
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImaging the reactivity and width of graphene's boundary region†
Huda S.
AlSalem
 abc,
Soha T.
Al-Goul
de,
Alejandro
García-Miranda Ferrari
fg,
Dale A. C.
Brownson
fg,
Luis
Velarde
d and
Sven P. K.
Koehler
*f
abc,
Soha T.
Al-Goul
de,
Alejandro
García-Miranda Ferrari
fg,
Dale A. C.
Brownson
fg,
Luis
Velarde
d and
Sven P. K.
Koehler
*f
aSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
bPhoton Science Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cSchool of Chemistry, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
dDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, New York 14260-3000, USA
eSchool of Chemistry, King Abdulaziz University, Rabigh, Saudi Arabia
fDepartment of Natural Sciences, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, UK. E-mail: s.koehler@mmu.ac.uk
gManchester Fuel Cell Innovation Centre, Manchester Metropolitan University, Manchester M1 5GD, UK
First published on 28th July 2020
Abstract
The reactivity of graphene at its boundary region has been imaged using non-linear spectroscopy to address the controversy whether the terraces of graphene or its edges are more reactive. Graphene was functionalised with phenyl groups, and we subsequently scanned our vibrational sum-frequency generation setup from the functionalised graphene terraces across the edges. A greater phenyl signal is clearly observed at the edges, showing evidence of increased reactivity in the boundary region. We estimate an upper limit of 1 mm for the width of the CVD graphene boundary region.
Ever since the discovery of graphene's unusual properties,1–5 determining whether and how these properties differ when moving from the basal plane of graphene towards its edge has been a recurring question.3,6–10 It is intuitive that some of these properties change at the graphene edge due to the termination of the continuous π-electron cloud. Graphene edges are effectively 1-dimensional defects, and simulations have confirmed an altered reactivity at defects such as edges.7,11–13
However, results from experimental studies aiming to establish changes in the reactivity of graphene at edges compared to terrace sites are contradictory. This confusion can in parts be explained by the different definitions of reactivity.10,14–16 The above mentioned computational studies (mainly density functional theory) agree that the density of states (DOS) on edge carbon atoms is increased;11–13,17,18 this would favour edge over terrace sites with respect to e.g. the catalytic reactivity towards the oxygen reduction reaction (ORR). This conclusion has in fact been confirmed experimentally by Brownson et al. (for graphene) and Shen et al. for highly-oriented pyrolytic graphite, who both observed a higher catalytic reactivity towards the ORR at edges as compared to the basal plane.7,18 This is in stark contrast to results by Unwin and co-workers who interpreted their elegant high-resolution electrochemical imaging experiments as the basal plane displaying a faster electron transfer.19,20
The interruption of the regular hexagonal structure at graphene's edges manifests itself through the emergence of the so-called D peak (∼1360 cm−1) in the Raman spectra, which indicates defects in the graphene lattice.10 Strano and co-workers even observed an increased D peak intensity around the edges after chemical functionalisation.21 However, Cançado et al. warn that due to momentum conservation, the D peak intensity in graphite is highly dependent on the exact structure of the edge (i.e. the D peak is more intense in the armchair structure, while absent or very weak along a perfect zigzag edge).22 This can lead to errors in the interpretation of Raman spectra. In addition, while Raman spectroscopy can highlight the presence of defects in graphene, it is limited as it cannot provide information about the type of defect or functionalisation, neither at the terraces nor at the edges.
Vibrational sum-frequency generation spectroscopy (vSFG), however, is an interface-selective spectroscopy technique with sub-monolayer sensitivity, and hence ideally suited to study carbon structures at interfaces and in particular functionalised graphene,23–27 with the potential to overcome the above noted limitations. In our own previous vSFG studies of phenyl-decorated graphene,28 we noticed that different quality graphene can lead to variations in the vSFG intensities of the functional groups, possibly due to different reactivities of the carbon atoms in vicinity to defects; in accordance with the results from other groups,7,11–13,29,30 graphene samples with a higher defect concentration typically yield higher coverages with surface functional groups.
We thus set out to directly image the reactivity of graphene terraces versus edges by functionalising single-layer graphene with phenyl groups in an azo-coupling reaction (i.e. testing for electrophilic reactions), and subsequently rastering the sample stage under the laser beams, gathering snapshot vSFG signal intensities due to the phenyl functionalisation as the laser beams move across a graphene edge. Our results therefore provide new insights into the long-standing issue of terrace vs. edge reactivity by mapping the vSFG signal intensity originating from phenyl groups grafted onto graphene across the boundary region of graphene.
Graphene samples were grown by CVD, the most promising method for large-scale graphene production, and transferred onto a gold substrate using the polymer-free transfer method.31,32 For details of the phenyl functionalisation through azo-coupling, see ref. 30 and the ESI.† Details of the custom-built infrared (IR)-visible (Vis) SFG spectrometer33,34 are also provided in the ESI,† but importantly, the IR beam was mildly focused to a spot size of ∼200 μm, thus determining our spatial resolution. The spatial resolution of the Raman maps is ∼0.5 μm.
The Raman spectra of a graphene terrace before and after functionalisation clearly show the well-known 2D and G peaks (∼2711 and 1585 cm−1, respectively) for pristine graphene, but with the addition of the prominent D peak around 1391 cm−1 due to defects after functionalisation, see Fig. 1(c) and (g). These peaks are slightly red-shifted and their intensity ratios altered due to the presence of a gold layer (∼50 nm) on the substrate,35,36 which may generate gold surface plasmons, see ESI.† The Raman maps show a homogenous pristine graphene sample, see Fig. 1(e), and a homogenous distribution of defects after functionalisation, Fig. 1(f). Maps of Raman peak positions in Fig. S3 (ESI†) indicate peak shifts up to 100 μm away from the edge.
 | ||
| Fig. 1 SFG and Raman spectra and Raman maps near the edge of a graphene sheet. vSFG spectra of pristine graphene and the gold substrate side in (a and b), respectively, both with hydrocarbon impurities visible. Single point spectra of the graphene side (c) and the gold substrate side (d) with no graphene contributions on the gold side. (e) Raman map of the intensity of the G peak before functionalisation; (f) Raman map of the D peak after functionalisation showing the presence of the defect activated D peak in functionalised graphene (g) and weaker graphene peaks on the gold substrate side (h) after functionalisation, suggesting some graphene flakes have shifted to the gold side. vSFG spectra of phenyl-functionalised graphene (i) recorded at a terrace site and at the edge and (j) on the gold substrate showing a change in intensity for the aromatic C–H stretch (∼3050 cm−1) in the SFG spectrum (with hydrocarbon impurities at <3000 cm−1 unaffected). Normalised and background subtracted raw vSFG data as dotted lines, with solid lines being the best fits to eqn (S1) in the ESI.† | ||
vSFG spectra of the pristine graphene and of functionalised graphene recorded on a terrace, at the edge, and past the edge (i.e. on the gold substrate where graphene should be absent) are shown in Fig. 1(a), (b), (i) and (j). The two peaks around 2915 cm−1 and 2960 cm−1 correspond to aliphatic hydrocarbon impurities on the substrate and appear before and after functionalisation.27 More importantly, however, the peak at ∼3080 cm−1 is due to the aromatic C–H stretch and only appears after the functional phenyl group is grafted onto graphene. Henceforth, we focus on this peak for our study of graphene's terrace vs. edge reactivity as this easily distinguishable band is clearly resolved and unique to surface phenyl groups.
In order to investigate the edges of CVD graphene, we first recorded a micrograph of graphene at its edge, see Fig. 2(a), where zero along the x-axis defines the edge of graphene. The micrograph clearly shows a fairly large amount of wrinkles which are introduced, but at the benefit of fewer polymer impurities on our graphene samples. Fig. 2(b) shows a Raman map of functionalised graphene at the edge as a faded background, and the D peak intensity from the Raman map averaged over all points recorded with the same x-position as an overlay. We fitted these data points to a hyperbolic tangent function typically used to describe density changes across interfaces, and establish an interface width (over which the Raman intensity changes from 90% to 10%) of 1.1 mm.
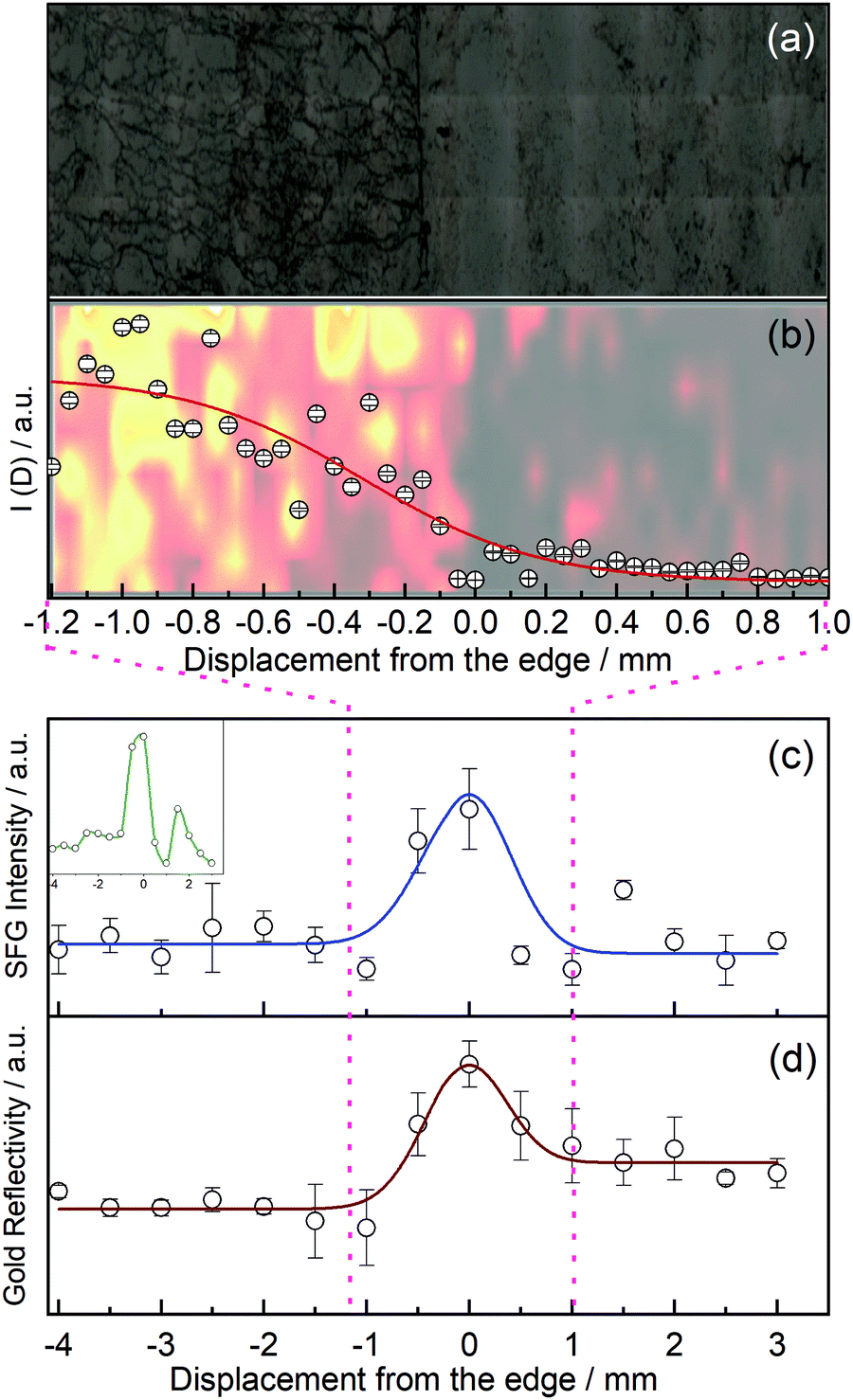 | ||
| Fig. 2 (a) Microscope image of the functionalised graphene sample highlighting the edge of the graphene, (b) intensity of the D peak as a function of distance from the edge extracted from Raman maps of functionalised graphene with fit as described in text, (c) vSFG intensity of the aromatic C–H stretch as a function of distance from the edge averaged over four measurements at four different positions (inset in green is the vSFG signal intensity from a typical single measurement), and (d) reflectivity of the sample while scanning across the surface. | ||
More importantly, we recorded the vSFG signal intensity of the phenyl C–H stretch at ∼3080 cm−1 as we translated our sample stage under the IR and Vis laser pulse pair with a raster increment of 500 μm from the terraces, across the edge, and onto the exposed neighbouring gold substrate, see Fig. 2(c). Each of the data points reported are the average of four separate measurements crossing the edge at different positions in the y-dimension. We started the sampling in the middle of the graphene terrace (negative values along the x-axis in Fig. 2) and scanned across the edge region to finish in an area of the gold substrate not initially covered by graphene, thus in situ mapping the reactivity of graphene towards electrophilic addition. The intensity of the aromatic C–H stretch changes as a function of displacement from the edge. It is worth noting that the vSFG signal on the basal plane is already higher than it would be on graphene without any wrinkles, as strain has been shown to increase the reactivity of graphene,37,38 and we have in our laboratory also noticed higher signal intensities on graphene samples with more wrinkles.28 Nonetheless, the graph still shows a clear increase in the vSFG signal intensity at the edge of the graphene sample compared to the basal plane. The vSFG intensity is related to the orientation of the molecular vibration that is probed with respect to the polarisation of the laser beams involved, and proportional to the square of the number density of surface functional groups. Due to the polarisation in our experiments (ppp for SFG/Vis/IR), and since we have previously established that the phenyl groups chemisorbed to graphene terraces are on average aligned along the surface normal,28 (any deviation from this perpendicular alignment would hence reduce the vSFG intensity; the experimental setup is hence not sensitive to any physisorbed benzene whose plane is parallel to the graphene), we conclude that molecular orientation alone cannot account for the significant intensity change in the boundary region. Instead, we attribute the origin of the signal increase to a higher phenyl concentration along the edges. These results, which indicate an enhanced reactivity in the boundary region, are in agreement with previous results,7,18 and strongly suggest that graphene is indeed more reactive at the edges than at its basal plane.
Fig. 2(d) shows the reflectivity of the gold substrate as a function of position relative to the edge, indicating different optical properties around the phenyl-rich edge region.
We independently fitted the vSFG intensity and reflectivity to modified Gaussian functions as a function of lateral displacement. The FWHM of both fits yield a width of the graphene edge of ∼(0.9 ± 0.4) mm, which correlates well with the width extracted from the Raman maps of 1.1 mm, see Fig. 2(b). We stress that both values are heavily averaged and thus overestimate the interface width, but at the advantage of vSFG imaging the reactivity across the boundary region directly.
Naturally, the width of ideal zigzag or armchair edges of graphene is on the Ångström scale. In contrast, our experiment measures the reactivity of CVD-grown and transferred graphene within our ∼200 μm diameter laser spot, therefore averaging over a relatively large area. If a straight zigzag or armchair edge would run through the centre of our laser spot of ∼200 μm diameter, then the number of basal plane carbon atoms would outnumber the edge carbon atoms by a factor of >105, and any increased reactivity of the edge atoms would be dwarfed by the sheer number of terrace atoms; we would also not detect an interface width of almost a millimetre. Instead, we speculate that the signal intensity and the width of the boundary region is increased because the edge of our supported CVD graphene is not straight, but rugged, similar to a coastline with inlets, peninsulas, and possibly small graphene islands. This can be seen in the inset of Fig. 2(c) which shows the SFG signal of a typical single scan across the boundary region (rather than averaged over four different positions along the y-axis). This inset rather shows features that are narrower than the averaged 0.9 mm width, and also indicates the likely presence of islands or peninsulas. The presence of such a ‘rugged’ edge including small islands increases the number of carbon edge atoms and folds. However, this is not to say that the edge atoms outnumber the terrace atoms; if that was the case, then there would be no graphene present, as even narrow graphene nanoribbons have more terrace than edge atoms. The presence of graphene in the boundary region is further confirmed by the Raman spectra recorded around the edge, showing the expected graphene peaks. Instead, we conclude that the number of carbon edge atoms is lower than the number of terrace atoms at any point as we scan our laser across the edge, but crucially, an increased reactivity of the edge atoms is responsible for the larger vSFG signal at the edge.
This enhanced reactivity may not only include atoms at the very edge, but as DFT calculations suggest,12,13 the reactivity of carbon atoms a few bond lengths away from the edge may still be enhanced when compared to true terrace atoms.
It is worth noting that we observe a non-negligible phenyl vSFG signal after functionalisation (but not before functionalisation) in areas of the substrate which were not originally covered by graphene. While one would expect the signal to decline completely after the edge, the reason for this non-zero signal is potentially two-fold: (1) the diazonium salt (see ESI†) used in the phenyl functionalisation binds to the gold substrate in areas which are not initially covered with graphene. In our azo-functionalisation, the phenyl cation attaches to the graphene in an electrophilic addition. It is well-known, however, that nitrogen can form bonds to Au atoms, and some of the diazonium salt may directly attach to the gold substrate, as shown in a blank test with a bare gold substrate, i.e. in the absence of graphene; the corresponding SFG spectrum is shown in the ESI† (Fig. S4b) and clearly shows an aromatic peak at 3071 cm−1, strongly indicating adsorption of the azo-compound in the absence of graphene. (2) We also find evidence for small graphene flakes to be lifted off the substrate during the azo-coupling reaction, only to adsorb again in areas of the substrate that were previously not covered by graphene. Raman spectra and maps underpinning this are shown in Fig. 1(d) and (h). One can clearly observe the absence of any graphitic peaks in the Raman spectra of areas past the edge of the graphene before functionalisation, but smaller graphitic structures are observed after functionalisation, see Fig. 1(h), and right half of the Raman map (f). These shifted graphene flakes display not only prominent G and 2D peaks, but also a D peak, indicating that these flakes themselves are functionalised.
In summary, we report spectroscopic imaging results of the reactivity of graphene edges vs. its basal plane and conclude that graphene's carbon atoms at the edges are more reactive (towards electrophilic azo-coupling) than those at graphene terraces. These results would be hard to detect on a straight graphene edge, but are enhanced in these experiments through the use of CVD graphene with a fairly rugged and wide edge. We estimate an upper limit of 1 mm for the averaged lateral width of the CVD graphene boundary region. This enhanced reactivity at the edges can be employed to create structures on graphene.
Conflicts of interest
There are no conflicts to declare.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- E. P. Randviir, D. A. C. Brownson and C. E. Banks, Mater. Today, 2014, 17, 426–432 CrossRef CAS.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS.
- K. S. Novoselov, V. I. Fal′ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- F. Liu, K. Huang, S. Ding and S. Dai, J. Mater. Chem. A, 2016, 4, 14567–14571 RSC.
- S. Casolo, O. M. Løvvik, R. Martinazzo and G. F. Tantardini, J. Chem. Phys., 2009, 130, 054704 CrossRef PubMed.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978 RSC.
- X. Jia, J. Campos-Delgado, M. Terrones, V. Meunier and M. S. Dresselhaus, Nanoscale, 2011, 3, 86–95 RSC.
- X. Zhang, J. Xin and F. Ding, Nanoscale, 2013, 5, 2556 RSC.
- C. Casiraghi, A. Hartschuh, H. Qian, S. Piscanec, C. Georgi, A. Fasoli, K. S. Novoselov, D. M. Basko and A. C. Ferrari, Nano Lett., 2009, 9, 1433–1441 CrossRef CAS PubMed.
- N. Ghaderi and M. Peressi, J. Phys. Chem. C, 2010, 114, 21625–21630 CrossRef CAS.
- R. Dettori, E. Cadelano and L. Colombo, J. Phys.: Condens. Matter, 2012, 24, 104020 CrossRef PubMed.
- F. T. Wang, L. Chen, C. J. Tian, Y. Meng, Z. G. Wang, R. Q. Zhang, M. X. Jin, P. Zhang and D. J. Ding, J. Comput. Chem., 2011, 32, 3264–3268 CrossRef CAS PubMed.
- L. Dai, Acc. Chem. Res., 2013, 46, 31–42 CrossRef CAS PubMed.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS.
- P. A. Denis and F. Iribarne, J. Phys. Chem. C, 2013, 117, 19048–19055 CrossRef CAS.
- A. Shen, Y. Zou, Q. Wang, R. A. W. Dryfe, X. Huang, S. Dou, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2014, 53, 10804–10808 CrossRef CAS PubMed.
- S. C. S. Lai, A. N. Patel, K. McKelvey and P. R. Unwin, Angew. Chem., Int. Ed., 2012, 51, 5405–5408 CrossRef CAS.
- G. Zhang, S. Tan, A. N. Patel and P. R. Unwin, Phys. Chem. Chem. Phys., 2016, 18, 32387–32395 RSC.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398–405 CrossRef CAS PubMed.
- L. G. Cançado, M. A. Pimenta, B. R. A. Neves, M. S. S. Dantas and A. Jorio, Phys. Rev. Lett., 2004, 93, 247401 CrossRef PubMed.
- T. Balgar, H. Kim and E. Hasselbrink, J. Phys. Chem. Lett., 2013, 4, 2094–2098 CrossRef CAS PubMed.
- J. L. Achtyl, I. V. Vlassiouk, S. Dai and F. Geiger, J. Phys. Chem. C, 2014, 118, 17745–17755 CrossRef CAS.
- C.-K. Lin, C.-C. Shih, Y. Niu, M.-Y. Tsai, Y.-J. Shiu, C. Zhu, M. Hayashi and S. H. Lin, J. Phys. Chem. C, 2013, 117, 1754–1760 CrossRef CAS.
- S. Xu, S. Xing, S.-S. Pei and S. Baldelli, J. Phys. Chem. B, 2014, 118, 5203–5210 CrossRef CAS PubMed.
- C. Holroyd, A. B. Horn, C. Casiraghi and S. P. K. Koehler, Carbon, 2017, 117, 473–475 CrossRef CAS.
- H. S. Alsalem, C. Holroyd, M. Danial Iswan, A. B. Horn, M. A. Denecke and S. P. K. Koehler, Phys. Chem. Chem. Phys., 2018, 20, 8962–8967 RSC.
- A. Eckmann, A. Felten, I. Verzhbitskiy, R. Davey and C. Casiraghi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 035426 CrossRef.
- B. J. Schultz, C. J. Patridge, V. Lee, C. Jaye, P. S. Lysaght, C. Smith, J. Barnett, D. A. Fischer, D. Prendergast and S. Banerjee, Nat. Commun., 2011, 2, 1–8 Search PubMed.
- G. Zhang, A. G. Güell, P. M. Kirkman, R. A. Lazenby, T. S. Miller and P. R. Unwin, ACS Appl. Mater. Interfaces, 2016, 8, 8008–8016 CrossRef CAS PubMed.
- L. A. Belyaeva, W. Fu, H. Arjmandi-Tash and G. F. Schneider, ACS Cent. Sci., 2016, 2, 904–909 CrossRef CAS PubMed.
- D. Elsenbeck, S. K. Das and L. Velarde, Phys. Chem. Chem. Phys., 2017, 19, 18519–18528 RSC.
- S. T. Algoul, S. Sengupta, T. T. Bui and L. Velarde, Langmuir, 2018, 34, 9279–9288 CrossRef CAS PubMed.
- B. G. Ghamsari, A. Olivieri, F. Variola and P. Berini, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 201408 CrossRef.
- M. Kalbac, V. Vales and J. Vejpravova, RSC Adv., 2014, 4, 60929–60935 RSC.
- X. Fan, R. Nouchi and K. Tanigaki, J. Phys. Chem. C, 2011, 115, 12960–12964 CrossRef CAS.
- P. Kovaříček, Z. Bastl, V. Valeš and M. Kalbac, Chem. – Eur. J., 2016, 22, 5404–5408 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02675a |
| This journal is © The Royal Society of Chemistry 2020 |