
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProof-of-principle for two-stage photodynamic therapy: hypoxia triggered release of singlet oxygen†
Seylan
Ayan‡
a,
Gurcan
Gunaydin‡
b,
Nisa
Yesilgul-Mehmetcik
c,
M. Emre
Gedik
b,
Ozlem
Seven
*c and
Engin U.
Akkaya
 *d
*d
aDepartment of Chemistry, Bilkent University, 06800, Ankara, Turkey
bDepartment of Basic Oncology, Hacettepe University, 06100, Ankara, Turkey
cUNAM-National Nanotechnology Research Center, Bilkent University, 06800, Ankara, Turkey. E-mail: ozlemseven90@yahoo.com
dState Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Road, 116024 Dalian, China. E-mail: eua@dlut.edu.cn
First published on 16th November 2020
Abstract
We propose to overcome oxygen deficiency and light attenuation problems in photodynamic therapy (PDT), by separating photoexcitation and singlet oxygen delivery of the PDT process into two distinct operations to be carried out sequentially, at different locations. We now demonstrate the viability of this approach, using 2-pyridone derivative which yields a relatively stable endoperoxide. The initial storage endoperoxide obtained is transformed enzymatically into a more labile compound when placed in hypoxic cell cultures, and releases singlet oxygen significantly faster. The potential of this approach in advancing PDT beyond its current limits is exciting.
More than 100 years after its initial discovery,1 clinical applications of photodynamic Therapy (PDT) are still highly limited. The fact that two of the critical components of PDT, namely oxygen and light, are very difficult to bring together inside a tumour, regardless of the wavelength of the irradiation are the two main reasons. While recent years witnessed an impressive rise in the interest in photodynamic action and its control,2 the troublesome issues of light penetration and tumour hypoxia continue to block further progress.
We are interested in transforming PDT into a broadly applicable therapeutic protocol.3 Our approach to achieve that is to separate the photosensitization event from the delivery of singlet oxygen, which is widely believed to be the primary cytotoxic agent of PDT. Thus, a “storage” compound for singlet oxygen has to be designed,4 which can react with molecular oxygen under typical photosensitization conditions, without any limitation on the wavelength of irradiation, since it is to be carried out ex situ, and then, the metastable product has to be transferred to the tumour site where it would release its cargo, preferably in response to a cancer related chemical or enzymatic cue. This approach postulates that singlet oxygen produced stoichiometrically (as opposed to catalytically through photosensitization) by the chemical transformation of the carrier molecule, would be sufficient to trigger apoptotic response in cancer cells. While to the best of our knowledge, there is no data concerning the value of intracellular quantum yields of singlet oxygen generation by any photosensitizer, a reasonable comparison of the photochemically generated singlet oxygen versus that from endoperoxide decomposition is to be found in one of our earlier studies.3b Using a bimodular compound (thus, ensuring equal concentrations of the photosensitizer and the endoperoxide), it was shown that the initial relative rate of the singlet oxygen generation by photosensitization is only 60-fold faster compared to the endoperoxide-released singlet oxygen, even in oxygen-saturated DMSO. The quantum yield of the intracellular photosensitized singlet oxygen generation is expected to rapidly approach zero, as cellular hypoxia becomes more severe.
As for the singlet oxygen storage in the first stage, we considered various options such as arenes, 2-pyridone and furan derivatives. They all form endoperoxides of varying thermal stabilities when reacted with singlet oxygen generated by photosensitization. Most of these endoperoxides release singlet oxygen when they undergo cycloreversion. A few years ago, we demonstrated that photothermally generated singlet oxygen from anthracene endoperoxides attached to gold nanorods clearly led to apoptosis in cancer cell cultures.3a Since endoperoxide decomposition rates generating singlet oxygen show a wide variation depending on the kind of arene, and substituent-related stereoelectronic factors, we believe that it is possible to control and alter singlet oxygen generation rates by structural (chemical) changes which can be induced in vivo. This could regenerate singlet oxygen, in principle, where it is needed without oxygen or light (Fig. 1).
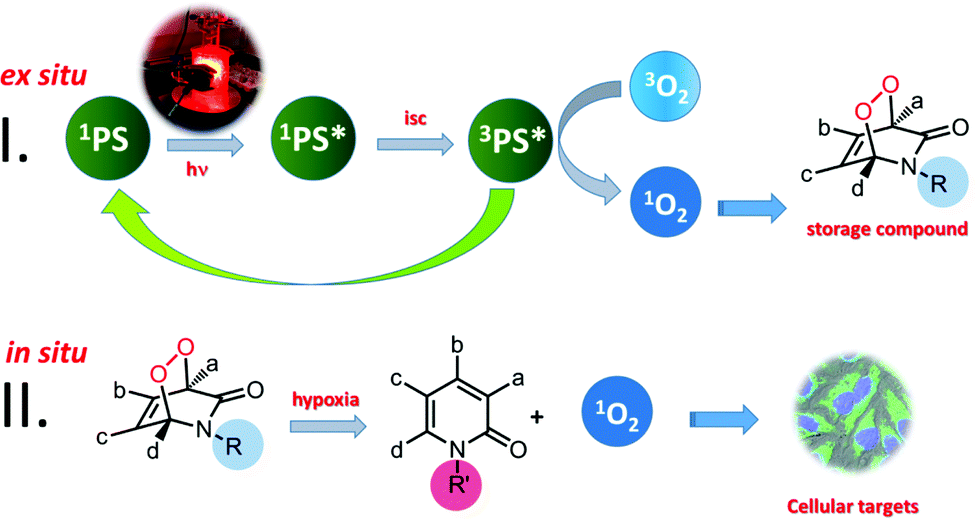 | ||
| Fig. 1 2S-PDT may circumvent the problems of light attenuation and oxygen deficiency in tumours, which currently limit clinical applications of PDT. Photosensitization (Stage I) is carried out ex situ, or in vitro, so the wavelength of excitation is not relevant, as long as the cycloaddition reaction proceeds with good yields. The endoperoxide product (storage compound) is then to be transferred to the target, where it is bioreductively changed (Stage II) into a more labile version of itself, at that point rapidly releasing singlet oxygen in the hypoxic region of a tumour. | ||
2-Pyridone endoperoxides were previously studied by us3b and others5 and known to be reliable sources for singlet oxygen. The cycloreversion is slower when the pyridone ring is substituted, and electron withdrawing substituents also decrease the reaction rate.5b A relatively straightforward approach to stabilize pyridine endoperoxides would be placing an acyl substituent on the pyridone nitrogen. It would be crucial to choose this substituent such that it could be removed under conditions unique to the tumour microenvironment. For example, hypoxia activated prodrugs6 or hypoxia probes7 make use of the reductive environment of the hypoxic tumour cells with the catalytic activity of nitroreductase enzymes.8 Nitroreductase was found to be upregulated and active in cancer cells under hypoxic conditions.9 It is well known that formation of a labile 4-aminobenzyloxy moiety, triggers a rapid bond cleavage via azaquinone methide elimination.10 With these considerations, we targeted the synthesis of N-(pNZ)-2-pyridone endoperoxide 1, with an expectation that the cycloreversion of the pNZ-protected endoperoxide should be slower (Fig. 2) compared to the parent compound due to the electron-withdrawing effect of the carbamate group. The pNZ protecting group can easily be removed under bioreductive conditions of tumour hypoxia.11 Synthesis procedures and additional data are available in the ESI.†
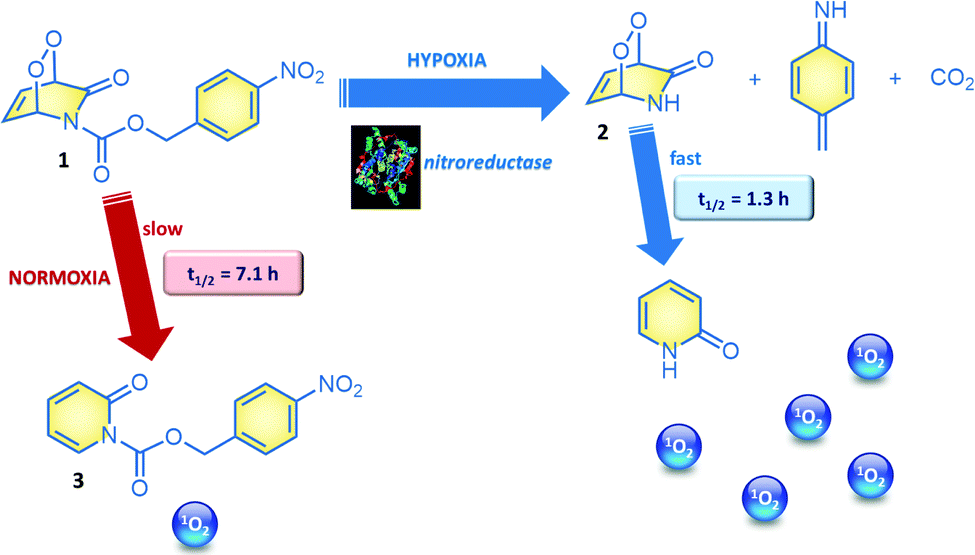 | ||
| Fig. 2 Hypoxia mediated activation of the endoperoxide 1. pNZ-protected 2-pyridone-endoperoxide 1 cycloreverts to the parent compound 3 with half-life of 7.1 hours at 37 °C. In cell cultures under hypoxic conditions, a pNZ group is reductively eliminated. The resulting pyridone endoperoxide 2, decomposes more than 5-fold faster to release singlet oxygen. | ||
In order to validate our design, we studied the rate of cycloreversion of endoperoxide 1 at 37 °C. Cycloreversion reaction can be followed by 1H NMR (ESI). We determined that the half-life of the pNZ-endoperoxide 1 was 5.5 times larger compared to the parent pyridone endoperoxide 2, which is to be produced by the action of nitroreductase on 1 under hypoxic conditions. This difference in the reaction rates is large enough to have a differential impact in their activity against the cancer cells. We also determined singlet oxygen mediated cytotoxicity of the target endoperoxide under normoxic and hypoxic conditions; MCF7 breast cancer cells were placed in a humidified modular incubator chamber containing 0.5% O2, 5% CO2 and 94.5% N2 (v/v).
Control cells were incubated under identical conditions for the same duration, but in normoxia. Subsequently, a hypoxic group of cells were incubated 24 h further under conditions of hypoxia, whereas, the normoxic group of cells was kept under normoxic conditions for the same period of time. Both the normoxia group and the hypoxia group (at the end of the pre-hypoxia period) of the cells were treated with varying concentrations of endoperoxide 1. The hypoxia group of cells was incubated further under conditions of hypoxia with endoperoxide 1; while the normoxia group of cells was incubated further under conditions of normoxia with endoperoxide 1 for 24 h. Glutathione (GSH) is a chemical and physical quencher of singlet oxygen12 and protects against its cytotoxic damage. L-Buthionine-sulfoximine (BSO) is an inhibitor of GSH synthesis, targeting γ-glutamylcysteine synthetase.13 Inhibition of its activity by BSO, sensitizes breast cancer cells to oxidative stress. Treatment of MCF7 cells with 100 μM BSO has been shown to reduce the glutathione content by approximately 55%.14 Therefore, we utilized BSO to inhibit GSH activity in MCF7 cells. For this reason, in another set of experiments, we also treated MCF7 cells with varying concentrations of endoperoxide 1 in the presence of 100 μM BSO under conditions of hypoxia, in order to better demonstrate selectivity to be achieved in hypoxic conditions in a BSO/singlet oxygen dual action scheme, mimicking multi-drug combination therapeutic protocols. The results of each of the groups (normoxia, hypoxia and hypoxia + BSO) were normalized to untreated negative control samples in either normoxic, hypoxic or hypoxic + BSO conditions (100% in Fig. 3). Therefore, the effects of hypoxic conditions or BSO treatment (without endoperoxide 1 treatment) alone on the cells could be eliminated, and the results revealed by the MTT assay data were just due to the action of endoperoxide 1. While there is some cytotoxicity of endoperoxide 1 even under normoxic conditions (at higher dose) (Fig. 3), the cytotoxicity is more pronounced under hypoxic conditions. At 66.7 μM endoperoxide 1 concentration, the cell death percentage is 23.1% under normoxia, whereas under hypoxia the cell death jumps to 34.2%, and with the BSO addition the cell death reaches 50.2% (Fig. 3). We determined the IC50 values of endoperoxide 1 under either normoxic or hypoxic conditions (with or without BSO) to be (IC50[normox]: 162 μM, IC50[hypox]: 91 μM, IC50[hypox + BSO]: 34 μM) (Fig. 3). We also had to eliminate any complications that may arise from a possible cytotoxicity of the compound 3 and any other reduction by-product. We demonstrated that (Fig. S11, ESI†) the control compound 3 had negligible toxicity either under hypoxic or normoxic conditions, even at very high concentrations of 1.6 mM. Also, no toxicity of compound 1 was observed with normal cells in the concentration range studied (Fig. S23, ESI†).
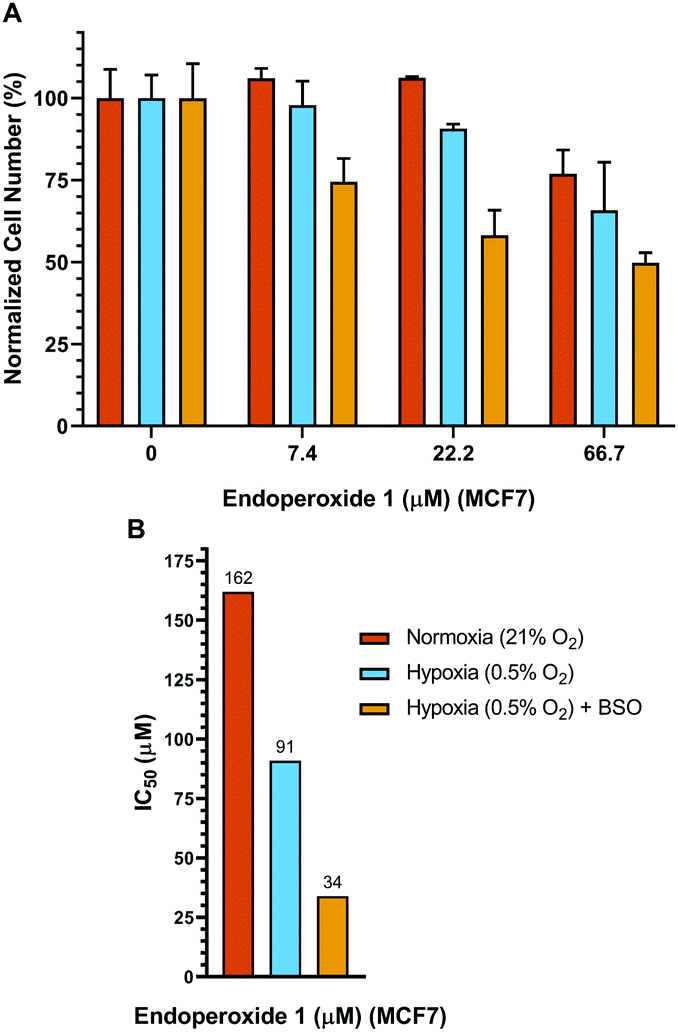 | ||
| Fig. 3 (A) Cell viabilities of MCF7 breast cancer cells were evaluated with the MTT assay after 24 hours of treatment with varying concentrations of compound 1 under either normoxia or hypoxia (with or without BSO); and normalized cell numbers are shown (mean ± SD). (B) IC50 values of compound 1 under either normoxic or hypoxic (with or without BSO) conditions are shown. Blue bar corresponds to normoxic conditions, red bar corresponds to hypoxic conditions; whereas orange bar corresponds to hypoxic conditions with 100 μM BSO. | ||
Flow cytometry is also useful in assessing differential cytotoxicity of the endoperoxide 1 under hypoxic and normoxic conditions (Fig. S20, ESI†). As singlet oxygen induces apoptosis, human suspension cancer cells (K562) were incubated with FITC-Annexin V (specifically targets and identifies apoptotic cells) and then the changes in the percentage of apoptotic fractions following endoperoxide 1 treatment were analysed. The number of cells labelled with Annexin V is greater under hypoxia, compared to normoxia (25.3% vs. 16.0% at 25 μM). This is yet another demonstration of higher cytotoxicity of the endoperoxide 1 under hypoxic conditions. In addition to the detection of apoptosis via Annexin V, cell viabilities of K562 cells were evaluated also with the MTT assay after 24 hours of treatment with varying concentrations of endoperoxide 1 under either normoxic or hypoxic conditions (Fig. S12, ESI†), utilizing a procedure similar to the one explained for MCF7 cells.
MTT assays and flow cytometry focus on the end point of the activity, and a time dependent response to the endoperoxide 1 should be more revealing for this study. This is important because processes like the establishment of the hypoxic conditions, reductive elimination of a pNZ group, release of singlet oxygen, and the initiation of apoptotic response have to take place sequentially, and it may be possible to observe a combined effect of these processes by recording time-dependent cellular impedance. To that end, cellular impedence analysis was performed: after a day of incubation under normoxic conditions, the hypoxic group of cells was incubated 24 h further under conditions of hypoxia (without endoperoxide 1 treatment); whereas, the normoxic group of cells was kept under normoxic conditions for the same period of time. At the end of this period, both groups of cells were treated with varying concentrations of endoperoxide 1. The hypoxia group of cells was further incubated under conditions of hypoxia with endoperoxide 1; while the normoxia group of cells was incubated further under conditions of normoxia with endoperoxide 1. The cell indices were measured by electrical impedance over an additional period of up to 24 hours. The results of each of the groups (hypoxia and normoxia) were normalized to relevant untreated control samples under either hypoxic or normoxic conditions, and the results were shown as a “percent of control” (t = 0 h corresponds to time of addition of endoperoxide; thus, 0 h values were used for the time point for temporal normalization). This approach provides a method for spatial and temporal dynamic view of the cell populations under the action of singlet oxygen as it is released by compounds 1 or 2 cycloreversions under normoxic or hypoxic conditions, respectively. The temporal profile of the effects of compound 1 on cancer cells (Fig. 4) demonstrates potent cytotoxicity under hypoxic conditions, and at 50 μM concentration of the compound 1, there is a significant dip in cell viability between 4 and 12 h region under hypoxia. The corresponding change under normoxic conditions is minimal. Real-time impedance-based analysis enables the observation of the time dependency of cytotoxicity due to compound 1. Singlet oxygen generation by endoperoxide 1 under normoxic and hypoxic conditions in vitro were analysed via a ROS sensor 2′,7′-dichlorofluorescein diacetate (DCFH2-DA), which generates green emission upon oxidation (Fig. 5). HeLa cells which were treated with compound 1 and incubated under hypoxic conditions demonstrated a bright green emission (Fig. 5B), in contrast to those kept under normoxia (Fig. 5A). The results show a noteworthy difference of ROS generation between the hypoxic and normoxic groups, and confirm the proof of principle concerning the hypoxia triggered intracellular singlet oxygen release. Since DCFH2-DA detects a variety of reactive oxygen species, a group of HeLa cells kept under hypoxia and a group of HeLa cells kept under normoxia were treated with NaN3, a known singlet oxygen quencher, together with endoperoxide 1, in order to prove that the generated ROS is indeed singlet oxygen. Cells treated with NaN3 as well as endoperoxide 1 demonstrated very weak emission, both under normoxic and hypoxic conditions (Fig. 5C and D), showing that endoperoxide 1 produces singlet oxygen as the primary cytotoxic agent. Control groups resulted in no emission (Fig S22, ESI†), confirming that compound 1 was responsible for the generation of singlet oxygen. Our microscopy results as well as our findings of the temporal effects of compound 1 clearly present a significant difference of singlet oxygen generation between hypoxic and normoxic conditions, thus confirming hypoxia triggered intracellular singlet oxygen release.
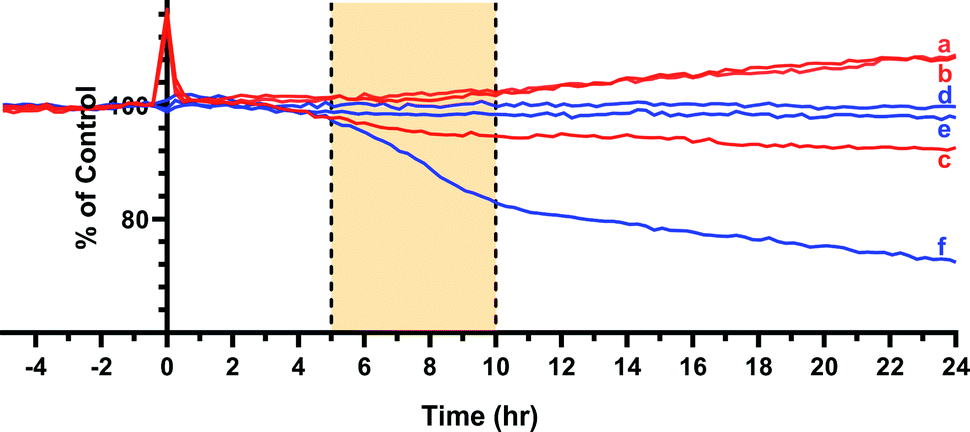 | ||
| Fig. 4 Cellular impedance analysis based cell viability assessment of MCF7 cells, as a function of time. Blue lines correspond to normalized cell numbers of MCF7 cells treated with endoperoxide 1 under hypoxic conditions (f: 50 μM, e: 25 μM, d: 12.5 μM). Red lines correspond to cells kept under identical conditions of incubation with endoperoxide 1, but under normoxic conditions (c: 50 μM, b: 25 μM, a: 12.5 μM). Control corresponds to cells incubated without endoperoxide 1 treatment, under either normoxic or hypoxic conditions. | ||
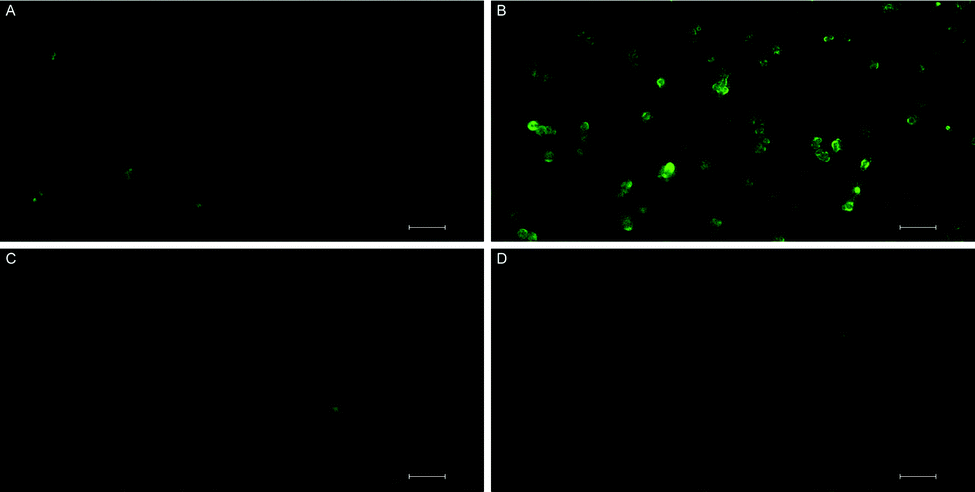 | ||
Fig. 5 Singlet oxygen generation by endoperoxide 1 under normoxic and hypoxic conditions in vitro was analysed using DCFH2-DA in HeLa cells. (A) Cells treated with 1 and incubated under normoxic conditions. (B) Cells treated with 1 and incubated under hypoxia. (C) Cells treated with NaN3, together with 1. (D) Cells that were treated with NaN3 together with 1, under hypoxic conditions (scale bar, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μm). μm). | ||
In conclusion, we presented an exciting new approach to circumvent persistent problems of PDT, while maintaining the advantages of a short-lifetime cytotoxic agent which is known to integrate very well into cellular apoptotic pathways. The fact that singlet oxygen release is conditional on the presence of hypoxic tumour states clearly enhances the potential of the approach. Just like hypoxia activated drugs, an endoperoxide can be transformed into a more labile form enzymatically through reductive elimination of the protecting group, which may also be an electronic/steric deactivator. The idea can easily be adapted to other cancer related parameters. We believe that this work represents a satisfactory proof-of-principle for a two-stage PDT (2S-PDT), a new therapeutic paradigm, where the benefits of PDT would be accessible without oxygen, or light in the second stage. Our work to establish this paradigm is in progress.
The authors acknowledge financial support from the Dalian University of Technology, Grant No. DUT18RC(3)062 (E.U.A.); and TUBITAK, Grant No. 1512 (2180228).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. v. Tappeiner, Muench. Med. Wochenschr., 1900, 47, 5–7 Search PubMed; (b) H. v. Tappeiner and A. Jodlebauer, Dtsch. Arch. Klin. Med., 1904, 80, 427–487 Search PubMed.
- (a) X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef; (b) X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC; (c) B. Li, L. Lin, H. Lin and B. C. Wilson, J. Biophotonics, 2016, 9, 1314–1325 CrossRef CAS; (d) M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS; (e) I. S. Turan, G. Gunaydin, S. Ayan and E. U. Akkaya, Nat. Commun., 2018, 9, 805 CrossRef; (f) S. Callaghan and M. O. Senge, Photochem. Photobiol. Sci., 2018, 17, 1490–1514 RSC.
- (a) S. Kolemen, T. Ozdemir, D. Lee, G. M. Kim, T. Karatas, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 3606–3610 CrossRef CAS; (b) I. S. Turan, D. Yildiz, A. Turksoy, G. Gunaydin and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 2875–2878 CrossRef CAS; (c) T. Ozdemir, Y.-C. Lu, S. Kolemen, E. Tanriverdi-Ecik and E. U. Akkaya, ChemPhotoChem, 2017, 1, 183–187 CrossRef CAS.
- E. Ucar, D. Xi, O. Seven, C. Kaya, X. J. Peng, W. Sun and E. U. Akkaya, Chem. Commun., 2019, 55, 13808–13811 RSC.
- (a) S. Benz, S. Notzli, J. S. Siegel, D. Eberli and H. J. Jessen, J. Med. Chem., 2013, 56, 10171–10182 CrossRef CAS; (b) M. Matsumoto, M. Yamada and N. Watanabe, Chem. Commun., 2005, 483–485 RSC.
- (a) R. M. Phillips, Cancer Chemother. Pharmacol., 2016, 77, 441–457 CrossRef CAS; (b) W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS.
- S. Luo, R. Zou, J. Wu and M. P. Landry, ACS Sens., 2017, 2, 1139–1145 CrossRef CAS.
- L. Cui, Y. Zhong, W. Zhu, Y. Xu, Q. Du, X. Wang, X. Qian and Y. Xiao, Org. Lett., 2011, 13, 928–931 CrossRef CAS.
- (a) J. Zhang, X. Chai, X. P. He, H. J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683–722 RSC; (b) Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407–6416 CrossRef CAS.
- J. Yan, S. Lee, A. Zhang and J. Yoon, Chem. Soc. Rev., 2018, 47, 6900–6916 RSC.
- B. Zhai, W. Hu, J. Sun, S. Chi, Y. Lei, F. Zhang, C. Zhong and Z. Liu, Analyst, 2017, 142, 1545–1553 RSC.
- T. P. Devasagayam, A. R. Sundquist, P. Di Mascio, S. Kaiser and H. Sies, J. Photochem. Photobiol., B, 1991, 9, 105–116 CrossRef CAS.
- O. W. Griffith and A. Meister, J. Biol. Chem., 1979, 254, 7558–7560 CAS.
- J. S. Lewis-Wambi, H. R. Kim, C. Wambi, R. Patel, J. R. Pyle, A. J. Klein-Szanto and V. C. Jordan, Breast Cancer Res., 2008, 10, R104 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section outlining synthetic procedures, characterization data, kinetic studies and cell studies as well as supplementary figures. See DOI: 10.1039/d0cc06031c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |