
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective 18F-incorporation into pyridyl acyltrifluoroborates for rapid radiolabelling of peptides and proteins at room temperature†
Aristeidis
Chiotellis‡
a,
Hazem
Ahmed‡
 a,
Thomas
Betzel
a,
Matthias
Tanriver
b,
Christopher J.
White
b,
Haewon
Song
b,
Sara
Da Ros
b,
Roger
Schibli
a,
Jeffrey W.
Bode
*b and
Simon M.
Ametamey
*a
a,
Thomas
Betzel
a,
Matthias
Tanriver
b,
Christopher J.
White
b,
Haewon
Song
b,
Sara
Da Ros
b,
Roger
Schibli
a,
Jeffrey W.
Bode
*b and
Simon M.
Ametamey
*a
aCenter for Radiopharmaceutical Sciences ETH-PSI-USZ, Institute of Pharmaceutical Sciences ETH, Vladimir-Prelog-Weg 4, 8093 Zurich, Switzerland. E-mail: simon.ametamey@pharma.ethz.ch
bLaboratory of Organic Chemistry, Department of Chemistry and Applied Biosciences, Vladimir-Prelog-Weg 3, 8093 Zurich, Switzerland. E-mail: bode@org.chem.ethz.ch
First published on 5th December 2019
Abstract
A new prosthetic group is reported for 18F-labelling of peptides and proteins based on the chemoselective ligation of potassium acyltrifluoroborates (KATs) and hydroxylamines without any detectable 18F/19F isotope exchange at the acyltrifluoroborate moiety. The new building block is appended via a common amide bond at room temperature with no need for protecting groups which enables an effective orthogonal 18F-radiolabelling.
Positron emission tomography (PET) can visualise, characterise and quantify biological processes at the cellular and molecular levels in vivo non-invasively, with high sensitivity and spatial resolution. It has evolved into a powerful imaging tool, contributing decisively in both basic research and medical decision-making.1 Fluorine-18 is the most frequently utilised radionuclide for diagnostic PET imaging since its decay properties (18F; β+ 0.635 MeV, 97% abundance, t1/2 = 109.8 min) provide significant advantages over other PET radionuclides.2 The ever-increasing use of biomolecular vector-based imaging agents has brought forth a high demand for 18F-radiosynthesis strategies that are fast, mild and selective. However, the harsh conditions required for direct 18F-labelling (high temperatures, basic pH, dry organic solvents), the extremely low concentration levels of 18F and its relatively fast radioactive decay make the development of 18F radiopharmaceuticals of complex molecules a challenging task.
Prosthetic groups have expanded the field of radioactive bioconjugation in recent years (Scheme 1).3 These are small bifunctional molecules that can be radiolabelled with a radionuclide and subsequently appended to biomolecules. The great majority of prosthetic groups have relied upon reactions with natural amino acids (most notably couplings between activated carboxyl groups and lysines and Michael additions between maleimides and cysteines).6–9 However, the complete loss of regiochemical control during the labelling of a biomolecule when more than one (unprotected) lysine or cysteine are present requiring time-consuming protection and/or purification steps prompted radiochemists to shift their attention towards ligations of higher specificity. As such, a wide panel of chemoselective ligation reactions have been used in PET radioligand development.10 Lately, the oxime-ligation methodology has gained increased attention.11
 | ||
| Scheme 1 Overview of work on boron-based 18F prosthetic groups.4,5 | ||
The fairly recent discovery of (almost) ideal orthogonal ligations gave a new impetus in the development of advantageous 18F-labelling prosthetic groups. Among these reactions, the Cu-catalyzed azide–alkyne cycloaddition (CuAAC),12 the strain-promoted azide–alkyne cycloaddition (SPAAC, azides-cyclooctenes)13 and the inverse electron-demand Diels–Alder reaction (IEDDA)14 are the most prevalent. The selectivity, ease and modularity of these ligations make them well-suited for the construction of complex 18F-labelled tracers. But despite their utility, these reactions exhibit several flaws that limit their universal application (e.g., use of toxic additives for CuAAC and formation of bulky hydrophobic linkages for SPAAC and IEDDA).15 Therefore novel approaches to facilitate 18F-labelling and diversify the pre-existing library of 18F-radiotracers are still needed.16
In 2012, Bode and Molander showed that the reaction between potassium acyltrifluoroborates (KATs) and various hydroxylamines leads to the formation of amide bonds under aqueous conditions at room temperature with a second-order rate constant of 20 M−1 s−1.17 The reaction shows excellent chemoselectivity and tolerates all common unprotected functional groups typically found on proteins and peptides. KAT ligation is faster than the majority of the ligations in common use and it has the distinct advantage of using chemically stable, easily handled functional groups.18 The Bode group successfully used this method for the modification of a synthetic GLP1 analogue19 and for the two-step modification of recombinant proteins.20 In order to harness the advantages of the KAT ligation for radiochemistry applications, we sought to develop a new 18F-labelling prosthetic group based on this chemistry.
Despite the advantages of KATs for rapid and chemoselective ligation, installation of the 18F could be complicated by 18F/19F isotopic exchange at the trifluoroborate group which would inevitably lower the overall 18F-incorporation yield. There is already an impressive body of work on 18F exchange with organotrifluoroborates. Most notably, Perrin and co-workers have employed 18F/19F isotopic exchange on trifluoroborates for PET imaging.21,22 Their studies showed that alkyl acyltrifluoroborates lose a fluoride with a long solvolysis half-life of >2000 min.23 We hypothesised that [18F]fluoride ions would favour nucleophilic aromatic substitution over 18F/19F isotopic exchange under non-aqueous solvents at higher pH, as these isotopic exchange reactions are usually carried out under aqueous acidic conditions or with strong Lewis acids.6,24,25
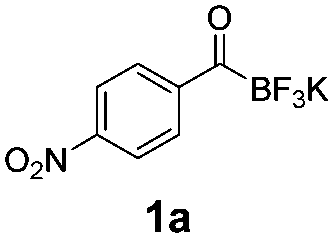
For the purpose of this study, five precursors were evaluated for 18F-incorporation (Table 1): KAT derivatives 1a–d and monofluoroacylboronate 1e. For the substrate scope, we selected targets likely to be metabolically stable and not prone to defluorination, in contrast to alkyl fluorides.26,27 Defluorination is particularly undesired in the context of in vivo PET imaging, due to bone uptake of free [18F]fluoride.
| Entry | Precursor | Labelled prosthetic group | 8F-Incorporation without DABCOa | 18F-Incorporation with DABCOa (140 mM) |
|---|---|---|---|---|
| a 10 μmol 1a–1e, [18F]CsF-K2.2.2, dry DMSO (300 μL), 150 °C, 10 min; n = number of trials. | ||||
| 1 |
|
|
0% | n/a |
| 2 |
|
|
3.5% ± 2.5, n = 3 | 35.5% ± 6.8, n = 5 |
| 3 |
|
|
2.3% ± 1.1, n = 3 | 3.2% ± 0.7, n = 3 |
| 4 |
|
|
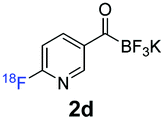
23.6% ± 1.1, n = 3 | 44.7% ± 6.2, n = 4 |
| 5 |
|
|
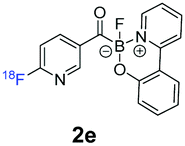
12.0% ± 3.1, n = 3 | n/a |
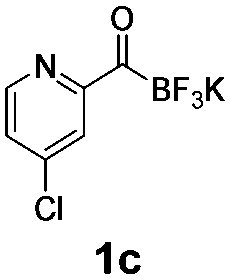
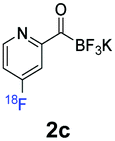
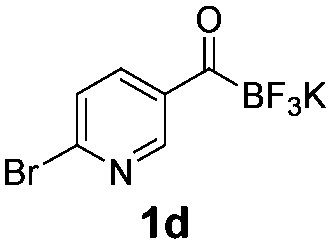
All precursors for the radiolabelling (Table 1, 1a–e) and their respective 19F-reference compounds were synthesised according to the protocols developed by the Bode group (see ESI†). The precursors were subjected to typical conditions for nucleophilic aromatic substitution – [18F]fluoride ions were eluted with a solution of Kryptofix®2.2.2/Cs2CO3 from an anion exchange cartridge then azeotropically dried with CH3CN. Precursors 1a–e in DMSO were added and the reaction mixture was heated at 150 °C for 10 min. 18F-Incorporation yield and product identity were determined by UPLC (see ESI†) and HPLC respectively, equipped with a radiodetector. KAT 1a with the electron-withdrawing nitro leaving group failed to afford the desired labelled compound since the electron-withdrawing effect of the KAT was not sufficient to activate the aromatic ring. Both precursors 1b and 1c bearing the KAT group in meta-position to the halogen showed less than 5% 18F-incorporation (entries 2 and 3). Precursor 1d afforded product 2d with 23.6% 18F-incorporation (entry 4), confirming a synergistic effect of the two activating groups. Acylmonofluoro-boronate 1e gave product 2e with 12% yield (entry 5).
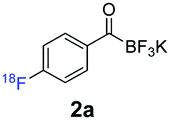
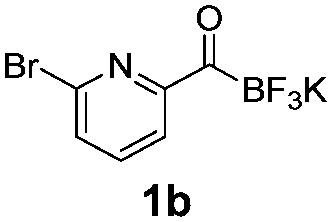
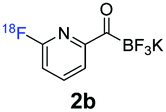
During the course of our experiments, Pike et al. reported the enhancing effects of 1,4-diazabicyclo[2.2.2]octane (DABCO) on 18F-radiolabelling of substituted 2-halopyridines.28 As anticipated, the addition of DABCO (140 mM) to the reaction mixture of the chloro-precursor 1c only marginally improved the 18F-incorporation (entry 3), as this method is effective only for 2-halopyridines. In contrast, a remarkable improvement was observed for precursor 1b and 1d (entries 2 and 4), with product 2d exhibiting the highest 18F-incorporation of 44.7%. In light of these findings, 1d became the precursor for our new prosthetic group 2d, termed [18F]FPAT (6-fluoropyridyl acyltrifluoroborate).
Subsequently, further optimisation experiments were performed, the results of which are summarized in the ESI.† We first established that the same high radiochemical conversions could be obtained at 120 °C with a lower amount of the precursor (1 mg, 3.4 μmol). Furthermore, aqueous DMSO (0.1–2% H2O v/v) had a positive impact on 18F-incorporation. Most importantly, the yield could be enhanced by avoiding the conventional azeotropic drying step. We were delighted to see that these optimisations significantly improved the conversion to 71.1% ± 12.5 (n = 11). This increase in yield can be attributed to less adsorption and higher availability of [18F]fluoride in the reaction medium. Moreover, the tolerance towards H2O could be rationalised by the strong charge interactions between the free [18F]fluoride ion and the positively charged quaternary nitrogen of DABCO at the expense of weaker interactions with water. Employing these conditions with NBu4HCO3 instead of Cs2CO3 further improved the radiochemical conversion to 90.5% ± 5.2 (n = 6). However, this caused broadening of the peaks and poor separation of [18F]FPAT during the HPLC purification step.
Exchanging the base to K2CO3 afforded [18F]FPAT with 84.2% ± 1.2 (n = 5) 18F-incorporation while maintaining a clean HPLC purification profile. This procedure was deemed optimal for [18F]FPAT, affording the prosthetic group in good molar activities of 81 ± 26 (n = 7) GBq μmol−1 and isolated RCYs of 58% ± 2.5 (decay corrected, n = 3). Unlike similarly structured 18F-nicotinamide based prosthetic groups (e.g., [18F]FPy-TFP)29 and other amine reactive building blocks (e.g., 18F-SFB),30 which all non-selectively react with Lys residues, [18F]FPAT reacts chemoselectively with hydroxylamines without the need for protecting groups.
We were also curious to see whether the acyltrifluoroborate engages in 18F/19F isotopic exchange. There was no observable radiosignal corresponding to the bromo-precursor 1d (10.3 min) on the HPLC after 18F-incorporation (Fig. 1). The high 18F-incorporation yields indicated that overall there was a strong preference for nucleophilic substitution over isotopic exchange under the radiolabelling conditions. The origin of the remarkably slow exchange of the fluorides on the KAT moiety is under further investigation.
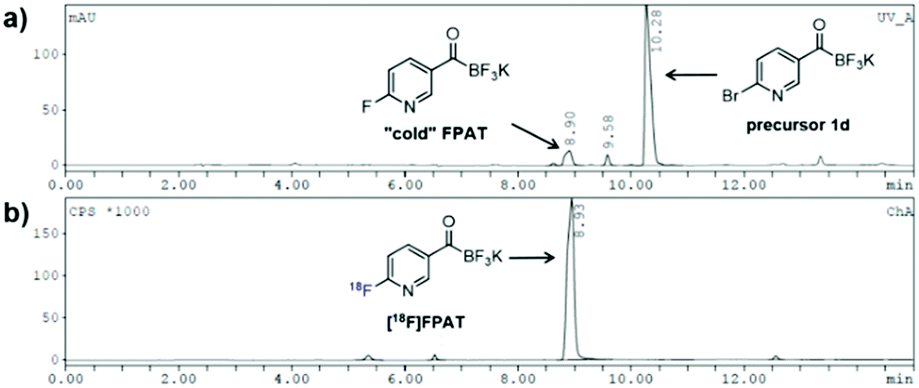 | ||
| Fig. 1 18F-Labelling of bromo-precursor 1d. (a) UV-HPLC chromatogram; (b) γ-HPLC chromatogram (crude). | ||
We next evaluated the ligation efficiency of [18F]FPAT at ambient temperature with various hydroxylamine substrates 3a–c (results summarised in the ESI†). For all succeeding experiments, [18F]FPAT was purified to avoid competing reactions with excess remaining precursor. These ligations were performed in aqueous medium with organic solvents (tBuOH or DMSO) as additives for solubilising the substrates. Aqueous oxalic acid was also added since KAT ligations are faster under acidic conditions.19 Complete conversion of [18F]FPAT to amide 4a was observed within 3 min at 10 mM and 1 mM concentrations of the precursor. Decreasing the concentration to 0.1 mM led to 87% conversion in 15 min and full conversion after 30 min (Scheme 2).
 | ||
| Scheme 2 Radiolabelling of hydroxylamine 3a with [18F]FPAT. Yields determined by γ-UPLC. | ||
In order to establish that this method could be used to introduce 18F into biologically relevant molecules, we selected a short eight-residue peptide. The peptide VSPTYRYL was synthesised by standard Fmoc-SPPS on a Rink Amide resin, and the N,N-diethylcarbamoylhydroxylamine functional group introduced at the N-terminus. 18F-Labelling of 3b was carried out with [18F]FPAT (1–4 GBq) using peptide concentrations typically reported for similar substrates31 (Scheme 3). At 800 μM and 400 μM, the ligation afforded the labelled peptide 4b quantitatively after only 15 min. Reducing the concentration to 200 μM resulted in 20% incorporation after 15 min.
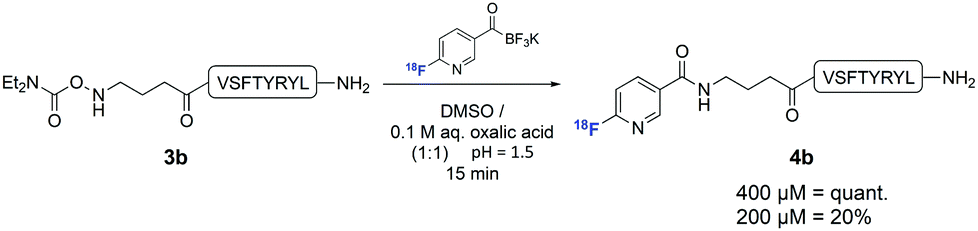 | ||
| Scheme 3 Radiolabelling of hydroxylamine peptide 3b with [18F]FPAT. Yields determined by γ-HPLC. | ||
To further extend this method towards larger biomolecules, we modified sfGFP(S147C)20 with a maleimide functionality bearing the hydroxylamine group at the exposed cysteine residue (Scheme 4). Due to the low concentrations of the protein used, small amounts of [18F]FPAT (50–100 MBq) were used respectively to avoid interference of the cold [19F]FPAT. At 70 μM, the reaction successfully afforded the labelled protein 4c in quantitative yields, while a three-fold lower concentration led to 80% conversion to the conjugation product.
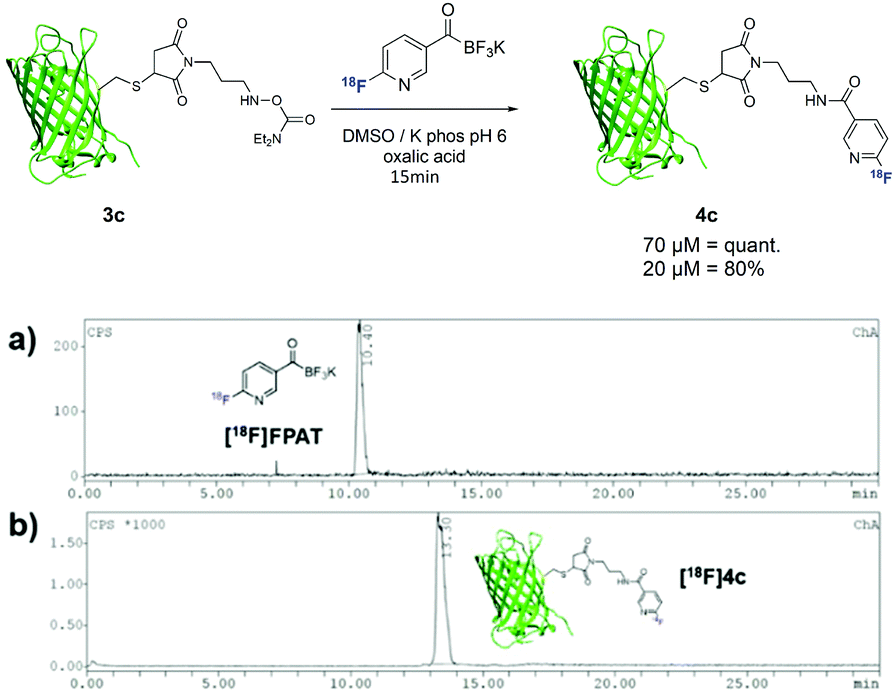 | ||
| Scheme 4 Radiolabelling of protein sfGFP(S147C) 3c with [18F]FPAT; (a) γ-HPLC chromatogram of [18F]FPAT; (b) γ-HPLC chromatogram of ligation (crude). | ||
In order to confirm that [18F]FPAT is resistant towards defluorination in vivo, which is particularly important for applications in pretargeting, [18F]FPAT was injected into a C57BL/6 mouse for a pilot PET/CT study (Fig. 2). No bone uptake was observed over the 90 minute time period, establishing that no defluorination of the prosthetic group was evident, indicating the high stability of the [18F]fluoride incorporated into this scaffold.
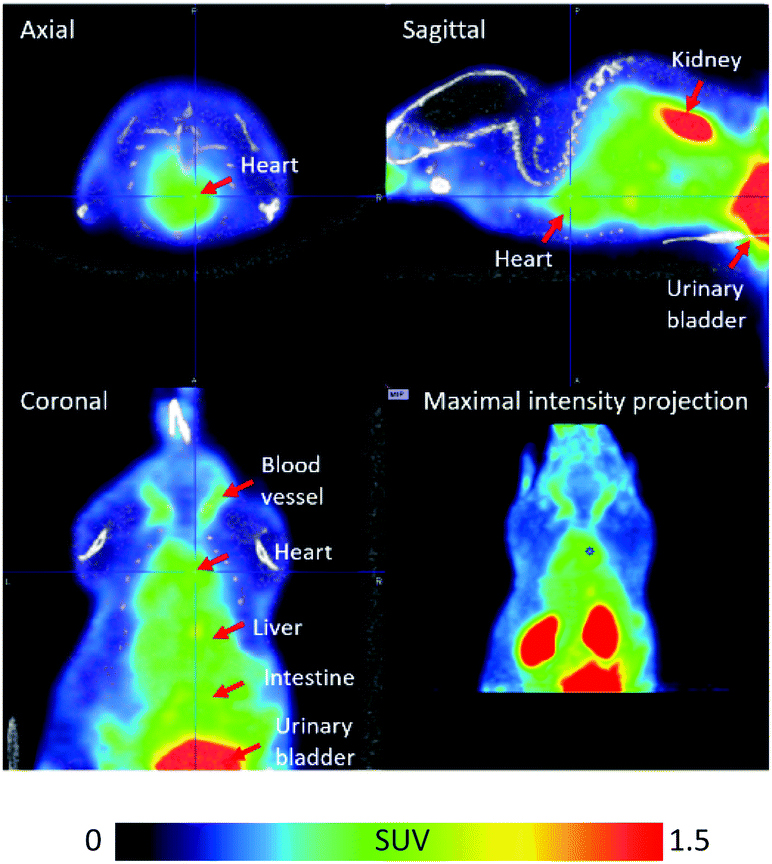 | ||
| Fig. 2 PET/CT images of [18F]FPAT averaged from 1 to 91 min post-tracer injection. The colour bar designates the standardised uptake value (SUV). | ||
In summary, we developed a new radioconjugation strategy for the 18F-radiolabelling of peptides and proteins based on KAT ligation. The novel prosthetic group [18F]FPAT was prepared in high radiochemical yield and good molar activity without any detectable 18F/19F isotopic exchange. [18F]FPAT coupled selectively within minutes in aqueous medium with O-diethylcarbamoylhydroxylamines at low concentrations with excellent conversions. Compared to modern orthogonal 18F-ligation approaches (e.g., SPAAC, IEDDA) where building blocks are appended to the biomolecules with bulky hydrophobic linkages, [18F]FPAT is tethered via a robust and innocuous amide bond which is expected to constitute a minimal perturbation of the native biomolecule. Moreover, compared to the increasingly used oxime ligation methodology, [18F]FPAT appears advantageous since its conjugation with hydroxylamine-functionalized biomolecules is unconditionally fast, and the amide adducts are expected to possess increased stability over a wide pH range in contrast to oxime conjugation products. Therefore, this approach meets all criteria of an effective orthogonal radiolabelling strategy and has the potential to become a method of choice for the 18F-radiolabelling of biomolecules. Implementation of this novel radiolabelling strategy to peptides and proteins of biological interest is currently under investigation, as well as for applications in pretargeting.
This work was supported by ETH Research Grant ETH-44 17-2. The authors thank Dr Adrienne Müller Herde and Ms Claudia Keller for performing the PET/CT scans, and Dr Hidetoshi Noda for his contributions in the early stages of this project.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Muehllehner and J. S. Karp, Phys. Med. Biol., 2006, 51, R117–137 CrossRef CAS PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- R. Schirrmacher, B. Wangler, J. Bailey, V. Bernard-Gauthier, E. Schirrmacher and C. Wangler, Semin. Nucl. Med., 2017, 47, 474–492 CrossRef PubMed.
- S. Liu, T. P. Lin, D. Li, L. Leamer, H. Shan, Z. Li, F. P. Gabbai and P. S. Conti, Theranostics, 2013, 3, 181–189 CrossRef CAS PubMed.
- Z. Liu, M. Pourghiasian, M. A. Radtke, J. Lau, J. Pan, G. M. Dias, D. Yapp, K. S. Lin, F. Benard and D. M. Perrin, Angew. Chem., Int. Ed., 2014, 53, 11876–11880 CrossRef CAS PubMed.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- Y. S. Chang, J. M. Jeong, Y. S. Lee, H. W. Kim, G. B. Rai, S. J. Lee, D. S. Lee, J. K. Chung and M. C. Lee, Bioconjugate Chem., 2005, 16, 1329–1333 CrossRef CAS PubMed.
- T. Toyokuni, J. C. Walsh, A. Dominguez, M. E. Phelps, J. R. Barrio, S. S. Gambhir and N. Satyamurthy, Bioconjugate Chem., 2003, 14, 1253–1259 CrossRef CAS PubMed.
- I. Koslowsky, J. Mercer and F. Wuest, Org. Biomol. Chem., 2010, 8, 4730–4735 RSC.
- C. Wangler, R. Schirrmacher, P. Bartenstein and B. Wangler, Curr. Med. Chem., 2010, 17, 1092–1116 CrossRef CAS PubMed.
- D. K. Kolmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef CAS PubMed.
- J. Y. Choi and B. C. Lee, Nucl. Med. Mol. Imaging, 2015, 49, 258–267 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- J. P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791–2807 CrossRef CAS PubMed.
- M. G. Campbell, J. Mercier, C. Genicot, V. Gouverneur, J. M. Hooker and T. Ritter, Nat. Chem., 2016, 9, 1–3 CrossRef PubMed.
- A. M. Dumas, G. A. Molander and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5683–5686 CrossRef CAS PubMed.
- F. Saito, H. Noda and J. W. Bode, ACS Chem. Biol., 2015, 10, 1026–1033 CrossRef CAS PubMed.
- H. Noda, G. Eros and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611–5614 CrossRef CAS PubMed.
- C. J. White and J. W. Bode, ACS Cent. Sci., 2018, 4, 197–206 CrossRef CAS PubMed.
- U. auf dem Keller, C. L. Bellac, Y. Li, Y. Lou, P. F. Lange, R. Ting, C. Harwig, R. Kappelhoff, S. Dedhar, M. J. Adam, T. J. Ruth, F. Benard, D. M. Perrin and C. M. Overall, Cancer Res., 2010, 70, 7562–7569 CrossRef CAS PubMed.
- D. M. Perrin, Acc. Chem. Res., 2016, 49, 1333–1343 CrossRef CAS PubMed.
- Z. Liu, D. Chao, Y. Li, R. Ting, J. Oh and D. M. Perrin, Chemistry, 2015, 21, 3924–3928 CrossRef CAS PubMed.
- Z. Li, K. Chansaenpak, S. Liu, C. R. Wade, P. S. Conti and F. P. Gabbaï, MedChemComm, 2012, 3, 1305–1308 RSC.
- Z. Liu, K. S. Lin, F. Benard, M. Pourghiasian, D. O. Kiesewetter, D. M. Perrin and X. Chen, Nat. Protoc., 2015, 10, 1423–1432 CrossRef CAS PubMed.
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed.
- V. W. Pike, Curr. Med. Chem., 2016, 23, 1818–1869 CrossRef CAS PubMed.
- G. R. Naumiec, L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2017, 6593–6603 CrossRef CAS PubMed.
- D. E. Olberg, J. M. Arukwe, D. Grace, O. K. Hjelstuen, M. Solbakken, G. M. Kindberg and A. Cuthbertson, J. Med. Chem., 2010, 53, 1732–1740 CrossRef CAS PubMed.
- G. Vaidyanathan and M. R. Zalutsky, Nat. Protoc., 2006, 1, 1655–1661 CrossRef CAS PubMed.
- A. Chiotellis, F. Sladojevich, L. Mu, A. Muller Herde, I. E. Valverde, V. Tolmachev, R. Schibli, S. M. Ametamey and T. L. Mindt, Chem. Commun., 2016, 52, 6083–6086 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc08645e |
| ‡ These authors made equal contributions to the paper. |
| This journal is © The Royal Society of Chemistry 2020 |