
The influence of intermolecular coupling on electron and ion transport in differently substituted phthalocyanine thin films as electrochromic materials: a chemistry application of the Goldilocks principle†
Thi Hai Quyen
Nguyen
a,
Marius
Pelmuş
 b,
Christopher
Colomier
b,
Sergiu M.
Gorun
*b and
Derck
Schlettwein
*a
b,
Christopher
Colomier
b,
Sergiu M.
Gorun
*b and
Derck
Schlettwein
*a
aInstitute of Applied Physics and Laboratory of Materials Research, Justus-Liebig-University Gießen, Heinrich-Buff-Ring 16, 35392 Gießen, Germany. E-mail: schlettwein@uni-giessen.de
bDepartment of Chemistry and Biochemistry and Center for Functional Materials, Seton Hall University, 400 South Orange Ave, South Orange, NJ 07079, USA. E-mail: sergiu.gorun@shu.edu
First published on 26th February 2020
Abstract
The transport of both electrons and ions in organic mixed ionic and electronic conductors such as phthalocyanines, is essential to allow redox reactions of entire films and, hence, to impart electrochromism. Thin films of a new type, tetrakis-perfluoroisopropyl-perfluoro phthalocyanine, F40PcCu of different thicknesses were obtained via vapor deposition. The extent of the intermolecular coupling within the F40PcCu films established by van der Waals interactions was investigated by in situ optical spectroscopy during film growth. The transfer of electrons and diffusion of counter cations in these films, as well as their electrochromic performance were characterized by electrochemical and spectroelectrochemical measurements with an aqueous solution of KCl as electrolyte. A moderate degree of intermolecular interaction of the F40PcCu molecules in the solid state was observed, compared to non-fluoroalkylated perfluoro phthalocyanine, F16PcCu and octakis-perfluoroisopropyl-perfluorophthalocyanine, F64PcCu, which exhibit stronger and weaker coupling, respectively. The replacement of F by perfluoroisopropyl is, thereby, established as a valuable approach to tune this coupling of chromophores and, hence, the transport coefficients of electrons and ions in the solid films. Reversible changes of the films upon reduction and intercalation of K+ counter ions and re-oxidation and expulsion of the counter ions were confirmed by simultaneously measured optical absorption spectra. Thin films of F40PcCu showed a well-balanced, equally fast transport of electrons and ions. The films provided a fast and reversible switching process over at least 200 cycles indicating the stability of these materials.
1 Introduction
Molecular thin films play an increasingly important role in electronic and optoelectronic devices as dielectric, conducting or semiconducting materials.1 One specific example is their use as electrochromic layers, i.e. layers which reversibly change their color and/or light transmission upon their reduction or oxidation. Such layers are of potential use in display applications or as smart windows or smart mirrors.2,3 When compared to classic electrochromic materials like WO3, organic materials offer faster switching due to their higher extinction coefficients which allow the use of thinner films.4,5 In order to establish such higher switching rates, the diffusion of ions and the mobility of electrons in the films have to be optimized since both are needed at a given molecular site in the films in order for the redox reaction to occur. As a consequence, organic mixed ionic and electronic conductors (OMIEC) are needed for this purpose, a group of materials recently established in the literature.6 For a number of such materials, namely polymer electronic (or hole-) conductors, water is needed in the films in order to provide channels of ionic conduction – polymers are chosen that are easily swollen with water.7,8 Electrochromic reactions, however, have also been established for pure molecular semiconductors, namely phthalocyanines (Pc).9In early studies, unsubstituted Pc (e.g., H16PcCu, Fig. 1a) showed partially reversible electrochromic oxidation.10 Substitution of aromatic hydrogen atoms by electron-withdrawing groups facilitate an electrochromic reduction characterized by a highly increased reversibility.11 Unsubstituted Pc, as well as suitably substituted Pc can be sublimed to form thin films by physical vapor deposition (PVD) on a variety of substrates. In the solid state, such Pc molecules typically form molecular crystals bound by van der Waals (vdW) interactions.12 The weak intermolecular interactions result in polycrystallinity for a given molecule, e.g. α- and β-phases of unsubstituted Pc13 or a variety of structures in thin films of, e.g. perfluorinated F16Pc.14 Different intermolecular orientations in these different crystalline phases leads to significant differences in the electronic coupling of the central aromatic chromophore systems and, hence, significant differences in their optical absorption spectra.15 The strength of intermolecular vdW interactions in Pc molecules in solids can further be rationally tuned by a judicious choice of the substituents of the aromatic macrocycles, detected in the absorption spectra of thin films.16,17 In a direct comparison of vapor-deposited thin films of a perfluorinated copper phthalocyanine, F16PcCu (Fig. 1b), with those of a copper phthalocyanine in which the eight non-peripheral 8 H-atoms of the parent H16PcCu had been substituted by F-atoms and the other, peripheral 8 H-atoms were substituted by perfluoroisopropyl groups, F64PcCu (Fig. 1d) it could be shown that the films of F16PcCu exhibit strong electronic coupling, also leading to their crystallinity, whereas those of sterically hindered F64PcCu showed very weak coupling, in line with amorphous growth.18
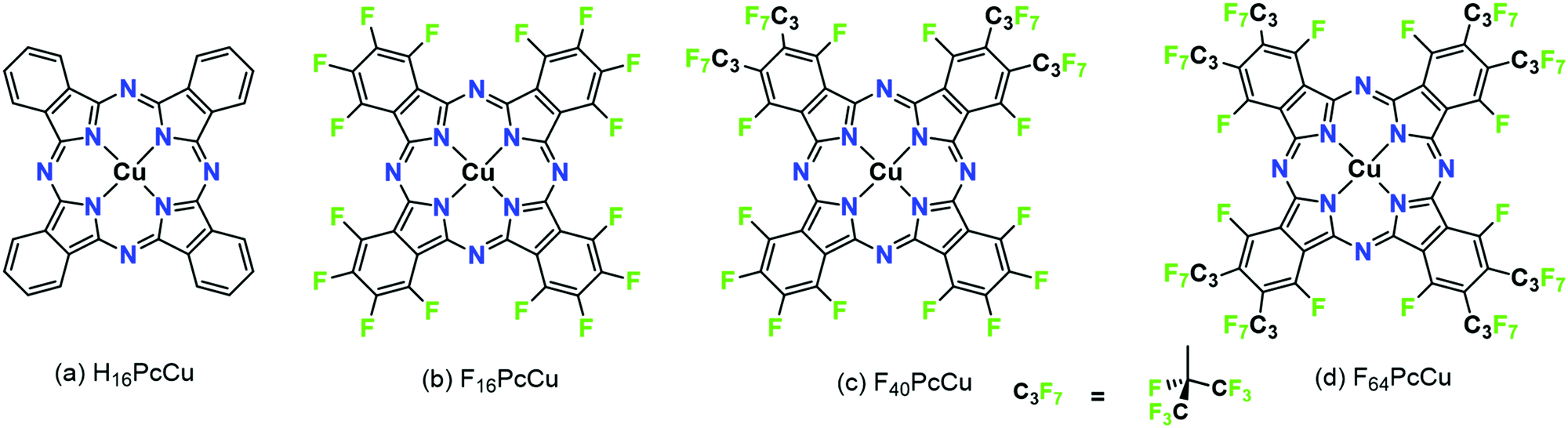 | ||
| Fig. 1 Structural formulae of copper phthalocyanines with different degree of fluorination. | ||
Films of both F64PcCu and F16PcCu exhibited electrochromic switching upon their reduction and subsequent re-oxidation. However, in accordance with the strength of vdW interactions and, hence, electronic coupling of the aromatic macrocycle, the rate of switching in films of F16PcCu was limited by ion diffusion (high electron mobility), as opposed to the switching rate in films of F64PcCu that was limited by electron conduction (fast ion diffusion). These limiting cases, linked to the limiting structural features imparted by the lack or presence of bulky fluoroalkyl substituents, respectively, suggested that the optimization of the substitution pattern and, thus, of the vdW interactions of Pc molecules in the solid state could result in a molecule exhibiting sufficiently strong interactions favoring a high electronic coupling and thus electron mobility, but also still sufficiently weak to facilitate fast diffusion of charge-compensating ions in films.
Following this strategy (the “Goldilocks principle”), F40PcCu was synthesized and characterized in the present work, Fig. 1c. In F40PcCu 12 H-atoms of H16PcCu are formally substituted by F but only 4 by perfluoroisopropyl groups. F40PcCu exhibits an intermediate degree of steric hindrance, electronic deficiency and likely an intermediate strength of vdW interactions, therefore, a likely optimized balance between electron mobility and ion diffusion. Fast, reversible electrochromic switching is indeed realized in this optimized OMIEC and the consequences for technical application of potential devices are discussed below.
2 Experimental
2.1 Synthesis
Tetrafluorophthalonitrile, 1, 0.016 g (0.08 mmol), perfluoro-(4,5-diisopropyl)phthalonitrile, 2, 0.1 g (0.2 mmol), and copper(II) acetate monohydrate, 0.015 g (0.08 mmol), were mixed with a few drops of nitrobenzene in a glass vial, sealed with a Teflon cap and heated in a microwave reactor at 185 °C for 12 minutes. The crude products were separated via gravity column chromatography using silica gel 60 (63–200 μm) and a 0–15% gradient of ethyl acetate in hexanes, followed by an isocratic 30% ethyl acetate/hexanes blend to yield a mixture of F64PcCu, F52PcCu and F40PcCu. The phthalocyanines were separated using flash chromatography in 0–50% CH2Cl2/hexanes. F40PcCu was isolated as a deep-blue solid in 17% yield. UV-Vis (TFT): λmax (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 686 (5.33), 616 (4.64), 360 (4.85) nm (L mol−1 cm−1); UV-Vis (ethanol): λmax (logε) 676 (4.95), 640 (4.74), 612 (4.49), 372 (4.62), 316 (4.52) nm (L mol−1 cm−1); HRMS (ESI, negative mode, Fig. S1 in ESI†): calculated for C44F40N8Cu + Cl− [M + Cl]− 1497.8586, found 1497.8611 (δ = 16.7 ppm), FT-IR (KBr disk) ν, cm−1 (intensity), Fig. S2 (ESI†): 2918.53 (w), 2850.76 (w); 1527.97 (w), 1491.21 (w) C
ε) 686 (5.33), 616 (4.64), 360 (4.85) nm (L mol−1 cm−1); UV-Vis (ethanol): λmax (logε) 676 (4.95), 640 (4.74), 612 (4.49), 372 (4.62), 316 (4.52) nm (L mol−1 cm−1); HRMS (ESI, negative mode, Fig. S1 in ESI†): calculated for C44F40N8Cu + Cl− [M + Cl]− 1497.8586, found 1497.8611 (δ = 16.7 ppm), FT-IR (KBr disk) ν, cm−1 (intensity), Fig. S2 (ESI†): 2918.53 (w), 2850.76 (w); 1527.97 (w), 1491.21 (w) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic; 1251.43 (s), 1171.29 (m), 1104.90 (w), C–F aliphatic and aromatic; 964.30 (w), 729.74 (w), 752.33 (w), 470.64 (w).
C aromatic; 1251.43 (s), 1171.29 (m), 1104.90 (w), C–F aliphatic and aromatic; 964.30 (w), 729.74 (w), 752.33 (w), 470.64 (w).
2.2 Chemicals and molecular characterization
All solvents (ACS grade or better) and reagents were purchased from commercial sources and used as received, unless stated otherwise. Copper acetate monohydrate and the fluorinated tetrafluorophthalonitrile, 1, (Fig. 2) were purchased from TCI Co., Ltd. Perfluoro-(4,5-diisopropyl)phthalonitrile, 2, was prepared as described in the literature.19F16PcCu was purchased from TCI in sublimed grade, F64PcCu was prepared as described in ref. 16.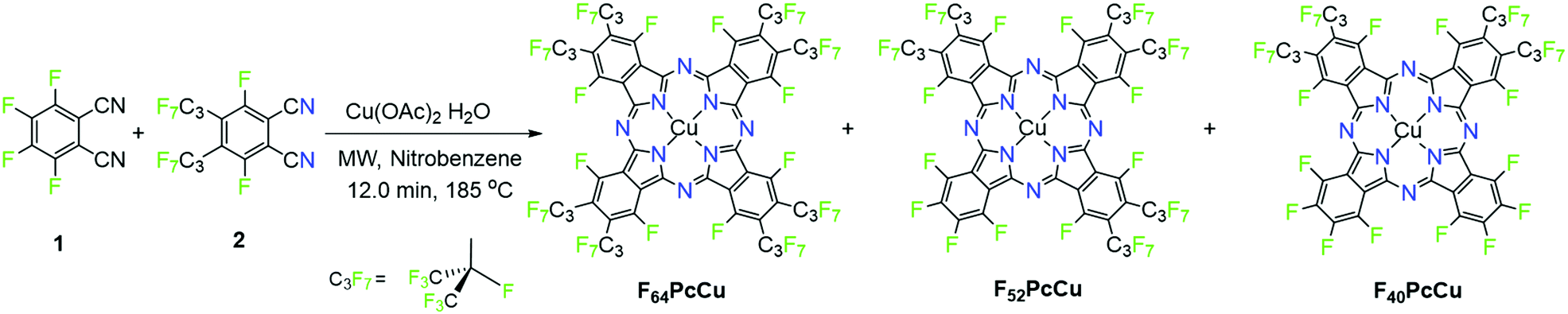 | ||
| Fig. 2 Reaction scheme for the synthesis of perfluorinated copper phthalocyanines. | ||
The microwave-assisted syntheses were performed using a CEM Discover system (CEM Corporation). Flash chromatography was carried out using a CombiFlash Rf+, (Teledyne ISCO). UV-Vis spectra were recorded on a Cary 500 Scan UV-Vis-NIR spectrophotometer. FT-IR spectra were recorded on a Nicolet 4700 FT-IR spectrophotometer using KBr pellets. High-resolution mass spectrometry (HRMS) data were obtained at Rutgers University (Newark, NJ) by direct injection of ethanolic solutions in an Apex-ultra 70 hybrid FT-MS. For the electrochemical investigations by CV (cyclic voltammetry) at a scan rate of 0.1 V s−1 and SWV (square wave voltammetry at a pulse amplitude of 50 mV, a frequency of 10 Hz and a potential step of 5 mV), solutions of the molecules in 0.1 M tetrabutylammonium tetrafluoroborate (TBATFB, Fluka, 99%, electrochemical grade) in trifluorotoluene (TFT, Sigma-Aldrich, ≥99%) were used. The measurements were carried out employing an IviumStat potentiostat/galvanostat, equipped with Pt wires as working electrode and counter electrode, respectively, and a leak-free Ag/AgCl reference electrode (LF-2, Innovative Instruments, Inc.) for which the potential was calibrated against ferrocene/ferrocenium (Sigma-Aldrich, 98%) at 0.4 V vs. Ag/AgCl20 in dimethylformamide (Sigma-Aldrich, 99.8%). All experiments were performed at room temperature in a glovebox under dry nitrogen.
2.3 Preparation of thin films
Glass substrates coated with FTO (Kaivo, <15 Ohm sq−1) were cut into 10 mm × 25 mm pieces. The FTO-coated glass pieces were cleaned for 15 min in RBS solution (Roth), acetone (Roth, ≥99.5%) and isopropanol (Roth, ≥99.8%), respectively, using an ultrasonic bath at room temperature. The cleaned substrates were dried with N2 gas. A piece of adhesive tape from TESA was attached to the shorter edge of the substrate in order to keep an area of the substrate uncovered for later electrical contacting.Thin films with different thicknesses of F40PcCu were prepared on FTO-coated glass by physical vapor deposition in a vacuum chamber with a quartz crystal microbalance which was calibrated as reported in ref. 16 for monitoring the deposited mass and calculating an average film thickness based on an estimated density (2.2 g cm−3) as an average value from the known values of F16PcCu and F64PcCu.13,16 The vapor deposition was carried out at a pressure of <10−6 mbar and an evaporation rate of about 0.5–1 nm min−1 by resistively heating the powder of F40PcCu in a BN crucible (Kurt J. Lesker Ltd). In order to analyze the thickness-dependent film growth of F40PcCu, an optical fiber was attached to the vacuum chamber and a pre-cleaned quartz glass (TED PELLA, INC.) was mounted into the chamber as a substrate. A lower deposition rate of 0.2 nm min−1 was chosen to allow measurements of the optical absorption spectra by a tec 5 diode array spectrometer during deposition at a pressure of <10−5 mbar. The sample was rotated between two positions to alternately vapor-deposit the film and measure the spectra as reported earlier.16
2.4 Characterization of thin films
The film coverage on the substrate and cross-sections of the films were analyzed by scanning electron microscopy (SEM) in a Zeiss MERLIN at an emission current of 100 pA and an acceleration voltage of 5 kV. The film morphology was investigated by atomic force microscopy (AFM) in air using a Smart SPM 1000 (AIST-NT) with NanoWorld Pointprobe SEIHR Non-Contact/Soft Tapping probes (tip radius < 12 nm) in an oscillating mode. Images were processed by the Gwyddion 2.55 program using the plane subtraction method and adjusting the individual lines by the median of differences. The crystallinity of the thin films was analyzed by grazing incidence X-ray diffractometry using a PANanalytical X’Pert Pro MRD instrument with Cu-Kα-radiation. The spectroelectrochemical analysis of the films was performed in a 1 M aqueous KCl (Aldrich, ≥99.5%) solution with a three-electrode setup. To remove the dissolved oxygen from the solution, N2 gas was flushed through the solution before the measurements. During the measurements, N2 gas was passed above the surface of the solution only, to avoid fluctuation of the solution. To contact the fluorinated phthalocyanine films for the spectroelectrochemical characterization, a Cu wire was attached with conductive Ag paste (Ferro GmbH) on the uncovered substrate area. Afterwards, the metal wire and the substrate area were sealed with Araldite Rapid epoxy resin. The prepared sample was mounted as working electrode in a glass cell (Starna) with a platinum wire counter electrode (Goodfellow, 99.995%) and a Ag/AgCl reference electrode (REF201 Red Rod, Radiometer analytical). Cyclic voltammetry between 0.6 V and −1.1 V at different scan rates and chronoamperometry between the bias potentials of 0.6 V and −1.0 V at a time delay of 3 s were carried out employing an IviumStat potentiostat/galvanostat. Optical spectra were simultaneously measured in situ by placing the working electrode into the beam of the tec5 diode array spectrometer. The charge measured during long-term switching experiments is underestimated by a factor of about 3.8 which is caused by a rather low sampling frequency (5 Hz compared to 1 kHz) needed to ensure reliable data acquisition by the spectrometer.3 Results and discussion
3.1 Synthesis and properties of F40PcCu
The synthesis method, Fig. 2, uses two different precursors with different propensities to form phthalocyanines. The ratio of precursors 1vs.2 is 2:2 in F40PcCu, but a ratio of 0.08/0.2 = 0.8:2 was used in order to minimize the formation of the 4:0 complex F16PcCu. The complexes 1:3, F52PcCu and 0:4, F64PcCu, Fig. 2, also form, but the reported 1vs.2 ratio seems to give F40PcCu in modest but maximum isolated yield. Further optimizations of either the reaction conditions or the purification method were not pursued. The structure of F40PcCu, Fig. 2, depicts the complex in its “cis” form, with the two coordinating, C3F7-substituted isoindole rings, labeled ‘isoR’ adjacent, i.e. the NisoR–Cu–NisoR angle is 90°. Alternatively the rings can be “trans”, the NisoR–Cu–NisoR angle being 180°. The assignment, in the absence of a single-crystal X-ray structure is based on the structure of the analogous F40PcCo complex, for which an X-ray crystal structure has been reported.21
The spectral position of the Q-band of F40PcCu in solution (Fig. 3) corresponds to that reported for F40PcZn or F40PcCo,22 for which the “cis” structure of the substituents was proven, in good agreement with literature reports on Q-band differences for complexes in either “cis” or “trans” structure.23,24 Our assignment of a “cis” structure for F40PcCu, therefore, is fully consistent with the literature.
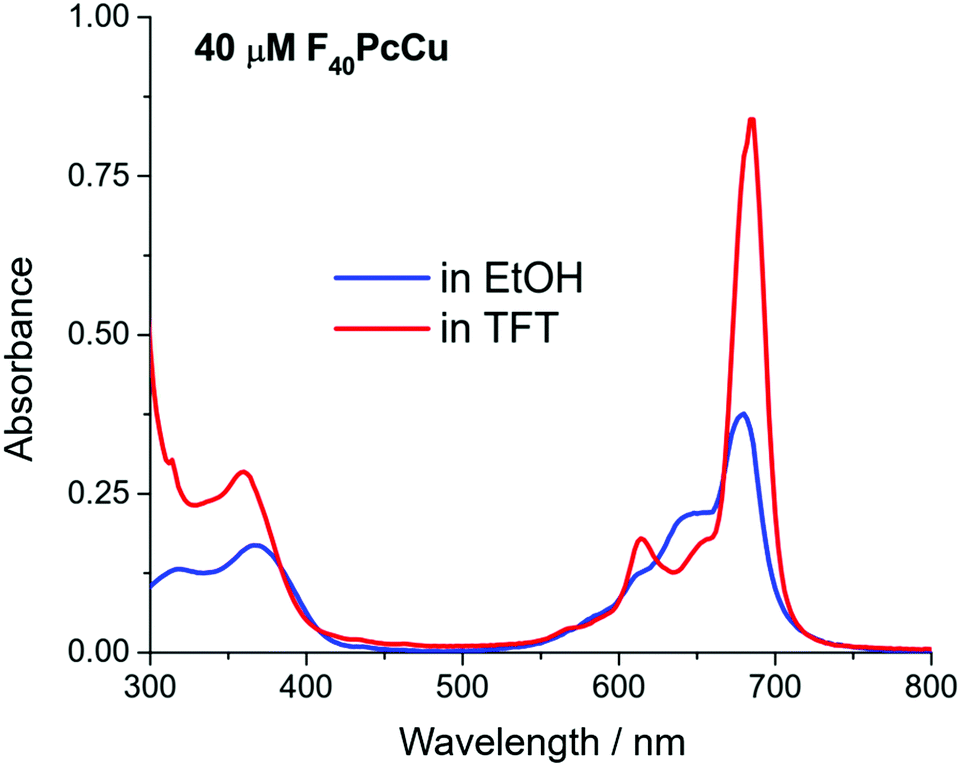 | ||
| Fig. 3 UV-Vis spectra of F40PcCu in ethanol (EtOH) and trifluorotoluene (TFT). | ||
F40PcCu exhibits solvent dependent UV-Vis spectra, Fig. 3. The main Q-band in TFT, 686 nm, appears at least twice as intense as the one in ethanol, 676 nm. The Beer–Lambert plots of concentration dependent spectra, Fig. S3 and S4 (ESI†), are linear, but the variation of absorbance of the Q-band with concentration is approximately double in TFT vs. ethanol. Taken together, the data suggest that a higher degree of aggregation exists in ethanol, consistent with ethanol axial coordination in F64PcCu.25
Cyclic voltammetry (CV) and square wave voltammetry (SWV) were performed for solutions of F40PcCu in direct comparison to solutions of F16PcCu and F64PcCu in order to discuss the influence of substituents on the redox potential of reduction reactions under otherwise identical conditions. The CV results for the new complex F40PcCu are shown in Fig. 4 and the main findings are summarized together with the SWV results for all complexes in Table 1. Values measured for F16PcCu reveal a potential for the first reduction (I) of −0.2 V and of the second reduction (II) of −0.9 V. The potential of I is found slightly less negative than the reported −0.6 V measured for the Zn-complex of F16Pc in dimethylformamide, whereas that of II is found in accordance with the reported value (−0.9 V).26 The values for F64PcCu are found similar to those reported under comparable conditions17 of I = −0.06 V, II = −0.56 V and III = −1.17 V.
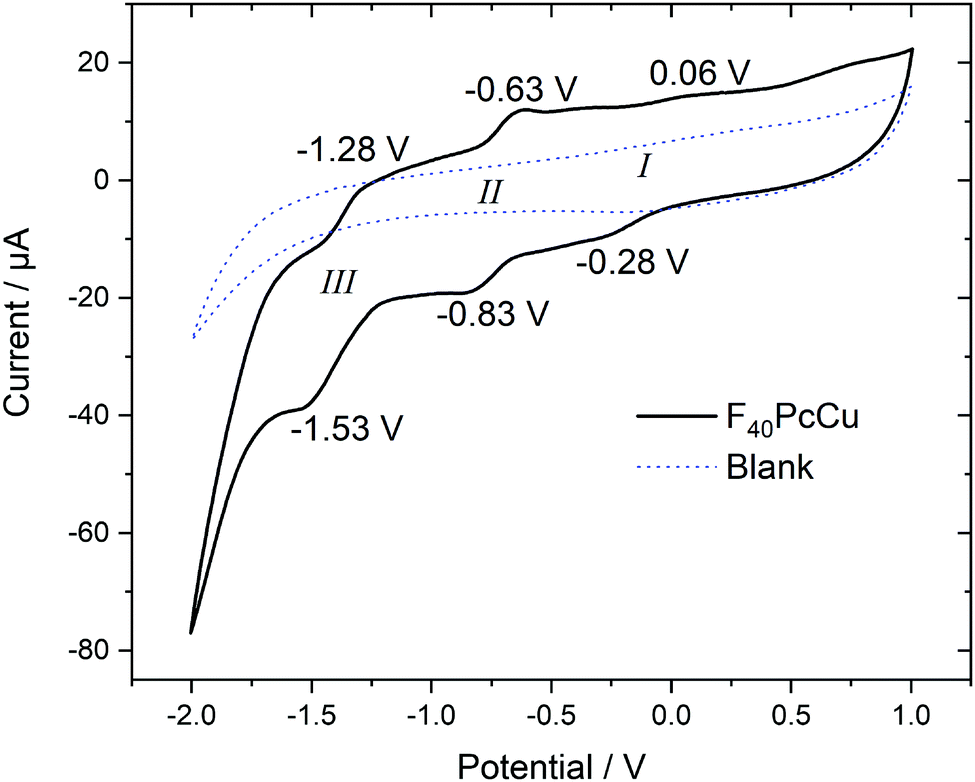 | ||
| Fig. 4 Cyclic voltammetry at 0.1 V s−1 of F40PcCu dissolved in 0.1 M TBATFB in TFT compared to a blank scan of the electrolyte in the absence of F40PcCu. | ||
| Wave | Method | |||||
|---|---|---|---|---|---|---|
| E/V | CV | SWV | ||||
| I | II | III | I | II | III | |
| F16PcCu | −0.21 | — | — | −0.19 | −0.88 | −1.64 |
| F40PcCu | −0.11 | −0.73 | −1.40 | — | −0.71 | −1.40 |
| F64PcCu | 0.05 | −0.53 | — | 0.02 | −0.49 | −1.40 |
The redox potentials indicate that F64PcCu is easiest to reduce, followed by F40PcCu, and F16PcCu. This is consistent with the stronger electron-withdrawing effect of the C3F7 substituents vs. F atoms. The broad characteristics in the region of I speak in favor of a parallel reduction of differently aggregated species of F40PcCu at this concentration needed to obtain reasonably high signals above background. Some small additional waves are detected which can be caused by solvent interaction with monomeric species, as also observed earlier for F64PcCu.17
3.2 Film growth of F40PcCu
Uniformly blue thin films, obtained after vapor deposition of F40PcCu showed a compact film morphology consisting of individually contacted grains around 60–100 nm lateral diameter, in a similar range compared to F64PcCu (200 nm)18 or F16PcCu (50 nm)18 as revealed by AFM (Fig. 5a). The covering film of F40PcCu on FTO is also observed in the SEM cross-section (Fig. 5b). The extent of intermolecular electronic coupling of F40PcCu in thin films was monitored by in situ measurements of the optical absorption spectra during film growth (Fig. 5c). For all spectra, the Soret band below 400 nm27 and the Q-band around 500–800 nm27,28 characteristic for phthalocyanines were found. When compared to solution spectra (Fig. 3 and dashed line in Fig. 5c), which is dominated by the characteristic vibronic fine structure, the films showed significant broadening and a splitting of the Q-band revealing more intense interaction of the molecules in the solid state compared to solutions.28,29 The splitting leads to a maximum at shorter wavelength which indicates dominance of face-to-face coupling of the molecules (H-aggregation) in solid state.28,29 The presence of an absorption maximum around 640 nm and a shoulder at about 690 nm implies a structure similar to the α-structure of unsubstituted phthalocyanines with a herringbone orientation of the molecules.15 Ultrathin films of F16PcCu in the range of a few monolayers30,31 showed similar features typical for the β-bilayer structure with a parallel stacking of the molecules. Since the F40PcCu molecule contains bulky perfluoroisopropyl substituents, a molecular orientation appears likely where the chromophores are not fully stacked as in the β-bilayer structure, but are oriented slightly inclined thereby approaching an orientation similar to the classic α-structure. The X-ray diffraction pattern (Fig. S5, ESI†) of a thin film of F40PcCu on FTO exhibits two reflections at around 6° and 19°. Their broad shape indicates the presence of films with a rather well-defined intermolecular orientation, but with still widely amorphous character. The positions of the peaks correspond to the (200) and (600) reflections of a herringbone orientation of the molecules parallel to the substrate as reported earlier for F16PcCu and PcCu thin films.32,33 These results, therefore, confirm the presence of an α-structure as also derived from the optical analysis. The films grew at constant band positions and constant band splitting from the monolayer regime up to the film thicknesses studied in this work (Fig. 5c) which yields proof for a constant intermolecular coupling of the F40PcCu molecules within the film and, hence, for a homogeneous structure of the film. Such an observation has also been reported earlier for F64PcCu.16 However, the spectra of solid F64PcCu matched well the solution spectrum indicating a very weak interaction of the molecules in the solid state caused by the strong suppression of electronic coupling by the bulky perfluoroisopropyl side groups.16 Thus, the lower degree of fluorination in F40PcCu provides a strength of intermolecular coupling in solid state that is stronger than that for F64PcCu, but weaker than that for F16PcCu.16,34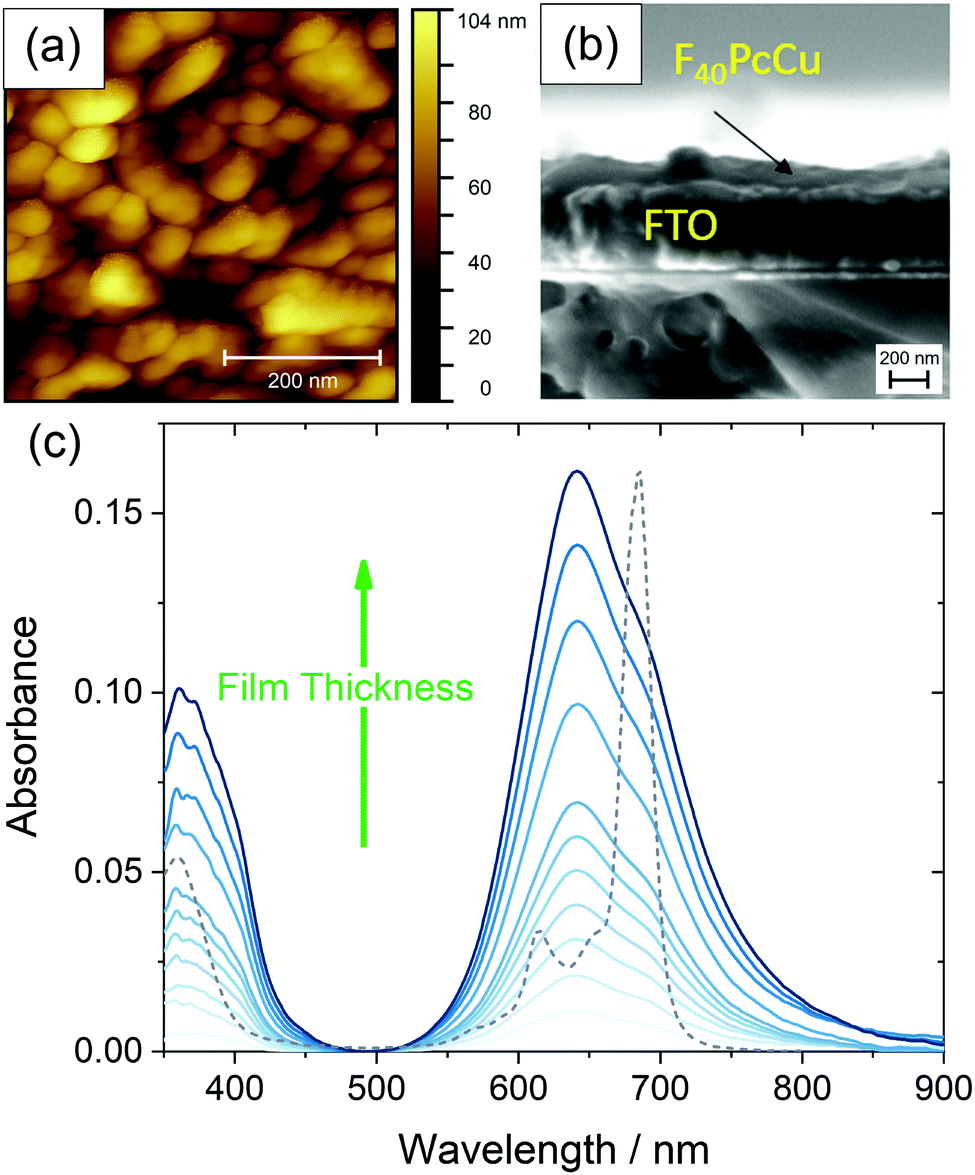 | ||
| Fig. 5 Morphology of a 35 nm thin film of F40PcCu on FTO measured by AFM (a), cross-section of a 35 nm thin film F40PCu on FTO observed by SEM (b) and optical absorbance spectra acquired during film preparation of F40PcCu with a film thickness d of 0–35 nm on quartz glass at room temperature, the optical spectrum of F40PcCu in TFT (see Fig. 3) is shown as a dashed line (c). | ||
3.3 Rate of the electron and ion transport in the F40PcCu films
In order to analyze the electron and ion transport within the F40PcCu films, cyclic voltammetry (CV) was performed at different scan rates ν. The initial cycle of the as-deposited films showed subtle differences in shape compared to subsequent cycles. Such conditioning, already reported for other fluorinated phthalocyanine films17,18 was assigned to an initial hindrance of counter ion intercalation into the van der Waals-bonded molecular crystals. However, after about 6 cycles, reproducible CV curves could be obtained for all the prepared F40PcCu films (Fig. 6a), a change which can be assigned to the reversible reduction and re-oxidation of the F40PcCu films with concurrent intercalation and expulsion of K+ ions. In Fig. 6b and c the current density j was divided by the scan rate to compare the CV curves for different scan rates in one plot. One main peak of reduction and one peak of re-oxidation were observed. The smaller reduction peak at less negative potentials (around −0.4 V) was mainly observed for lower scan rates and thinner films and is most likely indicative of a small concentration of molecules in a slightly different environment. With increasing scan rate, the peak potential of the main reduction process Ered shifted slightly towards more negative potentials, while the peak potential Ereox of the re-oxidation process underwent a larger shift towards less negative values, typical for kinetically hindered reactions. When the positions of the main reduction peak of around −1.04 V ≤ Ered(F40PcCu) ≤ −0.91 V and the re-oxidation peak of around −0.78 V ≤ Ereox(F40PcCu) ≤ −0.30 V measured for the stabilized CV curves of the F40PcCu films (10 to 50 nm average film thickness) are compared to other fluorinated Pc, some differences are observed. Thus, the reduction of F40PcCu requires a higher driving force than that of F16PcCu or F64PcCu with −0.92 V ≤ Ered(F16PcCu) ≤ −0.76 V and −0.79 V ≤ Ered(F64PcCu) ≤ −0.75 V.18 The values of Ereox for the re-oxidation process of F40PcCu, however, were comparable with the reported values for F16PcCu with −0.71 V ≤ Ereox(F16PcCu) ≤ −0.38 V, but in a slightly larger range compared to F64PcCu with −0.62 V ≤ Ereox(F64PcCu) ≤ −0.55 V.18 From the average value of Ered(F40PcCu) and Ereox(F40PcCu) the redox potential −0.91 V ≤ E(F40PcCu) ≤ −0.74 V was estimated depending on the scan rate and thickness of the films. The values are more negative compared to the redox potential for F16PcCu or F64PcCu with −0.78 V ≤ E(F16PcCu) ≤ −0.64 V18 and E(F64PcCu) ≈ −0.68 V,18 respectively suggesting that a slightly higher driving force is needed for reducing F40PcCu in solid state. This trend was found clearly more pronounced than in solution, speaking in favor of a higher intermolecular interaction energy in the neutral film of F40PcCu relative to the reduced film with intercalated K+ when compared to films of F64PcCu or F16PcCu. From the values of the main reduction and the re-oxidation peak of the films up to a scan rate of 2 V s−1 an average peak potential difference of ΔEp(F40PcCu) = 0.30 V was obtained, in the range of the values reported for films of F16PcCu and F64PcCu indicating (quasi-) reversibility of the redox processes35,36 within all studied perfluorinated Pc films.18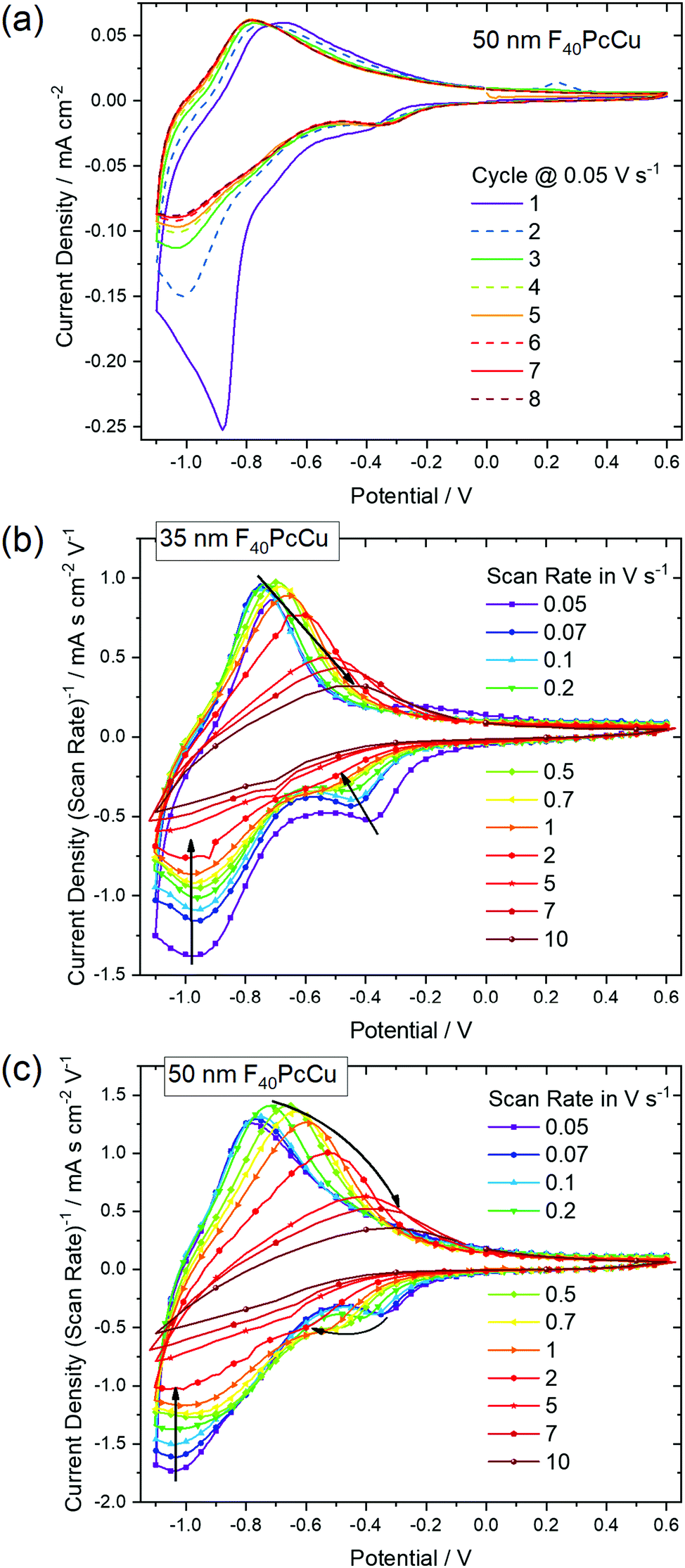 | ||
| Fig. 6 Cyclic voltammograms of F40PcCu films in contact with 1 M aqueous KCl (a) with a film thickness of 50 nm during conditioning at a scan rate of 0.05 V s−1 and at different scan rates after conditioning of (b) a 35 nm and (c) a 50 nm thin film. In (b) and (c) the current density is divided by the scan rate to represent the CV curves in one plot. | ||
To gain a deeper insight into the dependence of j on ν and, hence, the transport limitation within the films, the logarithm of the peak current densities of the cathodic (jpc) as well as those of the anodic branches (jpa) were plotted against the logarithm of the scan rates for different average film thicknesses (Fig. 7). A linear dependence of j on ν (slope of 1, solid black line) is assigned to films with reversible redox reactions without diffusion limitation, expected for an ideal adsorbed monolayer.35 For reactions which are limited by the diffusion rate of counter ions, a dependence of j on the square root of ν is expected (slope of 0.5, dashed black line).35 A slope of 0.6 (dotted black line) can be typically assigned to films where the reaction is limited by electron hopping between the redox centers.37–39 Despite a decreasing j/ν for higher ν in the CV of Fig. 6, a linear dependence of the peak current on scan rates up to 1 V s−1 can still be fitted for the thicker films (d ≥ 35 nm) in the reduction reaction and for all films in the re-oxidation reaction. Hence, the reduction of the films takes place with just a minor limitation by the transfer of electrons or counter ions and the re-oxidation reveals independence of any transport limitation, representing a high reversibility of the redox reactions of the films in this range of ν significantly larger than was observed for F16PcCu and F64PcCu films where a linear dependence could be observed only for ν < 0.2 V s−1.18 For F16PcCu this was only valid for d ≤ 20 nm and for F64PcCu for d ≤ 50 nm.18 For ν > 0.2 V s−1 the F64PcCu films thinner than 50 nm showed a dependence of j ∼ ν0.6 characteristic for a limitation by electron hopping in the films and sufficiently fast diffusion of the counter ions through the F64PcCu films, facilitated by the presence of the large perfluoroisopropyl substituents.18 At scan rates ν > 1 V s−1 the linear dependence of j on ν is no longer observed, but the reaction clearly becomes transport-limited. A clear assignment to a characteristic slope is no longer possible because for F40PcCu films with d ≥ 35 nm a dependence of j ∼ ν0.5 was indicated, whereas the 10 nm F40PcCu film approached j ∼ ν0.6. Therefore, limitation by either the counter ions (slope of 0.5) or by hopping of electrons (slope of 0.6) cannot be distinguished, the transport of both seems to be equally fast. It should be noted that F40PcCu films show no transport limitation up to a scan rate 5 times higher than that reported for F16PcCu and F64PcCu.18 Thus, the two isoindole units with two perfluoroisopropyl substituents in F40PcCu instead of four such units in F64PcCu16 (Fig. 1) lead to a film structure with optimized attributes of both F16PcCu and F64PcCu which allows a fast electron as well as a fast ion transport through the film.
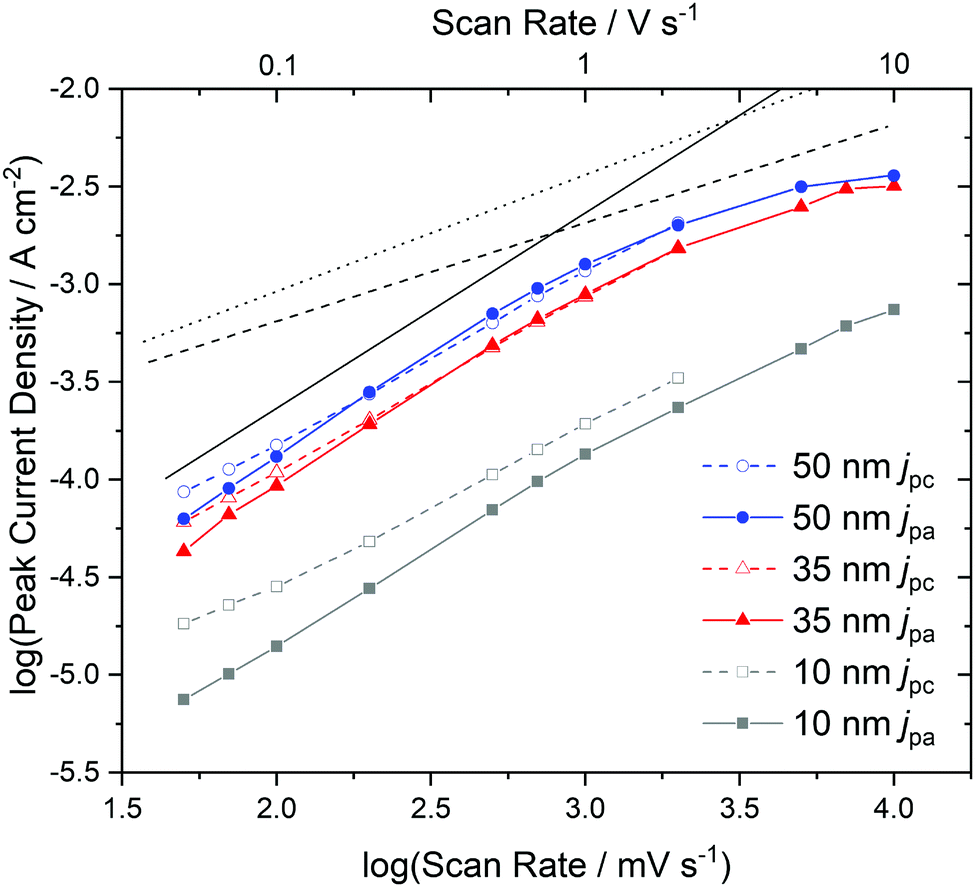 | ||
| Fig. 7 Dependence of the cathodic (jpc) and anodic (jpa) peak current densities on the scan rate, determined by cyclic voltammetry with F40PcCu films of different thickness. The black dashed line indicates a slope of 0.5, the dotted line a slope of 0.6 and the solid line a slope of 1. | ||
As a quantitative measure for the rate of transport in the F40PcCu films, an effective diffusion coefficient D was obtained from chronoamperometric measurements. After the initialization of the film during the first cycle, the subsequent cycles were invariable. The integrated current density yielded an average charge of 1.5 electrons for each deposited molecule of F40PcCu considering the molecular weight of 1464.01 g mol−1. The measured cathodic and anodic current densities of the second cycle were plotted against the reciprocal square root of time t−1/2 (Fig. 8) corresponding to the Cottrell eqn (1) valid for semi-infinite diffusion
| j = nFcD1/2π−1/2t−1/2 | (1) |
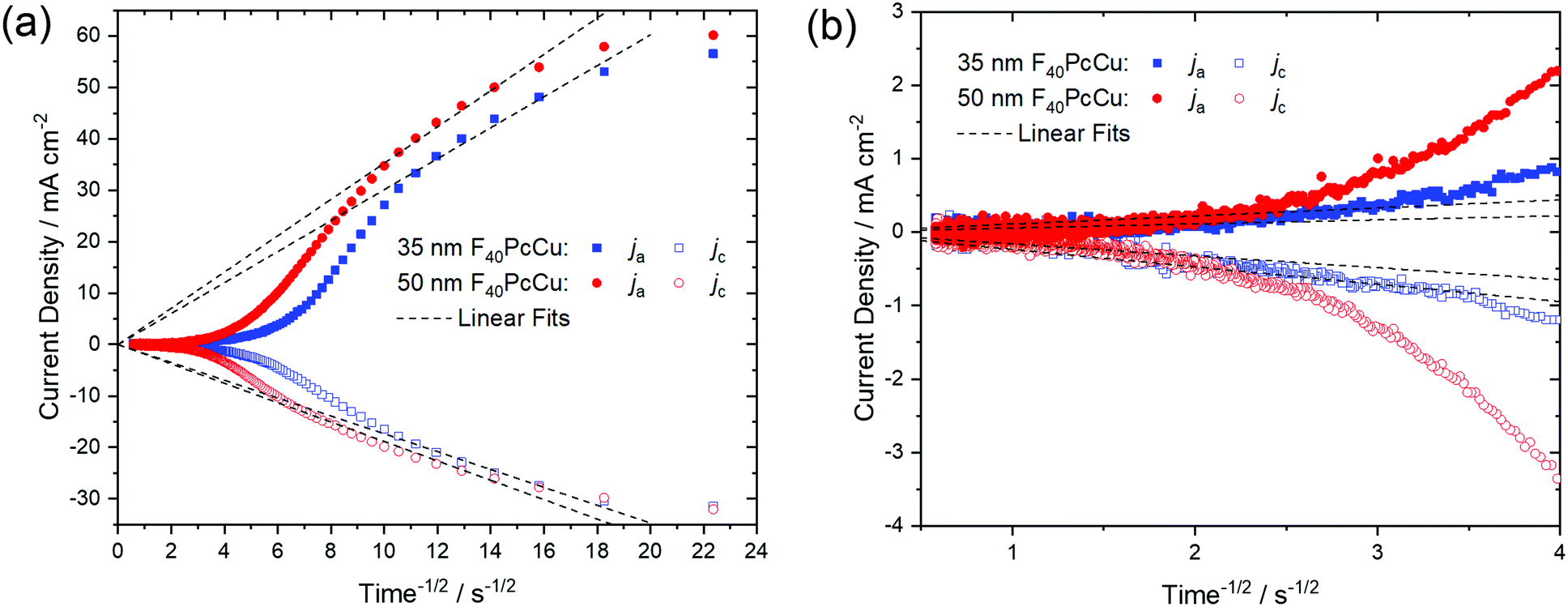 | ||
| Fig. 8 (a) Cathodic (jc) and anodic (ja) current densities dependent on the reciprocal square root of time obtained from chronoamperometric analysis of a 35 nm (blue) and 50 nm (red) thin F40PcCu film. (b) Plot of the data for long times. The linear fits according to the Cottrell equation are marked in dashed lines. | ||
For the 35 nm thin film, values for the effective diffusion coefficient of Dc(35 nm) = 4.4 × 10−10 cm2 s−1 and Da(35 nm) = 1.4 × 10−9 cm2 s−1 were obtained from the fits of the cathodic and anodic current densities, respectively, Fig. 8a. The values estimated for a 50 nm thin film are in the same range, with Dc(50 nm) = 5.3 × 10−10 cm2 s−1 and Da(50 nm) = 1.9 × 10−9 cm2 s−1. For both films, the values of Da for the re-oxidation were found higher than those of Dc for the reduction, as found earlier for F16PcCu.18 Such smaller effective diffusion coefficient upon reduction might be caused by a slow removal of the solvation shell42 before intercalation into the solid film of F40PcCu.
The values of the diffusion coefficient reported for F16PcCu, Dc(F16PcCu) = 8 × 10−11 cm2 s−1 and Da(F16PcCu) = 1.5 × 10−10 cm2 s−1, as well as Dc(F64PcCu) = Da(F64PcCu) = 5 × 10−11 cm2 s−1 for F64PcCu were significantly smaller18 compared to the values for F40PcCu. Thus, the faster diffusion in F40PcCu is a direct confirmation for the successfully enhanced overall diffusion of electrons and K+ within the film. By zooming into the range of the data at longer times around 4 s−1/2, as shown in Fig. 8b, another linear region was observed. The fits in this time regime yielded considerably smaller diffusion coefficients of D = 0.5–8.5 × 10−12 cm2 s−1, representing a lower limit for the diffusion into those parts of films which are least accessible, either because of poor electronic conduction or difficult intercalation of ions. However, for the overall yield of the reaction and, hence, prospective technical application of the reactions, the high values of D in the early stage of the reactions are most significant since they represent the highest currents and, hence the largest contribution. Along with the results from the CV measurements (Fig. 7) revealing a transport limitation in the films dominated by neither electrons nor ions, but given by a similarly fast movement of both with j ∼ ν up to 1 V s−1, it can be concluded that the moderate intermolecular coupling of the F40PcCu molecules provides facilitated pathways for both ions and electrons in the films.
3.4 Spectroelectrochemical characterization of F40PcCu films
Optical absorption spectra of the F40PcCu films were measured in situ during the electrochemical measurements to (i) investigate the changes of the electronic structure of the molecules in the solid state caused by the redox processes and (ii) to analyze the performance of these films as possible electrochromic smart windows.The changes in the electronic structure of the F40PcCu films observed in the stabilized CV curves are clearly reflected in the absorption spectra of the films, as shown in Fig. 9. As expected, the bands of the neutral films upon immersion into the aqueous KCl solution were found almost equal to those of the as-deposited films (Fig. 5). However, a small but significant shift of the Q-band maximum to about 630 nm was caused by this different environment. In contrast to earlier experiments with F16PcCu,18 the electrochemical conditioning of F40PcCu did not lead to any changes in the spectra, i.e. the intermolecular coupling was found unchanged following the first eight cycles of reduction and re-oxidation.
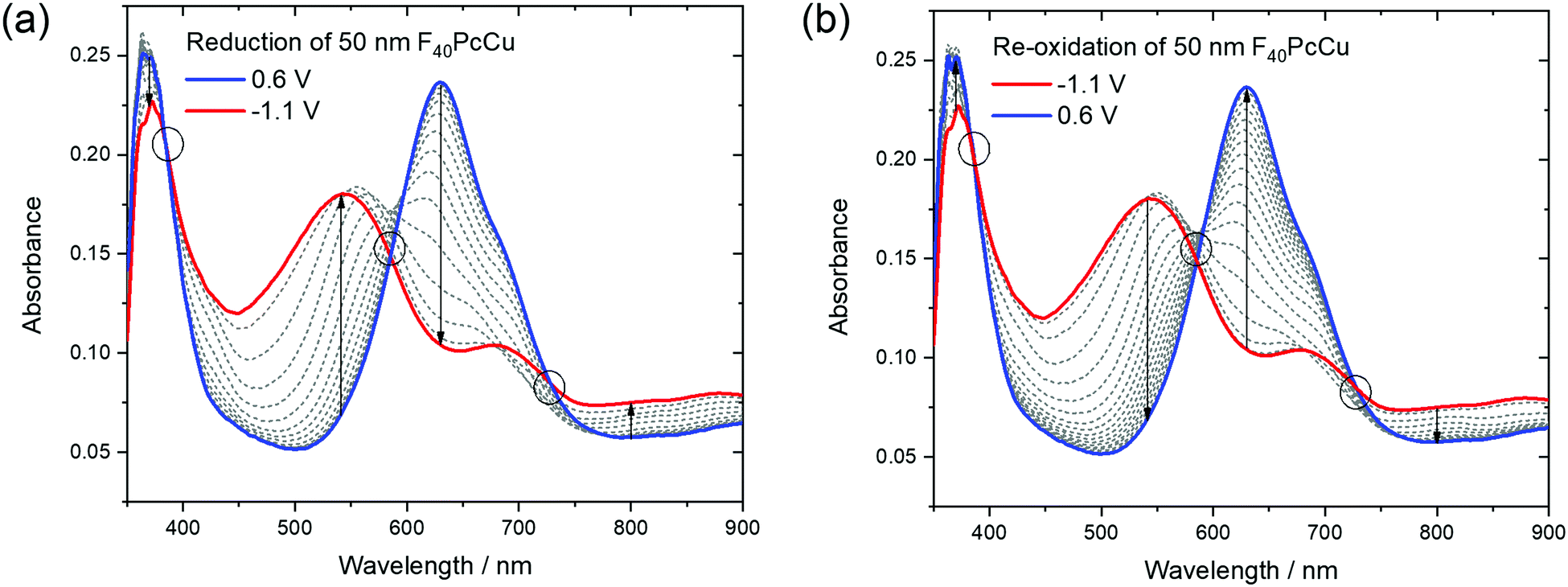 | ||
| Fig. 9 Optical absorption spectra of a 50 nm thin film of F40PcCu measured during cyclic voltammetry at 0.05 V s−1 upon (a) reduction and (b) re-oxidation of the film. The isosbestic points and ranges are marked by circles. | ||
The reduction of F40PcCu led to remarkable changes in the Q-band position as well as in the absorptions between the Soret and the Q-band, while the absorption of the Soret band was just slightly decreased without any remarkable shift. The absorption maximum at 630 nm decreased in intensity while the shoulder at around 690 nm transformed into a local maximum. A new absorption maximum at around 540 nm and a broad absorption in the NIR at 800 nm and beyond, both typical for phthalocyanine rings in their reduced state (Pc(−3)Cu(II)) arose (Fig. 9a) as also observed for F16PcCu18 and for F64PcCu.17,18 Upon re-oxidation these spectral changes were reversed (Fig. 9b). Well-defined isosbestic points were detected at 390 nm, characteristic of a uniformly smaller absorption by the Soret band in the reduced state. Small ranges of intersecting spectra (“isosbestic ranges”) were detected around 580 nm and around 730 nm upon re-oxidation of the films, indicating widely uniform transitions also in the Q-band range, which is known to be very sensitive to parameters of intermolecular coupling in Pc.28 These ranges were considerably wider upon reduction of the film, consistent with the two contributions in the cathodic wave of the CV (Fig. 6). The presence of isosbestic points and “isosbestic ranges” indicates the reversible A ↔ B transformation of neutral species A to a reduced species B with characteristic spectra for neutral (re-oxidized) A and reduced B species, similar to results for F64PcCu films reported earlier17,18 confirming the presence of a reversible redox reaction, with no permanent change in the material.
In order to check if a complete reduction and re-oxidation of the films was ensured even for faster scan rates, which would be beneficial for fast switching in electrochromic layers, the change in optical absorption between the reduced (−1.1 V) and the re-oxidized state (0.6 V) of the films was determined in dependence of the scan rate (Fig. 10). Absorption signals at 540 nm and 630 nm, for which most significant changes were detected upon reduction and re-oxidation were selected (marked with arrows in Fig. 9).
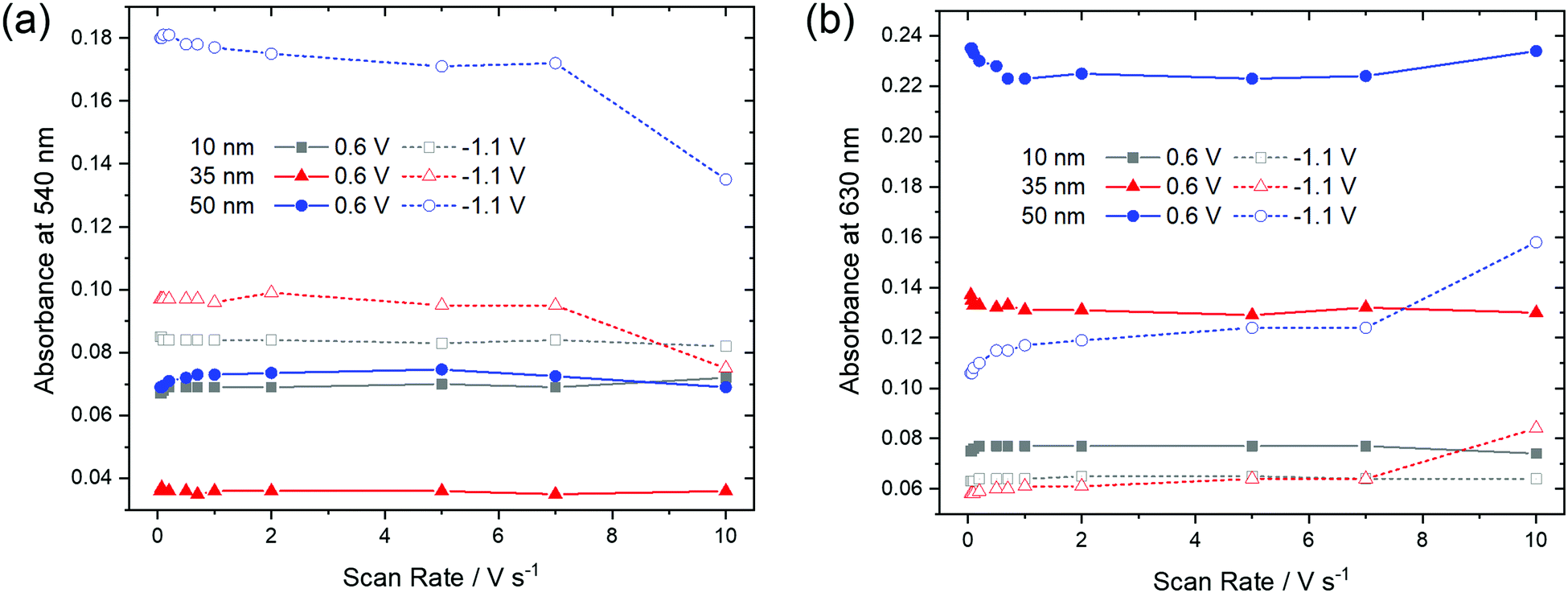 | ||
| Fig. 10 Optical absorbance at (a) 540 nm and (b) 630 nm as a function of scan rate of F40PcCu films of different thickness in the re-oxidized (0.6 V) or reduced (−1.1 V) states. | ||
For the thinnest, 10 nm film of F40PcCu the absorbances at 540 nm and 630 nm remained largely constant for all scan rates. The main observation, however, is that the thicker films showed significant changes at higher scan rates. Upon reduction (−1.1 V), the absorbance at 540 nm has a lower value, indicating incomplete reduction of the film. Correspondingly, a higher absorbance was found at 630 nm, characteristic of remaining neutral molecules. These features are most significant for the 50 nm thin film for which changes were noticeable at scan rates ν > 1 V s−1. Both trends clearly point at an incomplete reduction of the films at high scan rates. Upon re-oxidation (0.6 V), however, absorbance remained almost constant even for higher scan rates and thicker films, confirming the fast re-oxidation processes, also indicated by the higher effective diffusion coefficient determined during re-oxidation (Fig. 8).
3.5 Long-term, repeated switching characteristics of F40PcCu films
The stability of the time-dependent switching characteristics was studied by chronoamperometry of 50 nm thin films over 20 minutes (Fig. 11). It is remarkable that the absorbance, as shown in Fig. 11a for the 50 nm thin film in both the reduced and neutral states remained constant for all subsequent 200 cycles, thus defining a reversible switching process which, in turn, indicated a high stability of the films. Despite this stability, the total charge monotonously shifted to negative values indicative of a background current at the negative potential as already observed by CV (Fig. 6), in earlier studies at F16PcCu and F64PcCu films, and assigned to water reduction.17,18 Obviously, this side reaction had no influence on the spectral properties of the film, a feature also found for F16PcCu and F64PcCu films using Li+ or K+ as counter ions.17,18 The higher time resolution (Fig. 11b) indicates fast decrease of the absorbance at 630 nm upon reduction (−1.0 V) and recovery upon re-oxidation (0.6 V). The response times, 0.2 s ≤ tred ≤ 0.4 s and 0.3 s ≤ treox ≤ 0.4 s were determined, corresponding to the time needed to switch the absorption to 90% of the change to the reduced or the neutral state, respectively. The present response times (Fig. 11) are shorter than the 1.3 s and 0.6 s values, determined in earlier work for the reduction and re-oxidation of F64PcCu films,17 revealing a faster switching behavior of the present films caused by facilitated transport of both electrons and K+ counter ions in F40PcCu as directly seen in cyclic voltammetry (Fig. 7) and the chronoamperometric measurements (Fig. 8). The films provide fast and reversible switching capabilities, stable over many cycles, ideal for their use as electrochromic materials.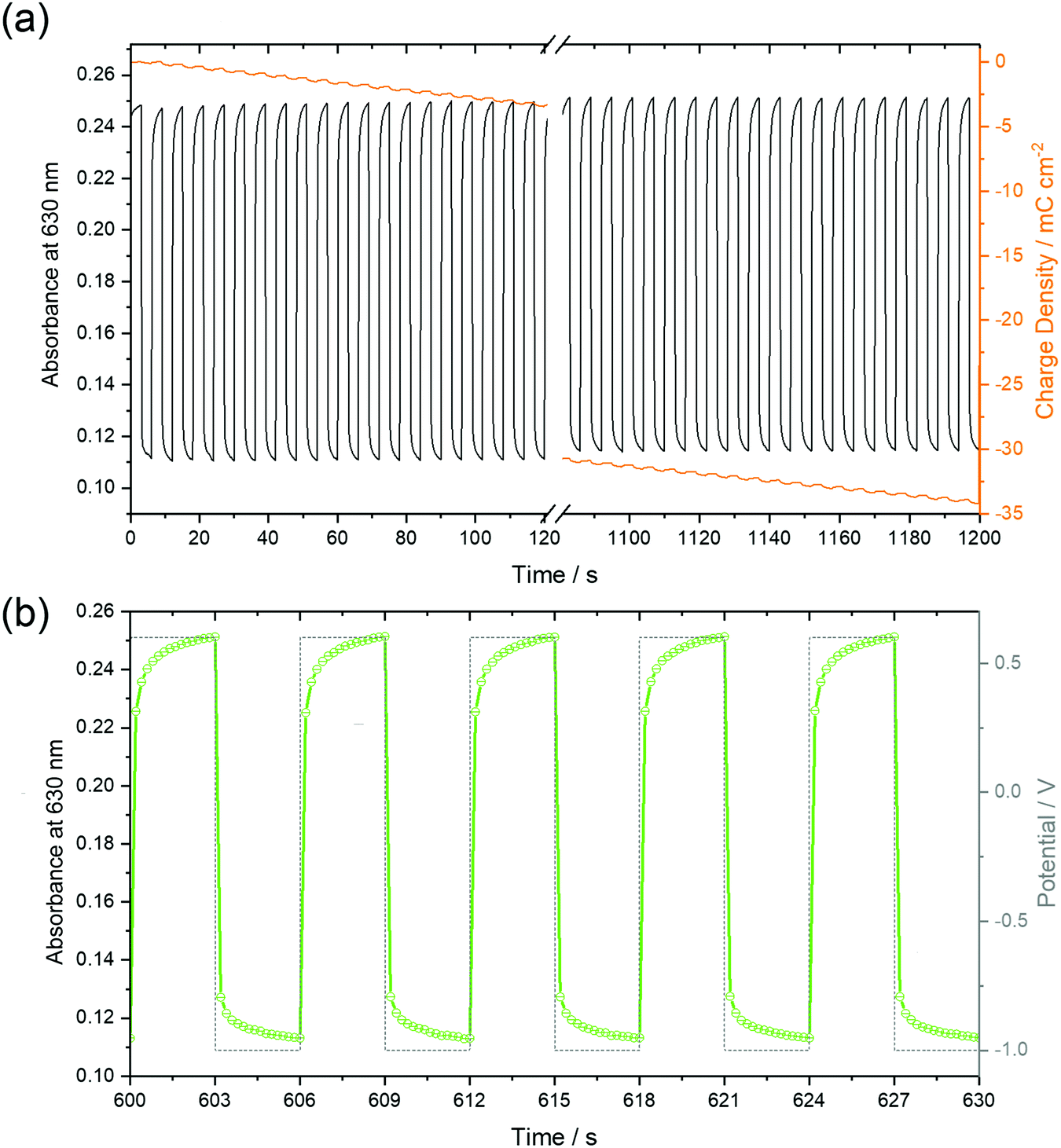 | ||
| Fig. 11 Optical absorption at 630 nm of a 50 nm thin F40PcCu film, recorded during chronoamperometry over 20 minutes (a) and at high resolution for an intermediate time interval (b) between the potentials depicted as dashed profiles in (b). The variation of the charge density within 200 cycles is shown as orange lines in (a). | ||
4 Conclusions
A new type of perfluorinated phthalocyanine, namely F40PcCu has been designed to exhibit simultaneously both efficient electron and ion transport properties. The molecule in solution shows the expected absorption bands of phthalocyanines and exhibits facile reduction and re-oxidation. Thin films, obtained by physical vapor deposition exhibited spectral broadening of the absorption spectra characteristic for thin films of weakly coupling chromophores in solid state. Electrochromic cells could be obtained by a contact of each individual grain of the films with the FTO support and the aqueous electrolyte. The reversible switching observed upon reduction and re-oxidation indicates a high stability of the films with no permanent change in the material. Changes of the absorption spectra in the reduced state typical for phthalocyanines (Pc(−3)Cu(II)) were noticed. The electrochemical characteristics of thin films showed that F40PcCu films exhibited intermolecular coupling within the grains, weaker than that for F16PcCu, but stronger than that for F64PcCu, leading to a facilitated transport of both electrons and counter ions up to scan rates of about 1 V s−1. Short response times during the switching process were determined for both the reduction and re-oxidation of the films. However, at very high scan rates the films could not be fully reduced, whereas a fast re-oxidation of the films was still observed. The apparent diffusion coefficient ranges from 10−10 to 10−9 cm2 s−1 depending on film thicknesses, a characteristic of fast reversible switching attractive for the use of the new material in electrochromic smart windows.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
THQN and DS are grateful for the financial support provided by the Deutsche Forschungsgemeinschaft (DFG) via the GRK (Research Training Group) 2204 “Substitute Materials for sustainable Energy Technologies” as well as to P. Schweitzer for the AFM measurements and to A. Ringleb for assistance with the GIXRD measurements. MP, CC and SMG are grateful for the support provided by the Center for Functional Materials (USA). CC thanks the New Jersey Space Grant Consortium (USA) for a research fellowship. R. Brukh is thanked for the MS data.References
- H. Klauk, Organic Electronics, Wiley-VCH, Weinheim, 2006 Search PubMed.
- C. G. Granqvist, Thin Solid Films, 2014, 564, 1–38 CrossRef CAS.
- R. Baetens, B. P. Jelle and A. Gustavsen, Sol. Energy Mater. Sol. Cells, 2010, 94, 87–105 CrossRef CAS.
- P. R. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2002, 77, 117–133 CrossRef.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 CrossRef CAS.
- M. Berggren, X. Crispin, S. Fabiano, M. P. Jonsson, D. T. Simon, E. Stavrinidou, K. Tybrandt and I. Zozoulenko, Adv. Mater., 2019, 31, e1805813 CrossRef PubMed.
- A. L. Dyer, A. M. Österholm, D. E. Shen, K. E. Johnson and J. R. Reynolds, in Electrochromic Materials and Devices, ed. R. J. Mortimer, D. R. Rosseinsky and P. M. S. Monk, Wiley-VCH, Weinheim, 2015, ch. 5, vol. 1, pp. 113–184 Search PubMed.
- L. Beverina, G. A. Pagani and M. Sassi, Chem. Commun., 2014, 50, 5413–5430 RSC.
- M. M. Nicholson, in Phthalocyanines: Properties and Applications, ed. C. C. Leznoff and A. B. P. Lever, VCH, Weinheim, 1993, ch. 2, vol. 3, pp. 71–118 Search PubMed.
- J. M. Green and L. R. Faulkner, J. Am. Chem. Soc., 1983, 105, 2950–2955 CrossRef CAS.
- B. Schumann, D. Wöhrle and N. I. Jaeger, J. Electrochem. Soc., 1985, 132, 2144–2149 CrossRef CAS.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- M. K. Engel, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, vol. 20, pp. 1–242 Search PubMed.
- T. Hosokai, A. Gerlach, A. Hinderhofer, C. Frank, G. Ligorio, U. Heinemeyer, A. Vorobiev and F. Schreiber, Appl. Phys. Lett., 2010, 97, 63301 CrossRef.
- L. Cornelius, M. Beu, C. Keil and D. Schlettwein, Phys. Status Solidi RRL, 2012, 6, 214–216 CrossRef CAS.
- C. Keil, O. Tsaryova, L. Lapok, C. Himcinschi, D. Wöhrle, O. R. Hild, D. R. T. Zahn, S. M. Gorun and D. Schlettwein, Thin Solid Films, 2009, 517, 4379–4384 CrossRef CAS.
- S. Nagel, M. Lener, C. Keil, R. Gerdes, Ł. Łapok, S. M. Gorun and D. Schlettwein, J. Phys. Chem. C, 2011, 115, 8759–8767 CrossRef CAS.
- J. Weissbecker, A. Loas, S. M. Gorun and D. Schlettwein, Electrochim. Acta, 2015, 157, 232–244 CrossRef CAS.
- S. M. Gorun, B. A. Bench, G. Carpenter, M. W. Beggs, J. T. Mague and H. E. Ensley, J. Fluorine Chem., 1998, 91, 37–40 CrossRef CAS.
- A. B. P. Lever, E. R. Milaeva and G. Speier, in Phthalocyanines: Properties and Applications, ed. C. C. Leznoff and A. B. P. Lever, VCH, Weinheim, 1993, vol. 3, ch. 1 Search PubMed.
- H. H. Patel, PhD thesis, Seton Hall University, 2015.
- E. N. Carrión, A. Loas, H. H. Patel, M. Pelmuş, K. Ramji and S. M. Gorun, J. Porphyrins phthalocyanines, 2018, 22, 371–397 CrossRef.
- K. J. M. Nolan, M. Hu and C. C. Leznoff, Synlett, 1997, 593–594 CrossRef.
- H. Konami, Y. Ikeda, M. Hatano and K. Mochizuki, Mol. Phys., 1993, 80, 153–160 CrossRef CAS.
- H. Moons, Ł. Łapok, A. Loas, S. van Doorslaer and S. M. Gorun, Inorg. Chem., 2010, 49, 8779–8789 CrossRef CAS PubMed.
- K. Hesse and D. Schlettwein, J. Electroanal. Chem., 1999, 476, 148–158 CrossRef CAS.
- D. Schlettwein, N. I. Jaeger and T. Oekermann, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, vol. 16, pp. 247–284 Search PubMed.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed.
- A. W. Snow, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, vol. 17, pp. 129–176 Search PubMed.
- C. Keil, PhD thesis, Justus-Liebig-University, 2012.
- D. G. de Oteyza, E. Barrena, J. O. Ossó, S. Sellner and H. Dosch, J. Am. Chem. Soc., 2006, 128, 15052–15053 CrossRef CAS PubMed.
- R. Ye, M. Baba, Y. Ohishi, K. Mori and K. Suzuki, Mol. Cryst. Liq. Cryst., 2006, 444, 203–210 CrossRef CAS.
- G. Liu, T. Gredig and I. K. Schuller, Europhys. Lett., 2008, 83, 56001 CrossRef.
- W. Michaelis, D. Wöhrle and D. Schlettwein, J. Mater. Res., 2004, 19, 2040–2048 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, New York, 2001 Search PubMed.
- J.-M. Savéant, Elements of Molecular and Biomolecular Electrochemistry, John Wiley & Sons, Hoboken, NJ, 2006 Search PubMed.
- E. Laviron, J. Electroanal. Chem., 1980, 112, 1–9 CrossRef CAS.
- E. Laviron, L. Roullier and C. Degrand, J. Electroanal. Chem., 1980, 112, 11–23 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1981, 122, 37–44 CrossRef CAS.
- M. D. Levi, R. Demadrille, A. Pron, M. A. Vorotyntsev, Y. Gofer and D. Aurbach, J. Electrochem. Soc., 2005, 152, E61–E67 CrossRef CAS.
- M. Tagliazucchi, D. Grumelli, C. Bonazzola and E. J. Calvo, J. Nanosci. Nanotechnol., 2006, 6, 1731–1740 CrossRef CAS PubMed.
- K. Xu, A. von Cresce and U. Lee, Langmuir, 2010, 26, 11538–11543 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectrometry, FT-IR spectra, concentration-dependent UV-Vis spectra, grazing incidence X-ray diffraction (GIXRD) pattern. See DOI: 10.1039/c9cp06709d |
| This journal is © the Owner Societies 2020 |