
Minimization of solid-state conformational disorder in donor–acceptor TADF compounds†
Tomas
Serevičius
 *a,
Rokas
Skaisgiris
a,
Jelena
Dodonova
b,
Karolis
Kazlauskas
a,
Saulius
Juršėnas
a and
Sigitas
Tumkevičius
b
*a,
Rokas
Skaisgiris
a,
Jelena
Dodonova
b,
Karolis
Kazlauskas
a,
Saulius
Juršėnas
a and
Sigitas
Tumkevičius
b
aInstitute of Photonics and Nanotechnology, Vilnius University, Sauletekio 3, LT-10257 Vilnius, Lithuania. E-mail: tomas.serevicius@tmi.vu.lt
bInstitute of Chemistry, Vilnius University, Naugarduko 24, LT-03225, Vilnius, Lithuania
First published on 27th November 2019
Abstract
Thermally activated delayed fluorescence (TADF) compounds with a flexible donor–acceptor structure suffer from conformational disorder in solid-state, which deteriorates their emission properties as well as OLED performance. Accordingly, TADF materials with predictable solid-state emission properties are highly desirable. In this work, we analyse the relation between the molecular rigidity and solid-state TADF properties. Two TADF compounds, 4,6-bis(2-methyl-4-(10H-phenothiazin-10-yl)phenyl)pyrimidine (PTZ-mPYR) and 1,2,3,4-tetrakis(carbazol-9-yl)-5,6-dicyanobenzene (4CzPN), with similar emission properties in toluene solutions but different rigidity of the molecular structure were systematically studied. The analysis was supplemented by comparison of solid-state TADF properties of PTZ-mPYR with its analogue 4,6-bis(4-(10H-phenoxazin-10-yl)phenyl)pyrimidine (PXZ-PYR), bearing a more sterically constrained planar electron-donor unit. All compounds showed conformational disorder in diluted polymer films; however its extent directly depended on the molecular structure. Large dispersion of singlet–triplet energy gaps resulted in remarkably prolonged TADF lifetime for PTZ-mPYR with a less sterically constrained donor unit. In contrast, weakened conformational disorder in rigid 4CzPN with sterically crowded donor units was shown to ensure rapid TADF decay with only a threefold lower solid-state rISC rate as compared to toluene. Similarly, selection of a more sterically constrained planar electron-donor unit was also shown to be preferable for lowering the conformational disorder. Our findings are important for the future design of compounds with efficient solid-state TADF as well as for the further application in OLEDs with low external quantum efficiency roll-off.
Introduction
The application of thermally activated delayed fluorescence (TADF)1–3 emitters allows one to boost the external quantum efficiencies (EQE) of organic light emitting diodes (OLEDs) far beyond the typical values for singlet-only emitters.4,5 The harvesting of triplet states in TADF compounds occurs via thermally activated reverse intersystem crossing (rISC). To achieve rapid rISC, generally a small energy gap (ΔEST) between the charge-transfer singlet (1CT) and the lowest triplet state, usually of local excited triplet (3LE) character, is required. The most usual approach to lower ΔEST is to minimize the spatial overlap of HOMO and LUMO.6 This is achieved by decoupling electron-donating (D) and electron-accepting (A) units in highly twisted molecular geometry with strong charge-transfer character. Besides the minimization of ΔEST, negligible energy splitting between the spin-vibronically coupled 3LE and charge-transfer triplet (3CT) states is also mandatory.7–10 As it was shown previously, 3CT and 3LE states are coupled through the torsion of the D–A angle,7–11 thus somewhat rotational flexibility of the D–A bond is required, otherwise the rISC rate would be minimized.12 On top of that, minimization of the nonradiative internal conversion rate13 together with decrease of the activation energy barrier for rISC,14 however, is achieved in compounds with suppressed structural deformation. All those contradictions can be well matched by comprehensive optimization of the molecular structure, and thus a great variety of different highly emissive TADF compounds, constructed of sometimes very different donor and acceptor units bound in diverse molecular geometries, have been presented.15,16 However, TADF behaviour becomes highly complicated in solid-state and these issues still are very scarcely studied. Regardless of the selected D and A units and their linking patterns, pronounced conformational disorder occurs in solid films.17–24 TADF molecules in solid polymer or small-molecule host films are “frozen” with a variation of dihedral angles between the D and A units, forming a set of solid molecular conformations, having different HOMO–LUMO overlap and thus different 1CT energies and singlet–triplet gaps.22,25 Such conformational disorder in solid state causes temporal shifts of the emission peak and, therefore, results in unwanted effects, like the broadening of the emission spectrum and the prolongation of TADF lifetime.19,20 All this makes the control of conformational disorder highly important for the successful application of TADF compounds, especially as emitters in OLED devices, since the EQE roll-off of OLEDs at high current densities directly depends on TADF lifetime.26 Recently it was demonstrated that compounds with large conformational disorder benefited from shortened TADF lifetime.20 Stable emission was achieved; however TADF decay was still remarkably slower than in solutions. Some control of delayed emission lifetime was also shown by tuning the polarity of the polymer host;19 however more efficient strategies are still required. As a solution, the enhancement of molecular core rigidity could be applied. Yersin et al.21 recently have demonstrated the benefits of a rigid molecular structure for avoiding the occurrence of prolonged TADF decay; however all the benefits were demonstrated in a rather specific molecular structure with rigid bridges.In this work, we followed the idea of the necessity of rigid molecular cores for the minimization of unwanted conformational disorder; however typical donor–acceptor TADF compounds with flexible single bonds were investigated. Time-resolved fluorescence was employed for the analysis of conformational disorder in dilute polymer films of two TADF compounds, with similar emission properties in toluene solutions but a completely different molecular structure and stiffness of the molecular core, PTZ-mPYR and 4CzPN. The analysis was supplemented by comparison of solid-state TADF properties of compound PTZ-mPYR and its analogue PXZ-PYR, bearing a more sterically constrained donor unit. The same trends were observed in both series of compounds, despite remarkable differences in the molecular structure. The rigid 4CzPN was shown to have almost alike TADF properties in solutions and solid-state due to minimized conformational disorder. Our study has shown that donor–acceptor TADF compounds with a consistently restricted molecular structure could be efficient emitters in both solutions and solid films.
Experimental details
4CzPN was synthesized according to Uoyama et al.3 Synthetic details of PTZ-mPYR and PXZ-PYR were published elsewhere.20,27 TADF compounds were analyzed in 1 × 10−5 M toluene solutions and in 1 wt% PMMA films. Polymer films were prepared by dissolving each material and PMMA at appropriate ratios in toluene solutions and then wet-casting the solutions on glass substrates. Time-integrated fluorescence spectra (TIFL), time-resolved fluorescence spectra (TRFL) and fluorescence decay transients were measured using a nanosecond YAG:Nd3+ laser NT 242 (Ekspla, τ = 7 ns, pulse energy 200 μJ, λex = 350 nm, repetition rate 10–1000 Hz) and time-gated iCCD camera New iStar DH340T (Andor). Time integrated prompt and delayed fluorescence spectra (TIPF and TIDF, respectively) were measured by using 0 ns delay and 200 ns (no prompt fluorescence was observed after 200 ns) exposure time for TIPF and 200 ns delay and 100 ms exposure time for TIDF. Exposure time for TIDF measurement was selected to be longer than TADF lifetime. Fluorescence quantum yields (ΦF) of the solutions and polymer films in an air atmosphere were estimated with the integrated sphere method28 by using an integrating sphere (Sphere Optics) connected to the CCD spectrometer PMA-11 (Hamamatsu) via an optical fibre. Fluorescence transients were obtained by exponentially increasing delay and integration time. This allows one to record up to 10 orders of magnitude in time and intensity of the fluorescence decay.29 Solid-state samples were mounted in a closed cycle He cryostat (Cryo Industries 204N) for all measurements (for oxygen-saturated and oxygen-free conditions). Toluene solutions were degassed by using the freeze–pump–thaw method (5 iterations).Quantum chemical calculations were performed by using density functional theory (DFT) as implemented in the Gaussian 09 software package at the B3LYP/6-31G(d) level.30 The Polarizable Continuum Model (PCM) was used to estimate the solvation behaviour of toluene surrounding in geometry optimization.
Results and discussion
Material selection
Three TADF compounds were selected to analyse the relation between the molecular geometry and conformational disorder in solid films (see Fig. 1). Compounds PTZ-mPYR and 4CzPN were selected to have completely different molecular geometry but similar TADF properties. PTZ-mPYR is composed of a diphenylpyrimidine electron-acceptor unit and two phenothiazine (PTZ) electron-donor fragments with nonplanar geometry, while 4CzPN is constructed of a dicyanophenyl electron-acceptor fragment, decorated with four adjacent carbazole electron-donating units. The nonplanar PTZ unit is found in two distinct conformer states, namely quasi-axial (QA) and quasi-equatorial (QE) with different corresponding absorption and emission properties.31,32 However, only the emission from QE states is of TADF nature20,33 and will be analysed in this paper. In the second set of compounds, two TADF compounds with a very similar structure were compared, PTZ-mPYR and its structural counterpart PXZ-PYR, constructed from the almost alike diphenylpyrimidine acceptor fragment but a different donor unit, planar phenoxazine (PXZ). As it was shown earlier, the presence of a methyl unit in the acceptor fragment of PTZ-mPYR has almost no effect on solid-state conformational disorder.20 | ||
| Fig. 1 Molecular structures of TADF compounds PTZ-mPYR, 4CzPN and PXZ-PYR. | ||
Initially, solid-state TADF properties of compounds PTZ-mPYR and 4CzPN, strongly differing in the molecular structure, will be investigated. Lastly, two very similar TADF emitters, PTZ-mPYR and PXZ-PYR, will be compared to reveal the impact of delicate structural changes on solid-state emission properties.
DFT simulations of PTZ-mPYR and 4CzPN
DFT simulations were performed to analyse the differences in the molecular structure. The optimized geometries of TADF compounds PTZ-mPYR and 4CzPN are shown in Fig. 2. The optimization revealed that the phenothiazine moiety in QE arrangement is almost perpendicular to the phenyl fragment (α1 ≈ 88.9°, see Fig. 2); however due to the twisted geometry, the PTZ units also are bent along the C–N bonds,34 leading to larger rotational flexibility than of planar donor moieties. Both phenyl fragments in the acceptor unit were twisted only for about α2 ≈ 46° with respect to the pyrimidine group due to the weak steric hindrance. Previously, it was shown that such a molecular structure is flexible with large conformational disorder in solid state.20 For 4CzPN, carbazole fragments are twisted with respect to the central phenyl moiety for about α3 ≈ 62–67°.35,36 Close proximity of the adjacent carbazole units ensures large steric hindrance and large stiffness of the molecular core.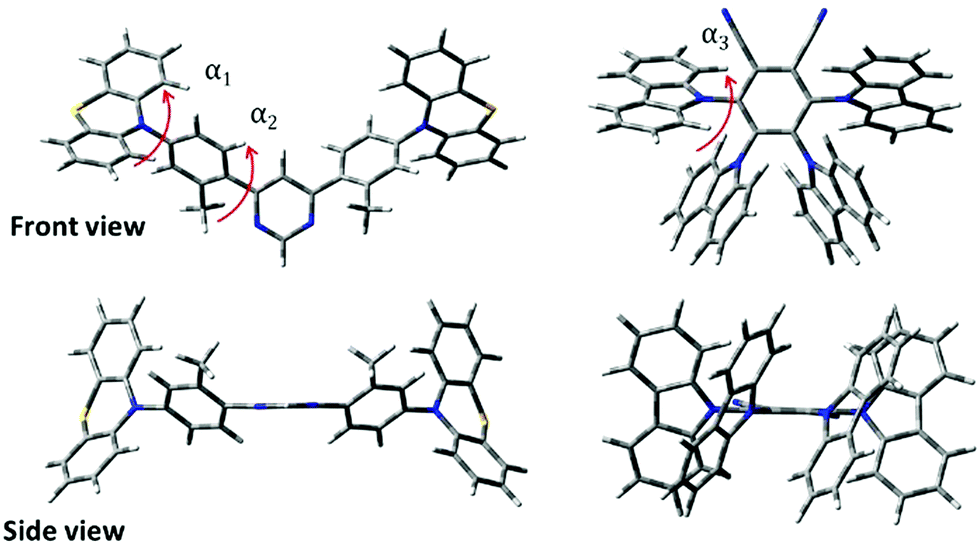 | ||
| Fig. 2 DFT-optimized molecular geometries of PTZ-mPYR and 4CzPN. Only quasi-equatorial conformation is shown for PTZ-mPYR. α1 is a twist angle between the phenothiazine unit and the phenyl fragment, α2 is a twist angle between the phenyl fragment and the pyrimidine unit and α3 represents the twist angle between carbazole and dicyanobenzene acceptor. | ||
Ground state dipole moments of 3.08 D and 7.38 D were estimated for PTZ-mPYR and 4CzPN, respectively.
In the context of this paper, molecular rigidity is described as rotational flexibility between the D and A units through C–N bonds. In such case, flexible molecules would have large dispersion of D–A twist angles, while the rigid ones lower dispersion.21 Differences in the structural rigidity of compounds PTZ-mPYR and 4CzPN with strongly different geometry were assessed by estimating the vibrational frequencies of the lowest vibrational modes in the harmonic approach (see Fig. 3). Ground-state optimized molecular structures were used for vibrational frequency calculations, as the ability of TADF compounds to reorient upon photoexcitation is strongly restricted in the solid films.34,37 Molecular compounds have a large number of various vibrational modes up to an energy of several thousand cm−1, like symmetric vinyl stretching modes observed at about 1400 cm−1, responsible for vibronic progression in absorption and emission spectra.38 However, in our case the most important are the lowest energy vibrations, usually arising from the vibration of the loose donor and acceptor molecular units, evidently altering the TADF properties.7,9–11 D–A twisting motions should occur at the lower energy for more flexible TADF compounds, rather for the rigid ones.39
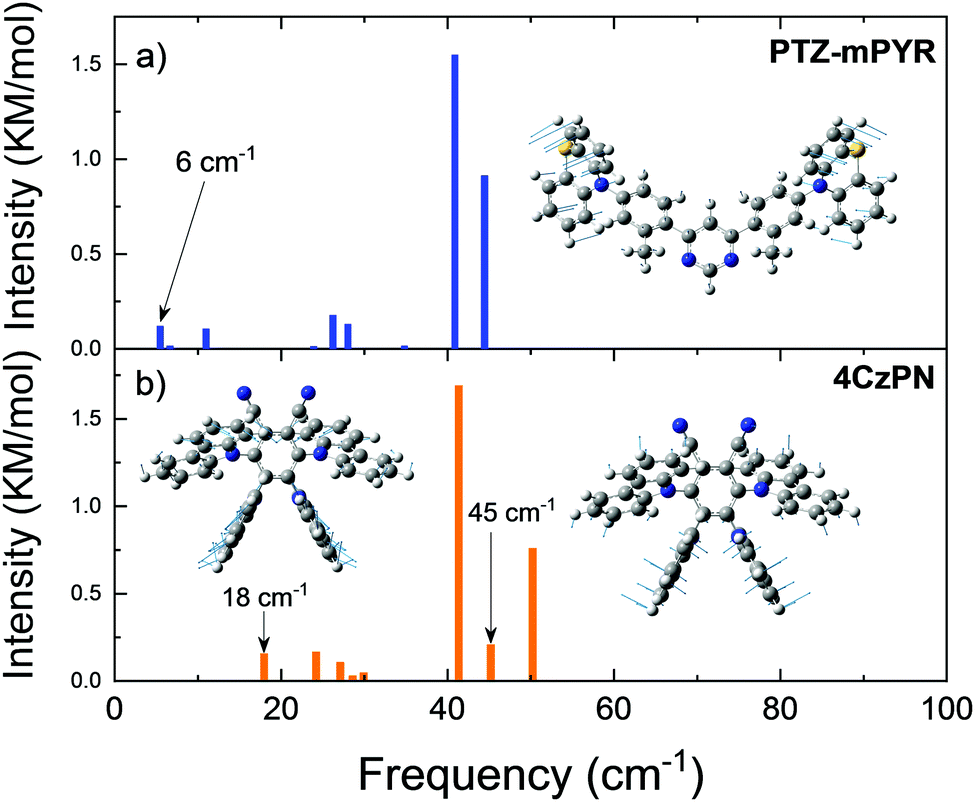 | ||
| Fig. 3 Calculated frequencies of the lowest vibrational modes (up to 50 cm−1) for compounds PTZ-mPYR (a) and 4CzPN (b). The B3LYP/6-31G(d) method was used. Nuclear displacement vectors, corresponding to the lowest-frequency vibrational normal modes at 6 cm−1 (for PTZ-mPYR), 18 cm−1 and 45 cm−1 (for 4CzPN), are shown. | ||
Clearly, the lowest vibrational modes of PTZ-mPYR and 4CzPN were of different nature. For PTZ-mPYR, the twisting motion of the donor unit was shown to be the lowest vibrational mode at only 6 cm−1, typical for TADF compounds.9,39 In contrast, the lowest vibrational mode for 4CzPN was found at the higher energy and was of different nature. The large steric hindrance between the carbazole units suppressed the twisting along the C–N bond, and thus the lowest energy vibration at 18 cm−1 was shown to be the waving motion of carbazole units, barely related to conformational disorder in solid state (see Fig. 3b). Only the higher energy vibration mode at 45 cm−1 was the twisting motion of the donor units, directly contributing to the rise of conformational disorder in solid state. Supposedly, larger energetic barriers for the twisting motion for donor units for 4CzPN should result in lower ΔEST dispersion and lower conformational disorder in solid state.40,41 Quantum mechanical and experimental analyses of similar to 4CzPN carbazole-benzonitrile TADF compounds, bearing several adjacent carbazole units, have also shown the large torsional barriers for twisting of carbazole units, ensuring a rigid molecular structure and subsequent highly efficient TADF.7,14,35
TADF properties in solutions: PTZ-mPYRvs.4CzPN
Despite the different molecular structure, compounds PTZ-mPYR and 4CzPN showed similar emission properties in toluene (see Fig. S1 in ESI† and Table 1). Both TADF compounds showed greenish fluorescence, peaking at 535 and 525 nm for PTZ-mPYR and 4CzPN, respectively, with comparable fluorescence lifetimes: the decay time for prompt fluorescence was 10 and 14 ns and TADF lifetimes were 11 and 15.5 μs for PTZ-mPYR and 4CzPN, respectively (see Fig. 5). Energy gaps between the lowest singlet and triplet states were also comparable, 25 meV vs. 10 meV, for PTZ-mPYR and 4CzPN, respectively. However, the fluorescence quantum yield of prompt and delayed fluorescence was lower for PTZ-mPYR with more rapid non-radiative decay39 (0.04 and 0.04 vs. 0.09 and 0.61, respectively).| λ FL (nm) | τ PR (ns) | τ TADF (μs) | Φ PF | Φ TADF | k ISC (×107 s−1) | k rISC (×105 s−1) | ΔESTh (meV) | |
|---|---|---|---|---|---|---|---|---|
| a Fluorescence wavelength. b Prompt fluorescence decay time. c Delayed fluorescence decay time. d Fluorescence quantum yield of prompt fluorescence. e Fluorescence quantum yield of delayed fluorescence. f Intersystem crossing rate. g Reverse intersystem crossing rate. h Singlet–triplet energy gap. | ||||||||
| PTZ-mPYR | 535 | 10 | 11 | 0.04 | 0.04 | 2.7 | 1.2 | 25 |
| 4CzPN | 525 | 14 | 15.5 | 0.09 | 0.61 | 6.5 | 7.1 | 10 |
TADF properties in polymer films: PTZ-mPYRvs.4CzPN
Although compounds PTZ-mPYR and 4CzPN showed similar properties in toluene, remarkable differences in solid-state emission properties were revealed. Firstly, flexible PTZ-mPYR showed strong TADF quantum efficiency enhancement in more rigid PMMA surrounding (0.04 vs. 0.32), whereas for rigid 4CzPN TADF quantum efficiency was rather the same (0.61 vs. 0.54). Secondly, typical for TADF compounds temporal shifts of prompt (PF) and delayed (DF) fluorescence spectra18–20,31 were observed in the time-resolved fluorescence spectra of 1 wt% PMMA films (see Fig. 4a). Initially, the redshift of PF was observed for about 100 ns, followed by the later blueshift of DF. No shift was observed from about 100 ns to 10 μs when emission originated from the conformer states with the lowest ΔEST.20 Evident differences in fluorescence shift trends were observed between PTZ-mPYR and 4CzPN. For PTZ-mPYR with a more flexible molecular structure, a redshift of 370 meV was observed, followed by 80 meV blueshift. For the rigid 4CzPN, both red and blueshifts were of the same value, at only about 100 meV (see Table S1 in ESI† for details). Obviously, lower conformational disorder and thus the smaller emission peak shifts for rigid 4CzPN were mediated by the lower dispersion of 1CT energies. The enhanced rigidity and lowered conformational disorder of 4CzPN had a remarkable advantage over PTZ-mPYR for emission wavelength stability (see Fig. 4b and c). Previously it was shown that the emission of compound PTZ-mPYR peaks at different wavelengths in oxygen-free and oxygen-saturated conditions due to the quenching of the latest delayed emission20 in oxygen-free conditions. Due to the wide distribution of singlet–triplet energy gaps, the latest emission spectra originate from conformations with very large singlet–triplet energy gaps and prolonged lifetime, which are largely susceptible to quenching by various quenching sites. Quenching of the latest emission with the largest 1CT energy disturbs the high-energy shoulder of the time-integrated delayed fluorescence spectrum in −O2 conditions, resulting in the redshift of its peak wavelength (see Fig. 4b) and the redshift of the overall time-integrated emission spectrum (see Fig. 4c). A completely different situation was observed for compound 4CzPN with low conformational disorder. In this case, the stable emission peak wavelength was observed. Due to the rigid molecular core, the latest delayed fluorescence of 4CzPN originated from the similar conformer states with rather low ΔEST and rapid rISC in both +O2 and −O2 conditions (see Fig. 4b and c).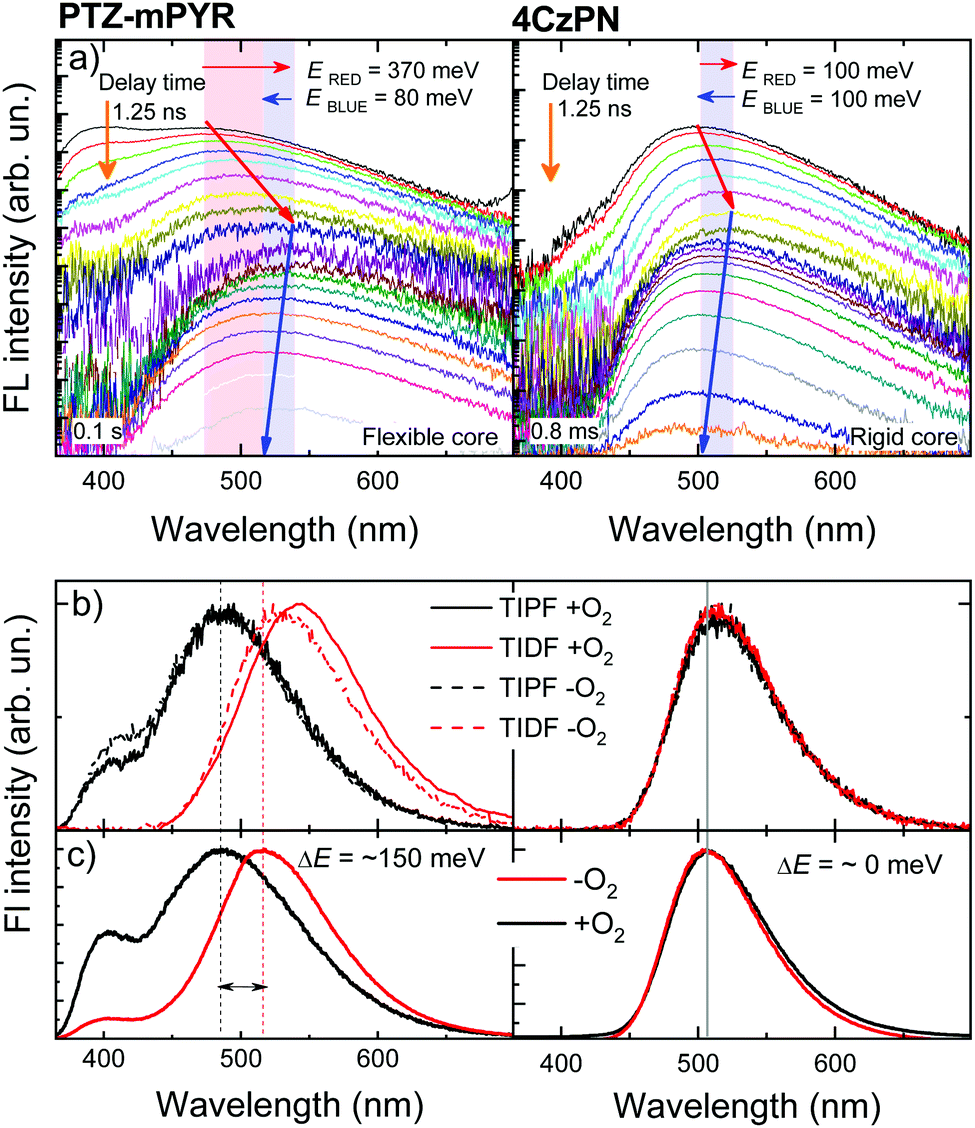 | ||
| Fig. 4 (a) Time-resolved fluorescence spectra of 1 wt% PMMA films of PTZ-mPYR (left picture) and 4CzPN (right picture). All measurements were performed in oxygen-free surrounding. (b) Time-integrated normalized prompt (TIPF) and delayed fluorescence (TIDF) spectra of 1 wt% PMMA films of PTZ-mPYR (left picture) and 4CzPN (right picture) in oxygen-saturated (+O2, solid lines) and oxygen-free (−O2, dashed lines) conditions. Experimental parameters are provided in the Experimental details section. (c) Time-integrated normalized fluorescence spectra of 1 wt% PMMA films of PTZ-mPYR (left picture) and 4CzPN (right picture) in oxygen-saturated (+O2, black lines) and oxygen-free (−O2, red lines) conditions. | ||
Likewise, FWHM values of time-integrated fluorescence spectra of polymer films also were molecular rigidity-dependent. For 4CzPN, FWHM values in both +O2 and −O2 conditions (400 meV) were only slightly larger than in toluene solution (370 meV). It again evidenced the high rigidity of the molecular structure of 4CzPN. In contrast, a moderate increase of FWHM of about 15% (from 400 meV in toluene to 460 meV in PMMA) was observed for the solid-state emission spectra of PTZ-mPYR in −O2 conditions. However, this was not mediated by small dispersion of 1CT peak energies, as it was for rigid 4CzPN, but specifically due to the large conformational disorder. PTZ-mPYR has large dispersion of 1CT emission energies and singlet–triplet gaps. In −O2 conditions, the latest long-lived delayed emission from the largest-energy conformers is quenched, and thus the emission spectrum mostly consists of the lowest-energy conformers with lower dispersion of 1CT energies. In +O2 conditions, even more delayed emission is quenched (see Fig. S2 in ESI†), and thus the time-integrated emission spectrum contains a larger fraction of prompt fluorescence, formed from nearly all possible conformer states with large dispersion of 1CT emission energies. In the case of −O2 conditions, a 46% increase of FWHM was observed (from 460 meV to 670 meV), and the FWHM of TIFL of polymer films in +O2 conditions was even 68% larger than that of toluene solutions (400 meV vs. 670 meV).
The presence of conformational disorder, increasing the spectral linewidth, as well as lower polarity of polymer surrounding resulted in an increase of ΔEST in polymer films, which represents the average gap of all conformer states (see Fig. S3 in the ESI†). A singlet–triplet gap of 187 meV was observed for PTZ-mPYR (with fluorescence spectra obtained in −O2 conditions as a reference) and 140 meV – for 4CzPN (see Fig. S4 in ESI†).
TADF decay transients: PTZ-mPYRvs.4CzPN
Pronounced differences in emission properties between compounds 4CzPN and PTZ-mPYR with different molecular core rigidity were also observed in solid-state fluorescence decay transients (see Fig. 5). The temporal profiles of emission decay transients in dilute solution and polymer films considerably differed. The presence of conformational disorder, as it was already shown, resulted in the dispersion of ΔEST values, when the later decaying states had larger singlet–triplet gaps and slower rISC. Compounds with a more flexible molecular core and larger conformational disorder had bigger ΔEST dispersion and more conformer states with inefficient rISC. It resulted in the extension of TADF lifetime in solid surrounding and the enhancement was remarkably larger for flexible PTZ-mPYR as compared to 4CzPN. This was evident from the comparison of fluorescence decay transients of compounds PTZ-mPYR and 4CzPN in toluene solution and PMMA films (see Fig. 5c and d). TADF lifetime enhancement for PTZ-mPYR was followed by subsequent boost of TADF efficiency from 0.04 in toluene to 0.32 in PMMA. The presence of a large number of conformer states with inefficient rISC increased the TADF lifetime and, concomitantly, the average number of ISC/rISC cycles.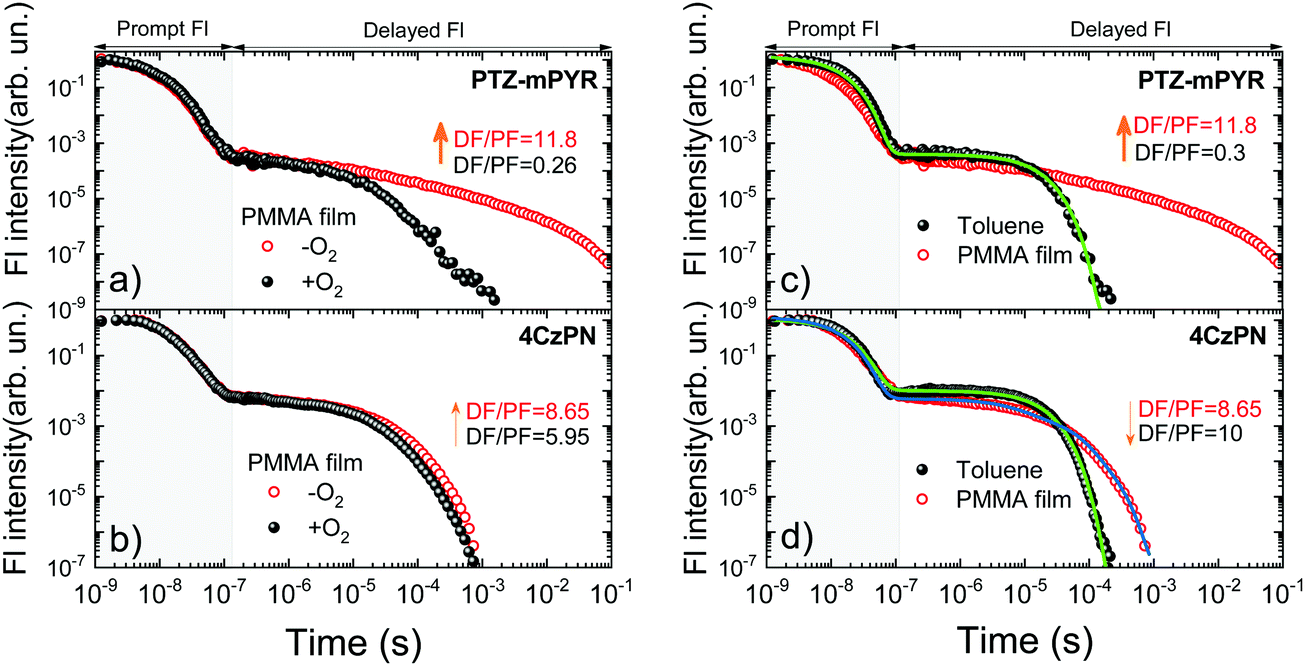 | ||
| Fig. 5 Fluorescence decay transients of 1 wt% PMMA films of PTZ-mPYR and 4CzPN in oxygen-saturated and oxygen-free conditions (a and b, respectively) and fluorescence decay transients of oxygen-free 1 wt% PMMA films and toluene solutions of PTZ-mPYR and 4CzPN (c and d, respectively). All fluorescence decay transients were normalized. Color lines are multiexponential fits. | ||
In an ideal case with negligible conformational disorder and highly efficient TADF, similar TADF properties should be observed in both surroundings. This was the case for rigid 4CzPN with similar TADF characteristics in toluene and PMMA films (see Fig. 5). In this case, low conformational disorder resulted only in small dispersion of ΔEST values, when the TADF lifetime from the conformer states with the largest singlet–triplet gaps was weakly increased. Therefore, rather a simple three-exponential fit of the delayed fluorescence decay allowed one to estimate the average TADF lifetime for 4CzPN in PMMA film. A three-fold increase of the average TADF lifetime from 15.5 μs to 46 μs was observed, resulting in a slight decrease of the rISC rate from 7.1 × 105 to 2 × 105 s−1. The negligible decrease of the DF/PF ratio in PMMA was due to the lower polarity of polymer surrounding19 and larger ΔEST. Certainly, the lower polarity of PMMA also could result in the lowered rISC rate, though the ΔEST dispersion and the subsequent increase of TADF lifetime still were the main reason. Secondly, fluorescence decay transients of PMMA films of compounds PTZ-mPYR and 4CzPN in +O2 and −O2 conditions followed different pathways (see Fig. 5a and b). A remarkable quenching of delayed fluorescence after about 10 μs was observed for compound PTZ-mPYR, though the initial TADF from the conformers with the lowest ΔEST was unquenched even in +O2 conditions. In this way, the intensity ratio of delayed and prompt fluorescence (DF/PF ratio) for PTZ-mPYR with large conformational disorder decreased from 11.8 in −O2 conditions to 0.26 in +O2 conditions and enabled the blueshift of the emission peak (see Fig. 4c). The delayed emission of compound 4CzPN with a rigid molecular core and low ΔEST dispersion was weakly sensitive to molecular oxygen. Only a modest quenching of the latest delayed emission from conformers with the largest ΔEST was observed, when the DF/PF ratio decreased about 30% from 8.65 to 5.95. This ensured the stable emission for rigid 4CzPN.
Phenothiazine vs. phenoxazine: the impact of donor planarity
To show the validity of our previous observations for various donor–acceptor TADF systems, the solid-state TADF properties of two compounds with a very similar molecular structure were compared (see Fig. 1, series II). In this case, compounds bearing a diphenylpyrimidine-based electron-acceptor unit, bound with two donor units of different geometry, phenothiazine (PTZ-mPYR) and phenoxazine (PXZ-PYR), were selected. As it was already mentioned, PTZ is of nonplanar geometry. Such orientation has less steric hindrance, leading to more perturbed TADF properties, especially in solid-state.42 Those shortcomings could be prevailed by selecting more sterically fixed planar donor units, like PXZ43,44 (see Fig. 6a), though the expected effect should not be as strong as for highly rigid 4CzPN.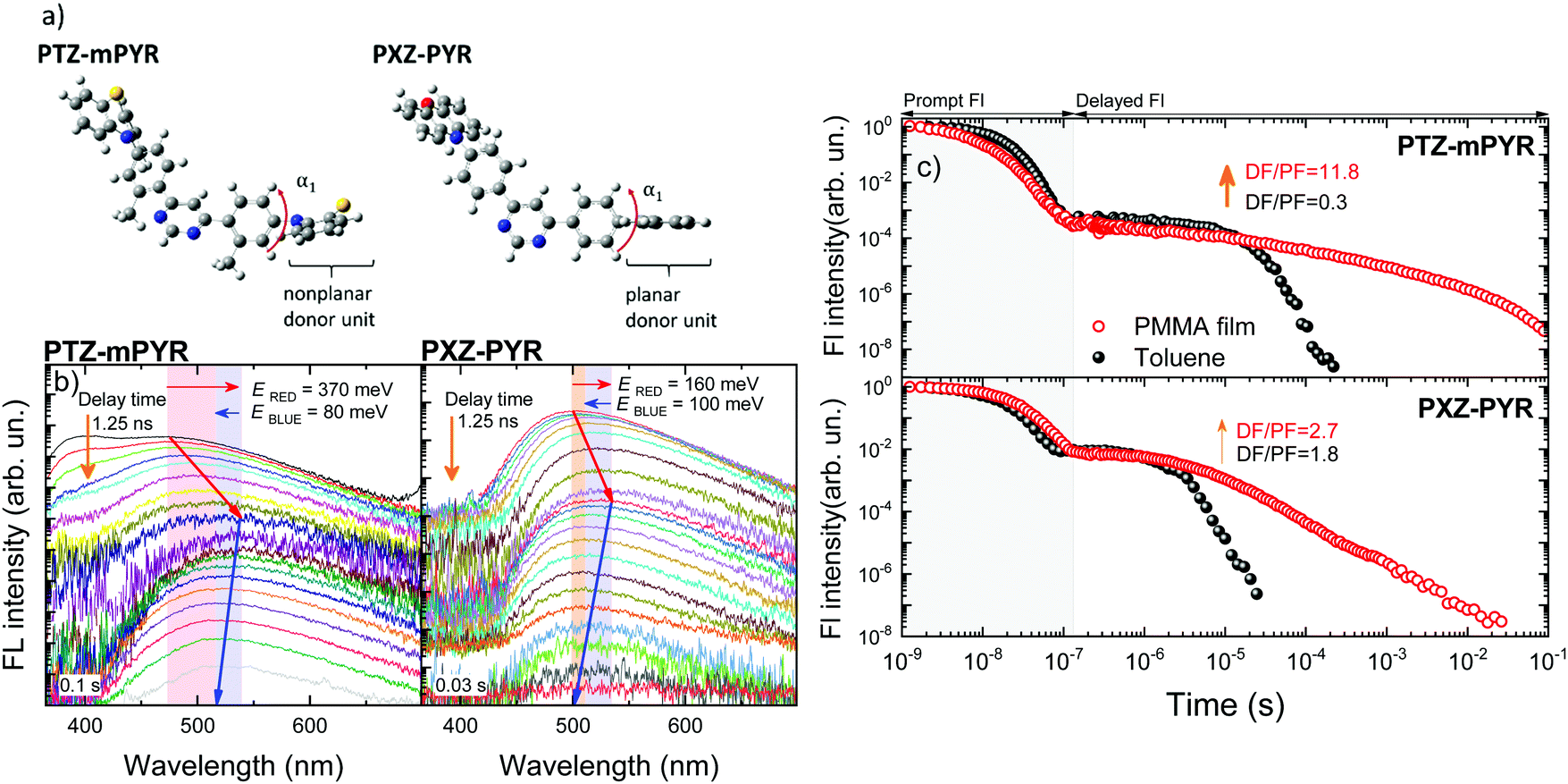 | ||
| Fig. 6 (a) Optimized molecular structures of compounds PTZ-mPYR and PXZ-PYR. α1 is the twist angle between the donor unit and the phenyl spacer. (b) Time-resolved fluorescence spectra of 1 wt% PMMA films of PTZ-mPYR (left picture) and PXZ-PYR (right picture). (c) Fluorescence decay transients of PTZ-mPYR (upper picture) and PXZ-PYR (lower picture) in toluene (red open dots) and 1 wt% PMMA films (closed black dots). All measurements were performed in oxygen-free surrounding. | ||
Despite the different donor unit and different ground-state dipole moment (0.4 D for PXZ-PYR), both compounds showed rather similar emission wavelength in toluene (see Fig. S5 in ESI†),20,27 though PTZ-mPYR showed lower prompt and TADF efficiency (0.04 and 0.04 vs. 0.15 and 0.27, respectively) together with slower TADF decay (11 vs. 1.6 μs, respectively). Simultaneously, rather different solid-state TADF properties were observed. The time-resolved fluorescence spectra of TADF compounds with different donor units in PMMA films are shown in Fig. 6b. As it was shown previously, PTZ-mPYR showed a pronounced redshift of the prompt fluorescence peak followed by a remarkably lower blueshift of delayed fluorescence at the larger time-scale, which was even lower in oxygen-saturated conditions. As a result, ambient-dependent emission colour was observed. In contrast, remarkably lower temporal shifts of TRPL spectra were observed for PXZ-PYR with a more sterically constrained planar donor unit (160 and 100 meV for redshift and blueshift, respectively). As both temporal shifts were comparable, stable emission colour was observed in both +O2 and −O2 conditions (see Fig. S6 in ESI†). A portion of the latest delayed fluorescence still was quenched, though this share was remarkably lower than for more flexible PTZ-mPYR. Lowered conformational disorder was especially beneficial for achieving lower TADF lifetime enhancement in solid-state (see Fig. 6c). Although the exact TADF decay time in PMMA film was unknown, a minor increase of the DF/PF ratio in solid-state indicated low TADF lifetime enhancement. On the contrary to PTZ-mPYR, lower dispersion of ΔEST values in PXZ-PYR indicates weaker enhancement on TADF lifetime as well as lower average number of ISC/rISC cycles.
Clearly, the rigid molecular structure is essential for minimization of solid-state conformational disorder, regardless of the selected donor and acceptor pair.
Conclusions
In this work, the importance of molecular rigidity for highly efficient delayed emission in solid-state was highlighted. Two TADF compounds with different molecular core rigidity were selected to showcase the impact of conformational disorder on solid-state TADF properties. Pronounced conformational disorder with large dispersion of singlet–triplet energy gaps was estimated for PTZ-mPYR with a more flexible molecular structure. It had a negative impact on the solid-state TADF properties. Firstly, the large ΔEST dispersion in polymer films resulted in the emergence of conformer states with large ΔEST and prolonged TADF lifetime. Secondly, the latest delayed emission with inefficient TADF was strongly quenched. The loss of delayed emission with the largest 1CT energies was the reason for the redshift of the solid-state emission peak in oxygen-free conditions. All the above-mentioned drawbacks were eliminated only by selecting a more rigid molecular structure of 4CzPN. The second TADF compound was selected to have similar TADF properties in solution but lower rotational flexibility of donor units. Minor conformational disorder was observed in solid-state with stable emission colour. More importantly, only a slight increase of TADF lifetime, as compared to that of solution, was observed. This is especially important for the production of TADF OLEDs with low external quantum efficiency roll-off, which is known to depend on the TADF lifetime. The reliability of the suggested design principle was evaluated in the second set of compounds, when two TADF compounds with an alike acceptor fragment but distinct donor units with different steric hindrance, PTZ-mPYR and PXZ-PYR, were compared. As it was expected, the minimized conformational disorder was evidenced for the compound with more sterically constrained planar donor unit. Our analysis clearly shows that the design of solid-state efficient TADF compounds requires more delicate approaches, when the stiffness of the molecular structure is as important as efficient separation of HOMO and LUMO overlap and strong coupling between the triplet states.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding from the Research Council of Lithuania (LMTLT), agreement no. S-MIP-17-73. We are grateful to Dr Alytis Gruodis and High Performance Computing Center ‘‘HPC Sauletekis’’ in Vilnius University Faculty of Physics for vibrational frequency calculations and fruitful discussions. We are also very grateful to the Professor Vytautas Getautis group in Kaunas University of Technology for the synthesis of 4CzPN.References
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- E. W. Evans, Y. Olivier, Y. Puttisong, W. K. Myers, T. J. H. Hele, S. M. Menke, T. H. Thomas, D. Credgington, D. Beljonne, R. H. Friend and N. C. Greenham, J. Phys. Chem. Lett., 2018, 9, 4053–4058 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- D. de Sa Pereira, C. Menelaou, A. Danos, C. Marian and A. P. Monkman, J. Phys. Chem. Lett., 2019, 10, 3205–3211 CrossRef CAS PubMed.
- Y. Olivier, J.-C. Sancho-Garcia, L. Muccioli, G. D’Avino and D. Beljonne, J. Phys. Chem. Lett., 2018, 9, 6149–6163 CrossRef CAS PubMed.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- Z.-P. Chen, D.-Q. Wang, M. Zhang, K. Wang, Y.-Z. Shi, J.-X. Chen, W.-W. Tao, C.-J. Zheng, S.-L. Tao and X.-H. Zhang, Adv. Opt. Mater., 2018, 6, 1800935 CrossRef.
- M. Saigo, K. Miyata, S. Tanaka, H. Nakanotani, C. Adachi and K. Onda, J. Phys. Chem. Lett., 2019, 10, 2475–2480 CrossRef CAS PubMed.
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- X. Liang, Z.-L. Tu and Y.-X. Zheng, Chem. – Eur. J., 2019, 25, 5623–5642 CrossRef CAS PubMed.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef PubMed.
- T. Northey, J. Stacey and T. J. Penfold, J. Mater. Chem. C, 2017, 5, 11001–11009 RSC.
- T. Serevičius, T. Buciunas, J. Bucevičius, J. Dodonova, S. Tumkevicius, K. Kazlauskas and S. Jursenas, J. Mater. Chem. C, 2018, 11128–11136 RSC.
- T. Serevičius, R. Skaisgiris, J. Dodonova, L. Jagintavičius, J. Bucevičius, K. Kazlauskas, S. Jursenas and S. Tumkevicius, Chem. Commun., 2019, 55, 1975–1978 RSC.
- H. Yersin, L. Mataranga-Popa, R. Czerwieniec and Y. Dovbii, Chem. Mater., 2019, 31, 6110–6116 CrossRef CAS.
- G. Méhes, K. Goushi, W. J. Potscavage and C. Adachi, Org. Electron., 2014, 15, 2027–2037 CrossRef.
- R. S. Nobuyasu, J. S. Ward, J. Gibson, B. A. Laidlaw, Z. Ren, P. Data, A. S. Batsanov, T. J. Penfold, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2019, 7, 6672–6684 RSC.
- S. Weissenseel, N. A. Drigo, L. G. Kudriashova, M. Schmid, T. Morgenstern, K.-H. Lin, A. Prlj, C. Corminboeuf, A. Sperlich, W. Brütting, M. K. Nazeeruddin and V. Dyakonov, J. Phys. Chem. C, 2019, 123, 27778–27784 CrossRef CAS.
- T. Northey, J. Stacey and T. J. Penfold, J. Mater. Chem. C, 2017, 5, 11001–11009 RSC.
- M. Inoue, T. Serevičius, H. Nakanotani, K. Yoshida, T. Matsushima, S. Juršėnas and C. Adachi, Chem. Phys. Lett., 2016, 644, 62–67 CrossRef CAS.
- K. Wu, T. Zhang, L. Zhan, C. Zhong, S. Gong, N. Jiang, Z.-H. Lu and C. Yang, Chem. – Eur. J., 2016, 22, 10860–10866 CrossRef CAS PubMed.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- C. Rothe and A. P. Monkman, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 075208 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., Int. Ed., 2018, 57, 16407–16411 CrossRef CAS PubMed.
- D.-G. Chen, T.-C. Lin, Y.-A. Chen, Y.-H. Chen, T.-C. Lin, Y.-T. Chen and P.-T. Chou, J. Phys. Chem. C, 2018, 122, 12215–12221 CrossRef CAS.
- Q. Peng, D. Fan, R. Duan, Y. Yi, Y. Niu, D. Wang and Z. Shuai, J. Phys. Chem. C, 2017, 121, 13448–13456 CrossRef CAS.
- S. Wang, Y. Zhang, W. Chen, J. Wei, Y. Liu and Y. Wang, Chem. Commun., 2015, 51, 11972–11975 RSC.
- H. F. Higginbotham, C.-L. Yi, A. P. Monkman and K.-T. Wong, J. Phys. Chem. C, 2018, 122, 7627–7634 CrossRef CAS.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- G. Kreiza, D. Banevičius, J. Jovaišaitė, K. Maleckaitė, D. Gudeika, D. Volyniuk, J. V. Gražulevičius, S. Juršėnas and K. Kazlauskas, J. Mater. Chem. C, 2019, 7, 11522–11531 RSC.
- J. Fan, Y. Zhang, Y. Zhou, L. Lin and C.-K. Wang, J. Phys. Chem. C, 2018, 122, 2358–2366 CrossRef CAS.
- J. Fan, L. Lin and C.-K. Wang, J. Mater. Chem. C, 2017, 5, 8390–8399 RSC.
- K. Wang, C.-J. Zheng, W. Liu, K. Liang, Y.-Z. Shi, S.-L. Tao, C.-S. Lee, X.-M. Ou and X.-H. Zhang, Adv. Mater., 2017, 29, 1701476 CrossRef PubMed.
- K. Wang, Y.-Z. Shi, C.-J. Zheng, W. Liu, K. Liang, X. Li, M. Zhang, H. Lin, S.-L. Tao, C.-S. Lee, X.-M. Ou and X.-H. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 31515–31525 CrossRef CAS PubMed.
- D.-G. Chen, T.-C. Lin, C.-L. Chen, Y.-T. Chen, Y.-A. Chen, G.-H. Lee, P.-T. Chou, C.-W. Liao, P.-C. Chiu, C.-H. Chang, Y.-J. Lien and Y. Chi, ACS Appl. Mater. Interfaces, 2018, 10, 12886–12896 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05907e |
| This journal is © the Owner Societies 2020 |