. S. Overkleeft and S. I. Van Kasteren, Bioconjugate Chem., 2014, 25, 1181–1191 CrossRef CAS PubMed.
- S. J. Williams, O. Hekmat and S. G. Withers, ChemBioChem, 2006, 7, 116–124 CrossRef CAS PubMed.
- L. M. Chauvigné-Hines, L. N. Anderson, H. M. Weaver, J. N. Brown, P. K. Koech, C. D. Nicora, B. A. Hofstad, R. D. Smith, M. J. Wilkins, S. J. Callister and A. T. Wright, J. Am. Chem. Soc., 2012, 134, 20521–20532 CrossRef PubMed.
- L. Wu, Z. Armstrong, S. P. Schröder, C. de Boer, M. Artola, J. M. Aerts, H. S. Overkleeft and G. J. Davies, Curr. Opin. Chem. Biol., 2019, 53, 25–36 CrossRef CAS PubMed.
- V. Bulone, J. G. Schwerdt and G. B. Fincher, Front. Plant Sci., 2019, 10, 1009 CrossRef PubMed.
- J. M. Fox, P. Jess, R. B. Jambusaria, G. M. Moo, J. Liphardt, D. S. Clark and H. W. Blanch, Nat. Chem. Biol., 2013, 9, 356–361 CrossRef CAS PubMed.
- I. Benoit, H. Culleton, M. Zhou, M. DiFalco, G. Aguilar-Osorio, E. Battaglia, O. Bouzid, C. P. J. M. Brouwer, H. B. O. El-Bushari, P. M. Coutinho, B. S. Gruben, K. S. Hildén, J. Houbraken, L. A. J. Barboza, A. Levasseur, E. Majoor, M. R. Mäkelä, H. M. Narang, B. Trejo-Aguilar, J. Van Den Brink, P. A. VanKuyk, A. Wiebenga, V. McKie, B. McCleary, A. Tsang, B. Henrissat and R. P. De Vries, Biotechnol. Biofuels, 2015, 8, 1–14 CrossRef CAS PubMed.
- O. Hekmat, Y. W. Kim, S. J. Williams, S. He and S. G. Withers, J. Biol. Chem., 2005, 280, 35126–35135 CrossRef CAS PubMed.
- O. Hekmat, C. Florizone, Y. W. Kim, L. D. Eltis, R. A. J. Warren and S. G. Withers, ChemBioChem, 2007, 8, 2125–2132 CrossRef CAS PubMed.
- S. Atsumi, H. Iinuma, H. Naganawa, H. Nakamura, T. Takeuchi, K. Umezawa and Y. Iitaka, J. Antibiot., 1990, 43, 49–53 CrossRef CAS PubMed.
- T. M. Gloster, R. Madsen and G. J. Davies, Org. Biomol. Chem., 2007, 5, 444–446 RSC.
- J. Jiang, C. L. Kuo, L. Wu, C. Franke, W. W. Kallemeijn, B. I. Florea, E. Van Meel, G. A. van der Marel, J. D. C. Codée, R. G. Boot, G. J. Davies, H. S. Overkleeft and J. M. F. G. Aerts, ACS Cent. Sci., 2016, 2, 351–358 CrossRef CAS PubMed.
- M. D. Witte, W. W. Kallemeijn, J. Aten, K. Y. Li, A. Strijland, W. E. Donker-Koopman, A. M. C. H. Van Den Nieuwendijk, B. Bleijlevens, G. Kramer, B. I. Florea, B. Hooibrink, C. E. M. Hollak, R. Ottenhoff, R. G. Boot, G. A. van der Marel, H. S. Overkleeft and J. M. F. G. Aerts, Nat. Chem. Biol., 2010, 6, 907–913 CrossRef CAS PubMed.
- M. Artola, L. Wu, M. J. Ferraz, C. L. Kuo, L. Raich, I. Z. Breen, W. A. Offen, J. D. C. Codée, G. A. van der Marel, C. Rovira, J. M. F. G. Aerts, G. J. Davies and H. S. Overkleeft, ACS Cent. Sci., 2017, 3, 784–793 CrossRef CAS PubMed.
- L. Wu, J. Jiang, Y. Jin, W. W. Kallemeijn, C. L. Kuo, M. Artola, W. Dai, C. Van Elk, M. Van Eijk, G. A. van der Marel, J. D. C. Codée, B. I. Florea, J. M. F. G. Aerts, H. S. Overkleeft and G. J. Davies, Nat. Chem. Biol., 2017, 13, 867–873 CrossRef CAS PubMed.
- S. P. Schröder, C. De Boer, N. G. S. McGregor, R. J. Rowland, O. Moroz, E. Blagova, J. Reijngoud, M. Arentshorst, D. Osborn, M. D. Morant, E. Abbate, M. A. Stringer, K. B. R. M. Krogh, L. Raich, C. Rovira, J. G. Berrin, G. P. Van Wezel, A. F. J. Ram, B. I. Florea, G. A. van der Marel, J. D. C. Codée, K. S. Wilson, L. Wu, G. J. Davies and H. S. Overkleeft, ACS Cent. Sci., 2019, 5, 1067–1078 CrossRef PubMed.
- L. I. Willems, T. J. M. Beenakker, B. Murray, S. Scheij, W. W. Kallemeijn, R. G. Boot, M. Verhoek, W. E. Donker-Koopman, M. J. Ferraz, E. R. Van Rijssel, B. I. Florea, J. D. C. Codée, G. A. van der Marel, J. M. F. G. Aerts and H. S. Overkleeft, J. Am. Chem. Soc., 2014, 136, 11622–11625 CrossRef CAS PubMed.
- A. R. A. Marques, L. I. Willems, D. Herrera Moro, B. I. Florea, S. Scheij, R. Ottenhoff, C. P. A. A. van Roomen, M. Verhoek, J. K. Nelson, W. W. Kallemeijn, A. Biela-Banas, O. R. Martin, M. B. Cachón-González, N. N. Kim, T. M. Cox, R. G. Boot, H. S. Overkleeft and J. M. F. G. Aerts, ChemBioChem, 2017, 18, 402–412 CrossRef CAS PubMed.
- N. G. S. McGregor, M. Artola, A. Nin-Hill, D. Linzel, M. Haon, J. Reijngoud, A. Ram, M. N. Rosso, G. A. Van Der Marel, J. D. C. Codeé, G. P. Van Wezel, J. G. Berrin, C. Rovira, H. S. Overkleeft and G. J. Davies, J. Am. Chem. Soc., 2020, 142, 4648–4662 CrossRef CAS PubMed.
- G. Davies, S. Tolley, K. Wilson, M. Schülein, H. F. Wöldike and G. Dodson, J. Mol. Biol., 1992, 228, 970–972 CrossRef CAS PubMed.
- L. F. Mackenzie, G. Sulzenbacher, C. Divne, T. A. Jones, H. F. Wöldike, M. Schülein, S. G. Withers and G. J. Davies, Biochem. J., 1998, 335, 409–416 CrossRef CAS PubMed.
- G. Winter, C. M. C. Lobley and S. M. Prince, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1260–1273 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- E. J. Van Rooden, B. I. Florea, H. Deng, M. P. Baggelaar, A. C. M. Van Esbroeck, J. Zhou, H. S. Overkleeft and M. Van Der Stelt, Nat. Protoc., 2018, 13, 752–767 CrossRef CAS PubMed.
- M. V. Aguilar-Pontes, J. Brandl, E. McDonnell, K. Strasser, T. T. M. Nguyen, R. Riley, S. Mondo, A. Salamov, J. L. Nybo, T. C. Vesth, I. V. Grigoriev, M. R. Andersen, A. Tsang and R. P. de Vries, Stud. Mycol., 2018, 91, 61–78 CrossRef CAS PubMed.
- S. P. Schröder, R. Petracca, H. Minnee, M. Artola, J. M. F. G. Aerts, J. D. C. Codée, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2016, 4787–4794 CrossRef.
- F. J. Weiberth, H. S. Gill, Y. Jiang, G. E. Lee, P. Lienard, C. Pemberton, M. R. Powers, W. Subotkowski, W. Tomasik, B. J. Vanasse and Y. Yu, Org. Process Res. Dev., 2010, 14, 623–631 CrossRef CAS.
- D. R. Dabideen and J. Gervay-Hague, Org. Lett., 2004, 6, 973–975 CrossRef CAS PubMed.
- K. Heller, P. Ochtrop, M. F. Albers, F. B. Zauner, A. Itzen and C. Hedberg, Angew. Chem., Int. Ed., 2015, 54, 10327–10330 CrossRef CAS PubMed.
- M. Verdoes, U. Hillaert, B. I. Florea, M. Sae-Heng, M. D. P. Risseeuw, D. V. Filippov, G. A. van der Marel and H. S. Overkleeft, Bioorg. Med. Chem. Lett., 2007, 17, 6169–6171 CrossRef CAS PubMed.
- J. Jiang, T. J. M. Beenakker, W. W. Kallemeijn, G. A. Van Dermarel, H. Van Den Elst, J. D. C. Codée, J. M. F. G. Aerts and H. S. Overkleeft, Chem. – Eur. J., 2015, 21, 10861–10869 CrossRef CAS PubMed.
- M. Schülein, J. Biotechnol., 1997, 57, 71–81 CrossRef.
- G. Sulzenbacher, M. Schülein and G. J. Davies, Biochemistry, 1997, 36, 5902–5911 CrossRef CAS PubMed.
- V. M. A. Ducros, C. A. Tarling, D. L. Zechel, A. M. Brzozowski, T. P. Frandsen, I. Von Ossowski, M. Schülein, S. G. Withers and G. J. Davies, Chem. Biol., 2003, 10, 619–628 CrossRef CAS.
- N. N. M. E. Van Peij, J. Brinkmann, M. Vršanská, J. Visser and L. H. De Graaff, Eur. J. Biochem., 1997, 245, 164–173 CrossRef CAS PubMed.
- M. M. C. Gielkens, E. Dekkers, J. Visser and L. H. De Graaff, Appl. Environ. Microbiol., 1999, 65, 4340–4345 CrossRef CAS PubMed.
- N. N. M. E. Van Peij, M. M. C. Gielkens, R. P. De Vries, J. Visser and L. H. De Graaff, Appl. Environ. Microbiol., 1998, 64, 3615–3619 CrossRef CAS PubMed.
- V. Ducros, S. J. Charnock, U. Derewenda, Z. S. Derewenda, Z. Dauteri, C. Dupont, F. Shareck, R. Morosoli, D. Kluepfel and G. J. Davies, J. Biol. Chem., 2000, 275, 23020–23026 CrossRef CAS PubMed.
- L. Lo Leggio, S. Kalogiannis, K. Eckert, S. C. Teixeira, M. K. Bhat, C. Andrei, R. W. Pickersgill and S. Larsen, FEBS Lett., 2001, 509, 303–308 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplemental figures and table, supplementary synthetic protocols and characterisation. See DOI: 10.1039/d0cb00045k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |