
Probing the peripheral role of amines in photo- and electrocatalytic H2 production by molecular cobalt complexes†
Dependu
Dolui
 a,
Ab Qayoom
Mir
a and
Arnab
Dutta
*ab
a,
Ab Qayoom
Mir
a and
Arnab
Dutta
*ab
aChemistry Discipline, Indian Institute of Technology Gandhinagar, Palaj 382355, India. E-mail: arnabdutta@chem.iitb.ac.in
bChemistry Department, Indian Institute of Technology Bombay, Powai 400076, India
First published on 2nd November 2020
Abstract
The incorporation of amine functionality in the periphery of a synthetic cobaloxime core induces excellent photo-(TON 180) and electrocatalytic H2 production (TOF 4330 s−1) in aqueous solution. The primary amine group displays a superior influence on the catalysis compared to a secondary amine group with an analogous cobaloxime template.
The emergence of H2 as an energy vector for the renewable energy economy has promoted the boom of synthetic H2 evolution reaction (HER) catalysts.1 Hydrogenase enzyme produces H2 in aqueous solution with remarkable pace and efficiency. The presence of a protein scaffold surrounding the active site plays crucial roles in achieving this feat by regulating the substrate (H+/e−) and product (H2) movement along with the structural dynamics of the metal core.2 Hence, this protein structure, also known as the outer coordination sphere (OCS), is key in imitating the enzymatic HER efficiency.3,4 Shaw and co-workers pioneered incorporating a synthetic OCS feature in the molecular catalyst design by precisely positioning amino acids and peptide motifs around the central metal framework to significantly improve the HER performance.5,6 The strategic arrangement of the basic functionalities and delicate modification of the primary coordination sphere allowed the evolution of synthetic nickel-catalysts capable of even reversible H+/H2 interconversion, a hallmark of enzyme catalysis.7,8 However, the intrinsic oxygen sensitivity of such catalysts has cast doubts on their long-term practical usability.
The inherent oxygen tolerance of cobaloxime[cobalt-(dimethylglyoxime)2] complexes prompted their usage as an alternative solution. Cobaloximes exhibit moderate H2 production when an N-heterocyclic axial donor (such as pyridine or imidazole) is appended to cobalt.9–11 The photo- or electrocatalytic HER activity of cobaloxime derivatives displays a promising improvement in catalysis in the presence of protein, metallopeptide, cyclodextrin, and even metal–organic framework-based OCS features.12–15 However, the bulkiness of the surrounding architectures has restricted the detailed analysis of their durable catalytic performance.16 We have recently anchored natural amino acids and their derivatives via the axial N-heterocycle to generate minimal but essential OCS features around a cobaloxime core, which significantly improved both its photo- and electrocatalytic H2 production in aqueous media.17,18 A close look into the structure–function relationship of these complexes revealed the plausible formation of water and the accompanying anion-mediated proton relay around the complexes to trigger a rapid proton movement under catalytic conditions.
Amine groups are considered vital factors in crafting an interactive proton relay in biology. The mercurial influence of the pendant amine on the [FeFe]-hydrogenase activity has motivated the evolution of synthetic Fe- and Ni-based analogs.19,20 In those complexes, variable protic nitrogenous functionalities such as N-heterocyclic motifs and secondary and tertiary amines were utilized to enhance the catalytic H2 production.21–23 However, the exclusive use of such functionalities for a cobalt-based H2 production catalyst template is rare. Our previous study highlighted the potential involvement of peripheral amines around a cobaloxime template, although their effect was masked in the presence of other fringe basic functionalities (carboxylate and phenol). Hence, cobaloximes containing only peripheral amines are required to unravel their impact as an OCS feature.
In the present study, we have developed a series of three complexes bearing the same cobaloxime template and varying only in the identity of the OCS feature (Scheme 1). Among them, the control complex contains an aliphatic ethyl group (C1), while the others include either a primary (C2) or a secondary amine group (C3) in the periphery.
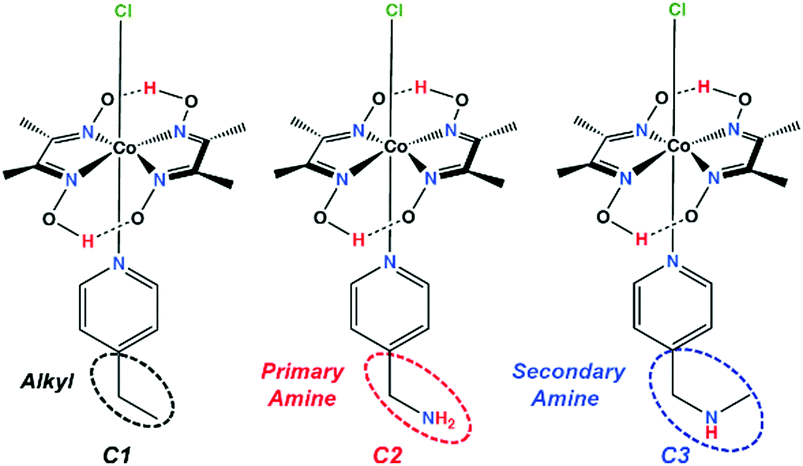 | ||
| Scheme 1 Schematic structures of the complexes C1–C3. The dotted ellipses highlight the outer coordination sphere (OCS) functionalities around the cobaloxime core. | ||
All these complexes were synthesized by the conventional stoichiometric addition of pyridine derivatives to the methanolic solution of [cobalt-(dimethylglyoxime)2] (ESI†).17 The optical spectra of these complexes exhibited the salient features of ligand-to-metal charge transfer (LMCT) (∼350 nm) and d–d transition bands (∼500–600 nm), which suggests the formation of an axial pyridine motif bound cobaloxime (Co–N5) core (Fig. S1A and Table S1, ESI†),24 which was supported further by the appearance of a characteristic absorption band at ∼500 cm−1 in the ATR-FTIR spectra (Fig. S1B and Table S1, ESI†).25 The corresponding 1H and 13C NMR spectral data of C1–C3 depicted that the axially bound pyridine derivatives remain intact even after generating the complexes (ESI†). The 1H NMR data also displayed a highly down-field shifted signal (∼18.5 ppm), indicating the formation of a hydrogen-bonded oxime network in the primary coordination sphere of the complexes (ESI†).17 The single-crystal structure of C2, grown in organic media, indicates the interaction between the primary amine with a cobaloxime core leading to the formation of a heterodimer. However, the protonation of that primary amine will ensure the generation of only an Npyridine-linked cobaloxime core in aqueous solution (pH ≤ 7) (Fig. S2 and Table S2, ESI†).
The electrochemical properties of the complexes were first inspected in polar aprotic acetonitrile media, where C1–C3 displayed two distinct signatures corresponding to quasi-reversible Co(III/II) and reversible Co(II/I) redox processes, respectively (Fig. S3 and Table S1, ESI†).9,25 The Co(III/II) reduction peak for C1 shifted by ∼100 mV towards a more negative potential (−1.11 V vs. FeCp2+/0) compared to amine-containing C2 and C3 (∼−1.0 V vs. FeCp2+/0) in acetonitrile. The weakened Npyridine→Co σ-donation for C2 and C3, due to the presence of electron-withdrawing amine groups, is presumably the reason behind this variation.16,24
The inclusion of the amine moiety improved the solubility of both C2 and C3, compared to moderately water-soluble C1. Hence, it was possible to probe the redox properties for all the complexes in an aqueous solution. The CV data for all the complexes (C1–C3) exhibited three distinct features during the reductive scan in pH 7.0 solution (Fig. 1A (inset 1) and Fig. S4, ESI†). The first reduction peak was noticed at −0.1 V (vs. SHE) during the scan, starting from a relatively positive direction. This signal belongs to a quasi-reversible stoichiometric Co(III/II) redox step analogous to the previously reported water-soluble pyridine-bound cobaloximes.17,24 An irreversible reductive response was observed next at −0.7 V (vs. SHE). This signal is catalytic in nature (for the HER activity), and it originated due to an ECEC catalytic mechanism that revolved around the Co(II)-centre (Fig. S5, ESI†).11,17,26 A relatively sharper catalytic H2 production signal was observed upon further reducing the potential starting from −0.85 V (vs. SHE). This dominant catalytic response originated from the Co(I)-centred EECC mechanism (Fig. S5, ESI†).17,26
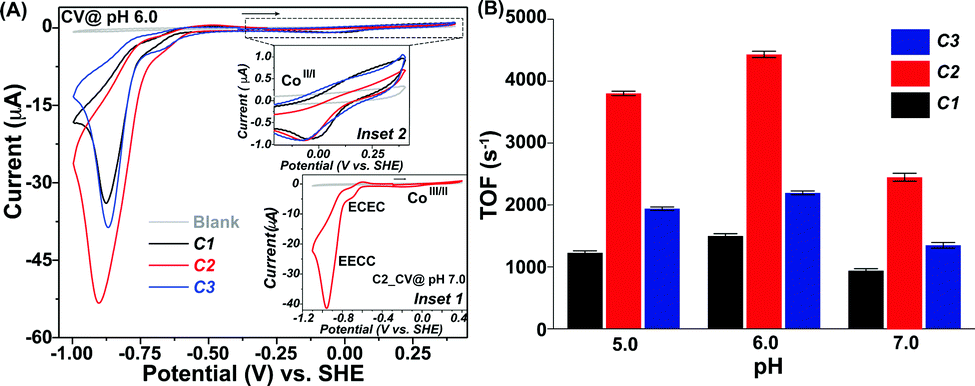 | ||
| Fig. 1 (A) Comparative cyclic voltammogram (CV) data for C1 (black trace), C2 (red trace), and C3 (blue trace), and blank solution (grey trace) recorded at pH 6.0, while the inset 1 and 2 exhibit the CV response for C2 (red trace) in pH 7.0 and the Co(III/II) stoichiometric signal for C1–C3 at pH 6.0 (scan rate 1 V s−1). (B) Variation of the electrocatalytic H2 production TOF values for C1 (black bar), C2 (red bar), and C3 (blue bar) recorded in aqueous solution ranging from pH 5.0 to 7.0. The horizontal arrows display the initial scan direction. The standard deviation values were calculated from at least three independent experimental data. | ||
The ratio of the catalytic current (icat) and the stoichiometric signal (from the Co(III/II) peak) (ip) was utilized (eqn (S1), ESI†) for evaluating the catalytic rate (kobs or the turn over frequency/TOF) of H2 production by these complexes. On the other hand, the parameter overpotential signifies the energy efficiency of these electrocatalysts that is measured as a difference between the thermodynamic H+/H2 couple (EH+/H2) and the potential of the half maxima of the catalytic current (Ecat/2) (eqn (S2), ESI†). The complexes exhibited weak H2 production via the ECEC mechanism at a relatively positive potential, while a rapid HER catalysis, proceeding through the EECC mechanism, was observed at a comparatively negative potential. Among the three complexes, the primary amine-containing C2 displayed the best EECC-mechanistic HER rate (2448 s−1) followed by secondary amine appended C3 (1350 s−1) in pH 7.0 aqueous solution (Fig. 1B and Table 1). The amine-deprived control complex C1 was the slowest H2 producer (TOF 930 s−1) under similar conditions, which is similar to the unsubstituted pyridine-coordinated cobaloxime.17 Although there was not much difference in the overpotential requirement for these complexes during the ECEC mechanism-based HER, they behaved differently during the fast EECC type catalysis. In the latter condition, C2 exhibited better energy efficiency than C1 and C3 (Table 1).
| Complex | Electrocatalytic HER | Photo-catalytic HER (TON vs. PS)c | |||||
|---|---|---|---|---|---|---|---|
| pH 5.0 | pH 6.0 | pH 7.0 | |||||
| TOFa (s−1) | OPb (mV) | TOF (s−1) | OP (mV) | TOF (s−1) | OP (mV) | ||
| a TOF: Turnover frequency. b OP: Overpotential. c PS: Photosensitizer.The standard deviation values were calculated from at least three independent experimental data. | |||||||
| C1 | 1228 ± 33 | 465 ± 5 | 1500 ± 37 | 459 ± 2 | 930 ± 41 | 476 ± 2 | 128 |
| C2 | 3800 ± 35 | 457 ± 5 | 4331 ± 52 | 448 ± 2 | 2448 ± 65 | 455 ± 2 | 180 |
| C3 | 1940 ± 28 | 488 ± 5 | 2195 ± 31 | 464 ± 2 | 1350 ± 45 | 496 ± 2 | 146 |
As the pH of the solution is lowered to 6.0, the ECEC-type HER signal became negligible for C1–C3 (Fig. 1A). On the contrary, the EECC-based H2 production signal improved under this condition. All the complexes displayed their fastest catalytic rate at pH 6.0 in the order of C2 > C3 > C1, analogous to the previous study at pH 7.0. Here, C2 reached the maximum TOF of 4331 s−1, which is one of the highest HER rates for cobaloxime derivatives containing only one basic functionality (primary amine for C2) in the OCS. The primary amine-containing C2 again demonstrated a lower overpotential requirement (448 mV) among all the complexes (Table 1).
Further reduction in the pH of the solution (pH 5.0) truncated the dominant EECC catalytic signal for all the complexes, while the catalytic response originating from the ECEC mechanism remained inconsequential (Fig. 1B and Fig. S6, ESI†). C2 continued to showcase its superior energy efficiency for H2 production over C1 and C3, even at pH 5.0 (Table 1). The complementary spectroelectrochemical experiments exhibited the continuous presence of the Npyridine→Co LMCT band during the reducing scan which suggested the stability of the Co–Npyridine axial bond even under catalytic conditions (Fig. S7, ESI†).
The catalytic response for the complexes decreased significantly below pH 5.0, largely due to the protonation-induced depletion of the oxime hydrogen-bonded network present in the primary coordination sphere.17 Hence, no HER electrocatalysis was performed for C1–C3 at pH < 5.0. Chronocoulometric experiments were performed for all the complexes at pH 6.0 (at their peak catalytic performance). They exhibited robust homogeneous catalytic activity with significant H2 production during this bulk electrolysis, which was detected by complementary gas chromatography (GC) experiments (Fig. S8, ESI†). The catalyst solution's optical spectra exhibited a minimal change which indicated the stability of C1–C3 during the chronocoulometric experiments (Fig. S9, ESI†). The plastic chip working electrode used during the experiment was further probed via field emission scanning electron microscopy in conjunction with energy-dispersive X-ray spectroscopy (FE-SEM/EDS).27 The results from this data displayed no cobalt deposition on the electrode material which indicated the robustness of the complexes under electrocatalytic HER conditions (Fig. S10, ESI†).
The electrochemical data established C2 as the best electrocatalyst for aqueous H2 production in this current series of complexes, followed by C3. The direct participation of the peripheral amine functionalities in H+ exchange during the catalysis is believed to be one of the chief reasons behind the improved reactivity of C2 and C3 over C1. Two-dimensional NMR technique exchange spectroscopy (EXSY) was performed to probe this hypothesis further. The C1 complex only displayed proton exchange between the hydrogen-bonded oxime groups with the bulk water with no participation from the alkyl-based OCS, analogous to the cobaloxime containing unsubstituted pyridine ligand (Fig. S11, ESI†).17 The presence of the primary amine group in C2 ensures a three-way proton exchange between the oximes, –NH2, and bulk water, which was evident from the appearance of multiple cross-peaks in the EXSY spectrum (Fig. 2). Analogously, the EXSY data of C3 depicted interactive proton hopping involving secondary amine, oxime, and external water molecules (Fig. S12, ESI†). This series of two-dimensional NMR experiments revealed the dynamic nature of the amine functionalities dangling from the axial pyridine motif, which play a vital role in amplifying the proton traffic between the bulk solution and the cobaloxime core. The EXSY experiment with the solution containing C2 and a stoichiometric amount of L2 ligand did not exhibit any new cross-peaks which indicated the intramolecular interaction between the protic functionalities present in the cobaloxime framework. This hypothesis was corroborated further by the lack of improvement in the catalytic HER behavior when the external L2 ligand was added to the unsubstituted pyridine-linked cobaloxime (Fig. S13, ESI†). The addition of D2O instead of H2O significantly affects the electrochemical behavior of C2 in organic media, which highlights the vital role of water molecules in inducing intramolecular proton exchange (Fig. S14, ESI†).
 | ||
| Fig. 2 Two-dimensional EXSY spectra recorded for C2 in d6-DMSO, containing a minute amount of water, with 350 ms mixing time at 298 K. The dotted lines connecting the cross peaks demonstrate the exchange between primary amine–oxime (sky blue trace), primary amine–water (red trace), and oxime–water (violet trace). | ||
Thus, the inclusion of amine groups in the OCS of cobaloximes resulted in improved electrocatalytic H2 evolution in aqueous solution. The complexes C1–C3 were probed further under photocatalytic conditions to gauge the effect of these peripheral amine functionalities beyond the electrochemical conditions. Cobaloximes, axially coordinated to pyridine derivatives, are known for photo-driven H2 production in conjunction with a photosensitizer (PS) Eosin-Y (Fig. S15, ESI† inset) and a sacrificial electron-donor triethanolamine (TEOA) in the presence of at least 30% of organic solvents.28 However, its photocatalytic HER activity was slow (TON ∼220 over 30 hours of irradiation), while significant photo-degradation of the complex was noticed. The inclusion of protic carboxylic acid groups around the pyridine improved the light-driven H2 production; however, the long-term stability of cobaloxime derivatives remained an issue.29 Here, the photocatalytic H2 production by complexes C1–C3 was investigated in pure aqueous solution (pH 7.0) in the presence of a typical setup consisting of a Eosin-Y and TEOA mixture.
All the complexes exhibited significant H2 production in neutral aqueous solution under visible light irradiation (λex 335–610 nm) (Fig. S15A, ESI†). During this experiment, the evolution of H2 was monitored periodically by GC (Fig. S15B, ESI†), which exhibited no lag period or photo-degradation of the sample during 6 hours of irradiation. Analogous to the electrochemical HER results, the same trend of C3 > C2 > C1 remained valid even for the photocatalytic H2 production performance. C2 exhibited the maximum TON of 180 over 6 hours of photo-irradiation, while C1 and C3 exhibited 128 and 146 turnovers, respectively (Fig. S15A, ESI† and Table 1). Thus, the presence of a primary amine proves to be critical in establishing one of the best photocatalytic HER performances by Npyridyl-linked cobaloximes under pure aqueous conditions.18 The complementary dynamic light scattering (DLS) experiments on either side of photocatalysis confirmed the homogenous nature of the catalyst during the study (Fig. S16, ESI†).
In summary, the current study of three cobaloxime derivatives depicts the importance of the strategic incorporation of amine functionalities as an OCS feature, which boosted the catalytic H2 production by an otherwise identical synthetic catalyst. The primary amine-decorated C2 illustrated one of the fastest electrocatalytic H2 production rates for the cobaloxime-pyridine template containing a solitary basic group in the periphery. The primary amine group (C2) proves to be superior in enhancing both the photo- and electrocatalytic HER performance compared to the secondary amine (C3), presumably due to the improved proton exchange. Hence, this set of complexes has allowed us to obtain a snapshot of the intricate role of peripheral amines in remotely influencing the catalytic active site to develop the next-generation artificial catalyst design strategies.
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Artero, Nat. Energy, 2017, 2, 1–6 CrossRef.
- B. Ginovska-Pangovska, M.-H. Ho, J. C. Linehan, Y. Cheng, M. Dupuis, S. Raugei and W. J. Shaw, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 131–138 CrossRef CAS.
- B. Ginovska-Pangovska, A. Dutta, M. L. Reback, J. C. Linehan and W. J. Shaw, Acc. Chem. Res., 2014, 47, 2621–2630 CrossRef CAS.
- J. A. Laureanti, M. O’Hagan and W. J. Shaw, Sustainable Energy Fuels, 2019, 3, 3260–3278 RSC.
- A. Jain, S. Lense, J. C. Linehan, S. Raugei, H. Cho, D. L. DuBois and W. J. Shaw, Inorg. Chem., 2011, 50, 4073–4085 CrossRef CAS.
- M. L. Reback, G. W. Buchko, B. L. Kier, B. Ginovska-Pangovska, Y. Xiong, S. Lense, J. Hou, J. A. S. Roberts, C. M. Sorensen, S. Raugei, T. C. Squier and W. J. Shaw, Chem. – Eur. J., 2014, 20, 1510–1514 CrossRef CAS.
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS.
- N. Priyadarshani, A. Dutta, B. Ginovska, G. W. Buchko, M. O’Hagan, S. Raugei and W. J. Shaw, ACS Catal., 2016, 6, 6037–6049 CrossRef CAS.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS.
- D. Dolui, S. Ghorai and A. Dutta, Coord. Chem. Rev., 2020, 416, 213335 CrossRef CAS.
- M. Bacchi, G. Berggren, J. Niklas, E. Veinberg, M. W. Mara, M. L. Shelby, O. G. Poluektov, L. X. Chen, D. M. Tiede, C. Cavazza, M. J. Field, M. Fontecave and V. Artero, Inorg. Chem., 2014, 53, 8071–8082 CrossRef CAS.
- S. R. Soltau, P. D. Dahlberg, J. Niklas, O. G. Poluektov, K. L. Mulfort and L. M. Utschig, Chem. Sci., 2016, 7, 7068–7078 RSC.
- M. Kato, K. Kon, J. Hirayama and I. Yagi, New J. Chem., 2019, 43, 10087–10092 RSC.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS.
- D. Dolui, S. Khandelwal, P. Majumder and A. Dutta, Chem. Commun., 2020, 56, 8166–8181 RSC.
- D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, ACS Catal., 2019, 9, 10115–10125 CrossRef CAS.
- D. Dolui, S. Das, J. Bharti, S. Kumar, P. Kumar and A. Dutta, Cell Rep. Phys. Sci., 2020, 1, 100007 CrossRef.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J.-M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS.
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS.
- M. E. Ahmed, S. Dey, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2018, 140, 12457–12468 CrossRef CAS.
- R. Zaffaroni, W. I. Dzik, R. J. Detz, J. I. van der Vlugt and J. N. H. Reek, Eur. J. Inorg. Chem., 2019, 2498–2509 CrossRef CAS.
- S. Lense, A. Dutta, J. A. S. Roberts and W. J. Shaw, Chem. Commun., 2014, 50, 792–795 RSC.
- D. W. Wakerley and E. Reisner, Phys. Chem. Chem. Phys., 2014, 16, 5739–5746 RSC.
- A. Panagiotopoulos, K. Ladomenou, D. Sun, V. Artero and A. G. Coutsolelos, Dalton Trans., 2016, 45, 6732–6738 RSC.
- E. S. Rountree, D. J. Martin, B. D. McCarthy and J. L. Dempsey, ACS Catal., 2016, 6, 3326–3335 CrossRef CAS.
- M. Perween, D. B. Parmar, G. R. Bhadu and D. N. Srivastava, Analyst, 2014, 139, 5919–5926 RSC.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS.
- J. Wang, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and X. Wang, Dalton Trans., 2015, 44, 17704–17711 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, experimental procedures, and additional figures. CCDC 2040107. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05786j |
| This journal is © The Royal Society of Chemistry 2020 |