
Metal–organic frameworks with solvent-free lanthanide coordination environments: synthesis from aqueous ethanol solutions†
Yulia M.
Litvinova
a,
Yakov M.
Gayfulin
 *a,
Konstantin A.
Brylev
ab,
Dmitry A.
Piryazev
ab,
Jan
van Leusen
c,
Paul
Kögerler
c and
Yuri V.
Mironov
a
*a,
Konstantin A.
Brylev
ab,
Dmitry A.
Piryazev
ab,
Jan
van Leusen
c,
Paul
Kögerler
c and
Yuri V.
Mironov
a
aNikolaev Institute of Inorganic Chemistry SB RAS, 630090, 3, Acad. Lavrentiev ave., Novosibirsk, Russian Federation. E-mail: gayfulin@niic.nsc.ru
bNovosibirsk State University, 630090, 2, Pirogova str., Novosibirsk, Russian Federation
cInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
First published on 6th October 2020
Abstract
A series of 12 new metal–organic frameworks based on lanthanide cations (Ln = Nd, Sm–Dy), octahedral cluster anions [Re6S8(CN)6]4− or [Re6Se8(CN)6]4− and adipamide (adp) were synthesized under mild conditions in aqueous ethanol solutions. Compounds of the general composition Cs[Ln(adp)2{Re6Q8(CN)6}]·3H2O (Ln = Nd, Sm–Dy; Q = S or Se) crystallize in the hexagonal space group P6422. Despite the presence of solvate H2O molecules in structure cavities, the crystal structures revealed that the coordination environment of lanthanide cations in these 3D polymers did not contain H2O ligands. This rare feature is attributed to the combination of a bulk cluster anion and flexible adp molecules, which occupy the lanthanide coordination sphere to yield a stable lanthanide-based building block. This assumption was confirmed by synthesis of a second crystalline modification of the compound Cs[Nd(adp)2{Re6S8(CN)6}]·3H2O (orthorhombic space group Cccm). Luminescence studies evidence the presence of the typical red luminescence of hexarhenium cluster complexes and the absence of observable luminescence of lanthanide cations for all the compounds except the Nd derivatives. Magnetic data indicate very weak antiferromagnetic exchange interactions between paramagnetic Ln3+ centers.
Introduction
Synthesis of metal–organic frameworks (MOFs) based on functional building blocks is an important goal of chemistry and materials science, since MOFs often combine the properties determined by the topology of the framework with the properties of discrete building units.1–3 In some cases, the crystal structure of a MOF can be proposed based on the well-defined coordination chemistry of metal centers (mostly d-metals) in combination with the appropriate selection of rigid organic linkers.4,5 This allows for rational design of structures of multifunctional MOFs and their properties.6–10 The situation is different for MOFs based on 4f-element cations. The coordination environment of lanthanide ions is very flexible resulting in unpredictability of the crystal structures of synthesized polymeric compounds.11,12 Additional complexity is created by the tendency of lanthanide ions to hydrolyse in aqueous solutions and the coordination of hard O-donor ligands, leading to the presence of a variable number of solvent molecules, such as H2O or DMF, or hydroxo ligands in the coordination sphere.13–15 Because of this, predictable synthesis of MOFs with a certain geometry based on lanthanide cations is difficult. However, lanthanide-based MOFs are very attractive because 4f-metal centers impart unique properties to the polymeric materials, which can be used in catalysis,16,17 bio-visualization,18,19 and magnetic20–22 and sensing23–27 applications. The search for ways to control the coordination environment of lanthanide ions is therefore important in order to obtain building blocks for construction of MOFs in a controlled manner.Over the past few years, the formation of coordination polymers and metal–organic frameworks based on 4f-metal cations and octahedral rhenium cyanide cluster complexes with the general formula [Re6Q8(CN)6]3−/4− (Q = S, Se or Te) has been studied.28–30 These cluster anions are based on six covalently bonded Re atoms, which form a stable octahedral Re6 metallocluster.31,32 The metallocluster is supported by a set of the inner μ3-Q ligands coordinating the trigonal faces of the octahedron and the apical CN ligands. Owing to the presence of ambidentate CN ligands, cluster anions are able to coordinate the cations of d- and f-metals, forming coordination polymers of various topologies.33,34 Clusters can act as structural analogues of mononuclear cyanide complexes of transition metals.35 However, due to their larger volume and specific electronic structure, they often form polymeric compounds with unique structures, possessing interesting functional properties, such as red photoluminescence and reversible redox transitions.36–39 This leads to sustained interest in the use of transition metal octahedral cluster complexes as building blocks for the preparation of functional coordination polymers.
Recently, we have shown that cyanoclusters [Re6S8(CN)6]4− and [Re6Se8(CN)6]4− can act as metallolinkers forming neutral frameworks with cations of 4f-elements in aqueous solutions in the presence of rigid polydentate organic ligands.40,41 We found that the cluster anions in the framework structures displayed luminescence and redox properties similar to those of discrete cluster complexes, which can be used for detection of strong oxidants in solution.41 In contrast, lanthanide cations in these compounds did not exhibit luminescence. Moreover, the luminescence of lanthanide cations was not reported previously for any of the obtained polymeric compounds based on octahedral rhenium clusters. The reason for that is supposed to be the presence of H2O molecules in the coordination sphere of lanthanide cations that effectively quench luminescence due to the excitation of O–H vibrations.42–44
Here we report on the synthesis and investigation of a series of new metal–organic frameworks based on [Re6Q8(CN)6]4− (Q = S or Se) cluster anions, Ln3+ cations and flexible linker adipamide (C6H12N2, adp). An important feature of the compounds with the general formula Cs[Ln(adp)2Re6Q8(CN)6]·3H2O is the absence of H2O molecules in the coordination sphere of Ln3+ cations, despite the fact that the compounds were obtained in aqueous solutions and contain solvate H2O molecules in the crystal structures. The structure-forming role of cluster anions and the luminescence and magnetochemical properties of the new compounds are analyzed in this article.
Results and discussion
Synthesis and crystal structures
Compounds Cs[Ln(adp)2Re6S8(CN)6]·3H2O (1–6) and Cs[Ln(adp)2Re6Se8(CN)6]·3H2O (7–12, Ln = Nd, Sm–Dy, respectively) were synthesized as crystalline solids by slow evaporation of water–ethanol solutions containing cluster anions [Re6S8(CN)6]4− or [Re6Se8(CN)6]4−, cations Ln3+ and adipamide (adp). Optimization of the synthesis conditions allowed obtaining compounds in preparative quantities. The preparation of single crystals of compounds 1–12 for X-ray diffraction analysis was complicated by the small crystal size and the formation of polycrystalline aggregates. Nevertheless, high-quality single crystals were obtained for compound 11, which allowed us to study its structure and confirm the structures of the phases 1–10 and 12 by powder X-ray diffraction.Compound 11 crystallizes in the non-centrosymmetric hexagonal space group P6422. It exhibits a framework polymeric structure based on the covalent bonding of Tb3+ cations with CN− ligands of cluster anions and adp molecules. The starting point and the most interesting feature of the crystal structure is the coordination environment of the Tb3+ ions. It includes four N atoms of cyano groups and four O atoms of adp (Fig. 1). The O and N atoms form a distorted square antiprism with ONON faces (Fig. 2a). The Tb–O and Tb–N distances are 2.33(2) and 2.49(2) Å, respectively. The coordination environment of Tb3+ cations does not contain H2O molecules. This is a rare case, although not unique,45–47 for Ln3+-based coordination polymers obtained in aqueous solutions. As shown in Fig. 2, adp molecules lie in the ab plane of crystal packing and form the cationic layers {Tb(adp)2}n3n+ by coordination with Tb3+ cations. The monodentate coordination of adp molecules is additional confirmation that amide groups did not hydrolyse during the reaction. The closest Tb⋯Tb distance in the cationic sub-lattice is 10.71 Å, while the closest Tb⋯Tb distance within the {Tb(adp)2}n3n+ layer is 10.83 Å. Most of the interlayer space is occupied by voluminous cluster anions [Re6Se8(CN)6]4− that bind Tb3+ cations of adjacent layers forming the negatively charged framework [Tb(adp)2Re6Se8(CN)6]− as shown in Fig. 2c and d. Adjacent layers {Tb(adp)2}n3n+ rotate relative to each other by 120° by the 64 axis lying the along c direction. Only four equatorial CN ligands of the cluster anion participate in the binding, while two CN ligands in the trans-position form weak interactions with Cs+ cations and –NH2 groups of adipamide. Visualization of the environment of Ln3+ ions, taking into account the van der Waals radii, shows that cluster anions occupy a large volume, forcing adipamide molecules to pack into layers and, apparently, preventing coordination of additional ligands to Ln3+ cations (Fig. S1†).
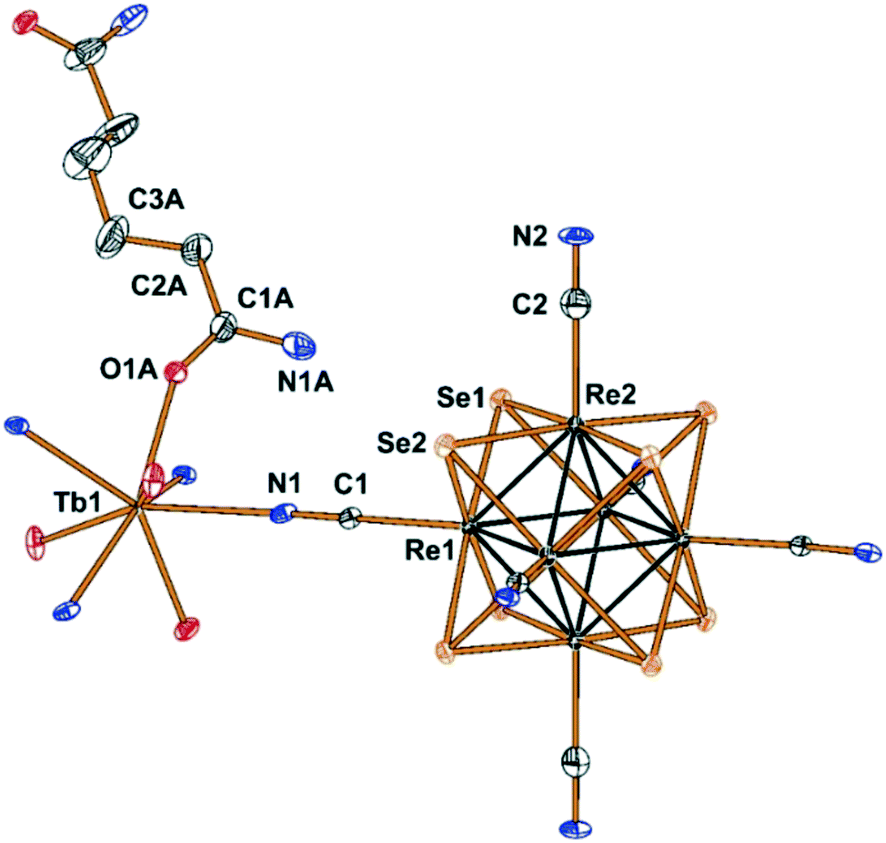 | ||
| Fig. 1 Fragment of the structure of compound 11 with numbered atoms of the asymmetric unit. Atoms are represented by thermal ellipsoids drawn at 60% probability. | ||
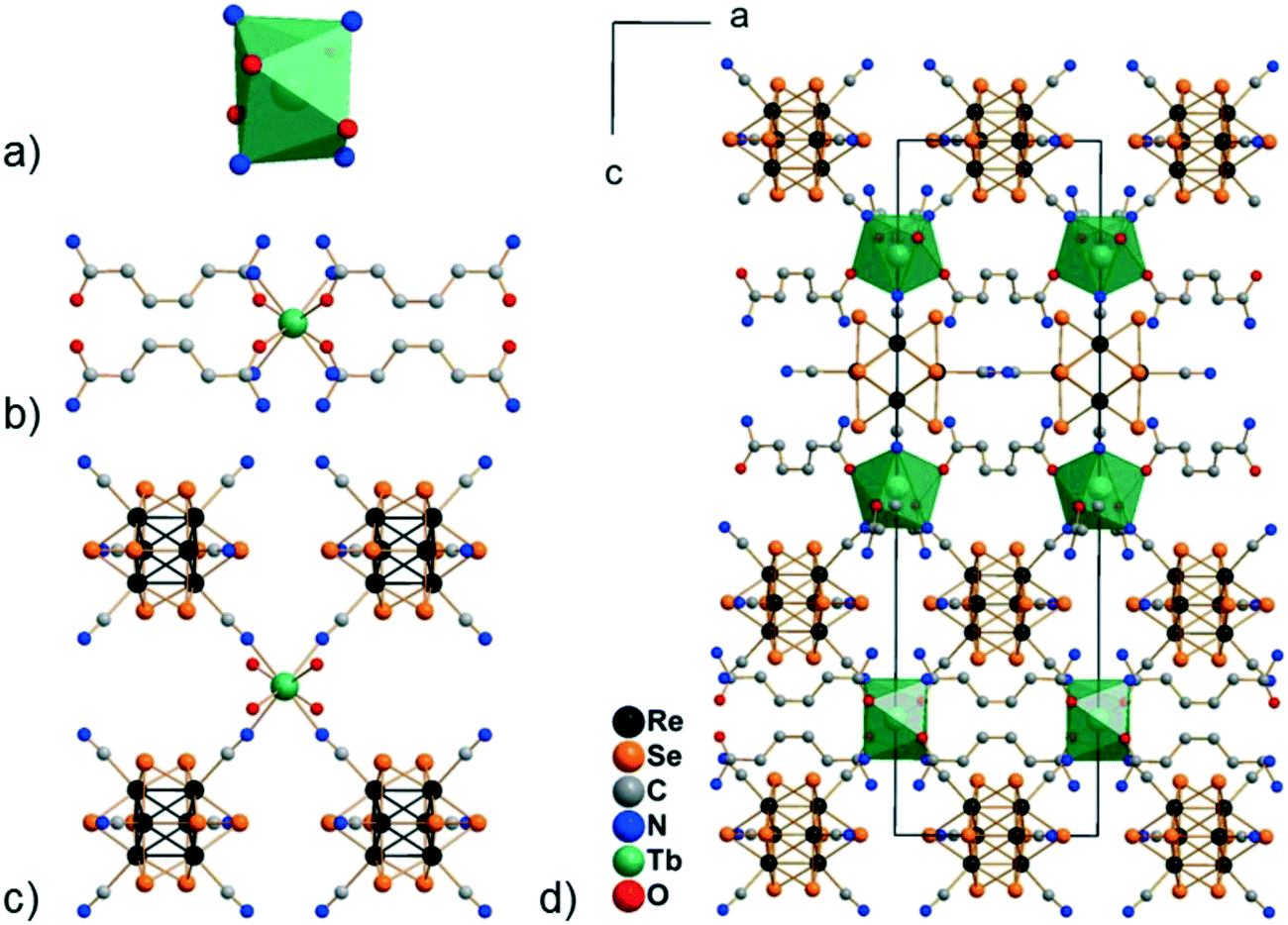 | ||
| Fig. 2 Coordination polyhedron of the Tb3+ cation in the structure of compound 11 (a); coordination of adipamide molecules to the Tb3+ cation in the structure of compound 11 (hydrogen atoms are not shown for clarity, only N atoms of cluster anions are depicted) (b); coordination of [Re6Se8(CN)6]4− cluster anions to the Tb3+ cation in the structure of compound 11 (only O atoms of adipamide molecules are depicted) (c); cell packing of the framework [Tb(adp)2Re6Se8(CN)6]− in the structure of compound 11 (view along the b axis; Cs+ cations, lattice H2O molecules and H atoms are omitted for clarity) (d). | ||
Analysis of the framework [Tb(adp)2Re6Se8(CN)6]− using the Mercury CSD program showed that it contains cavities with a volume of about 15% of the cell volume, elongated along the c axis and penetrating layers {Tb(adp)2}n3n+ (Fig. S2†). Disordered Cs+ cations are located in these cavities and compensate the negative charge of the framework. There are two O atoms of symmetry equivalent lattice H2O molecules in the coordination environment of Cs+ cations. The corresponding Cs⋯O distance is 2.76(5) Å. In addition, lattice H2O molecules form hydrogen bonds of about 3.0 Å with –NH2 groups of adipamide. Bridging μ3-Se ligands of cluster anions and N atoms of CN apical ligands as well as –NH2 groups of adipamide complement the coordination environment of Cs+ cations. Given the Cs+ cations and their coordination environment, the framework Cs[Tb(adp)2Re6Se8(CN)6]·3H2O contains less than 2% of solvent-accessible voids.
The structure of the other compounds belonging to the Cs[Ln(adp)2Re6Q8(CN)6]·3H2O series could not be characterized by single crystal X-ray diffraction analysis due to the low quality of the crystals. However, X-ray powder diffraction confirmed that all 12 compounds are isostructural (Fig. S3, Table S3†), while elemental analysis showed the compliance of the composition with the proposed formula. Trying to obtain single crystals of these compounds, we investigated a wide range of reaction conditions, including slow crystallization of the compounds from solutions with different concentrations of the reagents. As a result, we obtained several single crystals of a new crystalline modification of the compound Cs[Nd(adp)2{Re6S8(CN)6}]·3H2O (1a). This compound crystallizes in the orthorhombic space group Cccm. The structure demonstrates similar connection of lanthanide cations, adipamide molecules and cluster anions to the previously described structure with P6422 symmetry (Fig. S4†). The coordination environment of Nd3+ cations consists of four adipamide oxygen atoms and four nitrogen atoms of CN ligands forming a square antiprism. The Nd–O and Nd–N bond lengths are 2.396(3) and 2.576(3) Å, respectively. The most important difference in the structure of the frameworks 11 and 1a is the different rotation angles of the adjacent layers {Ln(adp)2}n3n+ and the cluster anions coordinated to them when viewed along the c axis. While the structure of 11 with P6422 symmetry demonstrates rotation of 120°, the structure of 1a with Cccm symmetry has a rotation angle of 180° due to the presence of an inversion center (Fig. 3). It should be noted that the compound in structural type 1a was obtained as several single crystals. The presence of Cccm type impurities was excluded for compounds 1–12 based on analysis of their X-ray powder diffraction patterns, which have well-distinguishable peaks at small angles (Fig. S5†). However, investigation of this structure showed that the tetragonal antiprismatic fragment {Ln(adp)4[Re6Q8(CN)6]4} is a secondary building block, which can be involved in formation of different types of structures.
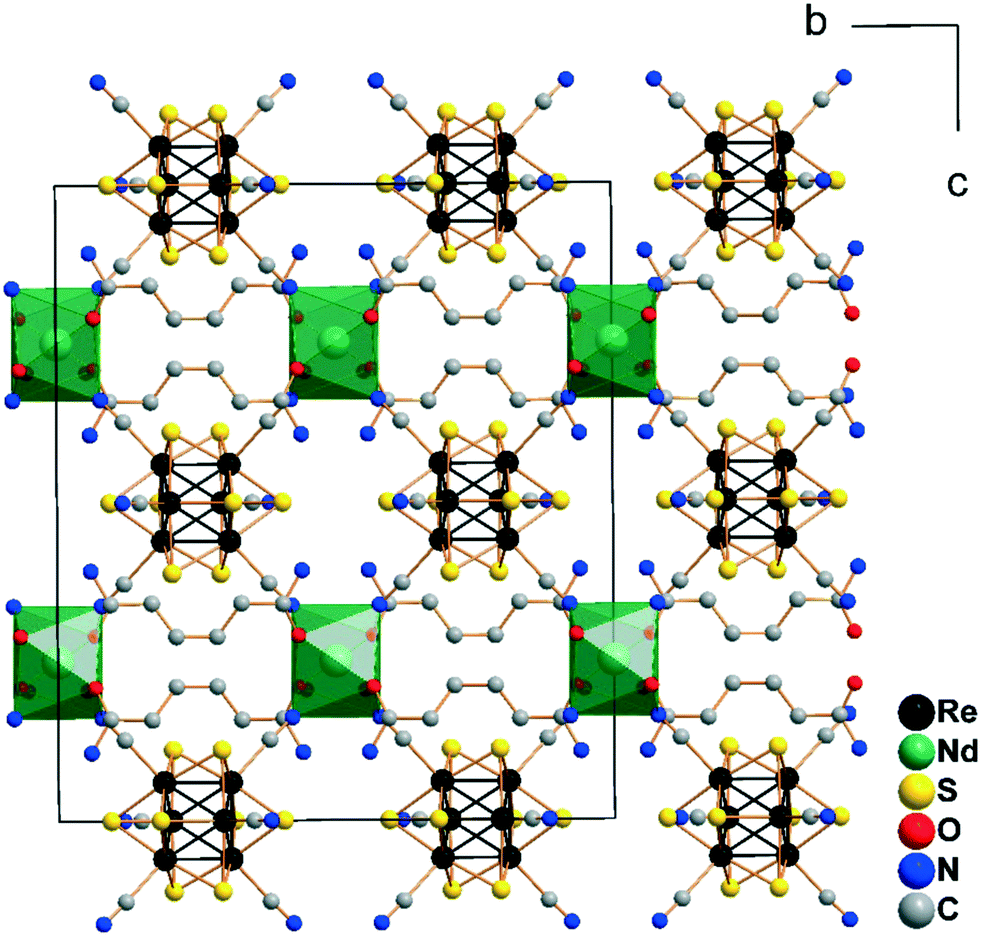 | ||
| Fig. 3 Cell packing of the framework [Nd(adp)2Re6S8(CN)6]− in the structure of 1a (view along the a axis; Cs+ cations, lattice H2O molecules and H atoms are omitted for clarity). | ||
Cluster anions and adipamide as building blocks
Lanthanide cations are known as strong Lewis acids and strong oxophiles, which exist in aqueous or water/non-aqueous solvent mixtures as solvates with high Ln–O bonding energies.14,48 The synthesis of compounds that do not contain solvent molecules in the coordination sphere of rare-earth cations requires the presence of bulk multidentate ligands, which promote the kinetic inertness of lanthanide complexes,49–51 or synthesis in molten organic compounds.52–54 Since removal of solvent molecules from the coordination sphere often allows one to enhance the luminescence characteristics of 4f metal centers,42 synthesis of such compounds is a subject of interest. The synthesis of compounds 1–12 takes place in a water–ethanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol.) mixture, so a specific combination of linkers surrounding the metal centers can prevent the binding of water molecules to Ln3+ and ensure the formation of coordination polymers based on water-free lanthanide cations. This assumption was confirmed by obtaining of two crystalline modifications of the compounds Cs[Ln(adp)2{Re6S8(CN)6}]·3H2O, namely 11 and 1a, crystallizing in the space groups P6422 and Cccm, respectively. The stoichiometry of the compounds, the coordination environment of the Ln cations and the geometry of the lanthanide coordination polyhedron remained the same despite the different packing of building blocks.
:1 vol.) mixture, so a specific combination of linkers surrounding the metal centers can prevent the binding of water molecules to Ln3+ and ensure the formation of coordination polymers based on water-free lanthanide cations. This assumption was confirmed by obtaining of two crystalline modifications of the compounds Cs[Ln(adp)2{Re6S8(CN)6}]·3H2O, namely 11 and 1a, crystallizing in the space groups P6422 and Cccm, respectively. The stoichiometry of the compounds, the coordination environment of the Ln cations and the geometry of the lanthanide coordination polyhedron remained the same despite the different packing of building blocks.
The chalcocyanide anions [Re6Q8(CN)6]4−/3− (S, Se or Te) are widely used for the synthesis of coordination polymers due to their chemical stability, rigid geometry and functional properties, the most known one is luminescence in the red and NIR range.55 Over the past few decades, a number of coordination polymers based on these cluster anions and transition metal cations including lanthanides have been characterized. It was shown that in most cases the cluster cores {Re6Q8}2+ act as nodes in the structures. Thus, the structure of inorganic cluster-based coordination polymers can be considered as a combination of two cationic nodes connected by bridging CN− ligands. Prussian blue-type structures like Fe4[Re6Te8(CN)6]3·27H2O or Ga4[Re6Se8(CN)6]3·38H2O are classical examples of such compounds.35 In the case of metal–organic coordination polymers, the octahedral cluster anion is able to act as a bulk spacer between cationic nodes connected by bridging organic ligands, as in the case of compounds [Cu2(threo-tab)3(NH3)][Re6Q8(CN)6]·nH2O (Q = S or Te, threo-tab = 1,2,3,4-tetraaminobutane)39 and K[Nd(μ-C4H10O4)(H2O)4Re6Se8(CN)6]·4H2O (C4H10O4 = butan-1,2,3,4-tetraol).28 Compounds 1–12 represent a new series of metal–organic coordination polymers based on hexarhenium cluster complexes. As in the above mentioned metal–organic frameworks based on octahedral clusters,40,41 the cluster anion can be described here as a bulk metal linker between layers of lanthanide cations connected by bridging organic ligands.
The bridging organic ligand that was used in the synthesis of compounds 1–12 is adipamide. The use of non-hydrolyzed adipamide derivatives as linkers is limited and, as a rule, there are bulk N,O-donor ligands.56,57 A product of its hydrolysis – adipate anion – is widely presented as a linker for coordination polymers with d- and f-metal cations.12,58–62 In contrast with adipate, adipamide demonstrates monodentate coordination and at the same time connects distant lanthanide centers, which allows one to place more voluminous ligands around the latter ones. It should be noted that our attempts to obtain coordination polymers based on hexarhenium clusters and lanthanide cations using adipic acid or adipates led to the obtaining of X-ray amorphous compounds. Thus, we can assume that the coordination sphere of lanthanide cations in compounds 1–12 does not contain water molecules due to the combination of bulk cluster anions and sterically flexible adipamide molecules, which form the stable and highly symmetrical node {Ln(adp)4[Re6Q8(CN)6]4}.
Hydrolytic stability and thermal decomposition
Coordination polymers based on lanthanide cations often have low hydrolytic stability, which is caused by the high stability of lanthanide aqua complexes in solution. Compounds 1–12 follow this tendency. Being stable in the mother solution, they show slow dissolution in distilled water at room temperature. However, after dissolution the compounds can be recrystallized by addition of EtOH and slow evaporation. Recrystallization of the compounds based on different cluster anions leads to different results. Compound 11 based on the [Re6Se8(CN)6]4− anion can be repeatedly recrystallized while retaining the crystal structure of type 11. At the same time, compound 1 based on the [Re6S8(CN)6]4− anion was recrystallized with the formation of a mixture of crystalline phases in structural types 1a and 11 (Fig. S6†). Thus, we can assume that the formation of compounds in structural type 1a is typical for compounds based on the cluster anion [Re6S8(CN)6]4−, and the possibility of forming these phases is determined by the concentration of the Ln3+ cation or the total concentration of reagents in solution.The thermal stability of the compounds in the solid state is characterized by a slow mass loss up to about 350 °C and a significant endothermic mass loss in the temperature interval of 350–550 °C (Fig. S7†). The slow mass loss at the initial stage is associated with the removal of solvate water molecules, which occurs in a very wide temperature range, possibly due to the intermediate formation of lanthanide oxo- and hydroxo-complexes. Further decomposition is associated with the pyrolysis of adipamide molecules and cyanide ligands of cluster anions. The products of the pyrolysis are stable up to ∼700 °C, when decomposition of the cluster cores occurs. Finally, the type of lanthanide cation or cluster anion has a negligible effect on the positions of the maxima of the mass loss rate indicating that the thermal stability of the compounds is determined mainly by the structure of the framework.
Luminescence properties
Chalcocyanide complexes [Re6Q8(CN)6]4− (Q = S, Se or Te) are one of the first hexarhenium cluster complexes for which the luminescence properties were studied in solution63 and then in the solid state.64 Despite the fact that since 1998 many coordination polymers have been synthesized based on these anionic complexes and various transition metal cations,34,39,65–68 the luminescence properties of cyano-bridged polymers comprising the {Re6Q8}2+ core have been described for the first time quite recently.36 In particular, it was shown that the polymeric compounds [{Ag(bpy)}{Ag4(bpy)4(μ-CN)}{Re6Q8(CN)6}] (Q = S or S) emit luminescence in the visible and near-infrared regions upon ultraviolet light excitation. The broad and structureless spectrum of the polymer is similar to the spectra of soluble salts of the starting complexes [Re6Q8(CN)6]4−. In addition, it was noted that the coordination of luminescent cyanide cluster units with such transition metal cations as Co2+, Ni2+, Cu2+, Zn2+, and Cd2+ or even lanthanides led to elimination of their photoluminescence abilities.36 Thus, the polymeric compounds with Ag+ cations were declared as the first luminescent CN-bridged coordination polymers based on [Re6Q8(CN)6]4− complexes. In our recent publications metal–organic frameworks based on lanthanide complexes and cyanide hexarhenium clusters have been described.40,41 In the luminescence spectra of the MOFs a broad band of the corresponding cyanide cluster complex was observed while bands of lanthanides were not shown. In the current study the luminescence spectra of the solid samples of all 12 synthesized compounds Cs[Ln(adp)2{Re6Q8(CN)6}]·3H2O as well as the initial salt Cs3K[Re6Q8(CN)6]·nH2O were recorded using an IR-sensitive detector. The spectra of all the compounds except the complexes with neodymium (1 and 7) show the band characteristic of [Re6Q8(CN)6]4− without noticeable bands from the lanthanides that make up them (Fig. 4). The emission intensity of the compounds with gadolinium (4 or 10) and terbium (5 or 11) is comparable with that of the powdered sample of the corresponding precursor Cs3K[Re6Q8(CN)6]·nH2O (Fig. S8a†) while the luminescence intensity of the complexes with samarium (2 or 8), europium (3 or 9) and dysprosium (6 or 12) is significantly lower. The spectra of the neodymium compounds (1 or 7) do not display a band of the Re6 cluster unit but show bands inherent to Nd3+ (Fig. S8b†). This can be attributed to the Förster resonance energy transfer (FRET) mechanism with the cluster complex acting as the donor and the Nd3+ cation as the acceptor69 because of the overlap of the excitation spectrum of Nd3+ with the emission spectrum of [Re6Q8(CN)6]4−.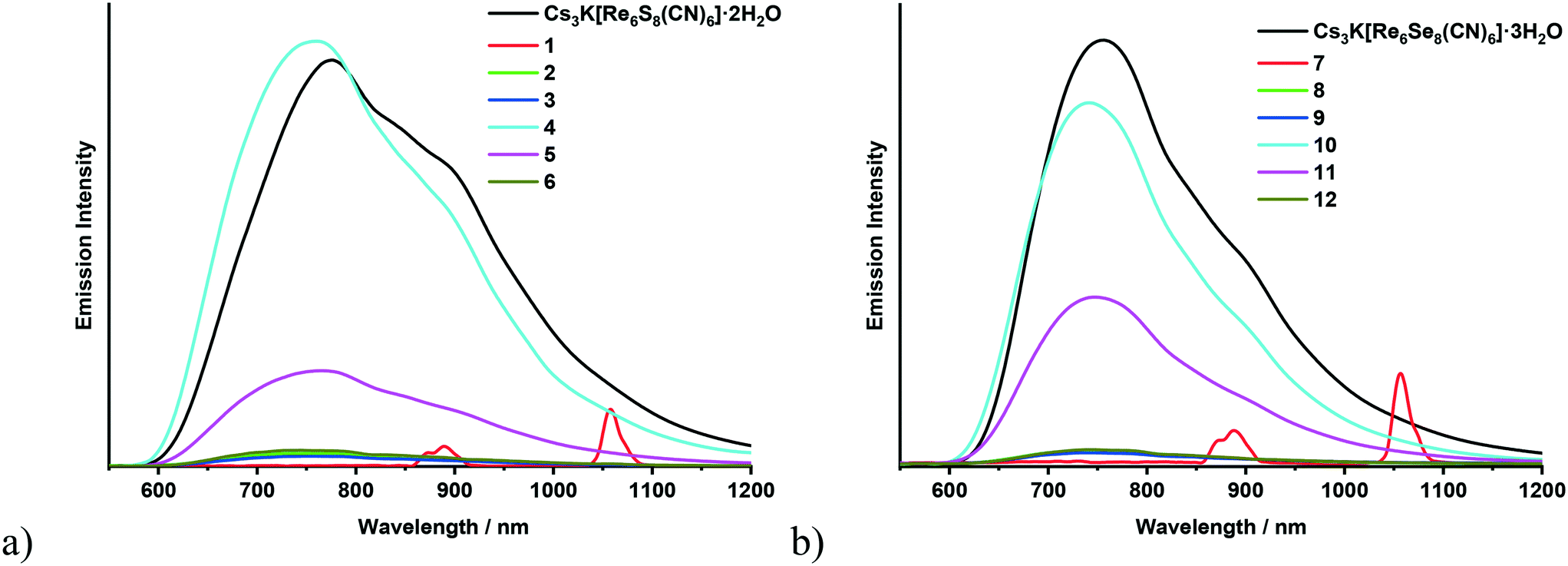 | ||
| Fig. 4 Emission spectra of Cs3K[Re6S8(CN)6]·2H2O and compounds 1–6 (a); emission spectra of Cs3K[Re6Se8(CN)6]·3H2O and compounds 7–12 (b). | ||
Magnetochemical properties
The SQUID magnetometry data of compounds 1–12 are shown in Fig. 5 as χmT vs. temperature T plots at 0.1 T and molar magnetization Mmvs. magnetic field B plots at 2.0 K. For 1–6, the values of χmT at 300 K are within the expected ranges70 of the respective lanthanide center (all values in cm3 K mol−1): 1: 1.47 (expected 1.4–1.5), 2: 0.35 (ca. 0.32), 3: 1.63 (ca. 1.5), 4: 7.84 (7.6–7.9), 5: 11.81 (11.7–12.0), and 6: 13.51 (13.0–14.1). These values are close to the values expected for the respective isolated Russell–Saunders ground terms of the free ions but for 2 and 3, for which the excited terms are energetically close to and distinctly mix into the corresponding ground term. Upon cooling the compounds, all values marginally decrease, and drop at temperatures below 150–50 K. In addition, there are increasing values of χmT observed for 4 and 5 at T < 7 K. At 2.0 K, χmT reaches the values of 0.34 (1), 0.03 (2), 0.01 (3), 7.64 (4), 7.65 (5) and 9.34 cm3 K mol−1 (6), respectively.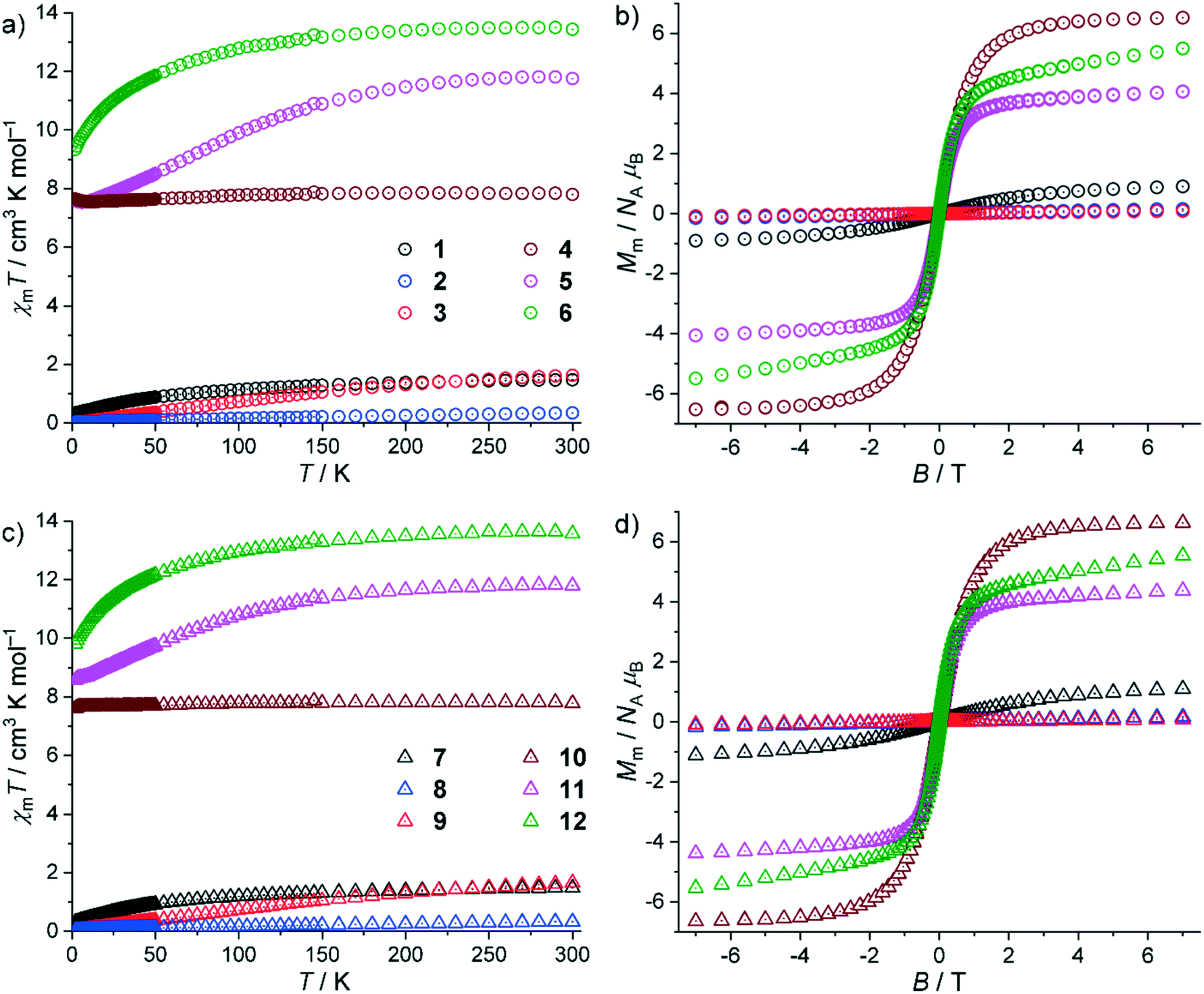 | ||
| Fig. 5 Magnetic properties of 1–12: (a) χmT vs. T at 0.1 T and (b) Mmvs. B at 2.0 K of 1–6; (c) χmT vs. T at 0.1 T and (d) Mmvs. B at 2.0 K of 7–12. | ||
For 4, the values only slightly decrease due to the spin-like behavior of Gd3+ centers, while for the other compounds the decrease is more pronounced due to the thermal population of the distinctly split mJ energy levels. Besides these single-ion effects, there are very weak antiferromagnetic exchange interactions between the lanthanide centers, which add to or induce (4) the decrease of χmT. The increases of χmT at low temperatures indicate also the presence of very weak ferromagnetic exchange interactions, at least for 4 and 5. At 2.0 K, the molar magnetizations Mm as a function of the applied field B are linear in the range from about −0.5 to 0.5 T, and at higher fields notably decrease the slope of the curve without being saturated at the highest applied fields of ±7.0 T. The values at 7.0 T are 0.9 (1), 0.2 (2), 0.1 (3), 6.5 (4), 4.1 (5) and 5.5 NAμB (6), which are either close to zero (2, 3), close to the saturation value (4, Mm,sat = 7.0 NAμB) or about half of the saturation value of about gJJNAμB (1, 3.3; 5, 9.0; 6, 10.0 NAμB). For 2 and 3, this is partially due to the aforementioned distinct mixing of energy states, and in particular for 3 due to the J = mJ = 0 ground term of Eu3+. The molar magnetization of 4 is close to the saturation value due to the almost isotropic Gd3+ center, while for 1, 5, and 6 (and partially 2) the observed features are essentially due to being the mean values of randomly oriented anisotropic centers (powder samples). Since all the curves additionally show a relevant slope at the highest applied fields, they are in agreement with the presence of very weak exchange interactions.
The magnetic properties of 7–12 are similar to those of the respective lanthanide compounds 1–6 with the following differences. At 300 K, the values of χmT are (all values in cm3 K mol−1): 7: 1.48, 8: 0.33, 9: 1.65, 10: 7.82, 11: 11.84, and 12: 13.65. These values are within the expected ranges and close to the values expected for the respective isolated Russell–Saunders ground terms of the free ions with the exception of 8 and 9, for the above-mentioned reasons. Upon decreasing the temperature, all values marginally decrease, and drop at temperatures below 150–50 K. In addition, there are further small drop offs observed for 7, 8, 10 and 11 at T < 4 K. At 2.0 K, χmT reaches the values of 0.31 (7), 0.04 (8), 0.02 (9), 7.61 (10), 8.57 (11) and 9.82 cm3 K mol−1 (12), respectively. As for 1–6, the observed behaviors are mainly due to single-ion effects. The presence of very weak, possibly antiferromagnetic exchange interactions between the lanthanide centers cannot be excluded. The drop offs below 4 K are, however, most likely due to the Zeeman splitting of the energy states. The molar magnetizations Mm at 2.0 K are approximately linear functions of the applied field B from −0.5 to 0.5 T, and notably change their slope at higher fields without reaching saturation at ±7.0 T. The values at 7.0 T are 1.1 (7), 0.2 (8), 0.1 (9), 6.6 (10), 4.4 (11) and 5.5 NAμB (12), which are slightly above the values measured for the respective analogs 1–6. This observation combined with the less pronounced decreases of χmT at T < 150 K most likely indicates overall slightly weaker antiferromagnetic exchange interactions in 7–12. The overall similar magnetic properties of 1–6 in comparison to those of 7–12 with small differences can be rationalized by the structural information of the compounds. They crystallize in the same space group yet with slightly different distances of the lanthanide centers (influencing the strength of interactions), slightly different angles between the lanthanide centers via the bridging ligands (strength and type of interaction) and slightly different ligand–center distances/angles (single-ion effects).
Conclusion
In conclusion, we have discovered that self-assembly reactions between various Ln3+ cations, [Re6Q8(CN)6]4− cluster anions (Q = S or Se) and adipamide (adp) in aqueous ethanol solution lead to the formation of 3D framework compounds 1–12. The structures of the frameworks consist of Ln3+ metal centers and {Re6Q8}2+ cluster units connected by CN− groups and adp molecules. The interesting feature of the frameworks is the presence of lanthanide centers coordinated only by linkers without the presence of water molecules in the coordination sphere. We propose that, due to the combination of cluster complexes and flexible adp molecules, a stable lanthanide-based building block {Ln(adp)4[Re6Q8(CN)6]4} was formed, which can be present in different types of structures. The combination of voluminous, highly symmetrical linkers with smaller and flexible ones could be used as a general approach for synthesis of Ln-based MOFs with solvate-free Ln centers.Investigation of the luminescence of the new compounds in the solid state using an IR-sensitive detector showed that all the compounds with the exception of the Nd-based ones had no photoluminescence from the Ln center, while a broad band of the corresponding cyanide cluster complex was observed. This suggested that quenching of the luminescence of lanthanides in metal–organic frameworks can be caused not only by the presence of solvate molecules in the first coordination sphere. However, the Nd-containing compounds 1 and 7 did not display a band of the Re6 cluster unit but showed bands inherent to Nd3+. This can be attributed to the FRET mechanism due to the overlap of the excitation spectrum of Nd3+ with the emission spectrum of [Re6Q8(CN)6]4−. Finally, the magnetic properties of the compounds are determined by the isolated lanthanide paramagnetic centers showing very weak antiferromagnetic exchange interactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The reported study was funded by the Russian Foundation for Basic Research (project 18-29-04007). The luminescence and magnetic measurements were supported by the grant of Russian Science Foundation (project 19-73-20196). The measurements were performed at the “Center for Optical and Laser Materials Research” (St. Petersburg State University, St. Petersburg, Russian Federation).References
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS.
- F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS.
- F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- K. Müller-Buschbaum, in The Chemistry of Metal–Organic Frameworks, ed. S. Kaskel, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, ch. 9, pp. 231–270, DOI:10.1002/9783527693078.ch9.
- C. L. Cahill, D. T. de Lill and M. Frisch, CrystEngComm, 2007, 9, 15–26 RSC.
- Z. Zhang, Y. Zhang and Z. Zheng, in Recent Development in Clusters of Rare Earths and Actinides: Chemistry and Materials, ed. Z. Zheng, Springer Berlin Heidelberg, Berlin, Heidelberg, 2017, pp. 1–49, DOI:10.1007/430_2016_12.
- J.-C. G. Bünzli, J. Coord. Chem., 2014, 67, 3706–3733 CrossRef.
- E. N. Rizkalla and G. R. Choppin, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1991, vol. 15, pp. 393–442 Search PubMed.
- C. Pagis, M. Ferbinteanu, G. Rothenberg and S. Tanase, ACS Catal., 2016, 6, 6063–6072 CrossRef CAS.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- G. A. Pereira, J. A. Peters, F. A. Almeida Paz, J. Rocha and C. F. G. C. Geraldes, Inorg. Chem., 2010, 49, 2969–2974 CrossRef CAS.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS.
- G. Huang, G. Fernandez-Garcia, I. Badiane, M. Camarra, S. Freslon, O. Guillou, C. Daiguebonne, F. Totti, O. Cador, T. Guizouarn, B. Le Guennic and K. Bernot, Chem. – Eur. J., 2018, 24, 6983–6991 CrossRef CAS.
- B. V. Harbuzaru, A. Corma, F. Rey, P. Atienzar, J. L. Jordá, H. García, D. Ananias, L. D. Carlos and J. Rocha, Angew. Chem., Int. Ed., 2008, 47, 1080–1083 CrossRef CAS.
- T. K. Prasad, M. V. Rajasekharan and J.-P. Costes, Angew. Chem., Int. Ed., 2007, 46, 2851–2854 CrossRef CAS.
- Y. Zheng, F.-Z. Sun, X. Han, J. Xu and X.-H. Bu, Adv. Opt. Mater., 2020, 8, 2000110 CrossRef CAS.
- A. Abdallah, S. Freslon, X. Fan, A. Rojo, C. Daiguebonne, Y. Suffren, K. Bernot, G. Calvez, T. Roisnel and O. Guillou, Inorg. Chem., 2019, 58, 462–475 CrossRef CAS.
- Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS.
- O. Guillou, C. Daiguebonne, G. Calvez and K. Bernot, Acc. Chem. Res., 2016, 49, 844–856 CrossRef CAS.
- J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- M. S. Tarasenko, A. Y. Ledneva, D. Y. Naumov, N. G. Naumov and V. E. Fedorov, J. Struct. Chem., 2011, 52, 172–179 CrossRef CAS.
- M. S. Tarasenko, N. G. Naumov, D. Y. Naumov, N. V. Kuratieva and V. E. Fedorov, Russ. J. Coord. Chem., 2006, 32, 494–503 CrossRef CAS.
- S. B. Artemkina, N. G. Naumov, A. V. Virovets, S. A. Gromilov, D. Fenske and V. E. Fedorov, Inorg. Chem. Commun., 2001, 4, 423–426 CrossRef CAS.
- V. E. Fedorov, Y. V. Mironov, N. G. Naumov, M. N. Sokolov and V. P. Fedin, Russ. Chem. Rev., 2007, 76, 529–552 CrossRef CAS.
- G. Pilet and A. Perrin, C. R. Chim., 2005, 8, 1728–1742 CrossRef CAS.
- Y. Kim, V. E. Fedorov and S.-J. Kim, J. Mater. Chem., 2009, 19, 7178–7190 RSC.
- V. E. Fedorov, N. G. Naumov, Y. V. Mironov, A. V. Virovets, S. B. Artemkina, K. A. Brylev, S. S. Yarovoi, O. A. Efremova and U. H. Peak, J. Struct. Chem., 2002, 43, 669–684 CrossRef CAS.
- M. P. Shores, L. G. Beauvais and J. R. Long, J. Am. Chem. Soc., 1999, 121, 775–779 CrossRef CAS.
- A. V. Ermolaev, A. I. Smolentsev, K. A. Brylev, N. Kitamura and Y. V. Mironov, J. Mol. Struct., 2018, 1173, 627–634 CrossRef CAS.
- M. Amela-Cortes, S. Cordier, N. G. Naumov, C. Mériadec, F. Artzner and Y. Molard, J. Mater. Chem. C, 2014, 2, 9813–9823 RSC.
- L. Xu, Y. Kim, S.-J. Kim, H. J. Kim and C. Kim, Inorg. Chem. Commun., 2007, 10, 586–589 CrossRef CAS.
- Y. V. Mironov, N. G. Naumov, K. A. Brylev, O. A. Efremova, V. E. Fedorov and K. Hegetschweiler, Angew. Chem., Int. Ed., 2004, 43, 1297–1300 CrossRef CAS.
- Y. M. Litvinova, Y. M. Gayfulin, J. van Leusen, D. G. Samsonenko, V. A. Lazarenko, Y. V. Zubavichus, P. Kögerler and Y. V. Mironov, Inorg. Chem. Front., 2019, 6, 1518–1526 RSC.
- Y. M. Litvinova, Y. M. Gayfulin, K. A. Kovalenko, D. G. Samsonenko, J. van Leusen, I. V. Korolkov, V. P. Fedin and Y. V. Mironov, Inorg. Chem., 2018, 57, 2072–2084 CrossRef CAS.
- D. Mara, F. Artizzu, P. F. Smet, A. M. Kaczmarek, K. Van Hecke and R. Van Deun, Chem. – Eur. J., 2019, 25, 15944–15956 CrossRef CAS.
- E. Kreidt, C. Kruck and M. Seitz, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2018, vol. 53, pp. 35–79 Search PubMed.
- T. Főrster, Spec. Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
- N. Ponjan, F. Kielar, W. Dungkaew, K. Kongpatpanich, H. Zenno, S. Hayami, M. Sukwattanasinittf and K. Chainok, CrystEngComm, 2020, 22, 4833–4841 RSC.
- Y. Pan, H.-Q. Su, E.-L. Zhou, H.-Z. Yin, K.-Z. Shaoc and Z.-M. Su, Dalton Trans., 2019, 48, 3723–3729 CAS.
- K. Zhang, X. Xie, H. Li, J. Gao, L. Nie, Y. Pan, J. Xie, D. Tian, W. Liu, Q. Fan, H. Su, L. Huang and W. Huang, Adv. Mater., 2017, 29, 1701804 CrossRef.
- T. Kimura, R. Nagaishi, Y. Kato and Z. Yoshida, J. Alloys Compd., 2001, 323–324, 164–168 CrossRef CAS.
- F. Artizzu, P. Deplano, L. Marchiò, M. L. Mercuri, L. Pilia, A. Serpe, F. Quochi, R. Orrù, F. Cordella, F. Meinardi, R. Tubino, A. Mura and G. Bongiovanni, Inorg. Chem., 2005, 44, 840–842 CrossRef CAS.
- A. Rodríguez-Rodríguez, D. Esteban-Gómez, R. Tripier, G. Tircsó, Z. Garda, I. Tóth, A. de Blas, T. Rodríguez-Blas and C. Platas-Iglesias, J. Am. Chem. Soc., 2014, 136, 17954–17957 CrossRef.
- B. El Aroussi, L. Guénée, P. Pal and J. Hamacek, Inorg. Chem., 2011, 50, 8588–8597 CrossRef CAS.
- A. Zurawski, M. Mai, D. Baumann, C. Feldmann and K. Müller-Buschbaum, Chem. Commun., 2011, 47, 496–498 RSC.
- J. C. Rybak, L. V. Meyer, J. Wagenhöfer, G. Sextl and K. Müller-Buschbaum, Inorg. Chem., 2012, 51, 13204–13213 CrossRef CAS.
- K. Müller-Buschbaum, Z. Anorg. Allg. Chem., 2005, 631, 811–828 CrossRef.
- S. Cordier, Y. Molard, K. A. Brylev, Y. V. Mironov, F. Grasset, B. Fabre and N. G. Naumov, J. Cluster Sci., 2015, 26, 53–81 CrossRef CAS.
- H. Wang, S. Zhao, P. Qian, J. Dai, Z. Cao and K. Yu, Polyhedron, 1995, 14, 407–412 CrossRef CAS.
- X.-L. Wang, F.-F. Sui, H.-Y. Lin, J.-W. Zhang and G.-C. Liu, Cryst. Growth Des., 2014, 14, 3438–3452 CrossRef CAS.
- X.-F. Tan, J. Zhou, H.-H. Zou, H.-P. Xiao, Q. Tang, T. Jiang and X. Zhang, J. Cluster Sci., 2016, 27, 2025–2033 CrossRef CAS.
- A. L. Pochodylo, J. A. Wilson, J. W. Uebler, S. H. Qiblawi and R. L. LaDuca, Inorg. Chim. Acta, 2014, 423, 298–306 CrossRef CAS.
- C.-G. Wang, Y.-H. Xing, Z.-P. Li, J. Li, X.-Q. Zeng, M.-F. Ge and S.-Y. Niu, Cryst. Growth Des., 2009, 9, 1525–1530 CrossRef CAS.
- Y.-Q. Zheng, D.-Y. Cheng, J.-L. Lin, Z.-F. Li and X.-W. Wang, Eur. J. Inorg. Chem., 2008, 2008, 4453–4461 CrossRef.
- D. T. de Lill, A. de Bettencourt-Dias and C. L. Cahill, Inorg. Chem., 2007, 46, 3960–3965 CrossRef CAS.
- T. Yoshimura, S. Ishizaka, Y. Sasaki, H.-B. Kim, N. Kitamura, N. G. Naumov, M. N. Sokolov and V. E. Fedorov, Chem. Lett., 1999, 28, 1121–1122 CrossRef.
- T. G. Gray, C. M. Rudzinski, E. E. Meyer, R. H. Holm and D. G. Nocera, J. Am. Chem. Soc., 2003, 125, 4755–4770 CrossRef CAS.
- L. G. Beauvais, M. P. Shores and J. R. Long, Chem. Mater., 1998, 10, 3783–3786 CrossRef CAS.
- K. A. Brylev, Y. V. Mironov, N. G. Naumov, V. E. Fedorov and J. A. Ibers, Inorg. Chem., 2004, 43, 4833–4838 CrossRef CAS.
- S. B. Artemkina, N. G. Naumov, A. V. Virovets and V. E. Fedorov, Eur. J. Inorg. Chem., 2005, 2005, 142–146 CrossRef.
- N. G. Naumov, M. S. Tarasenko, A. V. Virovets, Y. Kim, S.-J. Kim and V. E. Fedorov, Eur. J. Inorg. Chem., 2006, 2006, 298–303 CrossRef.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence Principles and Applications, Wiley-VCH, Weinheim, 2012 Search PubMed.
- H. Lueken, Magnetochemie, Teubner Verlag, Stuttgart, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional X-ray structural data, powder diffraction patterns, and TG data. CCDC 2011586 and 2011587. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01240h |
| This journal is © The Royal Society of Chemistry 2020 |