
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular catalysis of CO2 reduction: recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes
E.
Boutin
 a,
L.
Merakeb
a,
B.
Ma
a,
B.
Boudy
a,
M.
Wang
a,
J.
Bonin
a,
E.
Anxolabéhère-Mallart
a and
M.
Robert
*ab
a,
L.
Merakeb
a,
B.
Ma
a,
B.
Boudy
a,
M.
Wang
a,
J.
Bonin
a,
E.
Anxolabéhère-Mallart
a and
M.
Robert
*ab
aUniversité de Paris, Laboratoire d’Electrochimie Moléculaire, CNRS, F-75006 Paris, France. E-mail: robert@u-paris.fr
bInstitut Universitaire de France (IUF), F-75005 Paris, France
First published on 22nd July 2020
Abstract
Earth-abundant Fe, Ni, and Co aza macrocyclic and polypyridine complexes have been thoroughly investigated for CO2 electrochemical and visible-light-driven reduction. Since the first reports in the 1970s, an enormous body of work has been accumulated regarding the two-electron two-proton reduction of the gas, along with mechanistic and spectroscopic efforts to rationalize the reactivity and establish guidelines for structure–reactivity relationships. The ability to fine tune the ligand structure and the almost unlimited possibilities of designing new complexes have led to highly selective and efficient catalysts. Recent efforts toward developing hybrid systems upon combining molecular catalysts with conductive or semi-conductive materials have converged to high catalytic performances in water solutions, to the inclusion of these catalysts into CO2 electrolyzers and photo-electrochemical devices, and to the discovery of catalytic pathways beyond two electrons. Combined with the continuous mechanistic efforts and new developments for in situ and in operando spectroscopic studies, molecular catalysis of CO2 reduction remains a highly creative approach.
 Top row, from the left: Etienne Boutin, Lydia Merakeb, Bing Ma and Benjamin Boudy, Bottom row, from the left: Min Wang, Julien Bonin, Elodie Anxolabéhère-Mallart and Marc Robert | Etienne Boutin and Ming Wang obtained their PhD from Université de Paris in 2019 working on the heterogenization of molecular catalysts onto carbon materials for the CO2RR and the electrochemical conversion of CO2 into methanol, respectively. Benjamin Boudy and Bing Ma are currently working on the insertion of molecular CO2 reduction catalysts into flow cell electrolyzers and the design of hybrid systems for light-driven reduction of CO2 respectively, while Lydia Merakeb is developing new molecular catalysts for the electrochemical reduction of N2. All three are expected to defend their PhD by the end of the year 2020. |
Julien Bonin received his PhD in Physical Chemistry from the Université Paris-Sud XI in 2005. He then joined the Radiation Laboratory at the University of Notre Dame as a Postdoctoral Research Associate. Since fall 2006, he has been an Associate Professor of Chemistry at the Université Paris Diderot, now Université de Paris. His current research interests are related to CO2 reduction and photocatalysis. |
Elodie Anxolabéhère-Mallart received her PhD in Molecular Electrochemistry from the Université Paris Diderot in 1991. In 1992 she was appointed CNRS researcher at the Université Paris-Sud, where she first got interested in artificial photosynthesis. She was a visiting researcher at Stanford University (1998–1999) and Lawrence Berkeley National Laboratory (1997–1999) to study both natural and model systems using X-ray Absorption Spectroscopy. In 2008 she joined the Laboratoire d’Electrochimie Moléculaire (LEM), focusing her research on O2 and CO2 activation using transition metal complexes. |
Marc Robert obtained his PhD in 1995 from the Université Paris Diderot under the guidance of Jean-Michel Savéant and Claude Andrieux. Following a postdoctoral stay at Ohio State University with Matthew Platz, he started his academic career at the Université Paris Diderot in 1997. He is currently a Professor of Chemistry at the Université de Paris and a Senior fellow at the Institut Universitaire de France (IUF). His interests include electrochemical and photochemical approaches of electron transfer processes and catalytic activation of small molecules, mainly CO2 and N2. |
Introduction
CO2 may become the renewable feedstock for making fuels and commodity and pharmaceutical chemicals we need to sustain our societies. If we manage to solve the challenges in reaching this goal, we would achieve a circular economy based on the use of renewable energies and CO2. This goal remains elusive even in the most developed countries, and chemistry is poised to play a central role in addressing the key scientific challenges. A primary task is to develop selective, fast and efficient reduction processes of CO2 into valuable products, such as carbon monoxide (CO), formic acid (HCOOH), methanol (CH3OH), methane (CH4), ethanol (CH3CH2OH) and ethylene (C2H4), in a sustainable manner. Catalyst development is of key importance for this purpose, and various approaches, both by homogeneous and heterogeneous chemistry, are widely pursued. Achieving electrochemically or photochemically driven conversion of CO2 remains a grand challenge, especially if one considers that only abundant materials should be used in view of future large scale applications. Among various advantages, the use of molecular catalysts such as defined transition metal–ligand complexes allows for fine-tuning the chelating abilities and the steric, electronic and electrostatic effects of the ligands, thus opening a wide door towards thorough mechanistic and spectroscopic studies. Notably, abundant transition metals such as iron, manganese, cobalt, copper or nickel with activity for CO2 catalytic reduction were already investigated in the early 1970s, starting with phthalocyanines and porphyrins.1 The revival of these studies in the last 15 years has led to an enormous body of work and stimulating new results, even if molecular catalysts able to go beyond the 2-electron 2-proton traditional reduction products (CO and formic acid) remain scarce. Aza macrocycle ligands such as porphyrins and phthalocyanines, as well as polypyridine ligands are extensively investigated and show good results both under electrochemical conditions and light stimulation with appropriate sensitizers and sacrificial electron donors. A number of comprehensive and high level reviews have been published over the years notably concerning the use of abundant, cheap metals under electrochemical conditions.2–6 Less emphasis has been put on photochemically induced reduction of CO2 but related approaches are developing fast. Why publishing another review on the topic? Not only because the field is rapidly evolving, with more efficient and selective systems, but also because new perspectives are emerging, at the interfaces of homogeneous and heterogeneous catalysis, or of electrochemistry and photochemistry (molecular photoelectrodes, self-standing hybrid materials, etc.), as well as regarding mechanistic studies with in situ and in operando analytical methods. Moreover, the recent implementation of molecular catalysts into electrolyzers operating at large current densities (>150 mA cm−2) has opened new possibilities for the design of real devices that may be able to reach the market. Finally, first examples of molecular catalysts able to produce highly reduced products such as methanol and methane have recently been launched, opening also stimulating perspectives toward new chemistry. We have chosen to focus on compounds including mainly Co, Fe, and Ni porphyrin, phthalocyanine and polypyridine complexes so as to illustrate, discuss and highlight the above-mentioned electrochemical and light-driven processes for carbon dioxide catalytic reduction. Regarding electrochemical approaches, we have further focused the scope on typical examples rather than on an exhaustive description. In some cases, mechanistic studies have helped in designing the most efficient catalysts on a rational basis, and such examples will be described in more detail. Regarding visible-light-driven catalytic processes, mechanistic and spectroscopic in-depth studies are less numerous and we have rather chosen to give a more comprehensive overview of the field.1. Electrochemical approaches
| CO2 + e− → CO2˙−, E0′ = −1.90 V | (1) |
| CO2 + 2H+ + 2e− → HCOOH, E0′ = −0.61 V | (2) |
| CO2 + 2H+ + 2e− → CO + H2O, E0′ = −0.53 V | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O, E0′ = −0.48 V | (4) |
| CO2 + 6H+ + 6e− → CH3OH + H2O, E0′ = −0.38 V | (5) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O, E0′ = −0.24 V | (6) |
Ideally a proper understanding of the reaction pathways may lead to rational catalyst optimization. This is probably a definitive advantage of the above-mentioned catalysts, which are well-defined single metal catalytic sites, for which the tools of electrochemistry and spectroscopy, including in situ and in operando, are well established.
In most cases, the initially inactive form of the catalyst P is reduced to a low-valent oxidation state metal by outer sphere electron transfer at an electrode.7 In homogeneous catalysis, both substrate and catalyst are dissolved in the electrolyte solution and the chemical reactions take place within the reaction layer, a small fraction of the diffusion layer within which the concentration of the active catalyst Q is significant. Catalysis may be limited by diffusion of the substrate, concentration of the catalyst, of the substrate, of the product (inhibition) or by the intrinsic properties of the catalyst such as slow electron transfer kinetics or a chemical step of the catalytic process.8 The metrics for assessing catalyst performances include the selectivity for the target reaction (in electrochemistry, faradaic efficiency FE is also used to measure the fraction of electrons used for the target reaction), the rate or turnover frequency (TOF, s−1) which is defined by the mole amount of product formed divided by the mole amount of active catalysts per unit of time. The durability of the catalyst is measured through the turnover number (TON) given by the final mole amount of product once the catalysis has stopped because of the degradation of the catalytic system, divided by the initial mole amount of catalyst. Finally the overpotential, the thermodynamic cost for catalysis, is given by η = E0tr − E, where E is the applied potential and E0tr the apparent standard potential of the target reaction.
The intrinsic activity of a given catalyst may be represented by plotting the TOF as a function of the applied overpotential η (catalytic Tafel plot), as sketched in Fig. 1. The TOF is indeed dependent on η since the amount of active catalysts in the reaction layer increases when η increases, reaching a plateau value equal to kcat, the pseudo-first-order rate constant of the limiting step of the catalytic process (provided the experimental conditions allow reaching the pure kinetic regime; see the next section for details).
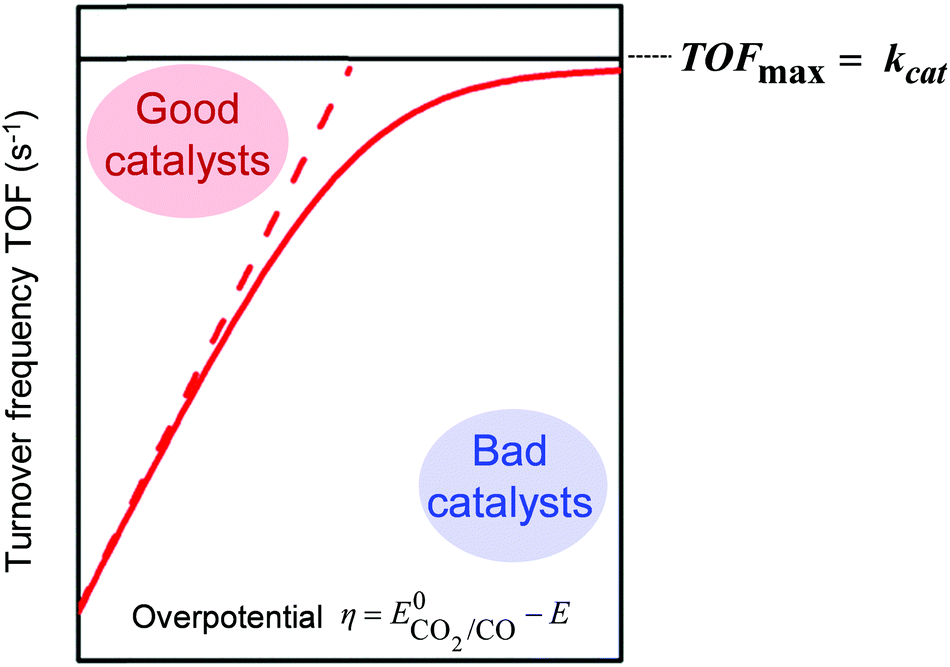 | ||
| Fig. 1 Benchmarking of molecular catalyst intrinsic properties for the CO2-to-CO reduction through the catalytic Tafel plot [TOF = f(η)]. | ||
Great care should be taken when estimating the TON (electrolysis experiment). In several publications, values are overestimated by orders of magnitude since authors use the mole amount of catalyst confined in the reaction layer, forgetting that the catalyst degrades over time and is progressively replaced by molecules from the bulk. As a consequence, the mole amount of product should be divided by the total mole amount of catalysts in the electrochemical reactor, once the reaction has ceased. This is the proper way to evaluate the turnover number.
Under supported conditions, the catalyst is immobilized at the electrode surface by various means, such as covalent linkage, non-covalent interactions such as electrostatic or π–π interactions or simply adsorption, or within periodic structures such as porous coordination polymers and metal–organic frameworks.9–12 The reaction then takes place within the electrode surface. The major benefits of heterogeneous catalysts are the low amount of catalyst needed and, the possibility of by-passing diffusion limitations when using micro-fluidic cells or gas diffusion electrodes13 and natural separation of catalyst and products (liquid or gaseous).
1.2.1 Cyclic voltammetry. Cyclic voltammetry (CV) can be used for mechanistic studies of systems in which electron transfer processes are coupled to chemical reactions. Advances in the studies of mechanisms, including catalytic multi-electron-multi-step ones, involved in homogeneous molecular catalysis of electrochemical reactions have been recently reviewed.14
Homogeneous catalysis. In the absence of catalyst, a substrate A can be reduced directly at the surface of an electrode upon applying a sufficiently negative potential (blue line in Fig. 2). When a catalyst P/Q is present in the medium, the reduced form Q of the catalyst reacts with the substrate A, giving B and thus regenerating the P form of the catalyst, which results in an increase of the recorded P-to-Q reduction current in cyclic voltammetry (red line in Fig. 2) and a loss in reversibility that is observed in the absence of substrate (green line in Fig. 2). In this case, transformation of A into B can be achieved at a more positive potential. The shape of the voltammogram depends on two dimensionless factors, the excess factor between substrate and catalyst concentration γ = C0A/C0P, and the kinetic parameter (λ = (RT/F) × kC0P/v) which accounts for the competition between kinetics of the catalytic reaction (rate constant k) and the diffusion rates (scan rate v), as illustrated in the catalytic zone diagram (Fig. 3). Analysis applies in the case of fast electron transfer to the catalyst and selective catalytic reaction.
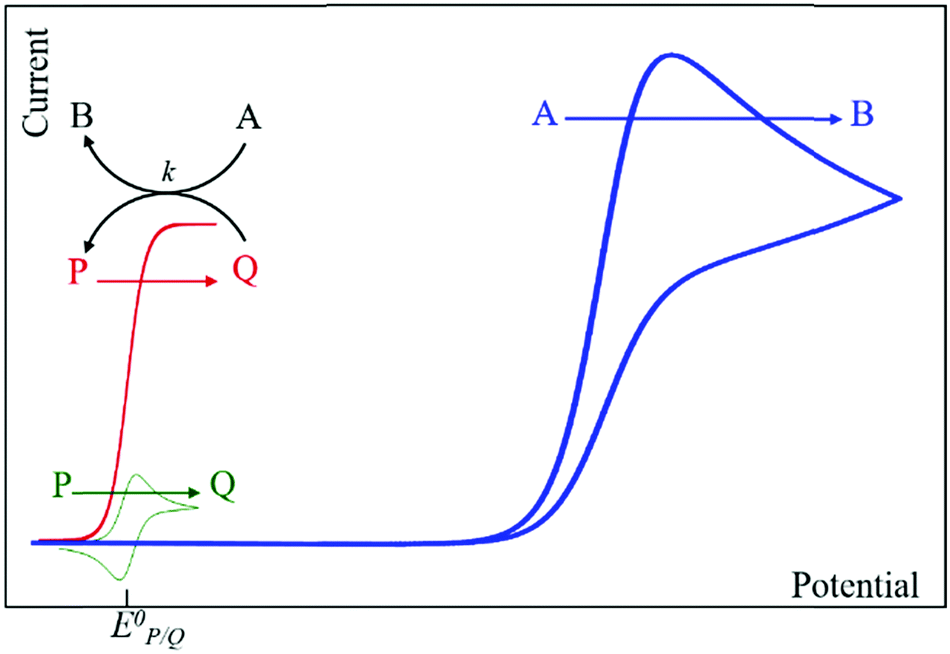 | ||
| Fig. 2 Canonical cyclic voltammograms for the molecular catalysis of a one-electron catalytic electrochemical reaction (blue: direct reduction of A; green: reversible one-electron reduction of P in the absence of A; red: catalytic reduction of A triggered by the reduced form of the catalyst Q). Adapted from ref. 15 with permission from Wiley. Copyright 2006. | ||
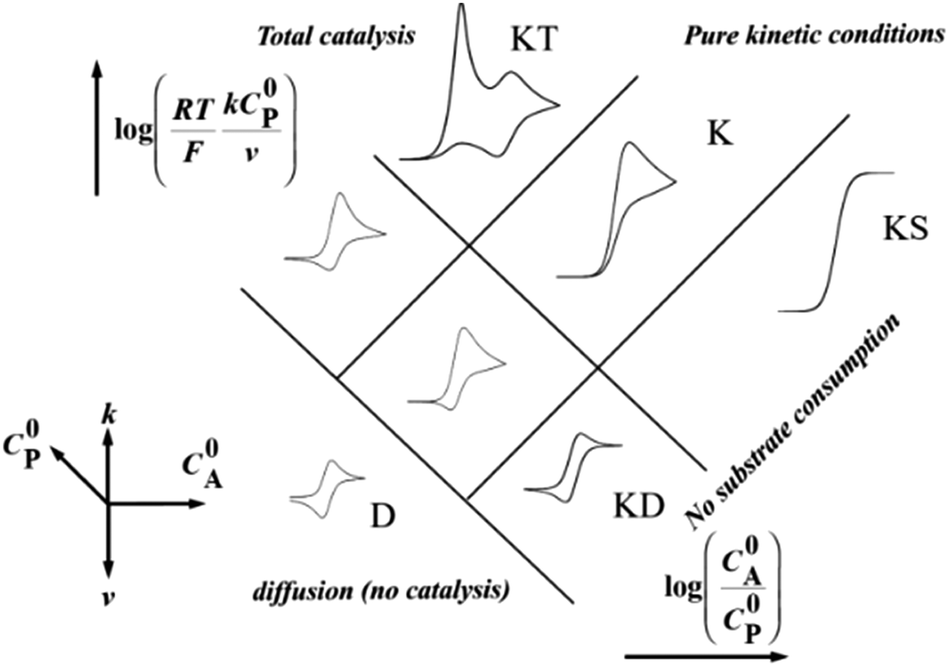 | ||
| Fig. 3 Zone diagram for a one-electron catalytic reduction. Adapted from ref. 15 with permission from Wiley. Copyright 2006. | ||
When the homogeneous catalytic reaction (rate constant k) is fast as compared to diffusion and concentration of the substrate is large enough to be constant (pure kinetic regime), a classical S-shaped CV is obtained as can be seen in the upper right part of Fig. 3. In this case, the rate constant k can be derived from the plateau current iplateau, which is independent of scan rate v (eqn (7)):
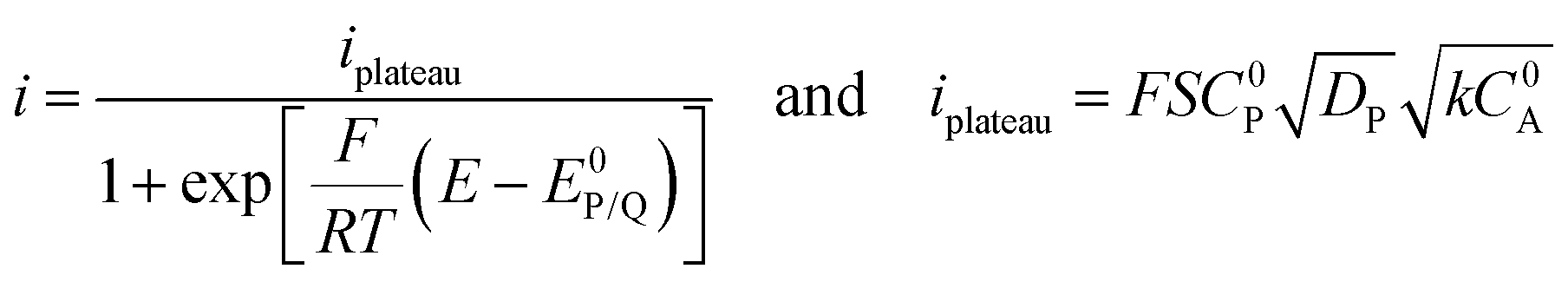 | (7) |
By dividing the current by the peak current i0 obtained in the absence of a substrate (eqn (8)), the diffusion constant DP and the electrode surface area S cancel (eqn (9)).
 | (8) |
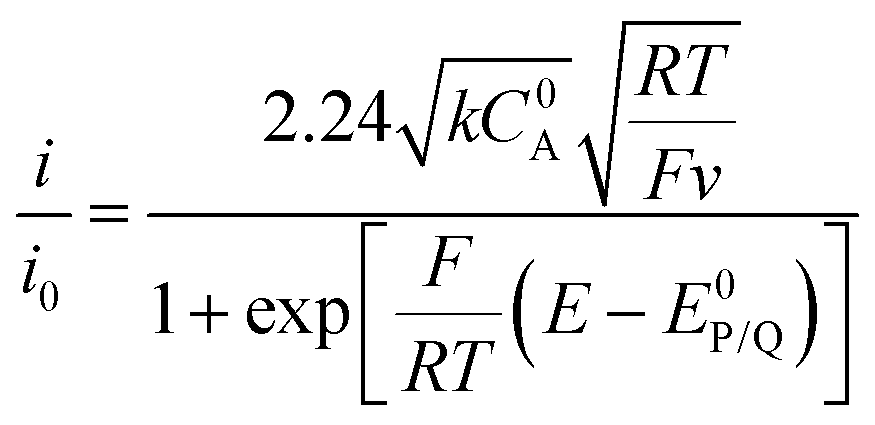 | (9) |
Special care should be taken when using these equations that are developed for the simple case of a one-electron reduction process. The number of exchanged electrons should be taken into account for more complex processes and different mechanisms will lead to different i/i0 values.16
Since depletion of the substrate in the reaction layer may occur for fast reactions and low substrate concentrations (e.g. in the case of efficient catalysis for CO2 reduction in water), the reduction current may be limited by substrate diffusion itself. Then the CV shape changes from a plateau one to a peak one (such as “total” catalysis (KT zone, Fig. 3) regime, obtained when the reaction layer is depleted from the substrate, while the remaining oxidized catalyst P is reduced at a more negative potential than the catalytic wave). Other phenomena can lead to a peak shaped CV, such as deactivation of the catalyst or inhibition by a product. In these latter cases, the above mentioned equations (7)–(9) do not apply anymore, but other strategies may be used in order to extract kinetic information from the voltammograms.
The first and most efficient strategy is to increase the scan rate (as long as the reaction rate allows it) so as to obtain the putative plateau-shaped CV. This strategy was followed by Azcarate et al. in the case of CO2-to-CO conversion using Fe porphyrins.17 Upon increasing the acid concentration, catalysis becomes stronger and the CV response changes from an S-shape to a peak-shape due to the interference of secondary phenomena (substrate consumption and/or partial inhibition of the electrode surface by gas bubbles). Increasing the scan rate (smaller charge transferred at the electrode surface) led back to plateau-shaped CVs and the rate constant for catalysis could be derived from the plateau current. It allowed studying the effect of successive phenyl perfluorination and of o,o′-methoxy substitution of Fe tetraphenylporphyrin (Fe1, Fe6–Fe9, Chart 3).
Another example is provided by Co quaterpyridine (Co20, Chart 1). The 2-electron reduction of the CoII complex in CH3CN generates a radical anion (ligand centered reduction). The reduced complex is then adsorbed onto the electrode surface, rendering the CV analysis and quantitative data collection difficult. However, by increasing the scan rate (Fig. 4), the amount of charge passed being smaller, this phenomenon was minimized and proper diffusion-controlled waves were obtained. This further allowed identification and characterization (from plateau current values) of two catalytic pathways for the electrochemical reduction of CO2 to CO mediated by cobalt quaterpyridine.18,19 The first one originates from the 2-electron reduction of the catalyst (at 3 M phenol concentration) while at lower acid concentration (1 M phenol) catalysis is triggered after 3-electron reduction of the catalyst, as illustrated in Fig. 4.
 | ||
| Chart 1 Cobalt complexes. | ||
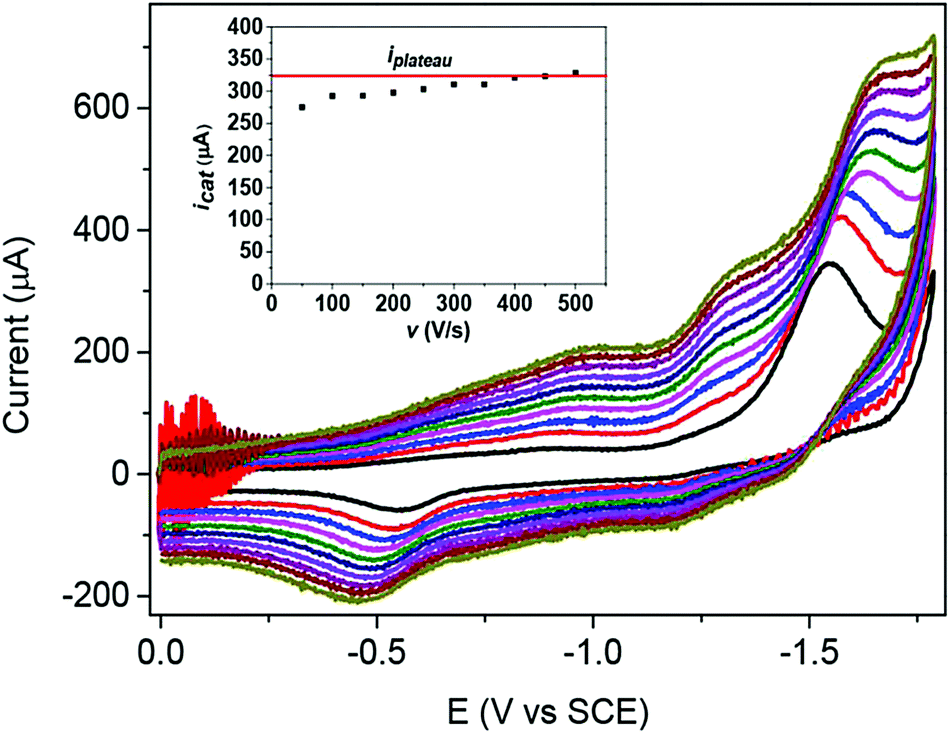 | ||
| Fig. 4 Plateau-shaped CV (most negative wave) establishment upon increasing the scan rate during CO2 electrochemical reduction with Co20 (0.5 mM) in CH3CN containing phenol (1 M) as a proton source and 0.1 M (tBu)4NPF6 as a supporting electrolyte. Scan rate v = 50 (black), 100 (red), 150 (blue), 200 (magenta), 250 (olive), 300 (navy), 350 (violet), 400 (purple), 450 (wine) and 500 V s−1 (dark yellow). Inset: Variation of the catalytic current obtained at the third reduction wave (calculated from subtraction of the background current value) as a function of the scan rate. Reprinted with permission from ref. 19. Copyright 2019 American Chemical Society. | ||
The impact of side phenomena on CVs increases as the passed charge increases. Analyzing the foot of the wave (FOWA), where the current is small (small charge), is another strategy minimizing these effects with i/i0 values being then again proportional to 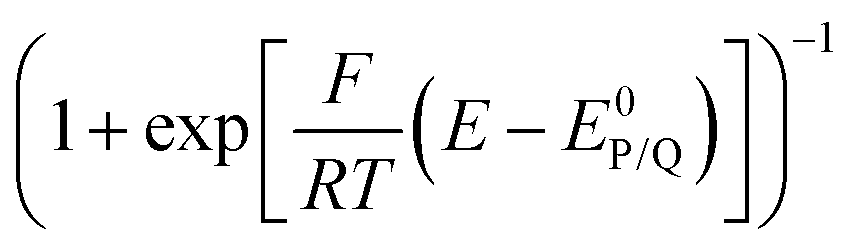 as shown from eqn (9).8 As an example, FOWA has allowed for the derivation of reaction orders toward various acid co-substrates in the reduction of CO2 to CO mediated by the electrogenerated Fe0 tetraphenylporphyrin in the presence of phenol as a co-substrate.20 It was further revealed that after the binding of CO2 to Fe0, catalysis involves an electron transfer from the Fe center concerted with proton transfer and C–O bond cleavage. In another example, using FOWA analysis, various Co terpyridine based complexes featuring different substituents on the ancillary ligand (Co21 and derivatives, Chart 1), were prepared, and studied as catalysts for CO2 reduction. It was shown that ligand modifications have a small effect on the kinetics of CO2 reduction while stronger effects were observed for the competitive proton reduction (HER). Higher rate constants for HER were measured with more electron rich ancillary ligand fields. Thus, slowing down the HER by carefully choosing the catalyst electronic properties resulted in improved selectivity for CO2 reduction.21
as shown from eqn (9).8 As an example, FOWA has allowed for the derivation of reaction orders toward various acid co-substrates in the reduction of CO2 to CO mediated by the electrogenerated Fe0 tetraphenylporphyrin in the presence of phenol as a co-substrate.20 It was further revealed that after the binding of CO2 to Fe0, catalysis involves an electron transfer from the Fe center concerted with proton transfer and C–O bond cleavage. In another example, using FOWA analysis, various Co terpyridine based complexes featuring different substituents on the ancillary ligand (Co21 and derivatives, Chart 1), were prepared, and studied as catalysts for CO2 reduction. It was shown that ligand modifications have a small effect on the kinetics of CO2 reduction while stronger effects were observed for the competitive proton reduction (HER). Higher rate constants for HER were measured with more electron rich ancillary ligand fields. Thus, slowing down the HER by carefully choosing the catalyst electronic properties resulted in improved selectivity for CO2 reduction.21
The above cases assume a fast electron transfer from the electrode to the oxidized form P of the catalyst. When electron transfer is in contrast slow (or if the follow up reaction is so fast that the initial electron transfer should be viewed as slow), foot of the wave analysis (FOWA) still applies but one should plot FIT(E − E0P/Q) vs. as (eqn (10)):22
as (eqn (10)):22
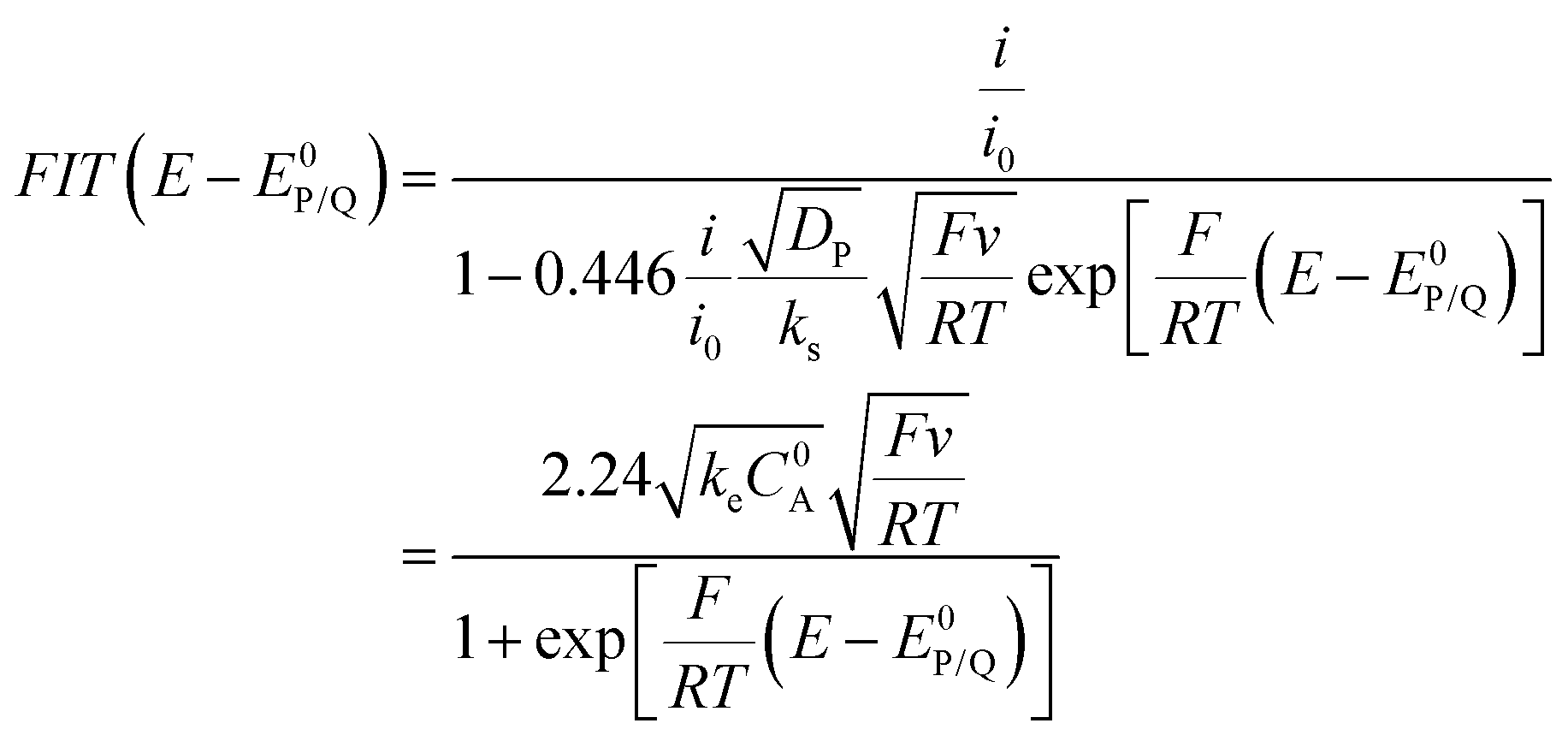 | (10) |
In the case of Fe2, a complete strategy combining analysis of all segments of the CVs has led to the full characterization of the catalytic cycle for CO2 reduction8 as shown in Fig. 5 (see Section 1.3 for a complete mechanistic discussion).
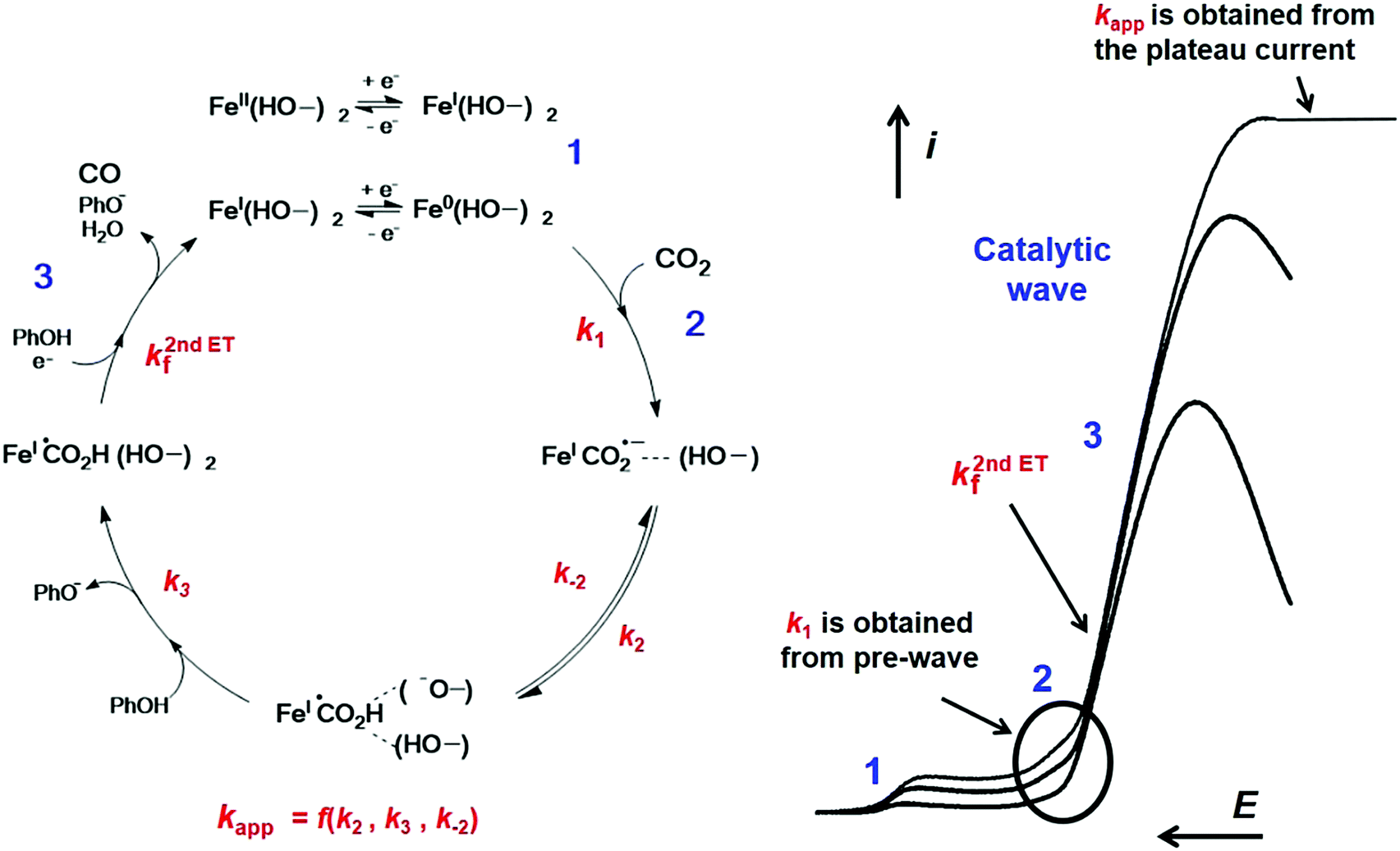 | ||
| Fig. 5 Molecular catalysis mechanism of CO2 reduction into CO with Fe2 in the presence of phenol (PhOH) as a co-substrate. Left: Mechanism for the catalysis obtained from CVs analysis. Right: Catalytic CVs as a function of increasing scan rate, with plateau current at high scan rate. Main kinetic parameters were obtained from the one-electron pre-catalytic wave peak position (k1), the rise of the current function (k2nd,ETf) and from the plateau current value (kapp), respectively. Adapted with permission from ref. 14. Copyright 2017, Elsevier. | ||
Regarding the total catalysis regime (KT zone, Fig. 3), a formal kinetic analysis has been developed in the case of 2-electron/2-step processes, allowing extraction of kinetic parameters and distinguishing between different mechanisms.23
Supported molecular catalysts. As convenient as homogeneous molecular catalysts are for mechanistic studies, it is difficult to conceive their implementation in devices since only a small fraction of catalyst close to the electrode surface is active. Attaching these molecular catalysts to an electrode may be a good strategy for efficient use while still benefiting from their good selectivity.24,25 While rotating disk electrode voltammetry (RDEV) has been widely used for the study of catalyst-containing films, CV is a good alternative when RDEV is hardly applicable (for example when using porous conductive materials as supporting electrodes). With an electrode surface coated with a porous film containing attached molecular catalysts (Fig. 6, top scheme), maximum current density for a fast catalytic reaction will be I = nFkcatΓcat where n is the number of electrons involved (n = 2 for CO or formate production), kcat the catalytic rate constant (assumed to be first order toward CO2kcat = κAk[CO2]) and Γcat the surface concentration of the catalyst (Γcat = C0pdf). Knowledge of the film thickness df and catalyst concentration can then lead to the value of kcat. Such a determination may be hampered by slow charge transport through the film (either in the context of a non-conductive support with charge hopping between catalytic redox sites, or of an electronically conductive support connecting the molecular catalysts).26,27
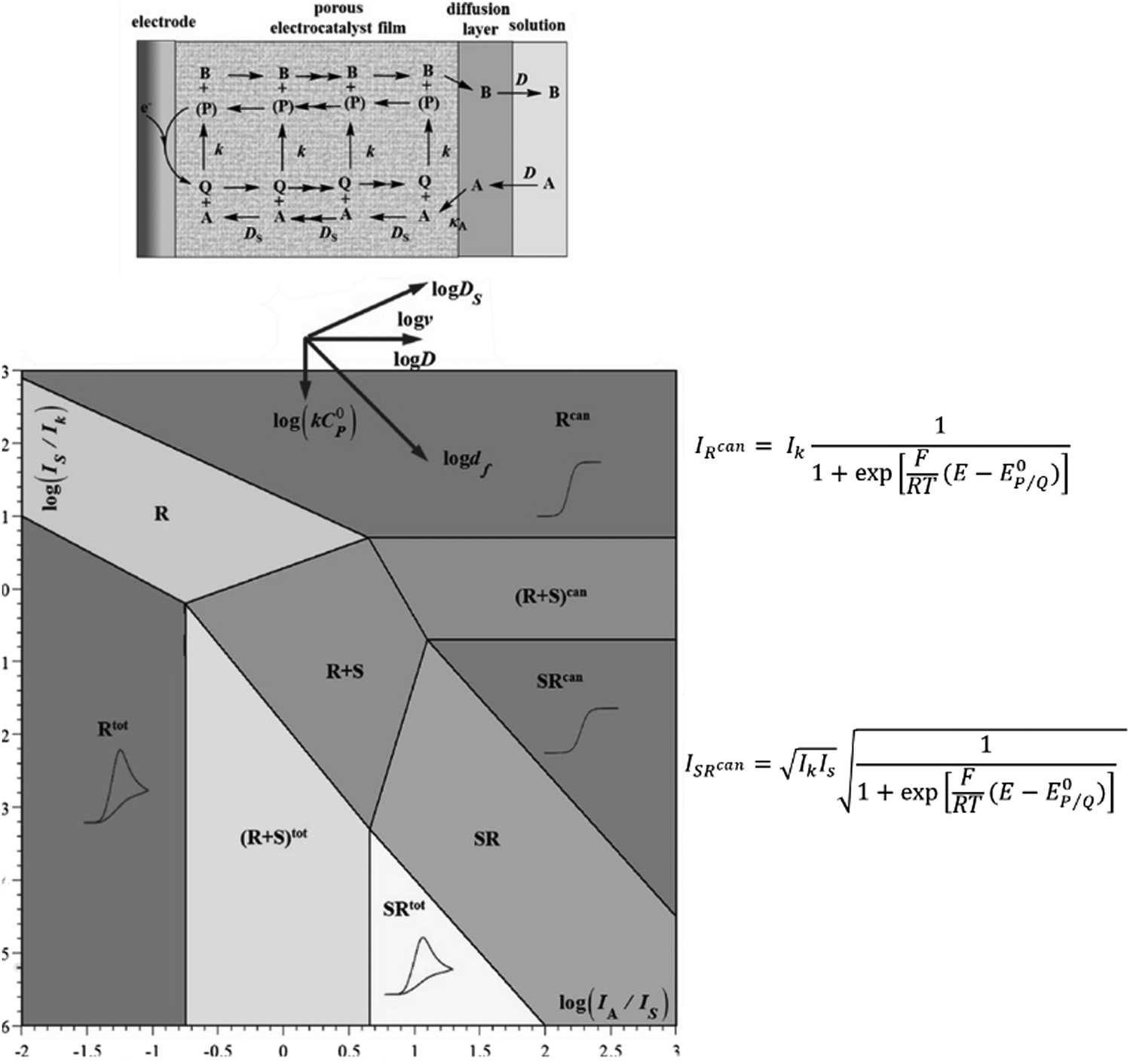 | ||
| Fig. 6 Kinetic zone diagram for fast conducting catalytic films in the case of a one-electron reaction. Top scheme illustrates a porous film in which the catalyst is attached and is in contact with a conductive support. Adapted from ref. 29 with permission from Elsevier. Copyright 2017. | ||
In the framework of fast charge transport and assuming that the molecular catalysts behave as molecular sites (i.e. with no strong conjugation of the catalyst molecular orbitals with the conductive support), the current density response for a fast one-electron catalytic reaction will depend on three parameters: the value of k (limiting current density Ik = FkκAC0AΓcat = FkC0pκAC0Adf), the diffusion rate of the substrate in the film (limiting current density 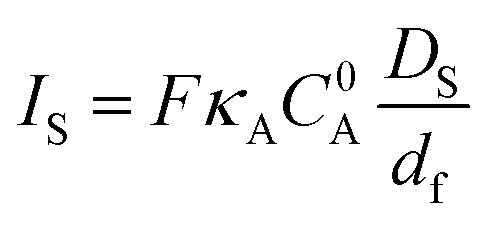 ) and the diffusion rate of the substrate outside the film (current density
) and the diffusion rate of the substrate outside the film (current density  ), as illustrated in Fig. 6. This kinetic zone diagram may be extended to multielectron/multistep processes. An appropriate stoichiometric factor should then be introduced.28,29 The rapid development of hybrid materials with immobilization of molecular catalysts into thin films at electrode surfaces calls for performing thorough mechanistic studies with the above tools. Such studies are currently missing.
), as illustrated in Fig. 6. This kinetic zone diagram may be extended to multielectron/multistep processes. An appropriate stoichiometric factor should then be introduced.28,29 The rapid development of hybrid materials with immobilization of molecular catalysts into thin films at electrode surfaces calls for performing thorough mechanistic studies with the above tools. Such studies are currently missing.
1.2.2 Spectroelectrochemistry. Spectroscopic techniques can be used as non-invasive (non-destructive) complementary tools to identify reaction intermediates and/or products. In this regard, the combination of “reaction oriented electrochemistry” and “species-focused spectroscopy” gives rise to spectroelectrochemistry (SEC), a technique aiming at the spectroscopic (UV-vis, IR, X-ray, etc.) detection of reaction intermediates generated in situ by application of a controlled potential at an electrode.30 While UV-vis, EPR and XAS spectroscopies can probe the redox state of catalysts, IR and Raman spectroscopies can monitor evolution of carbonyl groups involved in CO2 reduction. Additionally, labelled experiments are potentially fruitful since bond stretching and vibration are atom weight dependent. Although no standard setup exists, a suitable thin layer cell design is required for SEC experiments. Fig. 9 shows examples of UV-vis and IR-SEC cells.
One of the very first studies regarding SEC studies in the context of CO2 reduction identified the free anion radical CO2˙− to be the intermediate of CO2 reduction on lead in water and in some aprotic solvent using UV-vis reflectance spectroscopy.31 It was also shown that oxalate was produced through the coupling of CO2˙− with CO2 rather than through dimerization of the radical anion.31 Later, CO2 and CO2–adsorbates on platinum and in acetonitrile were identified using polarization modulation Fourier transform infrared reflection absorption spectroscopy.32 Reduction of CO2 under these conditions was shown to yield oxalate.33
Focusing on Fe porphyrins, Fe0 species was detected using in situ UV-vis and Raman SEC characterization.34 It was shown that the two-electron reduction of ironII tetraphenylporphyrin occurs on the metal center rather than on the porphyrin ring by comparison with data obtained for zinc tetraphenylporphyrin, which is known to yield a diradical anion porphyrin ring (ligand centered reduction). Such a description is still subject to extensive discussion as recent work suggests that reduction of iron tetraphenylporphyrin is not metal- but ligand-centered. Such a conclusion was supported by Mössbauer and X-ray absorption spectroscopy, with additional insight from computational methods.35 It was also supported by a resonance Raman spectroscopy study of reduced teraphenylporphyrin coupled with quantum chemistry calculations.36 However, these results do not account for the reactivity of the reduced porphyrin with CO2 at Fe and for the absence of ligand carboxylation. Additionally, stabilization of the reduced form of the ligand (diradical anion) is not comparable under the conditions usually reported in CO2 reduction studies (in DMF) and in spectroscopic studies, where much less polar solvents than DMF are used (ethers).37 Results obtained under these conditions are thus not directly comparable. This underlines the importance of performing in situ and in operando studies.
Illustrative examples include an iron porphyrin containing triazole substituents (Fe20, Chart 3) that facilitate H-bonding. Chemical reduction of the iron porphyrin in the presence of CO2 and various proton sources was performed at low temperature and some reaction intermediates were trapped and probed by Raman spectroscopy, as illustrated in Fig. 7.38 It was shown that the chemically generated Fe0 porphyrin gets oxidized by CO2, forming a FeIICO22− adduct, which is rapidly protonated to form a Fe–COOH species. At this point, introduction of a relatively strong acid (p-CH3C6H4SO3H) resulted in the cleavage of a C–OH bond and another intermediate was detected, which was shown to be FeII–CO. In the absence of acid, formate was produced by hydrolysis of the protonated intermediate. These results were qualitatively supported by density functional theory modelling of the Fe–C adducts.38
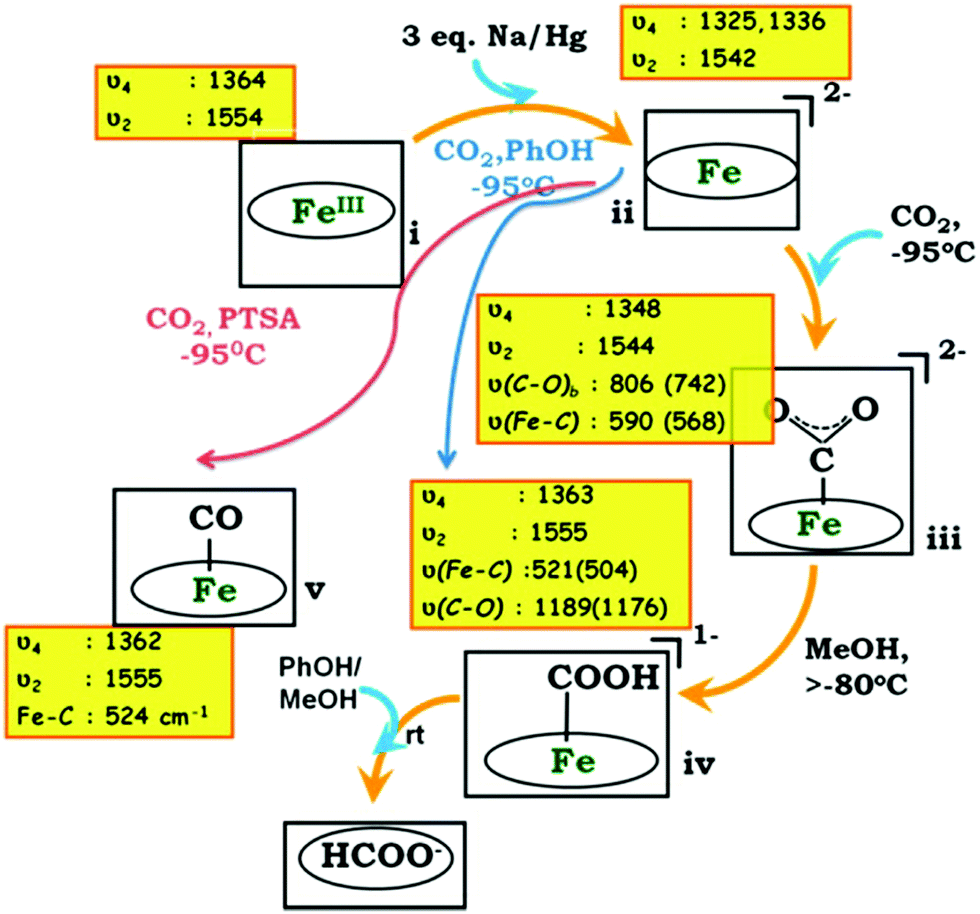 | ||
| Fig. 7 Proposed mechanism for CO2 reduction with Fe20, from Raman spectroscopic detection of intermediates. Reprinted with permission from ref. 38. Copyright (2015) American Chemical Society. | ||
Keeping with iron catalysts, IR-SEC was performed on an iron Schiff base complex (Fe21). It was shown that the formation of stable Fe–CO species with one or two CO molecules coordinated to the Fe center limits the activity of the complex under both protic and aprotic conditions and led to decomposition of the catalyst.39 This was also the case when Fe13 was used as a catalyst. In this case, the formation of the Fe0–CO adduct has been identified as a deactivation pathway, again using IR-SEC.18
The IR-SEC has been proven to be beneficial in the study of a binuclear Co complex bearing a bi-quaterpyridine (Co23, Chart 6) photo- and electrocatalyst for the reduction of CO2 to formate or CO selectively depending on the composition of the reaction medium (see Section 4, Fig. 14).40 A stable adduct between CO2 and the four-electron-reduced complex was evidenced by a band at 1635 cm−1 upon scanning the potential applied to the Pt grid working electrode from −0.35 to −0.85 V vs. Ag pseudo-reference. The configuration of this adduct was shown to involve C atom binding (of CO2) to one of the Co centers and one of the O atoms binding to the other Co center. Such a configuration with the CO2 molecule in between the 2 metal centers was supported by computational results. Upon setting the potential at the value of the catalytic wave, a signature related to the formation of a formato-complex or free formate was observed, in agreement with the fact that this complex's selectivity towards formate can be obtained with an appropriate reaction medium composition.
In the case of a cobalt aminopyridine catalyst (Co27, Chart 1), a new pathway for the CO2-to-CO conversion was identified, involving the non-catalytic CoII/CoI redox wave.41 Upon electrochemical generation of the CoI species in the presence of CO2, in situ IR-SEC shows a signal characteristic of a CoI–CO adduct. Labelled experiment as well as theoretical modelling confirmed the formation of the CoI–CO species. It was postulated that CO release is a key limiting step which prevents fast recovery of the catalytically relevant CoI species. Overall, this study has shown that CoI is nucleophilic enough to bind CO2 and that C–O cleavage occurs subsequently with no added protons, as represented in Fig. 8. This is not the case with Co porphyrins, for which it has been shown that the CoI complex is unreactive with CO2 and that the active species towards CO2 reduction is instead the formal Co0.42
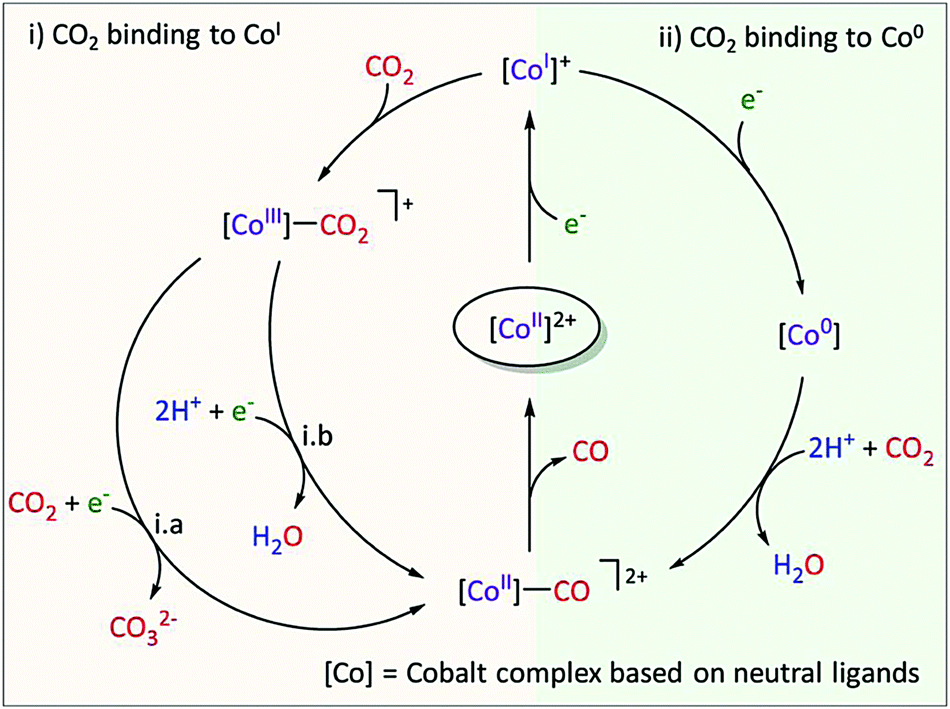 | ||
| Fig. 8 Proposed mechanism of CO2 reduction with Co27. During CPE at the CoII/I wave, trace amounts of CO were released. A formal oxidation state is given for the different Co species. Reprinted with permission from ref. 41. Copyright (2020) American Chemical Society. | ||
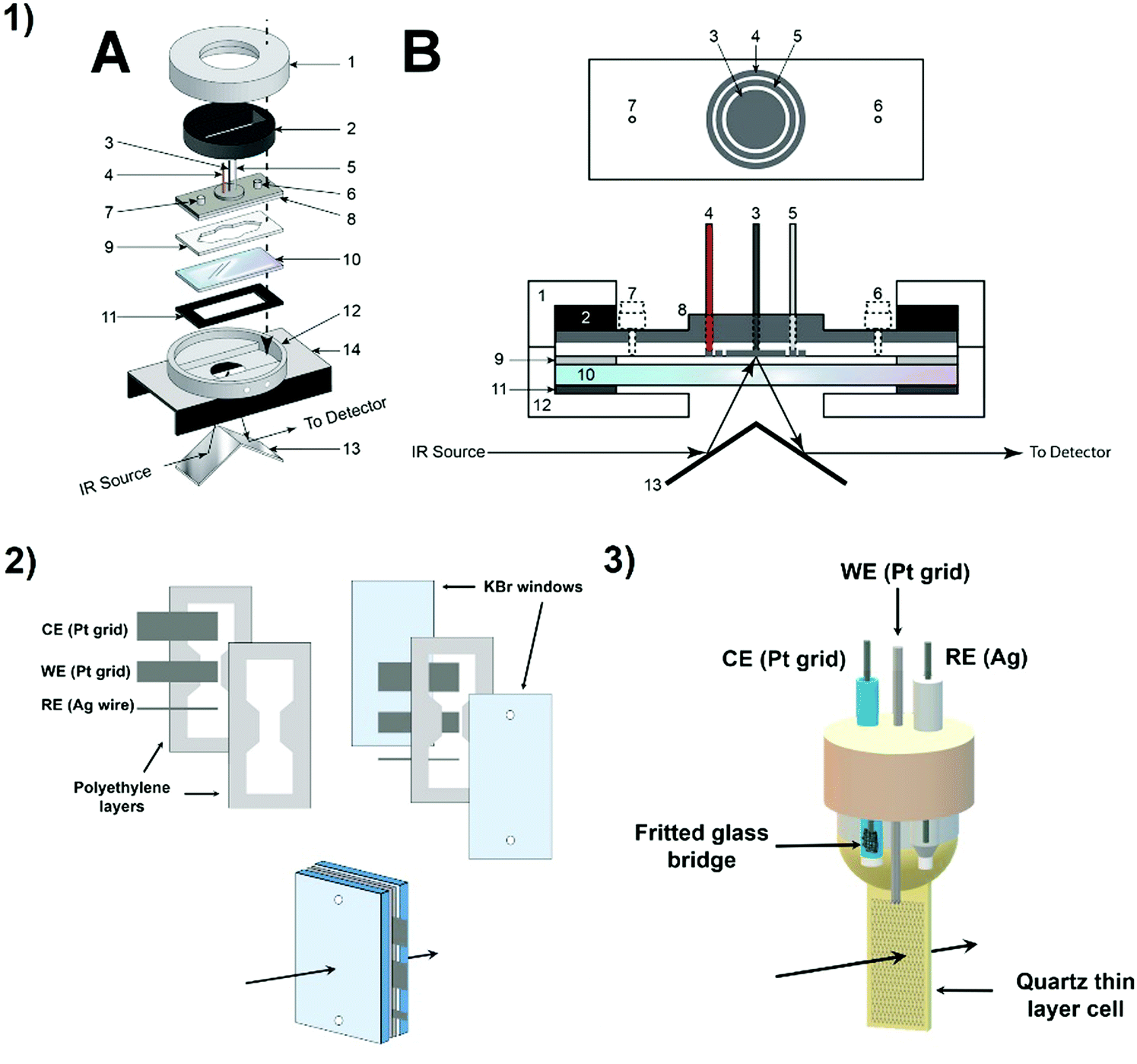 | ||
| Fig. 9 Examples of spectro-electrochemical cells. (1) IR specular reflectance – SEC cell: 1. tightening brass cap (threaded inside); 2. brass ring required to tighten the cell; 3. WE; 4. CE; 5. pseudo-RE; 6 and 7. injection ports; 8. cell body, top part aluminum, lower part Teflon; 9. Teflon spacer; 10. CaF2 window; 11. rubber gasket; 12. hollow brass cell body with threaded inlet and outlet ports (Swagelock) for connecting to the circulating bath; 13. mirrors; 14. two-mirror reflectance accessory; A: disassembled view, B: cross-sectional drawing of the cell;45 reproduced with permission from ref. 49. Copyright (2014) American Chemical Society. (2) IR transmission – SEC cell;46,47 (3) UV-Vis-SEC quartz cell.48 | ||
An in operando fast-scan FT-IR study of a cobalt catalyst reducing CO2 to CO under photochemical conditions (Co10, Chart 1) has shown that the ability of the one-electron reduced CoI complex to bind with CO2 plays a critical role. After injection of a second electron to the adduct, a new intermediate (CoI–CO2−) is formed, as evidenced by the fact that the IR signature of the macrocyclic ligand does not change, meaning that the second electron mainly resides on the bound CO2. The adduct then cleaves (rate-limiting step) to form CO and the CoII catalyst.43 Spectro-electrochemistry has also been used with supported catalysts, e.g. with cobalt porphyrin-based metal–organic frameworks (MOFs).44 In this case, UV-visible spectroscopy was used to probe the Co redox state during catalysis of CO2 reduction. The MOF was directly grown on a transparent electrode and it was shown that the majority of the Co centers were electrically addressed as the CoI active catalytic species was observed.
With Ni complexes (Chart 2), IR-SEC played a key role in the identification of nickel carbonyl species during CO2 reduction mediated by Ni(cyclam) (Ni1) at a glassy carbon electrode.50 The detection of species such as [Ni(cyclam)(CO)]+ and Ni(CO)4 helped confirming that catalyst deactivation is caused by carbon monoxide. This was further confirmed by in situ observation of [Ni(TMC)(CO)]+ (Ni6) (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) when [Ni(TMC)]2+ (Ni6) was used as a CO scavenger, allowing the catalyst to remain active. The same conclusions were driven when a [Ni(TPEN)]2+ (Ni13) was studied.51 Furthermore, the use of optical resonance Raman and electron paramagnetic resonance spectroscopies under catalytic conditions has led to the identification of different pathways for CO2 and proton reduction with Ni1, suggesting that a careful catalyst design can suppress the competing hydrogen evolution reaction and may thus lead to more selective catalysts.52
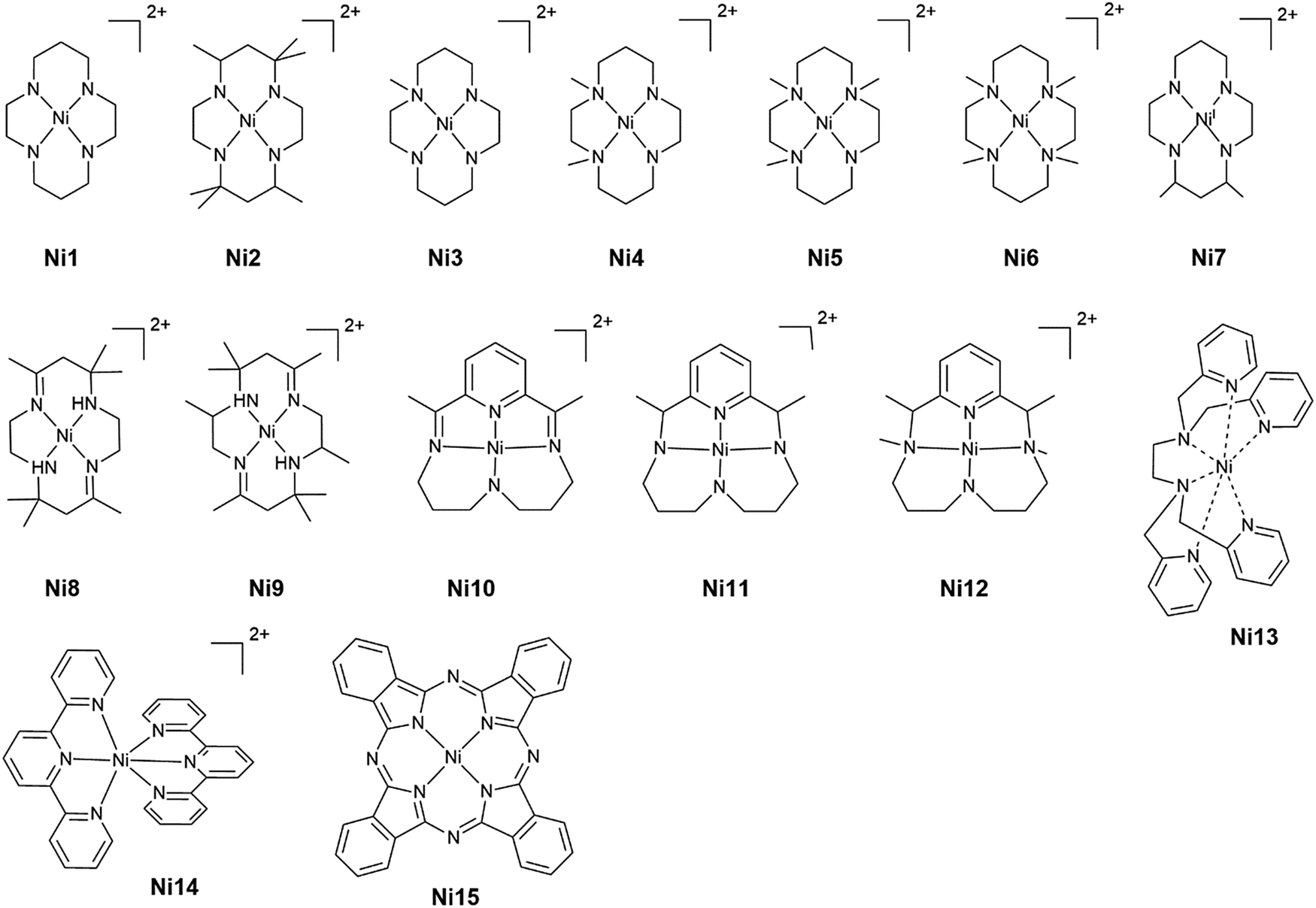 | ||
| Chart 2 Nickel complexes. | ||
1.2.3 X-ray absorption spectroscopy. X-ray absorption spectroscopy methods have recently emerged as a powerful source of information for investigating the redox state of the metal center as well as its coordination environment and geometry.53 For example, X-ray absorption near-edge spectroscopy (XANES) has shown that [CoIHMD]+ and [CoIIHMD]2+ (Co14, Chart 1) have a square planar geometry. It changes to a square pyramidal geometry upon axial coordination of CO2 to [CoIHMD]+. The resulting CoIII–carboxylate species was identified and indicated charge transfer from the Co center to CO2. Binding of a solvent molecule to the metal center was also evidenced.54 Binding of CO to an iron porphyrin (Fe10, Chart 3) has also been studied by XANES. The porphyrin was immobilized on a carbon material and analyzed in situ. Introduction of CO to the electrolyte solution resulted in binding to electrogenerated FeII porphyrin and formation of an L–Fe–CO adduct, L most probably being a water molecule. Charge transfer from iron to CO was further postulated to explain the shift of the Fe K-edge to higher energy values.55
Similar information has been obtained with Ni containing tetraazamacrocycles.56 The one-electron reduction/oxidation of the NiII starting complexes resulted in an energy shift of the absorption edge in XANES spectra. Moreover, the intensity of the pre-edge peak yielded information on the coordination environment. It was found that the starting NiII species is hexa-coordinated to the macrocycle and two solvent molecules that are lost upon reduction to NiI. After binding of CO as an axial ligand to NiI, the absorption edge is shifted to higher energy, showing that some charge transfer occurs from the metal center to the CO ligand.
Regarding catalytic studies, these techniques were used for the study of oxygen reduction catalysts.57–61 CO2 reduction studies with in situ XAS are now developing rapidly.41,62,63Ex situ XAS is a valuable complementary tool for analyzing immobilized molecular catalysts and their stability after performing CO2 reduction.64,65 Recently, catalysis with a cobalt aminopyridine (Co27) was investigated by combination of XAS, IR and UV-vis SEC experiments. Formation of a CoI species (from one-electron reduction of the starting complex) under an inert atmosphere was first evidenced, upon performing an electrolysis at a potential slightly more negative than the CoII/I redox wave in anhydrous deuterated acetonitrile at −40 °C. The frozen sample was then analyzed by XANES and EXAFS. Under a CO2 atmosphere, formation of a CoII–carbonate complex was evidenced by both XANES and EXAFS, upon comparing spectra with those of the chemically produced species upon mixing the starting CoII complex with tetrabutylammonium hydrogenocarbonate. However, the key CoI–CO species was not observed under these conditions. Nevertheless, these experiments helped identifying one of the key bottleneck intermediates for CO2 reduction. It was indeed confirmed by cyclic voltammetry that the CoII–carbonate species disfavors the catalytic reduction of CO2 at the CoII/I wave.41
It should also be emphasized that XAS experiments remain tedious and include selecting suitable synchrotron/beamline for a given experiment.66 As for the more practical side, and similarly to UV-vis and IR SEC, no standard setup exists. Using a thin layer cell comprising windows transparent to the incident light is mandatory, and experiments can be performed either in transmittance or fluorescence mode, although the latter is usually preferred.67Fig. 10 shows examples of typical XAS-SEC cells. Although X-ray absorption spectroscopy is not a routine technique, the field of electrocatalytic transformations using molecular complexes under homogeneous conditions and supported catalysts might strongly benefit from information it can provide. In operando studies with time-resolved techniques would be a valuable asset in the deciphering of reaction mechanisms. It may lead to a better understanding of catalytic systems and to their improvement.68
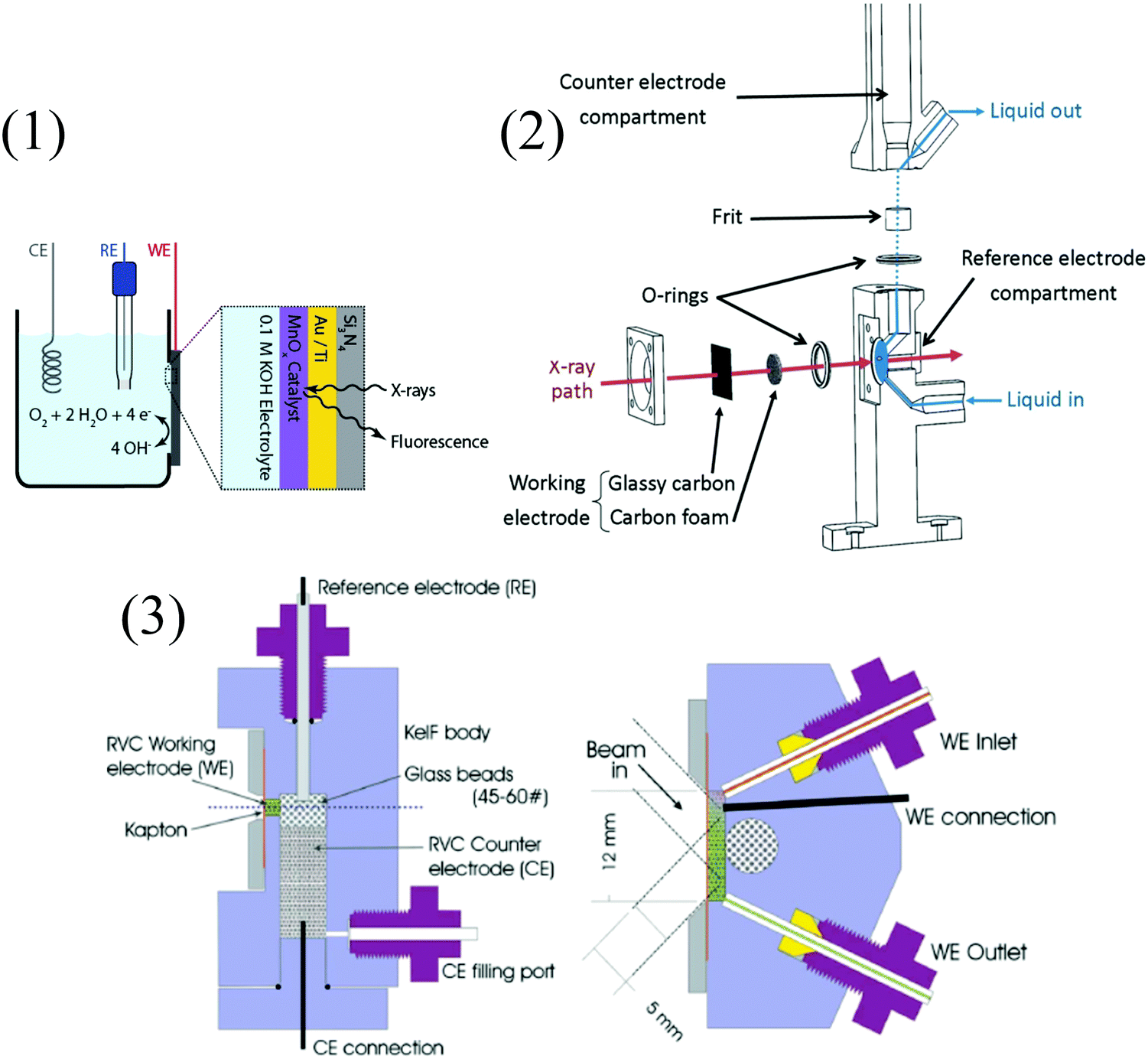 | ||
| Fig. 10 Examples of XAS-SEC cells. (1) In situ XAS setup, with the back side of the Si3N4 window facing the X-rays and the front side of the window covered with electrodeposited MnOx on a layer of Au/Ti facing the electrolyte. CE, RE, and WE stand for counter, reference, and working electrodes, respectively; reprinted with permission from ref. 69, copyright (2013) American Chemical Society. (2) Detailed view of a spectroelectrochemical cell developed for time-resolved data collection in transmission mode;70 and (3) vertical (left) and horizontal (right) sections through the working electrode chamber of a fluorescence X-ray absorption spectroscopy spectroelectrochemical (XAS-SEC) cell (RVC = reticulated vitrous carbon). Solution flow control is achieved using syringe pumps and Teflon tubing. The inlet and outlet tubes of the WE are sealed with the aid of Teflon ferrules; a flanged seal was used for the CE tubing.71 | ||
1.2.4 Differential electrochemical mass spectrometry. Differential electrochemical mass spectroscopy (DEMS) is an analytical technique combining in situ coupling of mass spectrometry to electrode processes. Gaseous and volatile compounds produced at an electrode by application of a controlled potential can be sampled through a microporous (mostly PTFE) membrane to a mass detection chamber upon application of high vacuum (Fig. 11). This technique allows for the fast characterization of reaction products as a function of the applied potential and it may be an interesting alternative to the tedious job of performing electrolysis at various potentials.72 Two cell setups have been mainly used in the context of CO2 electrochemical reduction: thin layer flow cells73–76 and cells using a capillary inlet, usually referred to as on line electrochemical mass spectrometry (OLEMS).77–82 The DEMS cell architecture has evolved over time. The technique was first demonstrated using a design where the electrode material was directly deposited onto a PTFE membrane that was supported on a stainless steel grid connected to vacuum for MS analysis. The rest of the electrochemical cell was rather conventional and was assembled by pressing the body of the cell on the working electrode.72 Although this design shows good collection efficiency and a fast spectrometer response, the choice of electrode materials that can be used is rather limited, and the study of bulk materials such as Cu electrodes is not allowed. Additionally, the electrode surface is positioned very close to the membrane (hence to the vacuum system). As a consequence, the concentration of volatile species at the electrode surface decreases, leading to depletion of CO2.83
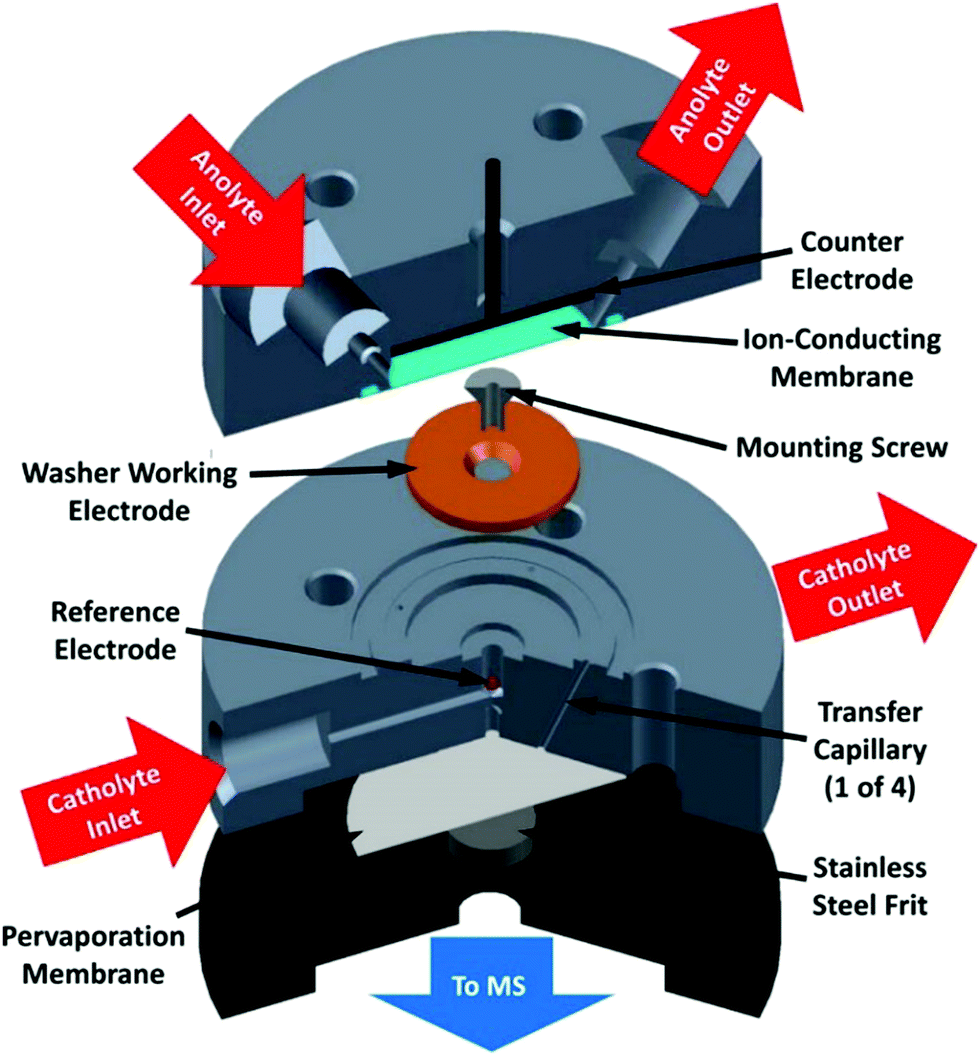 | ||
| Fig. 11 DEMS cell. Reprinted with permission from ref. 74. Copyright (2015) American Chemical Society. | ||
The thin layer design was first introduced in 1990, allowing processes occurring on smooth electrodes to be studied.84 In this design, the working electrode is separated from the PTFE membrane by a thin solvent layer. As a result, species produced at the electrode have to diffuse to the membrane. Consequently, slower spectrometry responses were observed. When the cell is under continuous flow, it results in low collection efficiency as diffusion of analytes to the membrane competes with the solvent flow.
A number of these flaws were addressed more recently with the design of a new DEMS cell.74 This design features parallel working and counter electrodes to ensure uniform potential distribution across the surface of the electrodes and a reference electrode located outside the working electrode chamber to prevent the formation of gaseous product bubbles that may affect potential referencing. Again, the working electrode is separated from the pervaporation membrane to prevent CO2 depletion, but the electrolyte volume between the working electrode and the pervaporation membrane is minimized to allow a short delay time between product generation and detection.78,85
In the OLEMS design, a classical 3 electrode cell configuration is adopted and a PTFE microporous membrane terminated capillary (pinhole inlet) is placed close to the working electrode (10–20 μm) in a hanging meniscus configuration. This particular configuration allows the study of CO2 reduction over single-crystal electrocatalysts. The capillary inlet can be attached to a 3D piezoelectric-driven positioning system (scanning DEMS) allowing the mapping of large electrodes and screening of multiple electrodes within a single experiment.86 However the OLEMS configuration has a number of flaws, notably a low collection efficiency.
Despite the possibilities offered by DEMS not only to study catalysts on a short time scale but also to follow both activity and selectivity in real time, the technique has not yet been widely used with molecular catalysts. Only one study performed on Ni(cyclam) (Ni1) was reported.87 CO (m/z = 28) production in the course of cyclic voltammogram recorded at a slow scan rate was evidenced, along with depletion in the signal corresponding to CO2 (m/z = 44). Hydrogen evolution at potentials more negative than the catalytic wave was also identified. More recently, the OLEMS configuration has been adopted for the study of Co protoporphyrin (Co26, Chart 1) immobilized on pyrolytic graphite (see Section 5).82 Special attention should be paid to the analysis of CO2 reduction products. It is usually admitted that CO is undetectable by DEMS because its ionization produces the same mass fragments as CO2 while this latter can be orders of magnitude more concentrated in the electrolyte. Moreover, under experimental conditions where formic acid is fully dissociated, it cannot pervaporate to the mass spectrometer, thus preventing its detection.74 In other words, DEMS should be used as a complementary experimental method.
| Catalyst | Concentration (mM) | Conditions | Duration (h) | Potential (V) | TOF or TOFmax (overpotential, method) | Product | FE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Triethylamine. | ||||||||
| Fe1 | 1 | DMF, 0.1 M PhOH, Hg pool | 1 | −1.46 vs. NHE | TOFmax = 104.2 s−1 (η = 800 mV; CV) | CO | 100 | 20 |
| Fe1 | 1 | DMF, 40 mM PrOH, 40 mM NEt3a | 4 | −2.4 vs. Fc+/Fc | — | Formate | 72 | 112 |
| Fe2 | 1 | DMF, 2 M PhOH, Hg pool | 4 | −1.16 vs. NHE | TOFmax = 103.8 s−1 (η = 800 mV; CV) | CO | 95 | 22 and 109 |
| Fe3 | 1 | DMF, 3 M PhOH | 3 | −1.28 vs. NHE | TOFmax = 104 s−1 (η = 800 mV; CV) | CO | 95 | 109 and 110 |
| Fe4 | 0.5 | H2O, pH 6.7 | 72 | −0.86 vs. NHE | TOFmax = 104.2 s−1 (η = 500 mV; CV) | CO | 98 | 109 |
| Fe5 | 0.5 | DMF, 0.1 M H2O, 3 M PhOH | 84 | −1.2 vs. SCE | TOFmax = 106 s−1 (η = 220 mV; CV) | CO | 100 | 111 |
| Ni1 | 0.17 | H2O, pH 4.1, Hg pool | 4 | −1.05 vs. NHE | TOF = 32 h−1 (η = 700 mV; CPE) | CO | 96 | 100 |
| Ni1 | 0.4 | DMF, 0.1 M NaClO4, Hg pool | 5 | −1.4 vs. SCE | TOF = 0.64 h−1 (η = 360 mV; CPE) | Formate | 75 | 26 |
| Ni2 | 1.2 | H2O/CH3CN 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v), 0.1 M LiClO4 :1 (v:v), 0.1 M LiClO4 |
1 | −1.6 vs. SCE | TOF = 6 h−1 (η = 640 mV; CPE) | CO | 65.3 | 90 |
| Ni14 | Nr | DMF/H2O 95:5 (v:v) |
3 | −1.71 to −2.14 vs. Fc+/Fc | — | CO | 18 | 22 |
| Co20 | 0.5 | CH3CN, 3 M PhOH, GC | 3 | −1.1 vs. SCE | TOF = 533 s−1 (η = 140 mV; CPE) | CO | 90 | 18 |
| TOFmax = 3.3 × 104 s−1 (η = 300 mV; CV) | ||||||||
| Co13 | 1.2 | H2O/CH3CN 2:1 (v:v), 0.1 M KNO3, Hg pool |
1 | −1.6 vs. SCE | TOF = 7.8 h−1 (η = 640 mV; CPE) | CO | 46.5 | 90 |
| Co18 | 1 | DMF | 1 | −1.5 vs. SCE | TOF = nr (η = 566 mV; CPE) | CO | 82 | 91 |
| Co19 | 1 | CH3CN, pyrolytic graphite | 0.5 | −1.4 vs. SCE | TOF = nr (η = 440 mV; CPE) | CO | 30 | 92 |
| Co5 | 0.5 | DMF,1.2 M TFE, 0.1 M (tBu)4NPF6 | 2 | −2.8 vs. Fc+/Fc | TOF = 170 s−1 (η = 850 mV; CPE) TOFmax = 1.7 × 104 s−1 (η = 510 mV; CV) | CO | 98 | 93 and 95 |
| Co21 | 2 | DMF/H2O 95:5 (v:v) |
3 | −1.93 vs. Fc+/Fc | — | CO | 20 | 97 |
1.3.1 Cobalt complexes. The use of earth abundant metal complexes for the electrochemical reduction of CO2 under homogeneous conditions was reported as early as 1974, when cobalt and nickel phthalocyanine catalytic activity was reported by cyclic voltammetry.1 They were shown to be active for the CO2-to-CO conversion.88 Co porphyrins were identified as effective catalysts in 1979 but no catalytic activity was noticed at that time for Fe and Cu porphyrins.89 In aqueous buffered solutions catalytic reduction of CO2 with Co2 (Chart 1) at a potential E = −1.3 V vs. NHE leads to formic acid. Catalytic activity was also observed with Co3 (Chart 1) but the products were not identified. It was suggested that both metal center and ligand structure played an important role in the catalytic process.
Cyclam Co14 (Chart 1) was also identified very early90 as a good CO2 catalyst with a faradaic efficiency of 46.5% for CO production in a mixture of water and acetonitrile (2:1, v:v) at a potential E = −1.60 V vs. SCE (mercury pool electrode). Related complex Co18 (Chart 1) shows good catalytic activity with 82% FE for CO production at −1.5 V vs. SCE in DMF (Table 1).91Co19 (Chart 1) is less selective with 30% FE for CO at a potential of −1.4 vs. SCE in acetonitrile (pyrolytic graphite electrode, 0.5 h).92 A methylated analog of Co19 was also reported to reduce CO2 to CO (45% FE) in wet CH3CN,93 as well as under photochemical conditions when associated with Ru(bpy)3 as a sensitizer.94
Aminopyridyl macrocycle Co5 (Chart 1) is in contrast highly selective, with 98% of FE for CO production at E = −2.46 V vs. Fc+/Fc and a TOF of 16900 s−1.95,96 The catalyst was suggested to undergo a two-electron metal-based reduction, Co0 being the catalytically active species. The N–H groups on the ring structure of the ligand were shown to provide assistance for creating an efficient H-bond network with the acid co-substrate, further accelerating the rate-determining C–O bond cleavage. Cobalt polypyridines are also highly active catalysts. Quarterpyridine Co20 (Chart 1) proved to be a very active catalyst for CO2 to CO reduction in acetonitrile solution, once doubly reduced at an electrode. In the presence of 3 M phenol as a weak acid co-substrate, high product selectivity (96%) and FE (90%) for CO were obtained at a very low overpotential of 140 mV.18 Moreover, Co20 proved to be among the most active molecular catalysts for CO2 reduction with a maximum TOF of 3.3 × 104 s−1, obtained at only 300 mV overpotential. Co21 (Chart 1) was reported to yield CO with 20% FE (E = −1.93 V vs. Fc+/Fc) in a DMF/H2O mixture (95/5, v:v) at a glassy carbon electrode.97 Catalytic reaction mechanism for Co21 was suggested to proceed through a metal centered reduction followed by a one electron ligand centered reduction, triggering loss of a neutral terpyridine ligand. It opens a coordination site for the low valent CoI species to coordinate CO2 and reduce it into CO in the presence of protons. The ligand centered reduction was in addition proposed to promote side reactions such as carboxylation or hydrogenation, giving a rational to the low FE. These contrasting results obtained with Co20 and Co21 illustrate how a simple change in the coordination environment can drastically affect both efficiency of the catalyst and its stability.
1.3.2 Nickel complexes. Ni cyclams such as Ni1–9 (Chart 2) were demonstrated to be catalytically active for CO2 reduction very early and were extensively studied since then.90,98 Initially, methylated Ni cyclam Ni2 was shown to yield CO and H2 in a 2/1 ratio (with a total FE of 98%) with a TOF of 6 h−1, at a potential of −1.6 V vs. SCE (mercury pool) in a CH3CN/H2O mixture (2
:1). A few years later, the simplest Ni cyclam Ni1 was shown to be a highly efficient catalyst.99–101 The ability of the cyclam ring to stabilize the Ni complexation and the acidic character of the N–H proton of the ligand were recognized as key factors to explain the remarkable reactivity. In acidic water (pH 4–5), high TON (116) and FE were obtained at an Hg electrode, with a TOF of 32 h−1 and a selective conversion of CO2 to CO with only 0.03% of H2 at the relatively negative potential of −1.05 V vs. NHE, corresponding to 700 mV overpotential.
A mechanism was proposed from these studies for the CO2 conversion to CO in water (Scheme 1), which accounts for the adsorption of the catalyst at the electrode surface and for the formation of a NiI–CO adduct. Further reduction of this adduct may lead to Ni0 species and deactivation of the catalyst. Following investigations confirmed that the negatively charged mercury surface can adsorb the NiI complex and may favor a trans-I structure (with N–H groups being all oriented toward the same axial coordination site) which can then effectively bind CO2 and catalyze its reduction at the electrode surface.102–104 Further DFT calculations suggested that the trans-III isomer, which possesses a chair like conformation (it accounts for 85% of Ni1), is in fact more strongly adsorbed at mercury. It leads to a NiI–CO adduct which is higher in energy, accelerating the CO loss and thus the catalysis.105,106 Recent studies at an inert glassy carbon electrode confirmed the formation of the NiI–CO species, as well as the CO loss as the rate limiting step.50,105 Remarkably, Ni6 could be used as a CO scavenger (stronger affinity for CO than Ni1) while having at the same time a lower reactivity towards CO2 reduction. In a CH3CN/water mixture (1/4, v:v), CO was then produced with 90% FE at −1.21 V vs. NHE (470 mV overpotential, 1 h electrolysis).
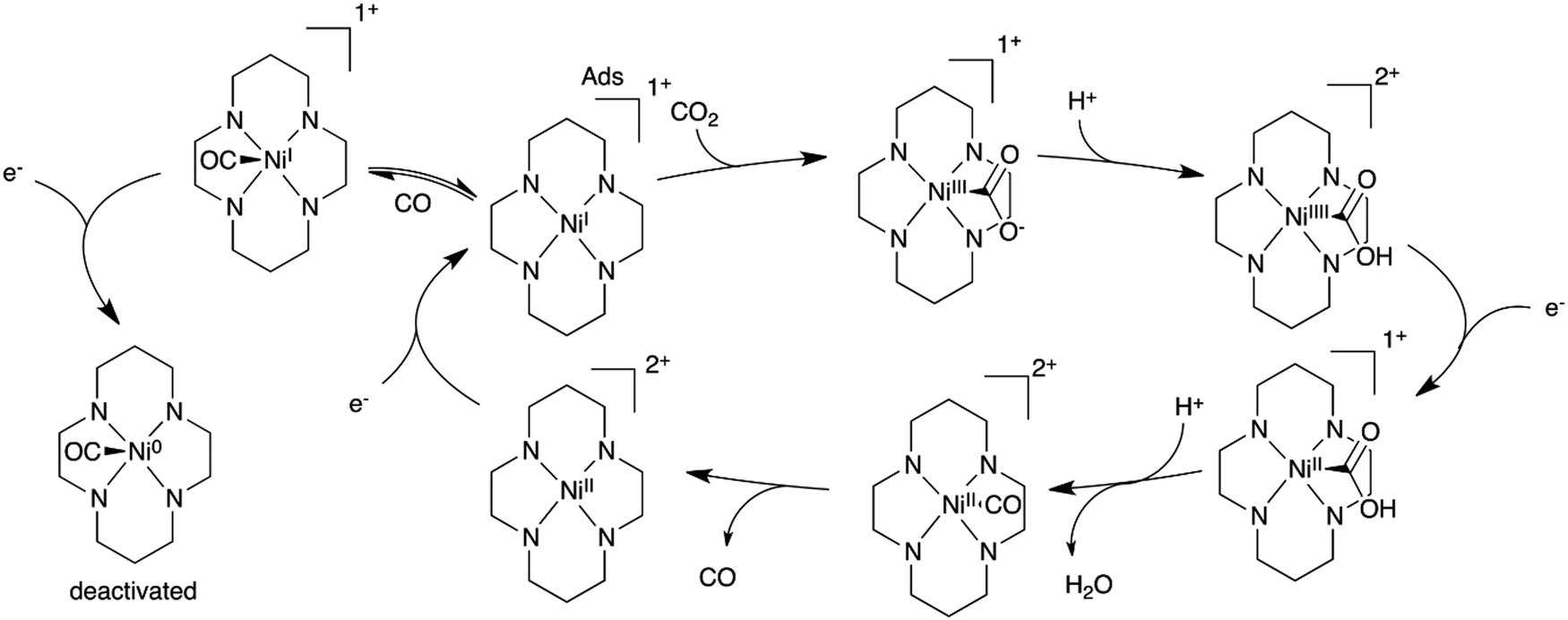 | ||
| Scheme 1 Proposed electrochemical reduction mechanism of CO2 to CO in water with the Ni1 catalyst adsorbed at a Hg electrode. | ||
CO2 reduction with Ni1 in DMF solution leads to the formation of formate with high selectivity. A typical electrolysis experiment showed 75% FE at E = −1.4 V vs. SCE, CO being the by-product.101 This change of product selectivity as compared to aqueous solutions was ascribed to preferred protonation on the carbon atom of CO2 and formation of a nickel-formato intermediate NiII–OCHO, and it was suggested that no hydride was formed. A related uncommon mechanism for formate production has been recently evidenced with Fe porphyrin (see below and Scheme 3). In contrast, nickel tetraaza macrocycles bearing pyridine functional groups (Ni10–12) were also carefully investigated but such systems have weak activity toward CO2 reduction.92
Finally Ni bis-terpyridine Ni14 was also investigated in a range of potentials (from −1.71 to −2.14 V vs. Fc+/Fc) in a DMF/H2O mixture (95/5, v:v) and led to only CO.97 Nevertheless, the FE for CO does not exceed 18% with most of the charge passed during electrolysis not related to CO2 reduction products. As in the case of the related cobalt compound Co21, the doubly reduced catalyst led to the loss of one tpy ligand, promoting side reactions and leading to limited efficiency.
1.3.3 Iron complexes. Variously substituted Fe tetraphenyl porphyrins (Chart 3) have shown to be among the most active molecular catalysts for the CO2-to-CO conversion. Under some experimental conditions, they have also been able to produce formate.2,112 Reactivity is triggered by electrogeneration of the Fe(0) active species, which can bind to CO2 efficiently, leading to an intermediate −FeI–CO2˙− ↔ FeII–CO22− adduct which upon protonation will lead to C–O bond cleavage and formation of an FeII–CO adduct that releases CO upon a one-electron reduction.
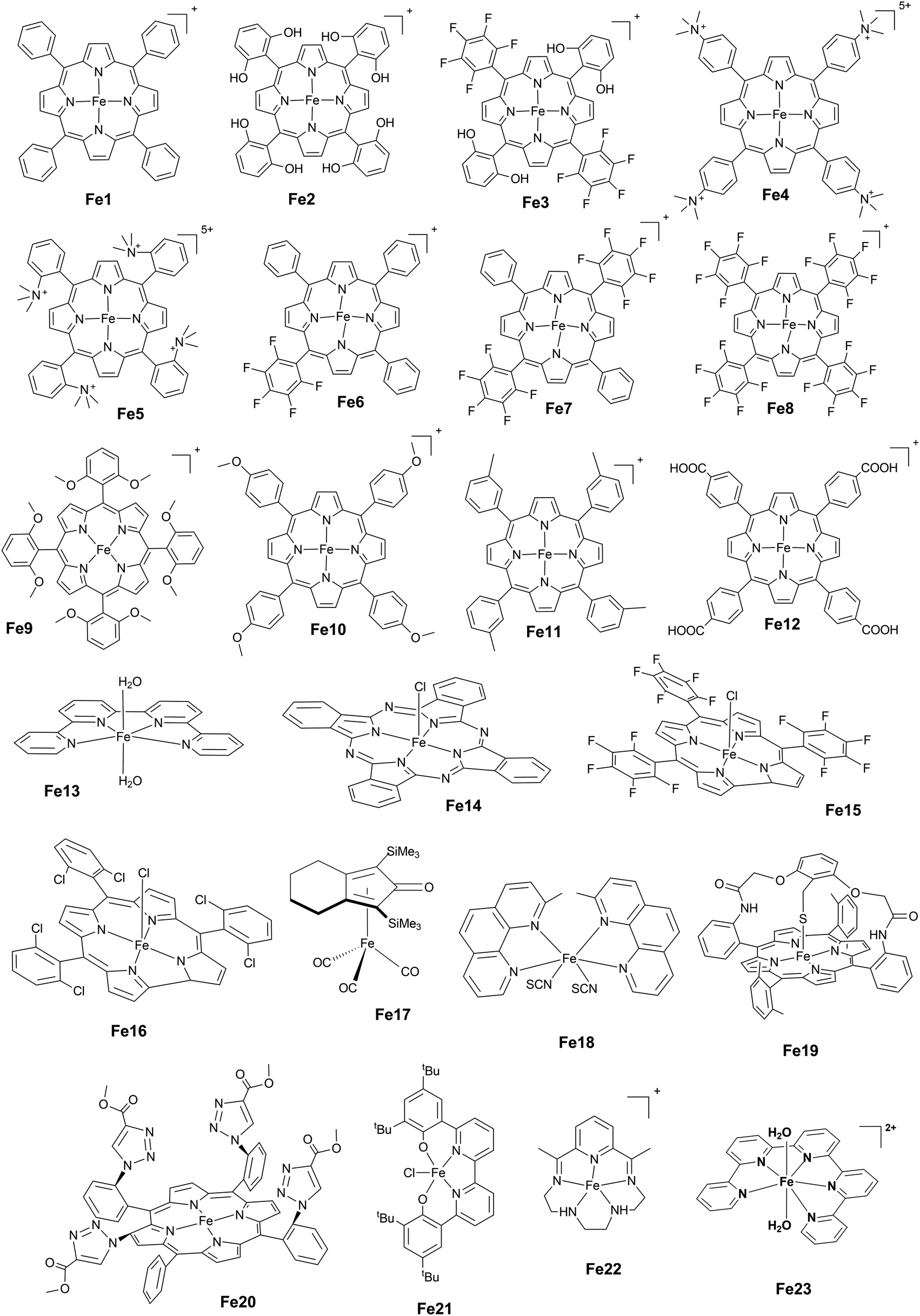 | ||
| Chart 3 Iron complexes. | ||
Weak Brønsted acids accelerate the catalysis107 and it was first observed that addition of CF3CH2OH to a solution of Fe1 led to 94% FE and a TOF of 40 h−1 in electrolysis experiments in DMF at a mercury pool (E = −1.70 V vs. SCE). The role of these weak acid co-substrates is to stabilize the Fe–CO2 adduct and promote bond cleavage. The use of a slightly more acidic acid such as Et3NH+ (pKa = 9.2 in DMF) promotes the formation of H2, demonstrating the importance of pKa control for such assisted catalysis. Lewis acids such as Ca2+ or Mg2+ also accelerate the CO2-to-CO conversion, again upon stabilization of the adduct Fe–CO2. CO cleavage is then fostered by a second CO2 molecule leading to CO32− and CO as reaction products.108 Installing the acid function directly on the phenyl ring of the porphyrin with phenol groups in ortho positions (Fe2) ensured proton availability (both for H-bonding stabilization properties of the Fe–CO adduct and proton relay assistance for protonation steps) and fast kinetics, with a TOF of 103.8 s−1 at an overpotential of 0.56 V in DMF solution as determined by cyclic voltammetry.22
The crucial role of the OH groups was further demonstrated upon comparison with Fe9 (substituted with methoxy groups in all ortho positions), which led to a TOF of only 101.3 s−1 at 1.04 V overpotential. Conversely, introduction of electron withdrawing fluorine moieties on the phenyl rings of the ligand (Fe6, Fe7 and Fe8) allowed decreasing the overpotential, but at the expense of a lower activity. This lower activity is the effect of a smaller electron density on the Fe(0) center produced by the fluorine withdrawing group and hence a less stabilized metal–CO2 adduct. Such an through-structure effect reflects a trade-off between activity and overpotential, and it has been the object of a thorough investigation.39 When the through-structure effect (electronic withdrawing groups on the ligand) was combined with the through-space effect of OH groups in the ortho position of the phenyl (Fe3), the catalytically active Fe0 species was generated at 200 mV less cathodic potential than Fe2, keeping the high activity and high selectivity of the Fe2 catalyst (maximum TOF at large overpotential in the range of 104 s−1, see Table 1, entries 3 and 4).109,110 With both Fe2 and Fe3, a mono-electronic pre-wave was observed at the foot of the catalytic wave and it was ascribed to the signature of an Fe0–CO2 adduct, which after protonation, requires a second electron reduction for the C–O bond cleavage to occur (concerted dissociative electron and proton transfer). It stands in contrast to the mechanism followed by Fe1, for which the second electron transfer occurs after bond breaking, and leads to CO release from the FeII–CO adduct.
Water soluble porphyrins109Fe4 and Fe5 bearing four positive charges (trimethylammonium groups) at the para- and ortho-positions of the phenyl rings respectively show high activity at very low overpotential. Both Fe4 and Fe5 show FE close to 100% for CO during electrolysis performed in DMF (0.1 M NBu4PF6, 0.1 M H2O) under 1 atm CO2 in the presence of 3 M PhOH.111 Mechanistic studies by cyclic voltammetry in DMF have shown a TOFmax = 104.2 s−1 at 500 mV overpotential for Fe4 and TOFmax = 106 s−1 at 220 mV overpotential for Fe5. A comparison with a series of porphyrins variously substituted at the phenyl rings showed positive effects on both thermodynamics (smaller overpotential) and simultaneously on kinetics (large rate constant) of the positive charges borne by Fe4 and Fe5, which was assigned to through-space stabilization of the Fe0–CO2 adduct (Scheme 2).17,111 This effect brought by positive charges allowed overcoming the intrinsic trade-off of the through structure effect. However, a full mechanistic picture is still missing in both aprotic and protic solvents.
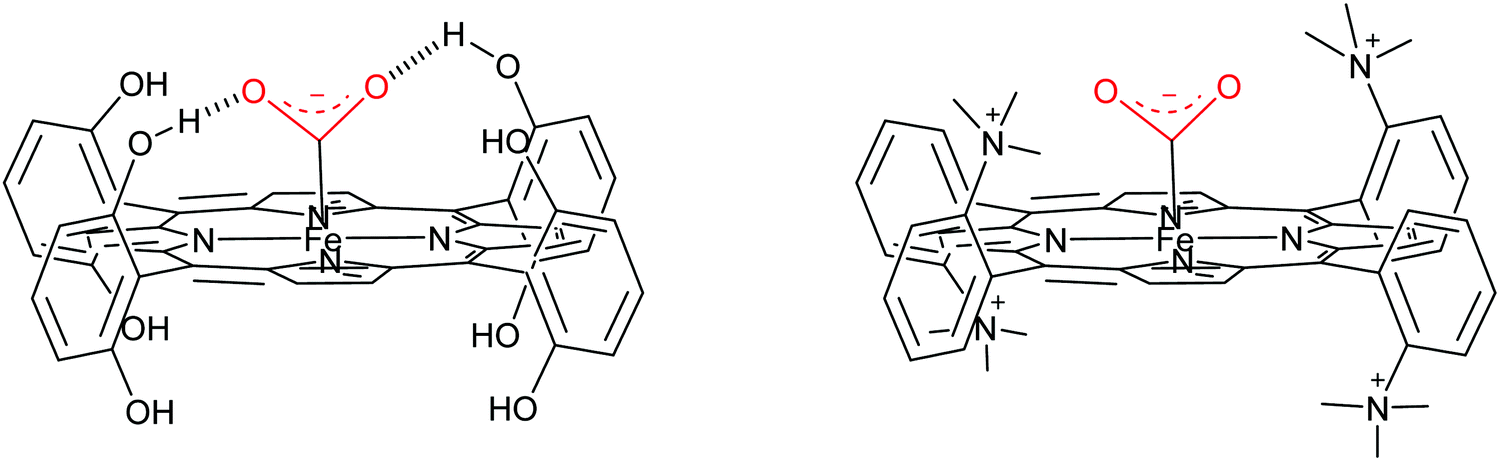 | ||
| Scheme 2 Contrasting H-bond (left, Fe2) and through space electrostatic effect (right, Fe5) stabilization in Fe(0) porphyrin–CO2 adducts. | ||
It was recently discovered that product selectivity for CO2 reduction with Fe1 as a catalyst could be tuned and changed to formate in DMF, upon addition of a tertiary amine such as triethylamine, with up to 72% FE. Upon careful mechanistic investigation by cyclic voltammetry, formation of an iron hydride intermediate could be ruled out and the proposed mechanism instead suggests that the amine acts as an axial trans ligand that enhances the basicity of the carbon atom of the Fe–CO2, directing the reaction toward formate (Scheme 3b).112 This new mechanism shed light on the under-investigated role of trans ligand in CO2 reduction with nitrogen containing macrocyclic complexes and will certainly lead to further developments.
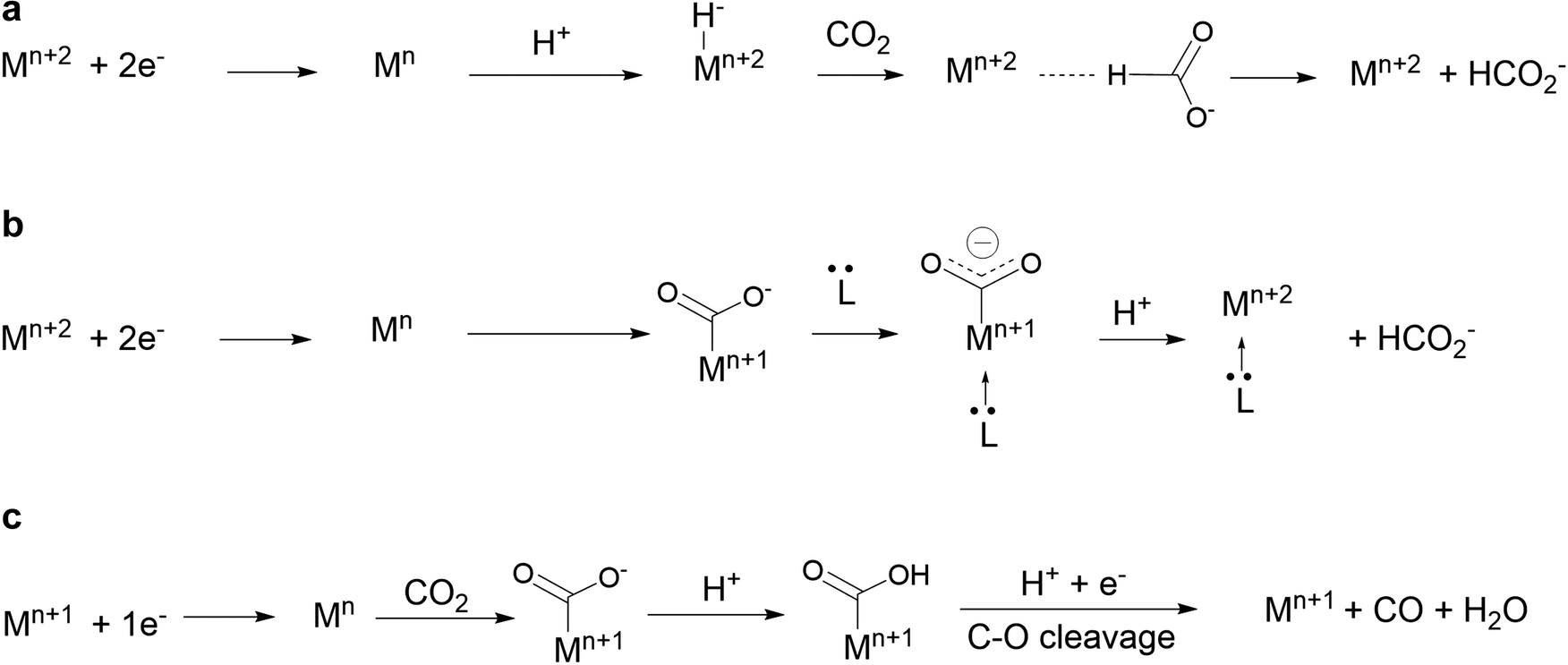 | ||
| Scheme 3 (a and b) Pathways for the two-electron CO2 reduction to formate involving a metal-hydride (a) or a metal carboxylate (b) as an intermediate. Pathway (b) has been shown to be followed with Fe1 in the presence of tertiary amines in DMF solutions.40 (c) Pathway for the two-electron CO2 reduction to CO. Mn is the active form of the catalyst. | ||
Co phthalocyanines have also been the focus of a lot of interest recently under supported conditions.12,64,116 Those systems may reach partial current density for CO up to 18 mA cm−2 in a H-cell, with faradaic efficiency in the range of 90–98% and potentials between −0.61 and −0.68 vs. RHE, which corresponds to overpotentials of 475 and 540 mV respectively (Table 2).116 Quaterpyridine Co20 was immobilized at the surface of multi-walled carbon nanotubes and proved to be an excellent CO2-to-CO catalyst in water at pH 7.3, with 100% FE and a partial current density of 19.9 mA cm−2 at an overpotential of 440 mV (E = −0.58 V vs. RHE).24 The performances of some supported catalysts are summarized in Table 2, although comparisons remain difficult due to the variety of set-ups and differences in catalytic film manufacture. Thorough mechanistic studies are necessary for these supported molecular catalysts as well as rigorous benchmark studies. As briefly alluded to in this section, these highly active Co and Fe catalysts for CO production in water have now been included into flow cells and assembled at gas diffusion electrodes (Table 2, entries 7–9). Full details and description of these systems are provided in the next section.
| Catalyst | Surface concentration (nmol cm−2) | E (V vs. RHE) | Electrolyte | j CO (mA cm−2) | TOF (s−1) | Selectivity (%) | Cell type | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Polymerized at carbon nanotubes. | ||||||||
| Co9 | 14.4 | −0.676 | NaHCO3 0.5 M | 18.1 | 6.8 | 93 | H cell | 115 |
| Co6 | 23.3 | −0.676 | NaHCO3 0.5 M | 13.1 | 4.1 | 92 | H cell | 115 |
| Co7 | 590 | −0.63 | KHCO3 0.1 M | 14.7 | 4.1 | 98 | H cell | 116 |
| Co6 | 440 | −0.61 | NaHCO3 0.5 M | 18 | 1.4 | 90 | H cell | 64 |
| Co20 | 8.5 | −0.58 | NaHCO3 0.5 M | 19.9 | 12 | 99 | H cell | 24 |
| Co26 | — | −0.60 | NaHCO3 0.5 M | 24.7 | 1.9 | 98 | H cell | 121 |
| Co9 | 1.5 | −0.92 | KOH 1 M | 165 | 1.67 | 94 | Flow cell | 115 |
| Co6 | 7 | 2.52 V (cell voltage) | KOH 1 M | 176 | 0.24 | 88 | Gas diffusion cell | 118 |
| Fe4 | 1.3 | −0.6 | KOH 1 M | 152 | 0.57 | 98.1 | Flow cell | 119 |
2. Molecular catalysts in electrochemical reactors
Electrochemical reactors employing gas diffusing electrodes have been developed to improve CO2 electrocatalytic reduction with the goal of reaching high efficiency and high durability at current densities in the range of several hundreds of mA cm−2 (geometric surface electrode area) which is not possible in typical H-cells due to mass transport limitations.122–126 Until recently, such performances were deemed to be only possible with solid state materials, precious metals such as Au and Ag being the two most investigated metals.125,127–129 For example, silver sputtered at PTFE or an anion exchange membrane onto a GDE can provide CO with about 90% FE and at a current density of about 200 mA cm−2. Using non-precious Ni single atoms at porous carbon membranes recently led to an impressive partial current density of 308 mA cm−2 for CO production.130 The progress achieved for the electrochemical catalytic reduction of CO2 with molecular catalysts have stimulated the insertion of these catalysts into lab-scale cell devices, in particular flow cells. Some examples for CO2-to-CO conversion have shown promising results, especially in terms of current densities and selectivity, opening new perspectives toward up-scaling and development of industrial devices. It is to be noted that pioneering studies have been done in the late 1980s and 1990s, in particular with the use of phthalocyanines and porphyrins (M = Co, Fe, Ni, Mn, Cu) deposited on a GDE.113,131,132 It led to partial current densities for CO production in water solutions in the range of a few tens of mA cm−2, with values up to 55 mA cm−2 for Fe tetraphenyl porphyrin in 0.5 M KHCO3 solution132 and 53 mA cm−2 with Co phthalocyanine.131 Another interesting facet of using a GDE is the possibility of performing mechanistic studies with molecular catalysts at surfaces, without being limited by mass transport of the reactant. Such studies, which are extremely tedious as with solid state catalysts, are still in their infancy but they are a key to improve performances.Recently, a micro-flow electrochemical cell including a GDE at which a modified phthalocyanine (Co7) mixed with multi-walled carbon nanotubes was deposited led to a maximum CO current density of 82 mA cm−2 (with ca. 60% FE for CO, full cell voltage 2.3 V) in 1 M KOH flowing electrolyte.133 Long term electrolysis (10 h) at 40 mA cm−2 could be sustained at about 90% CO selectivity. CO2 reduction selectivity and activity for CO production can be further improved even with commercially available compounds such as cobalt phthalocyanine Co6.
This catalyst, when mixed with porous carbon black and deposited at an electrode surface can mediate CO2-to-CO conversion in a zero-gap membrane flow reactor with selectivities >95% at 150 mA cm−2.118 As shown in Fig. 12, comparison with other molecular catalyst based systems (in H-cell or flow cell) illustrates the high performances of Co6 in terms of FECO and current density. It closely matches performances obtained with the most active noble metal-based, such as Au129 and Ag127 nanocatalysts both for cell voltage, energy efficiency and current density.
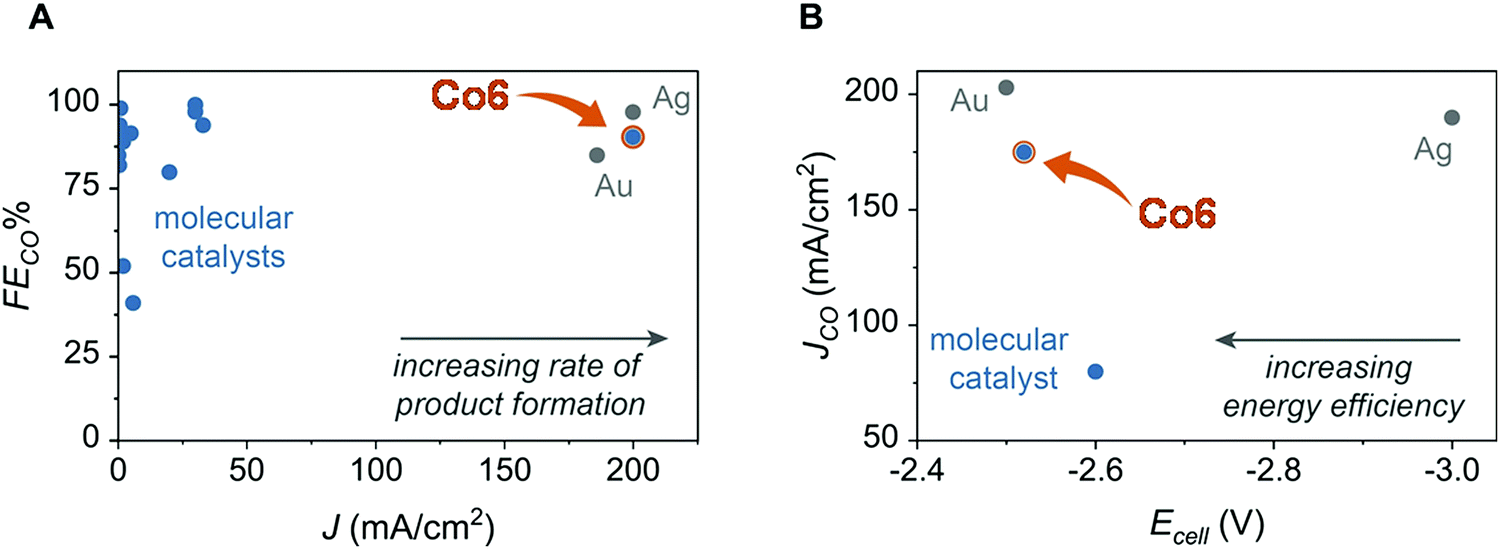 | ||
| Fig. 12 Comparison of selectivity (A) and activity (B, partial current density) for CO production with Co6 inserted in a zero-gap membrane flow reactor and previous molecular and heterogeneous CO2 reduction electrocatalysts (adapted from ref. 118 with permission from AAAS, copyright 2019). | ||
Tuning the ligand structure is a powerful approach to improve the activity of molecular catalysts, through electronic, steric and second sphere substituent effects, as already described in this review. Following this strategy, introducing a trimethylammonium group and three tert-butyl groups onto cobalt phthalocyanine (Co9) led to high activity toward CO2-to-CO conversion in water over a broad pH range (from 4 to 14).115 In that case, a flow cell electrolyzer operating under basic conditions was developed. Typical CO production with ca. 95% selectivity and good stability was obtained, with a maximum partial current density of 165 mA cm−2 (at an overpotential of 810 mV). Following a similar strategy for catalyst optimization, modified tetraphenyl iron porphyrin Fe4 was shown to achieve fast and selective electrocatalytic CO2 conversion to CO in a flow cell.119 The catalyst was mixed with carbon black and deposited at a carbon paper electrode (GDE). A current density for CO production as high as 152 mA cm−2 (>98% selectivity) was obtained at 470 mV overpotential in a basic solution (pH 14). Under the same conditions, jCO = 27 mA cm−2 was maintained for 24 h at only 50 mV overpotential and with close to perfect 99.7% selectivity. A maximum current density of 83.7 mA cm−2 was obtained at pH 7.3, with selectivity close to 98%. Very recently, a nickel catalyst (Ni1) was inserted in a continuous non-aqueous flow cell, giving CO with a selectivity close to 80% and a maximum current density of 50 mA cm−2 in CH3CN with NH4+ as a co-substrate.134 Examples described above illustrate the rich potentiality of using highly active catalysts into an electrolyzer comprising a GDE. As already mentioned, various set-ups may be used. In flow cells, a liquid electrolyte circulates on both sides of the membrane separator, which is usually a polymer electrolyte membrane type such as a cation-exchange membrane (CEM), anion-exchange membrane (AEM) and bipolar membrane (BPM). The designed cell architecture used for catalysts Co9 and Fe4 consists of a sandwich of flow frames, electrodes, gaskets and an AEM membrane as illustrated in Fig. 13a and b. The anode and cathode are assembled on both sides of the membrane. The CO2 gas flow is delivered from the back side of the cathodic compartment and flows through the GDE, while the catholyte solution is circulated in between the GDE and the anion exchange membrane (AEM). On the other side of the AEM, an anolyte electrolyte circulates between the AEM and the anode. Fig. 13c schematically illustrates the zero-gap membrane reactor (no electrolyte is flowing in the cathodic compartment) where the catalyst is directly sandwiched between the GDE and the membrane (MEA assembly type).
 | ||
| Fig. 13 (a) Scheme of the CO2 flow cell electrolyzer: cross-sectional view of the cell elements (adapted from ref. 115), (b) scheme of the experimental setup operating under alkaline conditions with catalysts Co8 and Fe4 (adapted from ref. 119 with permission from Wiley, copyright 2020). (c) Scheme of the zero-gap membrane reactor used for CO2 electroreduction of Co6 (left; the MEA is made up of the cathode and anode GDEs on either side of the AEM); image of the cell (right). Adapted from ref. 118, with permission from AAAS, Copyright 2019. | ||
There is still a lot of work to be conducted before molecular catalysts may be used in flow cell CO2 electrolyzers at an industrial scale. Long term stability remains an issue (as it is currently the case with solid catalysts too), which calls for vigorous efforts to decipher the catalytic mechanisms involving molecular catalysts inserted into thin films deposited at electrode surfaces, and to better identify possible degradation pathways for the catalysts. Theoretical tools have been recently devised and should be used.26,28 The understanding of local environment effects is also of prime importance. Such effects may manifest for example by high local pH due to fast proton consumption and may result in crystal growth at the GDE118 and/or membrane cross-over of ionic species during long term operation. Beneficial effects such as temperature and electrolyte cation optimization have also been identified. For example, it was observed for iron catalyst Fe4 that reaction kinetics could be boosted upon changing the temperature.119 Raising the temperature from 24 °C to 40 °C led to a 300 mV overpotential decrease at 50 mA cm−2 current density (pH ∼ 7, 0.5 M NaHCO3). Remarkably, it was also shown with the same iron molecular catalyst, as previously observed with solid electrocatalysts,135 that larger size cations could lead to better performance. At a fixed current density of 50 mA cm−2, a significant lowering of the overpotential (390 mV) was observed upon increasing cation size going from Na+ to K+ and finally Cs+. Such an effect may be ascribed to higher surface charge density and consequently increased affinity between CO2 and the cathode surface.135 Finally, optimization of the cell architecture (optimization of the membrane, of the catalytic film formulation, decrease of the cell resistance, etc.) is an additional challenge toward real devices. Further studies will certainly soon burgeon using related catalysts and approaches.
3. Light-driven and assisted approaches
Photocatalytic molecular systems for CO2 reduction comprise three components, i.e. a catalyst (CAT), a redox photosensitizer (PS) and a sacrificial electron donor (SD). The metal complex serves as a catalyst capable of accumulating multiple electrons needed to reduce CO2. Light-driven electron transfers can be classified as oxidative (Scheme 4a) or reductive quenching (Scheme 4b) according to the quenching reaction of the photosensitizer. In the first case, after excitation (Scheme 4a, step (1)), the lowest excited state of the PS (PS*) is a strong reductant and gives an electron to the electron acceptor (EA, being the CAT or an electron mediator). Emission of PS* (Scheme 4a, step (2)) is oxidatively quenched to give rise to the corresponding one electron-oxidized form PS+ (Scheme 4a, step (3)). When the standard redox potential of SD˙+/SD is more negative than that of PS+/PS, one electron can be donated to PS+ (Scheme 4a, step (4)) to recover PS. Alternative possibility involves the reductive quenching of PS* (Scheme 4b). In this case, PS* accepts an electron directly from SD (Scheme 4b, step (3)). The one-electron-reduced form PS− is thus produced. PS is regenerated through electron transfer from PS− to EA (Scheme 4b, step (4)). Favorable conditions for oxidative quenching include long lifetime and high reducing power of PS* as well as high concentration of EA because electron transfer between PS* (PS−) and EA is a bimolecular reaction.
 | ||
| Scheme 4 Photosensitizer (PS) emission quenching pathways. EA: electron acceptor (catalyst or electron mediator), SD: sacrificial electron donor. | ||
Back electron transfer (BET) is a drawback that may lower, sometimes dramatically, the reaction quantum yield. For example, in the case of oxidative quenching of PS* (Scheme 4a), the reduced acceptor EA˙− may quickly transfer back an electron to the oxidized sensitizer (PS+) since the reaction is endowed with a large driving force. Large concentration of donor (SD) may help in fighting BET.
Besides back electron transfer, many factors can affect the product yield, including light intensity, irradiation volume, concentration of CO2, photocatalysts and SD, as well as standard redox potential values of the photosensitizer and the sacrificial donor. Quantum yield (QY) and turnover number (TON) are typically employed to evaluate CO2 reduction efficiency. Quantum yield is a crucial parameter for assessing the performance of a photocatalyst or photocatalytic system. The internal quantum yield (IQY) and apparent quantum yield (AQY) are defined by eqn (11) and (12), respectively. IQY is calculated as the ratio of the mole amount of product multiplied by the number of electrons necessary for the product formation over the mole amount of absorbed photons, as shown in eqn (11), while AQY is calculated relatively to the number of incident protons (eqn 12).136 TON is defined, as under electrochemical conditions, by the ratio between the mole amount of product and the mole amount of catalyst once the reaction has stopped. Care should be taken when comparing data between studies since catalyst concentrations may vary in between a few tens of nM and a few hundreds of μM. In some cases, very high turnover numbers have been obtained but at very low concentration of catalyst (nM range), leading to small absolute quantities of CO2 reduction products and overstatement regarding the catalytic performances.
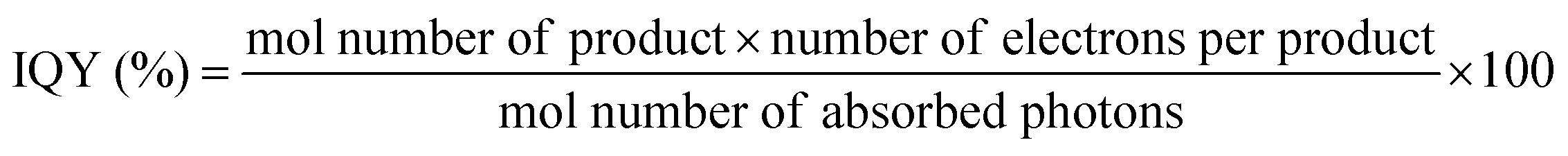 | (11) |
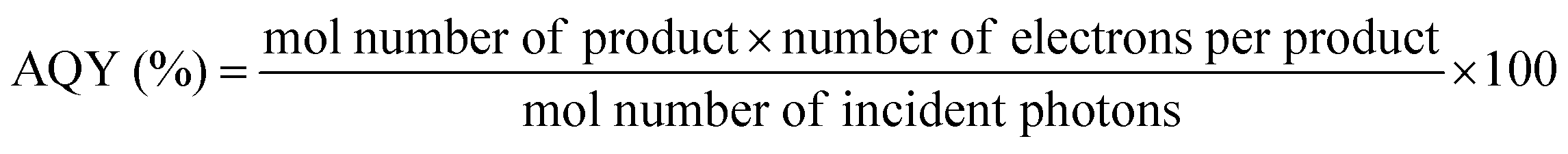 | (12) |
When evaluating the catalytic efficiency, the number of active catalytic sites is thus a crucial parameter but is difficult to evaluate in the case of supported systems. It indeed depends on the catalyst loading but also on active site accessibility. A determination of the catalyst content in supported systems can be made by calculating the amount of catalyst used in the synthesis process. Control of the remaining catalyst left in solution after preparation (through UV-Vis spectroscopy for example) can help in refining the previous estimation. More precise quantification can be obtained by using inductively coupled plasma atomic emission spectroscopy (ICP-AES, after treatment of the material with a strong acid solution) and X-ray photoelectron spectroscopy (XPS). Possibly, if the material is conductive and can be deposited at an electrode, electrochemical methods can also be employed to quantify the number of electrochemically active sites.
Molecular catalysts may be implemented at photoelectrodes following three strategies as depicted in Scheme 5. Each one can be deployed in various configurations (with a grafted or freely diffusing catalyst, irradiation from the front or from the back of the electrode, addition of protective layers on the semi-conductive material, etc.) and combination of several strategies in series can also be envisioned. For example, the p–n junction pictured in configuration b is usually protected from electrolyte corrosion by several layers with various doping and thickness that would in principle allow for optimizing the stability and the efficiency (e.g. charge transfer to the catalytic sites) of the device.137 This buried junction could eventually be doubled or tripled.138 In the following, we will not detail photoelectrode underlayer preparation.
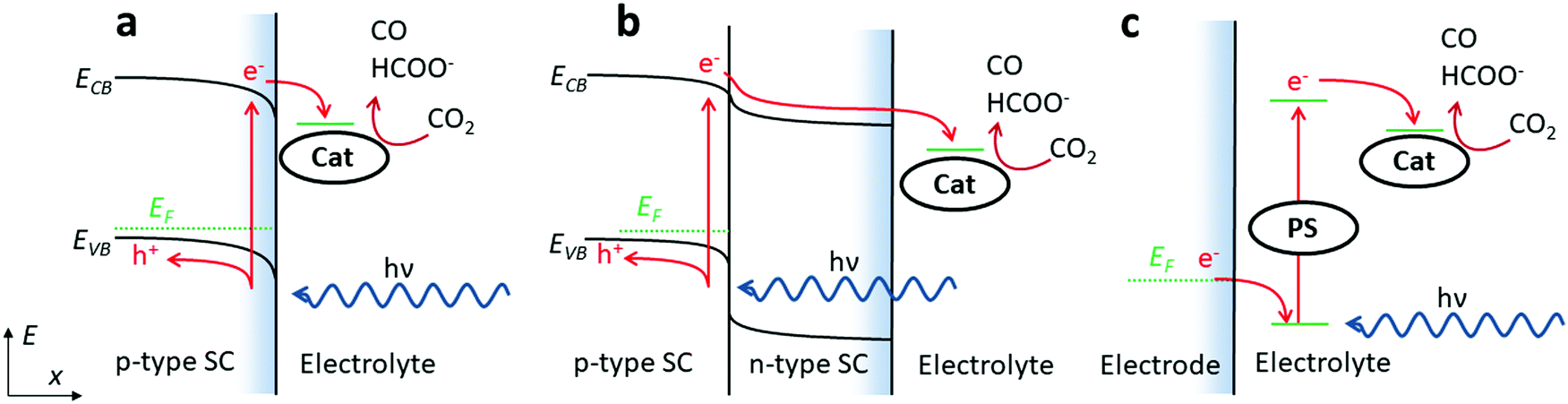 | ||
| Scheme 5 Possible photoelectrode configurations including molecular catalysts. (a) Catalyst at a p-type photocathode, (b) catalyst at a p–n photocathode, and (c) association between a catalyst and a sensitizer (PS) at a dark electrode surface. | ||
Regarding CO2 photoelectrochemical reduction with molecular catalysts, the three strategies a–c (Scheme 5) have been reported. Examples employing b–c type strategies are restricted to the last 10 years and early examples exclusively involved strategy a. The latter a was first reported in 1983, with the use of Ni and Co tetra-azamacrocyclic complexes (Co23, Ni2, Ni8 and Ni9) in a CH3CN/water (1:1, v:v) electrolyte at a p-Si electrode.139 Upon photo-electrolysis at −1.00 V vs. SCE, which corresponds to a negative overpotential of 50 mV estimated from 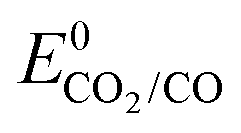 in acetonitrile/water (1:1, v:v) solvent,140 catalyst Ni2 showed a 64% FE for CO with H2 as the only by-product for more than 24 h. However, the absence of reported current density does not allow assessing the rate and efficiency of this process. The other three catalysts (Co23, Ni8 and Ni9) were mentioned to be less selective and required more negative potentials.
in acetonitrile/water (1:1, v:v) solvent,140 catalyst Ni2 showed a 64% FE for CO with H2 as the only by-product for more than 24 h. However, the absence of reported current density does not allow assessing the rate and efficiency of this process. The other three catalysts (Co23, Ni8 and Ni9) were mentioned to be less selective and required more negative potentials.
Several reports followed in the subsequent years where p-Si but also p-GaP, p-GaAs,141–143 p-CdTe144 p-WSe2145 were used as semi-conductive electrodes associated with nickel cyclam (Ni1)141–143 or phthalocyanines (MPc, with M being Co, Ni and Fe but also Zn, Mn, Cu and V)144 as molecular catalysts. Interestingly, upon associating Ni1 with p-GaAs, photo-electrolysis was performed in aqueous media (pH 4.5, 0.1 M KClO4) for more than 15 hours with FE for CO slightly below 50% at a quite negative potential E = −0.95 vs. NHE and gave 4 TON for CO.141 This low selectivity was attributed to catalysis of water reduction by p-GaAs itself. Replacement of this semi-conductor by p-GaP enabled enhancement of CO faradaic efficiency up to 85% in a 0.1 M NaClO4 aqueous solution and at a potential between −0.75 V and −0.2 V vs. NHE, although no turnover number was given and the photo-electrolysis duration time was also not specified.143,146 Interestingly nickel cyclam Ni1 could be employed without filtering the UV-light and catalytic activity was maintained for several hours. In terms of selectivity and activity, p-CdTe based electrodes coated with MPc were able to reach current density as high as 10 mA cm−2 in DMF (5% H2O) although such a current density does not match with the reported transient data.144 Selectivity for CO was high, close to 100% for CoPc, 92% for FePc and 77% for NiPc. Formate was systematically obtained with FE between 2 to 6%. At that time, only one other publication was using a precious complex, with Re(CO)3(bpy)Cl at p-Si and pWSe2 electrodes, also showing good selectivity.145
The field remained almost unattended for about 20 years until Kubiak et al. published a Re(CO)3(bpy)Cl catalyst at a p-Si electrode,147 attaining high selectivity for CO (up to 97%) at −1.35 V vs. Ag/AgCl (which corresponds to 500 mV overpotential relatively to 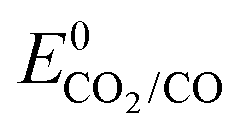 in CH3CN).22 Large current density, up to 31 mA cm−2, was reported under transient conditions (at a scan rate of 100 mV s−1), but the steady-state current density over photo-electrolysis was not specified. Estimation of the latter from the mole amount of electrons (7 × 10−5) passed for 3 hours at a 0.27 cm2 electrode yields a current density close to 2.3 mA cm−2. A few weeks later, Arai, Sato et al. published a study in which a ruthenium complex was used as a CO2 reduction catalyst at a InP photoelectrode.148 Contributions from various groups were then focused on precious metal complexes149–152 or other transition metals such as manganese.153 Concerning Fe and Co aza macrocycles, Fe porphyrins (Fe1 and Fe19) have been associated with p-Si in acetonitrile, with 650 mV photovoltage that allowed catalytic activity at only −1.1 V vs. SCE bias (210 mV overpotential).154 Under these conditions, 90% FE for CO was obtained, with a current density of 3 mA cm−2. Cobalt tris[((2-pyridylmethyl)amine)Cl]Cl (Co24) was also used at a p-Si nanowire electrode in CH3CN solution, but with a lower current density of 1 mA cm−2 and 69% FE for CO at −1.57 V vs. Fc+/Fc, corresponding to a large 520 mV overpotential.155 Recently, strategy a has been much less investigated, likely because of the instability of narrow bandgap semiconductors under irradiation and the fast conversion of the p-Si interface with electrolyte into SiO2 with only traces of oxygen.147,154,156,157 Such instability has also been noticed as a limiting factor for p-GaAs materials.158 These drawbacks lead to a tedious experimental methodology for electrode preparation that is not compatible with technological upscale.141,147
in CH3CN).22 Large current density, up to 31 mA cm−2, was reported under transient conditions (at a scan rate of 100 mV s−1), but the steady-state current density over photo-electrolysis was not specified. Estimation of the latter from the mole amount of electrons (7 × 10−5) passed for 3 hours at a 0.27 cm2 electrode yields a current density close to 2.3 mA cm−2. A few weeks later, Arai, Sato et al. published a study in which a ruthenium complex was used as a CO2 reduction catalyst at a InP photoelectrode.148 Contributions from various groups were then focused on precious metal complexes149–152 or other transition metals such as manganese.153 Concerning Fe and Co aza macrocycles, Fe porphyrins (Fe1 and Fe19) have been associated with p-Si in acetonitrile, with 650 mV photovoltage that allowed catalytic activity at only −1.1 V vs. SCE bias (210 mV overpotential).154 Under these conditions, 90% FE for CO was obtained, with a current density of 3 mA cm−2. Cobalt tris[((2-pyridylmethyl)amine)Cl]Cl (Co24) was also used at a p-Si nanowire electrode in CH3CN solution, but with a lower current density of 1 mA cm−2 and 69% FE for CO at −1.57 V vs. Fc+/Fc, corresponding to a large 520 mV overpotential.155 Recently, strategy a has been much less investigated, likely because of the instability of narrow bandgap semiconductors under irradiation and the fast conversion of the p-Si interface with electrolyte into SiO2 with only traces of oxygen.147,154,156,157 Such instability has also been noticed as a limiting factor for p-GaAs materials.158 These drawbacks lead to a tedious experimental methodology for electrode preparation that is not compatible with technological upscale.141,147
Strategy b offers an answer to the instability of narrow bandgap semiconductors, by adding protective layers on top of the semi-conductive surface and forming at the same time a complete p–n junction. The first report for such an approach for CO2 reduction with molecular catalyst is recent137 and involved the use of copper oxide Cu2O, known for its instability under irradiation,156 as a light harvesting material with protective layers made of AZO (aluminum doped zinc oxide) and TiO2. In this, the classical Re(CO)3(bpy)Cl catalyst was used, first in solution and then attached through phosphonate groups to the top TiO2 oxide layer.159 A current density as high as 2.5 mA cm−2 was obtained in chopped light experiments (light being turned on and off at short regular intervals in order to distinguish photocurrent from background dark current). Photoelectrolysis was performed for 1.5 h with FE for CO decreasing from 95% to 80%, at a very large overpotential (1000 mV, E = −2.05 V vs. Fc+/Fc). This publication was followed by reports using precious metal complexes such as Ru complex grafted at a Fe2O3 cathode.160 The first studies using Co, Ni and Fe aza macrocycles only appeared in 2019, with a phosphonated Co bis(terpyridine) catalyst (Co25) grafted onto a TiO2 layer covering a p-Si cathode. At a bias potential of −1.0 V vs. Fc+/Fc (negative overpotential of −200 mV),140 CO2 was catalytically reduced into CO in CH3CN:water mixtures, with a low current density of ca. 100 μA cm−2 and an optimal FE for CO of 48%.161 The photocathode was also operated in water (pH 6.7, 0.1 M KHCO3), producing a small amount of CO (9.5% FE, partial current density of ca. 29 μA cm−2) but also HCOO− (13% FE, partial current density of ca. 40 μA cm−2) at E = 0 V vs. RHE (−115 and +110 mV overpotential for CO and HCOO−, respectively).
Strategy c was first reported in 2013 with a molecular Re catalyst162 and was followed by several studies involving noble metal based catalysts and sensitizers.163–167 This strategy has also been associated in series with strategy b. For example, a Ru–Re molecular dyad anchored at a CuGaO2 photocathode through a phosphonate linkage allowed reaching 72 and 81% FE for CO at −0.3 and −0.1 V vs. Ag/AgCl respectively (overpotential of −400 to −600 mV respectively) under illumination with visible light (λ > 460 nm).166 However current densities remained low, typically in the range of a few tens of μA cm−2. More recently higher current densities were obtained for CO2 conversion to formate (0.7 mA cm−2 at E = −0.25 V vs. RHE, pH 6.8), using a Ru sensitizer and a Ru catalyst assembled at a Si|GaN|NiO photocathode.167 This approach is certainly worth being further investigated upon employing earth abundant metal complexes and cheap sensitizers, such as organic dyes (see Chart 4 for examples).
 | ||
| Chart 4 Typical molecular photosensitizers including metal-based (Ru, Ir) complexes and organic aromatic molecules. | ||
Considerations on relative energy levels between the semi-conductor, photosensitizer, electron relays and catalyst are of major importance to better understand the different approaches. In strategy a, and for an ideal case, the energy at which the catalyst is reduced to the catalytically active species has to lie below the conduction band (CB) of the illuminated semi-conductor.157 Nevertheless, due to surface states that pin the Fermi level at the interface, the semi-conductor/electrolyte interface may rather be considered as a semi-conductor/metal/electrolyte interface so that the photovoltage is determined by the difference in work function between the semi-conductor and the surface state.168 Consequently, photovoltages have been obtained even for catalysts with redox energy levels lying above the conduction band of the semi-conductor.141 Such considerations do not apply for configuration c since the energy levels of the photosensitizer and the catalyst have to be adjusted to enable electron transfer from the former to the latter. But contrary to configuration a, excited electrons in the photosensitizer are not driven toward the catalyst by an internal electric field. It usually results in higher electron–hole recombination efficiency and it may explain the low current density typically reported with strategy c. A larger current density may be obtained by increasing the driving force between the photosensitizer and the catalyst, but at the expense of a reduced photovoltage.167 In strategy b, a key factor is the energy difference between p- and n-type semiconductors which will define the maximum photovoltage similarly to a PV cell. Such a device, where the p–n junction is buried may be regarded as a photovoltaic cell in series with an electrochemical electrode.169 Consequently, meaningful comparison could be made with other systems, for example when a (photo)cathode is coupled with a photoanode,150,165,166,170 or when both the anode and the cathode are two sides of a multiple-junction cell138 or either when the cathode includes both configurations b and c in series.166,167
In the cases of buried junctions, the interfacial layer could be conductive.169 However, to enable the passage of light the use of such conductive layer is precluded and large band gap material with high electron conductivity are instead preferred. These layers also need to be stable under reduction and to allow for molecular functionalization. So far only TiO2, which complies with these requirements, has been employed as a top-cathode layer. Nevertheless, this metal oxide is electrochemically active,171,172 and FE for CO2 reduction is significantly below unity. Anchoring strategies also deserve special attention, especially regarding pH range stability.6,173 Finally, a different strategy would be to cover the buried junction by a conductive and electrochemically inert material while back irradiating the photocathode.169,174,175 Catalytic performances obtained with Co, Fe, and Ni aza macrocycle complexes are summarized in Table 4.
Ni, Co and Fe aza macrocycles have not been investigated under photoelectrochemical conditions as much as precious metal complexes, despite some early promising performances. CO2 reduction products have been almost restricted to CO, in fewer cases to formate, while no example with more reduced compounds has yet been reported. Looking at the most recent investigations, it seems that strategies b–c are favored over a, and that aqueous electrolytes are systematically targeted despite CO2 lower solubility. It should however be kept in mind that due to intrinsic limitations of photovoltaic devices under more concentrated sunlight, a current density of a few tens of mA cm−2 is likely to be an upper limit, so that a first priority should be put on stability of both the catalyst and the anchoring groups so as to get robust photocathodes. Fundamental challenges further include control of the proton environment at the catalytic sites and synchronization of the various steps of the process (charge separation and transport to the active sites, substrate and co-substrate diffusion, catalytic reaction). Finally, the future of photoelectrochemistry may not only rely on the performances of the catalyst, but will also depend on advances in photovoltaics and photoanodes.
| Entry | Catalyst | PS | SD | Solvent | Light source | Irrad. time | Product (TON) | Selectivity | Φ (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Co10 0.08 mM | [Ru(bpy)3]2+ 0.5 mM | 0.5/0.5 M H2A/HA− | H2O (pH = 4) | Daylight lamp | 18 h | CO (1.2) | <1% (CO) | — | 158 |
| 2 | Co11 0.08 mM | [Ru(bpy)3]2+ 0.5 mM | 0.5/0.5 M H2A/HA | H2O (pH = 4) | Daylight lamp | 18 h | CO (28.7) | 71% (CO) | — | 158 |
| 3 | Ni1 2 mM | [Ru(bpy)3]2+ 0.5 mM | H2A | H2O (pH = 5) | Xe lamp 1000 W, λ = 440 nm | 24 h | CO (0.1) | — | 0.06% (CO) | 176 |
| 0.003% (HCOO−) | ||||||||||
| 4 | Ni1 0.1 mM | [Ru(bpy)3]2+ 0.5 mM | H2A | H2O (pH = 4) | Xe lamp 1000 W 340 < λ < 600 nm | 8 h | CO (4.8) | 12% (CO) | — | 176 |
| 88% (H2) | ||||||||||
| 5 | Ni21 0.25 mM | [Ru(bpy)3]2+ 0.5 mM | H2A | H2O (pH = 4) | Hg lamp 60 W, λ = 435 nm | 1 h | CO (4.4) | 89% | — | 176 |
| 6 | Co12 1.7 mM | p-Terphenyl 2 mM | TEOA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (10.2) | — | 15 (CO) | 177 and 178 |
| HCOO− (6.7) | 10 (HCOO−) λ = 313 nm | |||||||||
| 7 | Co3 1.7 mM | p-Terphenyl 2 mM | TEA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (4.7) | 60.5% (CO) | — | 177 and 178 |
| HCOO− (2.3) | 31.8% (HCOO−) | |||||||||
| 8 | Co13 1.7 mM | p-Terphenyl 2 mM | TEOA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (5.3) | 51.6% (CO) | — | 177 and 178 |
| HCOO− (3.5) | 34.4% (HCOO−) | |||||||||
| 9 | Co14 1.7 mM | p-Terphenyl 2 mM | TEOA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (2.0) | 52.4% (CO) | — | 177 and 178 |
| HCOO− (0.3) | 7.9% (HCOO−) | |||||||||
| 10 | Co15 1.7 mM | p-Terphenyl 2 mM | TEOA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (4.9) | 58.5% (CO) | — | 178 |
| HCOO− (2.5) | 30.4% (HCOO−) | |||||||||
| 11 | Co11 1.7 mM | p-Terphenyl 2 mM | TEOA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (1.0) | 44% (CO) | — | 178 |
| 12 | Co16 1.7 mM | p-Terphenyl 2 mM | TEOA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (2.7) | 30.3% (CO) | — | 178 |
| HCOO− (0.4) | 4.8% (HCOO−) | |||||||||
| 13 | Co17 1.7 mM | p-Terphenyl 2 mM | TEOA | CH3CN/CH3OH (4:1 v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | — | — | — | 178 |
| 14 | Co12 10 mM | Phenazine | TEA | CH3CN/CH3OH/TEA (2:1:1, v:v:v) |
Hg lamp 500 W high-pressure λ > 290 nm | 1 h | CO (0.08) | 92.2% (HCOO−) | — | 179 |
| HCOO− (1.1) | 7.12% (CO) | |||||||||
| 15 | Co20 0.005 mM | [Ru(bpy)3]2+ 0.3 mM | BIH | CH3CN/0.5 M TEOA | LED λ = 460 nm | 80 min | CO (2660) | 98% (CO) | — | 258 |
| 16 | Co20 0.05 mM | [Ru(bpy)3]2+ 0.3 mM | BIH | CH3CN/0.5 M TEOA | LED λ = 460 nm | 80 min | CO (497) | 98% (CO) | 2.8 (CO) | 258 |
| HCOO− (5) | ||||||||||
| H2 (3) | ||||||||||
| 17 | Co20 0.005 mM | Purpurin 2 mM | BIH | DMF | LED λ = 460 nm | 11 h | CO (790) | 95% (CO) | 0.8 (CO) | 258 |
| HCOOH (78) | ||||||||||
| H2 (11) | ||||||||||
| 18 | Fe13 0.005 mM | [Ru(bpy)3]2+ 0.2 mM | BIH | CH3CN/TEOA (4:1 v:v) |
LED λ = 460 nm | 3 h | CO (3844) | 85% (CO) | 1.45 (CO) first 45 min | 258 |
| HCOO− (534) | ||||||||||
| H2 (118) | ||||||||||
| 19 | Fe13 0.05 mM | [Ru(bpy)3]2+ 0.2 mM | BIH | CH3CN/TEOA (4:1 v:v) |
LED λ = 460 nm | 3 h | CO (1879) | 97% (CO) | 8.8 (CO) | 258 |
| HCOO− (48) | ||||||||||
| H2 (45) | ||||||||||
| 20 | Fe13 0.005 mM | Purpurin 0.02 mM | BIH | DMF | LED λ = 460 nm | 12 h | CO (1365) | 92% (CO) | 1.1 (CO) | 258 |
| HCOOH (115) | ||||||||||
| 21 | Co1 10 μM | — | TEA | CH3CN | Xe lamp λ > 320 nm | 20 h | HCOO−/CO (300) | — | — | 42 |
| 22 | Co4 0.05 mM | p-Terphenyl 3 mM | TEA | CH3CN | Xe lamp λ > 300 nm | 20 h | CO (62) | — | — | 185 |
| 23 | Fe11 0.034 mM | p-Terphenyl 3 mM | TEA | CH3CN | Xe lamp 300 W, λ > 300 nm | 20 h | CO (61.8) | — | — | 186 |
| 24 | Fe1 0.01 mM | — | TEA | DMF | Xe lamp 300 W | 15 min | CO (70) | — | 5 | 187 |
| 25 | Fe1 0.01 mM | — | TEA | CH3CN | VIS light (BG40-type optical filter) | 10 h | CO (17) | 8% (CO) in 1 h | — | 188 |
| 26 | Fe14 0.15 mM | — | TEA | CH3CN | Xe lamp λ > 310 nm | 6 h | CO (9) | — | — | 189 |
| 27 | Fe14 0.15 mM | p-Terphenyl 3 mM | TEA | CH3CN | Xe lamp λ > 310 nm | 6 h | CO (90) | — | — | 189 |
| 28 | Co8 0.15 mM | p-Terphenyl 3 mM | TEA | CH3CN | Xe lamp λ > 310 nm | 6 h | CO (47.5) | — | — | 189 |
| 29 | Co22 0.042 mM | p-Terphenyl 3 mM | TEA | CH3CN | Xe lamp λ > 310 nm | 6 h | CO (86) | — | — | 190 |
| 30 | Fe15 0.048 mM | p-Terphenyl 3 mM | TEA | CH3CN | Xe lamp λ > 310 nm | 6 h | CO (43.8) | — | — | 190 |
| 31 | Fe16 0.045 mM | p-Terphenyl 3 mM | TEA | CH3CN | Xe lamp λ > 310 nm | 6 h | CO (51.1) | — | — | 190 |
| 32 | Fe2 0.01–0.05 mM | — | TEA | CH3CN | VIS light BG40 filter | 10 h | CO (28) | 93% (CO) in 1 h | — | 188 |
| 33 | Fe2 2 μM | 9-CNA 0.2 mM | TEA | CH3CN | Xe lamp λ > 420 nm | 45 h | CO (40–60) | 100% (CO) | 0.08 (CO) | 191 |
| 34 | Fe2 2 μM | fac-Ir(ppy)3 0.2 mM | TEA | CH3CN | Xe lamp λ > 420 nm | 55 h | CO (140) | 93% (CO) | 0.13 (CO) | 191 |
| 35 | Fe4 2 μM | — | BIH 20 mM | CH3CN | Solar simulator λ > 420 nm | 47 h | CO (63) | 100% (CO) | — | 192 |
| 36 | Fe4 2 μM | Purpurin 2 mM | TEA | CH3CN/H2O (1:9 v:v), 0.1 M NaHCO3 |
Solar simulator λ > 420 nm | 47 h | CO (60), H2 (5) | 95% (CO) | — | 193 |
| 37 | Fe4 2 μM | fac-Ir(ppy)3 0.2 mM | TEA | CH3CN | Solar simulator λ > 420 nm | 102 h | CO (367) | 17% (CO) | — | 231 |
| CH4 (79) | 78% (CH4) | |||||||||
| H2 (26) | 5% (H2) | |||||||||
| 38 | Fe4 2 μM | fac-Ir(ppy)3 0.2 mM | TEA | CH3CN/H2O (3:7 v:v) |
Solar simulator λ > 420 nm | 47 h | CO (24) | 9% (CO) | — | 194 |
| CH4 (3) | 75% (CH4) | |||||||||
| H2 (5) | 16% (H2) | |||||||||
| 39 | Fe31 3–6 μM | Ir(ppy)3 0.25 mM | TEOA | (N-Methyl-2-pyrrolidone) NMP | Hg lamp 2.5 W 400 < λ < 700 nm | 5 h | CO (100) | 50% (CO) | — | 195 |
| 40 | Fe17 5 μM | Ir(ppy)3 1.67 mM | TEOA | NMP/TEOA (5:1, v:v) |
Hg lamp 2.5 W 400 < λ < 700 nm | 1 h | CO (600) | 50% (CO) | — | 196 |
| 41 | Fe17 1 μM | [Cu(CH3CN)4] PF6 5 μM | BIH | NMP/TEOA (5:1, v:v) |
Hg lamp 1.5 W 400 < λ < 700 nm | 5 h | CO (487) | 99% (CO) | 13 | 197 |
| 42 | Fe18 0.05 mM | Cu–PS 0.25 mM | BIH | CH3CN/TEOA (5:1 v:v) |
Hg lamp λ = 435.8 nm | 12 h | CO (273) | 78% (CO) | 6.7 | 198 |
| 43 | Fe23 0.05 M | Ru(phen)32+ 0.2 mM | BIH | CH3CN/H2O (1:1 v:v) |
LED λ = 460 nm | 68 h | CO (14095) |
98% (CO) | 0.8 (in 24 h) | 184 |
| 44 | Fe4 10 μM | PS7 1 mM | TEA 0.1 M | DMF | Solar simulator λ > 435 nm | 102 h | H2 (23) | 12% (H2) | — | 232 |
| CO (140) | 73% (CO) | |||||||||
| CH4 (29) | 15% (CH4) |
| Catalyst | Concentration or loading | Electrode | Solvent | η (mV) overpotential | j (mA cm−2) | Time (h) | FECO (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Ni2 | 180 mM | p-Si | CH3CN:H2O (1:1; v:v) |
−50 | — | 24 | 64 | 139 |
| Ni1 | 0.52 mM | p-GaAs | H2O | 580 | 0.063 | 15 | 44 | 141 |
| Ni1 | 6.75 mM | p-GaP | H2O | 380 | ≈0.250 | 15 | 85 | 143 |
| Co6 | — | p-CdTe | DMF:H2O (95:5; v:v) |
— | 11.94 | — | 104 | 144 |
| Ni15 | — | p-CdTe | DMF:H2O (95:5; v:v) |
— | 7.2 | — | 77 | 144 |
| Fe14 | — | p-CdTe | DMF:H2O (95:5; v:v) |
— | 8 | — | 92 | 144 |
| Fe1 | 0.2 mM | p-Si | CH3CN:DMF (95:5; v:v) |
210 | 3 | 6 | 92 | 154 |
| Co24 | 1 mM | p-Si NWs | CH3CN (1% H2O) | 520 | 1 | 5 | 69 | 155 |
| Co25 | 45 nmol cm−2 | TiO2/p-Si | CH3CN:H2O (6:4; v:v) |
−200 | 0.1 | 8 | 48 | 161 |
Co and Fe cyclams and polypyridines have shown interesting performances (Chart 1). This has led to mechanistic studies, notably with the identification of some reaction intermediates involved in the processes. In order to avoid the use of noble metal-based sensitizers, some light-absorbing aromatic organic molecules have also been used. The main catalytic product is CO with p-terphenyl (PS2, Chart 4 and Table 3, entries 6–13)177,178 while it changes to formate with phenazine (PS3, Table 3, entry 14).179 It was shown that phenazine is able to donate both electron and proton during catalysis. In that latter case, a Co–H hydride species, whose formation is enhanced by proton transfer from the sensitizer, was proposed to lead to formate upon CO2 insertion. Note that in all these examples, catalytic activity is moderate with a low TON. Brunschwig, Fujita et al. have investigated several Co cyclams (Co12–14, Co16) to unveil possible intermediates under photocatalytic conditions.180–182 Using a combination of transient absorption, Fourier-transform infrared spectroscopy (FTIR) and X-ray absorption near edge structure (XANES) analysis, a 5-coordinated CO2 adduct intermediate [CoIIIL(CO22−)]+ (in which L stands for the cyclam type) was identified for the four studied cyclams. In this adduct, CO2 is stabilized by the NH protons in the macrocycle ligand. A solvent molecule (CH3CN in this case) can also coordinate the metal center to lead to a more stable six-coordinated intermediate [(CH3CN)CoIIIL(CO22−)]+ by internal charge transfer from the metal center to the CO2. The latter intermediate is then further reduced, with assistance of a proton or another CO2 molecule, to afford CO.54,183 Lau and Robert have reported two cobalt and iron complexes (Co18, Fe22) exhibiting different selectivity with CO and formate production respectively (Table 3, entries 15–17 and 18–20).91,258 Such a difference was ascribed to slower C–O bond cleavage with the iron catalyst and concomitant higher basicity of the bound carbon atom, facilitating formate production at the expense of CO (no hydride was postulated on the reaction pathway). Recently a robust Fe-based quinquepyridine complex [Fe(qnpy)(H2O)2]2+ (Fe23) that can reduce CO2 to CO in aqueous acetonitrile solution was reported. Using Ru(phen)3Cl2 (phen = 1,10-phenanthroline) as the photosensitizer and BIH as the sacrificial reductant, CO was produced with a TON of 14095 and a high selectivity of 98% under visible light irradiation (Table 3, entry 43).184
During the same period, Co porphyrin Co142 and its derivative Co4,185 as well as the iron analogue Fe11186 were found to be active CO2 catalysts (Table 3, entries 21–23). Fe-porphyrin Fe1 was first used for the photoreduction of CO2 in 1997 (Table 3, entries 24 and 25).187,188 In a DMF/TEA solution of Fe1, photoinduced ligand-to-metal charge transfer generated FeII from the initial FeIII species. FeII was further converted to FeI upon light excitation and reductive quenching by TEA. Finally, the catalytically active species Fe0 was proposed to be formed by disproportionation of two FeI species. Fe and Co phthalocyanines (Fe14, Co8, Table 3, entries 26–28)189 and corroles (Co22, Fe15, Fe16, Table 3, entries 29–31)190 were also investigated for photocatalytic reduction of CO2. In the presence of p-terphenyl, TONs for CO of Fe and Co phthalocyanine complexes are comparable to those of porphyrin analogs, such as Fe1 and Co1.185
Earth-abundant metal complexes were again put on the spotlight ten years later. A simple modified Fe tetraphenylporphyrin with OH groups in all ortho, ortho′ positions of the four phenyl groups (Fe2, Table 3, entry 32) was investigated under photochemical conditions.188 As demonstrated in electrochemistry,110 the OH groups have a dual role, first providing an H-bonding pattern to stabilize the Fe–CO2 adduct and then playing the role of proton relay to boost the C–O bond cleavage. In the absence of an external sensitizer, good selectivity was already obtained for CO (85%), significantly higher than with Fe1. Higher CO selectivity was further obtained, reaching 93% and 100%, upon using an organic sensitizer (PS5, 9-CNA) and a strongly reducing Ir complex (PS6, Ir(ppy)33+) respectively (Table 3, entries 33 and 34).191
Self-sensitized CO2 reduction was also investigated with Fe4, the porphyrin bearing four positively charged trimethylammonium groups at all para positions of each phenyl ring (Table 3, entry 35).192 Both the catalyst efficiency and selectivity were increased as compared to Fe2 under similar conditions. Fe4 could indeed achieve 100% selectivity using BIH as a sacrificial agent in CH3CN. Moreover, the catalyst was able to perform CO2 reduction in aqueous solutions (acetonitrile/water 1:9, v/v) with PS4 as an organic photosensitizer, showing 95% selectivity (Table 3, entry 36).193 Moreover, it has been found that Fe4 can achieve CH4 production in a CH3CN solution containing up to 70% H2O (Table 3, entry 38).194
Fe-carboxyl complexes are also active catalysts for CO2 reduction to CO and formate. The commercially available Fe3(CO)12 (Fe31) was reported to reduce CO2 in (N-methyl-2-pyrrolidone) NMP solvent, affording a 50% selectivity and TON ca. 100 for CO (Table 3, entry 39).195 (Cyclopentadienone)iron-tricarbonyl (Fe(cpd)(CO)3, Fe17) was reported by the same group and could accomplish CO2 reduction with a TON of 600 in 1 hour (Table 3, entry 40).196 Together with a Cu complex as a sensitizer and BIH as a sacrificial donor, the selectivity for CO could reach 99% with a high quantum yield of 13.3% (Table 3, entry 41).197 Similarly, Ishitani et al. have reported an efficient photocatalytic system using a Fe complex as a catalyst in which the metal is chelated by two di-methyl 1,10-phenanthroline molecules (Fe(dmp)2(NCS)2, Fe18) and a Cu-based complex as PS that can convert CO2 to CO under visible light with a TON of 273 and a quantum yield of 6.7% (Table 3, entry 42).198
Supramolecular assemblies (often called dyads, see Chart 5), formed by the covalent linkage of the catalyst and the photosensitizer units via bridging ligands, also showed interesting performances, by potentially facilitating energy/electron transfers between subunits. A first structure was reported in 1992 by Kimura et al. (Ru(phen)2[phen-Ni(cyclam)](ClO4)4, RuNi1). It included a Ru(phen)32+ (phen = 1,10-phenanthroline) as a sensitizer unit and [Ni(cyclam)]2+ as a catalyst but CO2 reduction was not catalytic (TON < 1). Improved performance for CO production was obtained with a mixed system of Ni(cyclam)2+ and Ru(phen)32+ (Table 6, entry 4).199 Aukauloo et al. reported the supramolecular assembly RuNi2, including a ruthenium trisbipyridyl-like unit covalently attached to a nickel cyclam via a triazole ring. RuNi2 was able to convert CO2 to CO in aqueous solution with ascorbate as a sacrificial donor under 450 nm illumination with a few TON (Table 6, entry 5).200 Cobalt catalysts were also linked to a ruthenium photosensitizer for CO2 reduction (RuCo1 and RuCo2, Chart 5). After 29 hours of visible light irradiation in the mixed DMF:H2O:triethanolamine (3:1:1) solution, the selectivity is improved (TONCO = 3, TONformate = 31, TONH2 = 1 for RuCo1, TONCO = 5, TONformate = 34, TONH2 = 1 for RuCo2, Table 6, entries 6 and 7). The yields for CO2 were comparable with those obtained with [Co-tris(bpy)]2+ simply mixed with [Ru(bpy)3]2+ (TONCO = 9, TONformate = 28, TONH2 = 16) (Table 6, entry 8), although the selectivity was slightly better.201
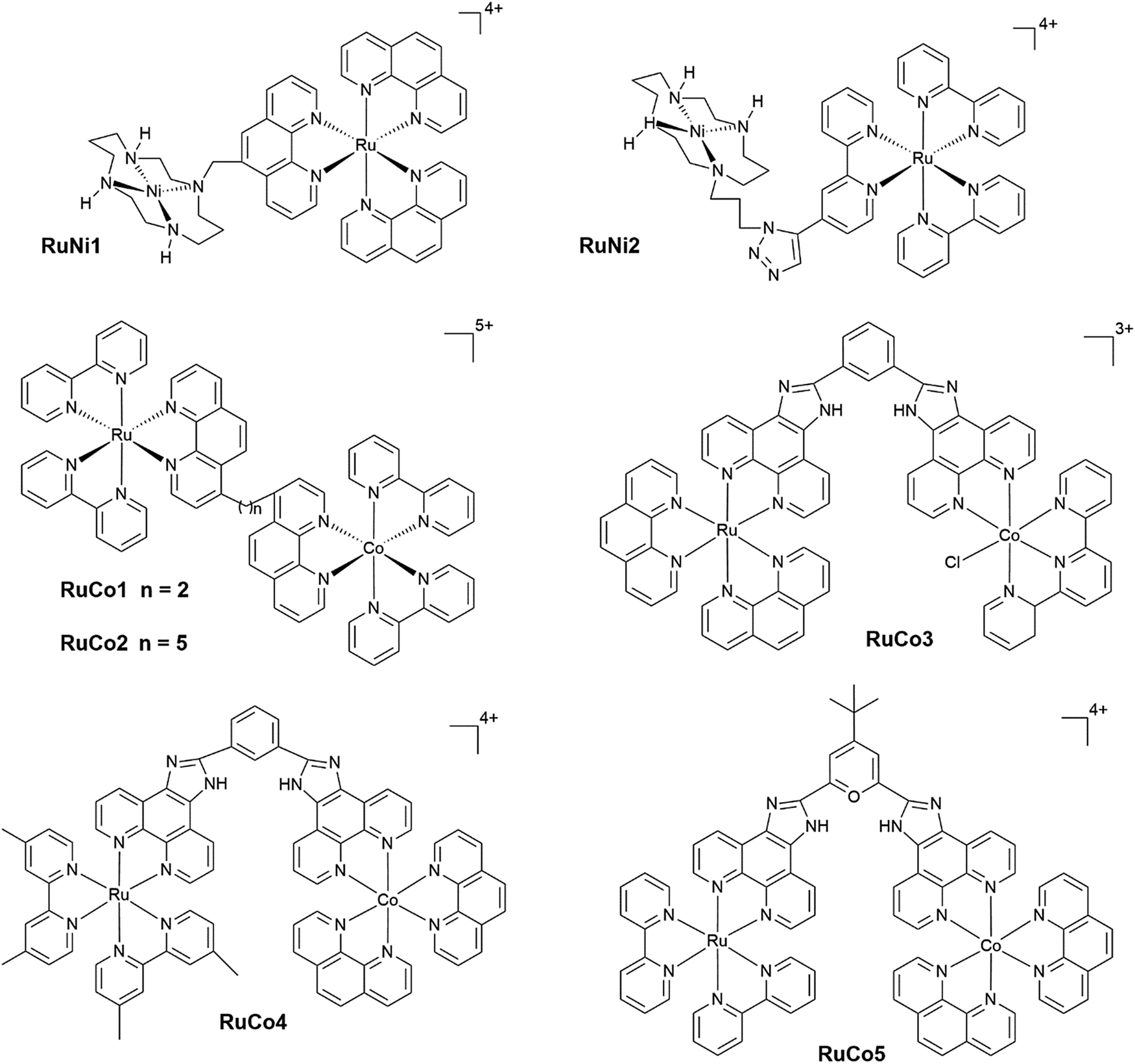 | ||
| Chart 5 Molecular structures of sensitizer–catalyst molecular dyads. | ||
Performances could be significantly enhanced upon introducing conjugation in the linkage between the PS center and catalyst, and thus better electronic interactions between the subunits. Using this strategy, selectivity for CO2 reduction and stability of the systems were improved. With the three catalysts RuCo3, RuCo4, RuCo5, TON values for CO production in the range of 50 were obtained with selectivity ranging from 67% to 87% in CH3CN/TEOA (5:1 v:v) using BIH as a sacrificial donor under Xe lamp irradiation (λ > 415 nm, Table 6, entries 9–11).202
3.4.1 Inorganic bulk materials as scaffolds. Adsorbing or grafting molecules onto inorganic materials, such as silicon, zeolite and kaolin is the simplest approach. Hybrids consisting of CoIII cyclam (1,4,8,11-tetraaza-cyclotetradecane) complexes and silica SBA-15 via a refluxing and microwaving method have been prepared. By associating Co cyclam and SBA-15, higher catalytic activity (TONCO = 160) and selectivity were obtained than free cyclam molecules (TONCO = 142).205 Further grafting of the Co-cyclam onto silica by a microwave-assisted process afforded a uniform distribution of the CoIII active sites in the silica mesopores, boosting the activity for CO production (TONCO = 312) under similar conditions, and TON was increased over 500.206 In a different strategy, a [Mn(CO)5Br] complex was attached onto bipyridyl-containing periodic mesoporous silica (bpy-PMO, Scheme 6), resulting in isolated Mn-carbonyl centers coordinated to the bipyridine functionalities of bpy-PMO. Employing [Ru(bpy)3]Cl2 as PS and BIH as SD, the hybrid can complete the photo-stimulated reduction of CO2 with TONs of 484 and 239 for formate and CO respectively.207 Such an approach may be extended to other earth-abundant metals.
 | ||
| Scheme 6 Synthetic pathway to [Mn(bpyPMO)(CO)3Br]. Reproduced from ref. 207 with permission from the Royal Society of Chemistry, copyright 2017. | ||
Most metal complexes grafted on these inorganic porous matrices yet display lower activity than under homogeneous conditions due to limitations either in charge transfer steps or mass transport. Metal–organic frameworks (MOFs) have also attracted attention as a scaffold for developing high specific area hybrids able to host molecular catalysts. Significant progress has been made in constructing MOF-based systems for CO2 reduction. A series of amine-functionalized Ti-, Fe-, and Zr-based MOFs showed good absorption in the visible domain and promising photocatalytic performance for CO2 reduction.208–210 A cobalt-based zeolitic imidazolate framework (Co-ZIF-9) led to a TON of 52.2 for CO in photoreduction of CO2 employing TEOA as the sacrificial agent under visible light illumination.211 Unfortunately, the weak coordination bonds in the framework commonly result in poor stability of the hybrid which may lead active species to leach out.212 Overcoming these limitations may lead to new breakthrough in this rapidly emerging topic.
3.4.2 Particulate semi-conductors as supports. Semi-conductors including carbon-based materials (graphene, carbon nitride, carbon nanotube and their derivatives)213–217 as well as inorganic metal sulfides (MoS2, CdS, CdSe)218,219 and metal oxides (TiO2 and ZrO2)220–223 have been extensively used in constructing heterogeneous electro- and photocatalysts. In these catalytic systems, the semiconductor works as a photosensitizer, as co-catalyst and/or as an electronic channel.
Niu et al. have reported a thin film photocatalyst constructed by merging Co tetrahydroxyphenyl porphyrin (CoTHPP) and graphene. The hybrid film was suggested to reduce CO2 to hydrocarbons under visible light, affording CH4 and C2H2 through switching the electrons transfer direction from graphene–H2O to graphene–CO2, albeit no 13C labelled experiments were shown to confirm the origin of the products.224 By providing spatially and temporally separated centers for electrons and protons, the iron tetra(4-carboxylphenyl) porphyrin chloride molecular catalyst (Fe12) assembled with a carbon nitride nanosheet reduced CO2 into CO at a rate of 6.52 mmol g−1 in 6 h with a selectivity of 98%.225 Mesoporous graphitic carbon nitride (mpg-C3N4) has also been employed as a redox photosensitizer in association with iron quaterpyridine (Fe13), thus forming a hybrid system fully made of abundant elements.226 It was shown that under visible light irradiation (λ ≥ 400 nm), in a mixed CH3CN/triethanolamine (4:1, v:v) solvent, CO was obtained with 97% selectivity, reaching a TON of 155 after 17 h, and with an apparent quantum yield of ca. 4.2%. A polymeric cobalt phthalocyanine (Co6) catalyst (CoPPc) coupled with mesoporous carbon nitride was also developed.216 Photocatalytic activity was observed to be highly dependent on the catalyst loading and also on irradiation conditions. In CH3CN solution with TEOA as an electron donor, CO was obtained with TONCO = 84 and a selectivity of 85% under full solar spectrum irradiation (>300 nm), whereas under visible light, TONCO = 51 and selectivity decreased to 76% with identical catalyst loading. Recently, Co quaterpyridine (Co20) modified with an acid carboxylic group was covalently attached to the NH2 pendent groups of a mesoporous C3N4 material through an amide linkage. It led to enhanced stability and selectivity for CO production, thanks to the robust connection and efficient charge transfer between the material and the catalyst.227 In acetonitrile as a solvent with BIH as a sacrificial donor and phenol as a co-substrate, a 98% selectivity for CO was maintained over 4 days of irradiation with a TON of ca. 500.
In a similar approach, Reisner et al. also explored the association of molecular catalysts with semi-conducting materials. In a first example, they reported the immobilization of nickel terpyridines (Ni14 and derivatives) previously employed as CO2-to-CO electrocatalysts, adsorbed onto CdS quantum dots. These hybrids showed moderate photochemical activity under visible light in aqueous solutions for the reduction of CO2 into CO with a selectivity of above 90% for the thiol derivative (terpyS).218 They later reported the association of a phosphonic acid-functionalized Ni(cyclam) (Ni1) catalyst with ZnSe quantum dots.228 The hybrid photocatalyst reduced CO2 to CO (Ni-based TONCO = 121, selectivity 8% after 20 h) in aqueous solution with ascorbic acid as the sacrificial agent. Interestingly, ZnSe surface modification with 2-(dimethylamino)ethanethiol (MEDA) resulted in the partial suppression of H2 generation and thus enhanced CO production (Ni-based TONCO = 283, selectivity 33%). Recently, Ni terpyridine (Ni14) was associated with halide perovskite nanocrystals (CsPbBr3) and the light-driven CO2 was studied in ethyl acetate as a solvent. Although no sacrificial donor was mentioned, CO was produced along with a small amount of methane, whose origin will likely be further investigated.229 Weiss et al. employed CuInS2/ZnS quantum dots in colloidal DMSO solution to photosensitize the catalytic conversion of CO2 to CO with Fe1 as a homogeneous catalyst under visible laser light (450 nm).230 Efficient sensitization was ascribed to ultrafast (<200 fs) electron transfer between the quantum dot and Fe1, enabled by the formation of QD/catalyst complexes. The CO2 reduction to CO was achieved with a selectivity for CO of 84% (16% for H2) and a TON of ca. 50. Further studies led to combining the negatively charged CuInS2/ZnS quantum dots with the positively charged Fe4 thus creating a strong electrostatic assembly.219 The photochemical reduction of CO2 to CO was then achieved in water, under 450 nm irradiation, reaching a TONCO of 450 after 30 h, with a selectivity of 99%.
These examples illustrate the remarkable potential of combining molecular catalysts to well defined semi-conductive nanomaterials and quantum dots. Controlling electronic interactions between the two sub-units and properly adjusting the catalyst choice may lead to high catalytic activity. Further development of complete Z-scheme systems, upon association of two sub-systems, one for CO2 reduction and one for water oxidation, may soon lead to stimulating discoveries.
4. Bi-metallic systems with synergy between metals
If molecular catalysts mainly involve monometallic species, both natural and synthetic ones may also include two metals in close proximity, which may lead to a beneficial effect on CO2RR through synergy (cooperativity) between the metals. Ni–Fe carbon monoxide dehydrogenase (CODH) is a natural metalloenzyme that can reversibly convert CO2 into CO.237 CO2 binds to both metal centers, with the C atom interacting with the NiI nucleophilic site and one of the O atoms bound to the FeII electrophilic site. Thus the two metals work synergistically to facilitate the cleavage of the C–O bond and achieve high selectivity. Many efforts have been made to mimic this sophisticated catalytic process. But regarding synthetic catalysts, only a few bimetallic centers have been shown to act in concert to reduce the CO2 in molecular catalysts.Solar-driven CO2 reduction with dinuclear bimetallic complexes have attracted quite intensive research efforts. In 1996, Mochizulki et al. reported two dimerized Ni-cyclam complexes. Compared to monometallic macrocyclic complex Ni7 (Table 6, entry 1),176 the dimerized Ni-cyclam Ni22 (Chart 6) shows enhanced efficiency and selectivity in CO2 to CO conversion with a TON of 4.4 in aqueous solution at pH 4 (Table 6, entry 2). When the Ni-cyclam dimer Ni21 (Chart 6) without methyl groups on the ligand was employed, the yield for CO was smaller than that for Ni22. Zhu et al. synthesized a dinuclear Co complex Co21 (Chart 6) containing an oxygen atom at the cis-coordination site.238 In CH3CN/H2O (5:1) solution with TEA as a sacrificial donor, Co21 can achieve photocatalytic CO2 reduction to both CO and formate under 450 nm irradiation, with TON values of 57 and 64 respectively (Table 6, entry 3). In that case however, the major product obtained was H2 (TON 182). However, for all the above cases, geometry constraints likely prevent cooperativity effects between the metal atoms. One recent example related to photocatalytic reduction of CO2 was obtained with a binuclear cobalt cryptate catalyst [Co2(OH)L](ClO4)3 (L = N[(CH2)2NHCH2(m-C6H4)CH2NH(CH2)2]3N, Co22, Chart 6). It affords CO with a TON of 16896 and TOF of 0.47 s−1 under 450 nm LED light irradiation for 10 hours (Table 6, entry 12), while the corresponding monomer gave a TONCO = 1600 with selectivity for CO of 85% in 10 hours.239 Based on experimental data and DFT calculations, authors concluded that during the photocatalytic process, the two cobalt atoms work synergistically, with the CO2 molecule bound to the 2 Co atoms. After replacing one of the Co centers by Zn (CoZn1, Chart 6), the photocatalytic performance of the hetero bimetallic complex [CoZn(OH)L](ClO4)3 (CoZn1) was largely boosted to a TON of 65000 and a TOF of 1.8 s−1 (Table 6, entry 13).240 The improved catalytic efficiency can be ascribed to the enhanced dinuclear metal synergistic catalysis effect between CoII and ZnII. ZnII plays the role of a Lewis acid center, helping C–O bond cleavage from an O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–OH intermediate, thus significantly decreasing the activation barrier of the catalysis rate determining step.
C–OH intermediate, thus significantly decreasing the activation barrier of the catalysis rate determining step.
 | ||
| Chart 6 Molecular structures of various bi- and multi-metallic complexes. | ||
Recently, a binuclear Co complex (Co23, Chart 6) bearing a bi-quaterpyridine ligand was shown to selectively reduce CO2 to formate or CO under visible light.40 Formate was produced selectively (maximum of 97%) in basic acetonitrile solution with a TON of up to 821 (Table 6, entry 14). Conversely, in the presence of a weak acid, CO2 reduction affords CO with high selectivity (maximum of 99%) and a maximum TON of 829 (Table 6, entry 15). Spectro-electrochemistry (SEC) in the infrared region revealed an absorption band at 1635 cm−1 (Fig. 14), which was assigned to the formation of a stable adduct between CO2 and the four-electron-reduced complex. Together with DFT calculations, these results indicate that CO2 was sandwiched by the two cobalt atoms of the binuclear complex with the C atom from CO2 binding to one Co atom, and one O atom interacting with the second Co atom. The catalytic process is controlled by the synergistic action of the 2 Co atoms, each playing a different, complementary role, and the selectivity can be oriented towards one or the other product by simply changing the acidity of the co-substrate to form different active intermediates. Upon protonation at the oxygen atom, CO is evolved while protonation at the C atom furnishes formate.
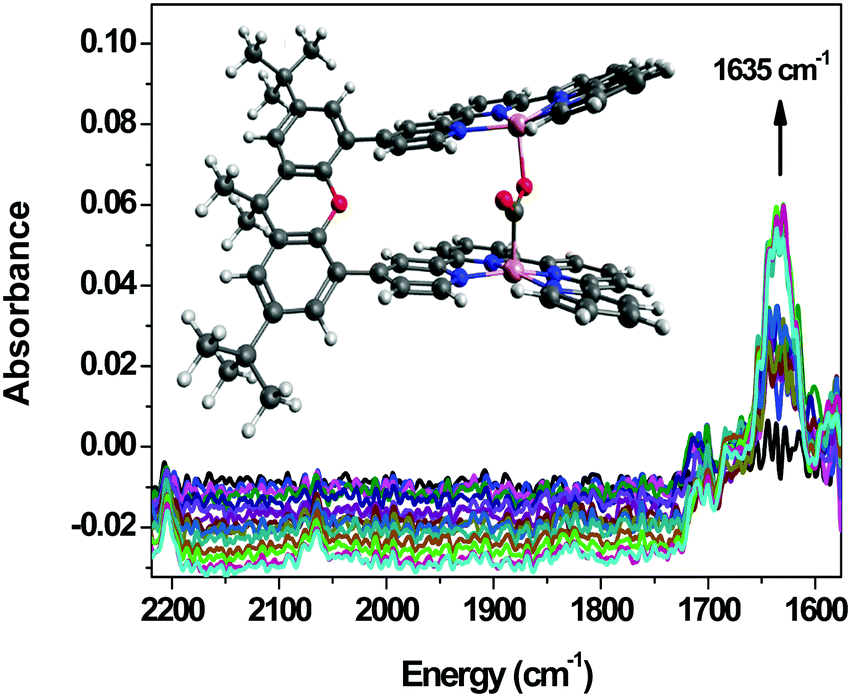 | ||
| Fig. 14 Infrared spectro-electrochemistry data on a 1 mM solution of Co23 in CH3CN (0.1 M nBu4NPF6, 0.5 M TEA) under CO2 at potentials between −0.35 V and −0.85 V vs. Ag pseudo-reference. Inset: Calculated structure of the CO2-reduced complex adduct identified at 1635 cm−1. Adapted with permission from ref. 40. Copyright 2019, Springer Nature. | ||
Going to electrochemical experiments, it was found that linking two Ni1 monomers to form a Ni21 dimer can lead CO2 to HCOOH conversion while the monomer complexes get CO. Potentiostatic polarization at −1.4 V vs. SCE in DMF containing 0.1 M H2O led to the formation of formate with a good selectivity of 81% and FE of 68% after 5 hours (Table 5, entry 2).101 In such a case however and due to geometry constraints there is likely no cooperation between metal sites, as noted previously in the case of light-driven processes. Such cooperation was triggered upon using copper-based catalysts. A remarkable example is given by binuclear copper complex Cu21 (Chart 6) possessing two tripodal tris(2-pyridylmethyl)amine ligands, able to reduce CO2 into oxalate spontaneously. Upon CPE (7 h) at −0.03 V vs. NHE in CH3CN containing LiClO4 (to precipitate Li2C2O4), oxalate was obtained with an FE of 96% and a TON of 6 (Table 5, entry 3).234 The related complex Cu22 (Chart 6) gave similar results.241 A CuICuI species was formed after reduction with sodium ascorbate in DMF, which further combined with two CO2 molecules, resulting in C2O42− generation (Table 5, entry 4). In these two cases, appropriate complex geometry and cooperativity between the two metal centers allowed for C–C bond formation.
| Entry | Catalyst | Solvent | Electrode | Time | TON product | TOF product | Potential (V) overpotential η (V) | FE% product | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Fe21 0.5 mM | DMF:H2O (9:1) |
GC | 10 h | 188 CO | — | −1.55 V vs. Ag/AgCl η = 0.66 V | 88 CO | 235 |
| 2 | Ni21 0.2 mM | DMF + 0.1 M H2O | HME (Hg) | 5 h | 3.2 HCOO− | — | −1.4 V vs. SCE | 68 HCOO− | 101 |
| 0.76 CO | 16 CO | ||||||||
| 3 | Cu21 0.5 mM | CH3CN + LiClO4 | GC | 7 h | 6 | — | 0.03 V vs. NHE | 96 | 234 |
| C2O4H2 | C2O4H2 | ||||||||
| 4 | Cu22 0.3 mM | DMF + Na2A | — | — | C2O42− | — | — | — | 241 |
| 5 | Fe41 0.1 mM | H2O (pH 7) | GC | 50 min | 28 | — | −1.20 V vs. SCE | 98 | 233 |
| HCOO− | HCOO− | ||||||||
| 6 | Fe42 0.1 mM | CH3CN:H2O (95:5) |
GC | 50 min | 5.4 HCOO− | — | −1.40 V vs. SCE | 61 | 236 |
| 3.3 H2 | HCOO− | ||||||||
| 7 | Fe43 0.1 mM | CH3CN:H2O (95:5) |
GC | 50 min | 40 H2 | — | −1.40 V vs. SCE | <3 | 236 |
| HCOO− |
| Entry | Cat. | PS | SD | Solvent | Light source | Irrad. time (h) | Product (TON) | Selectivity | Φ (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ni7 0.25 mM | [Ru(bpy)3]2+ 0.5 mM | H2A | H2O (pH = 4) | Hg lamp 460 W | 1 | CO (0.7) | — | — | 176 |
| 2 | Ni22 0.25 mM | [Ru(bpy)3]2+ 0.5 mM | H2A | H2O (pH = 4) | Hg lamp 460 W | 1 | CO (4.4) | 94% CO | — | 176 |
| 3 | Co21 10 μM | fac-Ir(ppy)3 0.4 mM | TEA | CH3CN/H2O (5:1 v:v) |
LED λ = 450 nm | 60 | CO (56.9) | 18.8% CO | — | 238 |
| HCOO− (64.2) | 21.2% HCOO− | |||||||||
| H2 (181.6) | 60% H2 | |||||||||
| 4 | RuNi1 0.5 mM | — | H2A | H2O (pH = 4) | Xe lamp 500 W λ > 450 nm | 44 | CO (<1) | 72% CO | — | 199 |
| 5 | RuNi2 0.035 mM | — | H2A | H2O (pH = 6.5) | Xe lamp 500 W λ > 450 nm | 60 | CO (5.2) | — | — | 200 |
| H2 (2) | ||||||||||
| 6 | RuCo1 0.5 mM | — | TEOA | DMF:H2O (3:1 v/v) |
Xe lamp 500 W 400 < λ < 750 nm | 29 | CO (3) | 9.3% CO | — | 201 |
| HCOO− (31) | ||||||||||
| H2 (1) | ||||||||||
| 7 | RuCo2 0.5 mM | — | TEOA | DMF:H2O (3:1 v:v) |
Xe lamp 500 W 400 < λ < 750 nm | 29 | CO (5) | 12.7% CO | — | 201 |
| HCOO− (34) | ||||||||||
| H2 (1) | ||||||||||
| 8 | [Co(bpy)3]3+ 0.5 mM | [Ru(bpy)3]2+ 0.5 mM | TEOA | DMF:H2O (3:1 v:v) |
Xe lamp 500 W 400 < λ < 750 nm | 29 | CO (9) | 17% CO | — | 201 |
| HCOO− (28) | ||||||||||
| H2 (16) | ||||||||||
| 9 | RuCo3 0.1 mM | — | BIH | CH3CN:TEOA (5:1 v:v) |
Xe lamp 300 W λ > 415 nm | 8 | CO (51) | 79.6% CO | — | 202 |
| H2 (13) | ||||||||||
| 10 | RuCo4 0.1 mM | — | BIH | CH3CN:TEOA (5:1 v:v) |
Xe lamp 300 W λ > 415 nm | 8 | CO (54) | 87.2% CO | — | 202 |
| H2 (8) | ||||||||||
| 11 | RuCo5 0.1 mM | — | BIH | CH3CN:TEOA (5:1 v:v) |
Xe lamp 300 W λ > 415 nm | 8 | CO (70) | 64.3% CO | — | 202 |
| H2 (39) | ||||||||||
| 12 | Co22 0.025 μM | [Ru(phen)3]2+ 0.4 mM | TEOA | CH3CN:H2O (4:1 v:v) |
LED λ = 450 nm | 10 | CO (16896) |
98% CO | 0.04% CO | 239 |
| 13 | CoZn1 0.025 μM | [Ru(phen)3]2+ 0.4 mM | TEOA | CH3CN:H2O (4:1 v:v) |
LED λ = 450 nm | 10 | CO (65000) |
98% CO | 0.15% CO | 240 |
| 14 | Co23 0.05 mM | [Ru(phen)3]2+ 0.2 mM | TEOA/BIH | CH3CN | LED λ = 460 nm | 60 | CO (221) | 20.4% CO | 2.6% HCOO− | 40 |
| HCOO− (821) | 75.9% HCOO− | |||||||||
| H2 (40) | 3.7% H2 | |||||||||
| 15 | Co23 0.05 mM | [Ru(phen)3]2+ 0.2 mM | PhOH/BIH | CH3CN | LED λ = 460 nm | 1 | CO (829) | 96% CO | — | 40 |
| HCOO− (12) | 1.5% HCOO− | |||||||||
| H2 (22) | 2.5% H2 |
Dinuclear Fe porphyrin molecule Fe21 with a Fe–Fe distance of 3.4–4.0 Å (Chart 6) was used to reduce CO2 in DMF solution. It was shown that the CO2 likely binds to the FeIIFeII species. Catalytic reduction is further triggered upon generating a FeIFe0 species, at a potential ca. 100 mV more positive (∼−1.25 V vs. NHE in DMF) than for the monomeric Fe1. Upon adding 10% H2O, the catalytic current was increased by a factor 6 as compared to the monomer catalyst under similar conditions. Ten hours of electrolysis at −1.55 V vs. Ag/AgCl yielded 88% FE for CO with H2 as a by-product (Table 5, entry 1).235 The conversion of CO2 to formate has also been reported using an iron cluster Fe41.233 By using organic acids with different pKa, the selectivity of the reaction to formate or hydrogen can be controlled, with strong acids favoring hydrogen generation. CV and IR-SEC allowed identifying [Fe4N(CO)12]2− as a catalytic species. Formate was produced in aqueous solution after electrolysis at −1.20 V vs. SCE with 98% selectivity, with a postulated bridging hydride between two Fe centers. This selectivity remained above 85% for pH between 5 and 13, and no CO was detected (Table 5, entry 5). Replacing one CO ligand with PPh3 (Fe42) led to lower selectivity with formate as the main product still (Table 5, entry 6). Finally, upon replacing one CO ligand with PPh2CH2CH2OH (Fe43) almost no formate was obtained, with mainly H2 as a product (Table 5, entry 7).236,242
Examples of synthetic homo or hetero bi(multi)-metallic molecular catalysts displaying increasing performances and/or specific reactivity, thanks to the synergistic effects exerted by the metal centers, remain scarce. It may however be an interesting pathway to precisely control the distance between the metals and the environment of each center so as to trigger new reactions, such as C–C bond coupling. Such a strategy is briefly described in the next section.
5. Beyond 2e− reduction of CO2
A growing number of articles have recently reported the reduction of CO2 beyond two electrons using molecular electro- or photo-catalysts, starting directly from the gas or sometimes using CO or HCOOH as a reactant. Such stimulating studies may reveal completely new catalytic pathways. A first mandatory requirement is to track for the carbon source, which necessitates careful labeled studies. Looking at reports in this area indicates that many studies using metal-based complex catalysts for the (photo)electrochemical reduction of CO2 to HCHO,243 CH3OH,243–245 CH4131,132,246–249 or even C2H4247,248 do not yet confirm their origin. It is an even more critical issue when molecular catalysts lead to stoichiometric or sub-stoichiometric processes.244 Another hurdle lies in the fact that reporting on labeled experiments may lead to ambiguous conclusions regarding the origin of the catalysis products. An illustrative example is provided in Fig. 15. In this contribution, the authors have employed Co corroles deposited on carbon electrodes for the electrochemical reduction of CO2 into CH3OH in aqueous solution at pH 6.250 They compared GC/MS data under 12CO2 and 13CO2 to assess the carbon source for the observed methanol. The fragmentation pattern for CH3OH displayed m/z peaks shifted up by one unit under 13CO2, as expected; however, the relative intensity of the various peaks drastically changed upon labeling the carbon dioxide, casting doubt on the conclusion that the methanol is issued from CO2.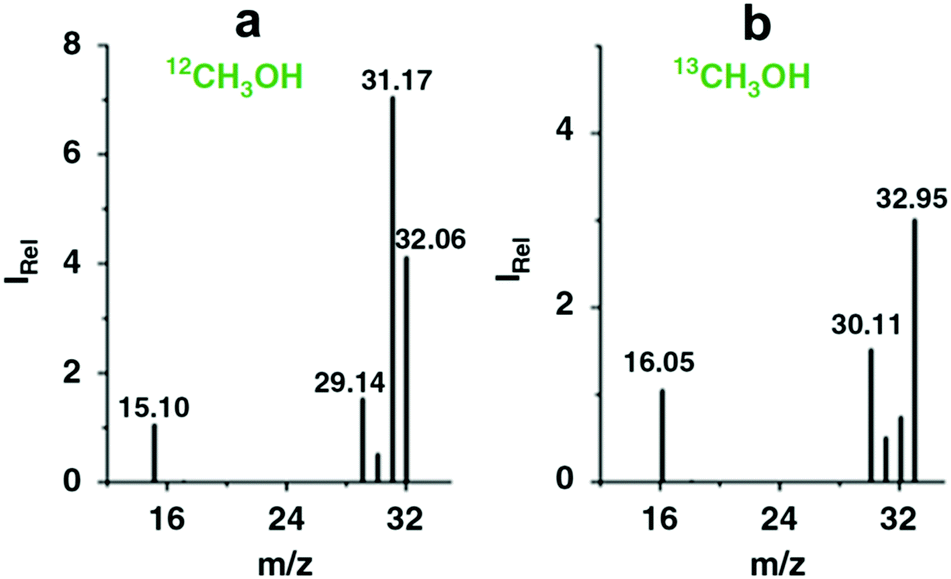 | ||
| Fig. 15 GC/MS spectra and fragmentation pattern for the electro-catalyzed CO2 conversion into CH3OH. Graphs show the mass fragmentation pattern of CH3OH (m/z 29 to 33) produced employing (a) 12CO2 or (b) 13CO2 as a reactant substrate. Reproduced with permission from ref. 250. | ||
Another example concerns the electrochemical reduction of CO2 with cobalt protoporphyrin using on-line mass spectroscopy detection (OLEMS).82 Labeled experiments are obscured by the presence of a mixture of products, solvent and electrolyte in the fragmentation pattern. Mass peak at m/z 17 could not be used to assess 13CH4 formation from 13CO2 as a reactant in water since the corresponding peak is dominated by the solvent signature. The authors instead used the peak at m/z 21 to track for the formation of 13CD4 using 13CO2 as a reactant in D2O solvent. A blank experiment using 12CO2 as a reactant for which no m/z = 21 signal would have been detected was however missing in the study, leaving the proof incomplete. In the following, we will focus our survey to cases where the source for products has been demonstrated without any ambiguity.
The first evidence of molecular electrochemical reduction of CO2 beyond 2e− was recently obtained using cobalt phthalocyanine Co6 as a catalyst.65 The complex was mixed with multi-walled carbon nanotubes and deposited as a thin film onto carbon paper. Upon electrochemical reduction of CO2 and CO intermediate, in neutral or basic solutions, formaldehyde was formed and further reduced into methanol with a maximum 14% FE at pH 13 and a partial current density for methanol of 0.68 mA cm−2 (E = −0.64 V vs. RHE). The origin for methanol and the molecular nature of the catalysis were demonstrated by 1H NMR and XANES analysis respectively. Interestingly in this catalytic process, no methane was formed. Shortly after this publication, highly dispersed Co6 on MWCNTs has been shown to improve the catalytic activity of this catalyst for methanol, starting from CO2,251 with up to 44% FE at −0.94 V vs. RHE (pH 6.8). It should be noted that it was not proved in this work that methanol is formed from CO2 reduction; however, similarity to the previous paper makes the case consistent. It may also be noted that reduction of CO2 and CO into methanol involving molecular complexes, such Fe, Ni and Co porphyrins, has been reported by Ogura et al. in a series of papers published in the 1980s. However, these systems involve the use of Everitt's salt on a platinum electrode with the initial introduction of a primary alcohol and the molecular complex rather played the role of a co-catalyst.252–254
The eight-electron–eight-proton reduction of CO2 to methane has rarely been achieved. Under photochemical conditions, some iron porphyrins (Fe2 and Fe4, Table 3, entries 37, 38, 44) have been reported to convert CO2 into CH4 in acetonitrile or DMF solution containing an appropriate sensitizer and an amine as a sacrificial electron donor.194,231,232 Both highly reducing Ir complex PS6 and phenoxazine PS7 were employed as sensitizers with visible light above 420 nm and 435 nm respectively. A TON of ca. 80 was typically obtained using CO2 as a reactant with a selectivity for CH4 around 17%. CO has been identified as a key intermediate in this process and could even be used as a starting reactant, leading to a maximum value of 160 for methane with a selectivity in the range of 82–85% (H2 being the sole by-product) and an apparent quantum yield of up to 0.47%. A full mechanistic picture is still missing, although the bottleneck for the process is likely the reduction of the FeIICO species formed upon reduction of CO2 to CO.
Another approach to get such a highly reduced product is to use metal complexes as pre-catalysts for in situ electrodeposition of highly active material catalysts. For example, starting from Fe13, Fe nanoparticles (NPs) can be formed upon electrolysis in CH3CN at a glassy carbon electrode in the presence of TEOA. These Fe NPs are able to achieve up to 2.6% FE for methane, being more active than particles electrodeposited from an iron salt or an iron based electrode.255 In this study, proper proof for the carbon source was brought about by the observation of an m/z 17 peak in GC/MS for methane when 13CO2 was used as the reactant, along with correct fragmentation pattern and retention time as shown in Fig. 16.
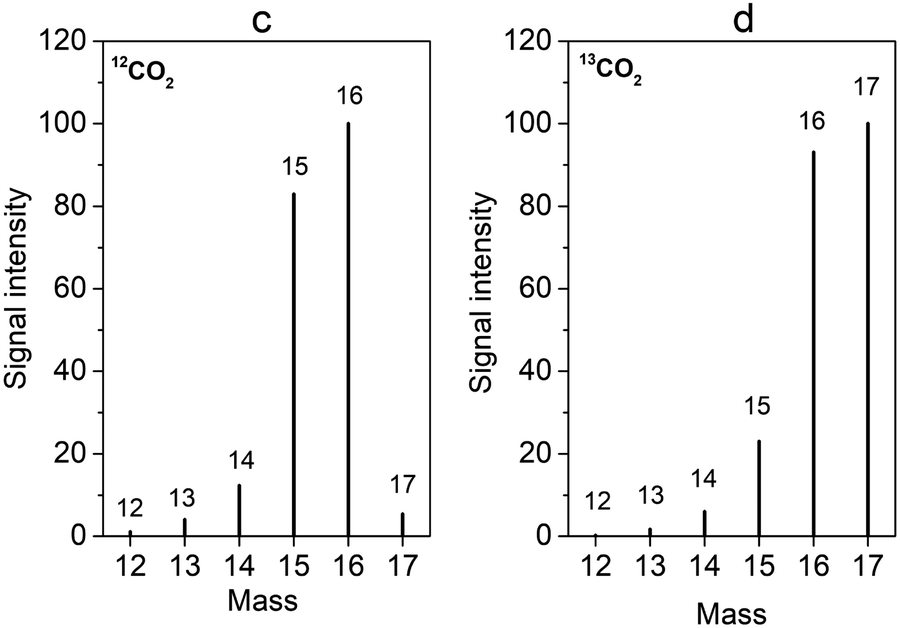 | ||
| Fig. 16 Mass spectra labeling experiment demonstrating the carbon source for CH4 production during CO2 electrolysis, using Fe13 as a molecular precursor catalyst. Reproduced with permission from ref. 255. Copyright 2019, Wiley. | ||
The same phenomena were observed for copper based porphyrin and phthalocyanine Cu1 and Cu2 catalysts (Chart 7) that decompose into Cu NPs upon electrolysis in water (0.5 M KHCO3) when deposited as thin films on carbon paper electrode or carbon electrodes. The nano-catalysts have higher activity for CH4 and C2H4 than a bare Cu surface, with partial current density in the range of ca. 8.5 and 13 mA cm−2 respectively, at a potential close to −1 V vs. RHE.256,257 These spectacular cases call for extreme vigilance not only about the carbon source but also about the actual state of the catalyst. For Cu1, catalysis was indeed originally claimed to be a molecular process.256 In all these above reported examples (Fe13, Cu1, Cu2) the ligand might have an effect on NPs shaping upon decomposition.
 | ||
| Chart 7 Copper-based molecular complexes as pre-catalysts for CO2 reduction. | ||
Overall, these molecular pre-catalysts certainly deserve to be vigorously investigated since it may lead to unexpected catalytic properties and high activity.
The next challenge will be to design molecular catalysts able to create C–C bonds so as to produce highly reduced compounds such as alkenes and alcohols. It probably comes with no surprise that all examples investigated so far for multi-electron multi-proton (n > 2) CO2 reduction, involving monometallic complexes under homogeneous conditions or deposited in thin films, have not led to dimerization or coupling of partially reduced substrates (e.g. CO, HCOOH) bound at the two closely spaced metal centers. Such coupling has indeed been shown to be a key step in heterogeneous catalysis, for example at copper materials. The development of multimetallic molecular complexes able to bind several CO2 molecules at a time in a controlled environment may open interesting perspectives toward this challenge.
Concluding remarks
Multi-facet mechanistic and spectroscopic studies have allowed making progress in the understanding of catalytic processes involved during CO2 reduction with molecular complexes. Much remains to be done, both in electrochemical and light-driven catalysis. Further progress will certainly emerge from advanced mechanistic and in situ/in operando spectroscopic studies allowing not only deciphering the elementary steps of the processes but also clear identification of the degradation pathway(s). It will then open a path toward rational tuning of the next generation of catalysts. Nevertheless, Co, Fe and Ni coordinated by azamacrocycles and polypyridine ligands have led to high reactivity towards electrochemical and visible-light driven CO2 reduction to CO and formate, both in organic solvents and in water. Robust catalysts, once immobilized onto conductive supports, have shown remarkable stability, extending over a day. The achievement of high current densities in electrochemical cells has opened a door to the design of devices at the industrial scale. Progress has also been made recently with hybrid materials in which molecular catalysts are connected to semi-conductive particles or bulk electrodes. Studies of such materials are certainly an area that could strongly benefit from the synergistic combination of homogeneous and heterogeneous catalysis. Finally, the ability of molecular catalysts to drive the multi-electron multi-proton reduction of CO2 beyond CO and formate, even if still limited to a few examples, opens new exciting perspectives for the synthesis of more complex molecules, with the creation of carbon–carbon bonds. Development of such processes using well defined metal complexes will lead to a breakthrough in CO2 reduction chemistry.Abbreviations
| P | Oxidized form of the catalyst (inactive) |
| Q | Reduced form of the catalyst (active) |
| A | Substrate |
| B | Product |
| i | Catalytic current |
| i 0 | Peak current in the absence of substrate |
| i plateau | Scan rate independent catalytic plateau current |
| F | Faraday constant |
| S | Electrode geometric surface area |
| C 0A | Substrate concentration in the bulk solution |
| C 0P | Catalyst concentration in the bulk solution |
| D P | Catalyst diffusion coefficient |
| D A | Substrate diffusion coefficient in solution |
| D S | Substrate diffusion coefficient in a film |
| d f | Catalytic film thickness |
| κ A | Substrate partition coefficient in a film |
| v | Scan rate |
| R | Gas constant |
| T | Temperature |
| E | Applied potential |
| E 0P/Q | Standard potential of the P/Q redox couple |
| k s | Standard electron transfer rate constant |
| k cat | (Pseudo first order) rate constant of the catalytic process |
| η | Overpotential |
| TOF | Turnover frequency |
| TON | Turnover number |
| NHE | Normal hydrogen electrode |
| RHE | Reversible hydrogen electrode |
| SCE | Standard calomel electrode |
| GC | Glassy carbon (electrode) |
| GDE | Gas diffusion electrode |
| CPE | Controlled potential electrolysis |
| CV | Cyclic voltammetry/cyclic voltammogram |
| QD | Quantum dot |
| MWCNT | Multiwalled carbon nanotube |
| EA | Electron acceptor |
| SD | Sacrificial electron donor |
| PS | Photosensitizer |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by Université de Paris, CNRS, Air Liquide, SATT Erganeo and the French National Agency for Research (ANR-16-CE05-0010-01). M. W. and B. M. thank the China Scholarship Council for their PhD fellowship (CSC student number 201606220034 and 201707040042, respectively) and partial financial support to M. R. from the Institut Universitaire de France (IUF) is also gratefully acknowledged.References
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159 RSC.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- C. Costentin, M. Robert and J. M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- C. Jiang, A. W. Nichols and C. W. Machan, Dalton Trans., 2019, 48, 9454–9468 RSC.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- B. M. L. Dioos, I. F. J. Vankelecom and P. A. Jacobs, Adv. Synth. Catal., 2006, 348, 1413–1446 CrossRef CAS.
- D. M. Chisholm and J. Scott McIndoe, Dalton Trans., 2008, 3933–3945 RSC.
- X.-M. Hu, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2019, 15, 148–154 CrossRef CAS.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Curr. Opin. Electrochem., 2017, 2, 26–31 CrossRef CAS.
- J.-M. Savéant, Elements of Molecular and Biomolecular Electrochemistry, John Wiley & Sons, 2006, pp. 78–181 Search PubMed.
- C. Costentin and J.-M. Savéant, ChemElectroChem, 2014, 1, 1226–1236 CrossRef CAS.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, ACS Catal., 2018, 8, 3411–3417 CrossRef CAS.
- C. Cometto, L. Chen, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, Organometallics, 2019, 38, 1280–1285 CrossRef CAS.
- C. Costentin, S. Drouet, G. Passard, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2013, 135, 9023–9031 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- C. Costentin, D. G. Nocera and C. N. Brodsky, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11303–11308 CrossRef CAS PubMed.
- M. Wang, L. Chen, T. C. Lau and M. Robert, Angew. Chem., Int. Ed., 2018, 57, 7769–7773 CrossRef CAS PubMed.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, ChemElectroChem, 2015, 2, 1774–1784 CrossRef CAS.
- C. P. Andrieux, C. Costentin, C. Di Giovanni, J.-M. Savéant and C. Tard, J. Phys. Chem. C, 2016, 120, 21263–21271 CrossRef CAS.
- C. Costentin and J.-M. Savéant, J. Phys. Chem. C, 2015, 119, 12174–12182 CrossRef CAS.
- J.-M. Savéant, ChemElectroChem, 2016, 3, 1967–1977 CrossRef.
- W. Kaim and A. Klein, Spectroelectrochemistry, The Royal Society of Chemistry, 2008 Search PubMed.
- A. W. B. Aylmer-Kelly, P. R. C. A. Bewick and A. M. Tuxford, Faraday Discuss. Chem. Soc., 1973, 56, 96–107 RSC.
- K. Chandrasekaran and J. O. M. Bockris, Surf. Sci., 1987, 185, 495–514 CrossRef CAS.
- J. Desilvestro and S. Pons, J. Electroanal. Chem., 1989, 267, 207–220 CrossRef CAS.
- E. Anxolabéhère, G. Chottard and D. Lexa, New J. Chem., 1994, 18, 889–899 Search PubMed.
- C. Romelt, J. Song, M. Tarrago, J. A. Rees, M. van Gastel, T. Weyhermuller, S. DeBeer, E. Bill, F. Neese and S. Ye, Inorg. Chem., 2017, 56, 4746–4751 CrossRef PubMed.
- C. Romelt, S. Ye, E. Bill, T. Weyhermuller, M. van Gastel and F. Neese, Inorg. Chem., 2018, 57, 2141–2148 CrossRef PubMed.
- C. Costentin, J. M. Savéant and C. Tard, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9104–9109 CrossRef CAS PubMed.
- B. Mondal, A. Rana, P. Sen and A. Dey, J. Am. Chem. Soc., 2015, 137, 11214–11217 CrossRef CAS PubMed.
- A. W. Nichols, S. Chatterjee, M. Sabat and C. W. Machan, Inorg. Chem., 2018, 57, 2111–2121 CrossRef CAS PubMed.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W.-L. Man, S.-M. Yiu, K.-C. Lau, T.-C. Lau and M. Robert, Nat. Catal., 2019, 2, 801–808 CrossRef CAS.
- S. Fernandez, F. Franco, C. Casadevall, V. Martin-Diaconescu, J. M. Luis and J. Lloret-Fillol, J. Am. Chem. Soc., 2020, 142, 120–133 CrossRef CAS PubMed.
- D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, J. Phys. Chem. A, 1998, 102, 2870–2877 CrossRef CAS.
- H. Sheng and H. Frei, J. Am. Chem. Soc., 2016, 138, 9959–9967 CrossRef CAS PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- I. S. Zavarine and C. P. Kubiak, J. Electroanal. Chem., 2001, 495, 106–109 CrossRef CAS.
- M. Krejčik, M. Daněk and F. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef.
- C. Cometto, Doctoral thesis, Université Paris Diderot, 2018.
- C. Gueutin and D. Lexa, Electroanalysis, 1996, 8, 1029–1033 CrossRef CAS.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565–3573 CrossRef CAS PubMed.
- L. E. Lieske, A. L. Rheingold and C. W. Machan, Sustainable Energy Fuels, 2018, 2, 1269–1277 RSC.
- S. L. Behnke, A. C. Manesis and H. S. Shafaat, Dalton Trans., 2018, 47, 15206–15216 RSC.
- M. Wang, L. Árnadóttir, Z. J. Xu and Z. Feng, Nano-Micro Lett., 2019, 11 CrossRef CAS.
- E. Fujita, L. R. Furenlid and M. W. Renner, J. Am. Chem. Soc., 1997, 119, 4549–4550 CrossRef CAS.
- I. T. Bae and D. A. Scherson, J. Phys. Chem. B, 1998, 102, 2519–2522 CrossRef CAS.
- L. R. Furenlid, M. W. Renner and E. Fujita, Physica B, 1995, 208–209, 739–742 CrossRef.
- S.-K. Lee, S. D. George, W. E. Antholine, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 6180–6193 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dincă, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- P. S. Miedema, M. M. van Schooneveld, R. Bogerd, T. C. R. Rocha, M. Hävecker, A. Knop-Gericke and F. M. F. de Groot, J. Phys. Chem. C, 2011, 115, 25422–25428 CrossRef CAS.
- E. M. Erickson, M. S. Thorum, R. Vasic, N. S. Marinkovic, A. I. Frenkel, A. A. Gewirth and R. G. Nuzzo, J. Am. Chem. Soc., 2012, 134, 197–200 CrossRef CAS PubMed.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- E. E. Benson, M. D. Sampson, K. A. Grice, J. M. Smieja, J. D. Froehlich, D. Friebel, J. A. Keith, E. A. Carter, A. Nilsson and C. P. Kubiak, Angew. Chem., Int. Ed., 2013, 52, 4841–4844 CrossRef CAS PubMed.
- D. Karapinar, A. Zitolo, T. N. Huan, S. Zanna, D. Taverna, L. H. Galvao Tizei, D. Giaume, P. Marcus, V. Mougel and M. Fontecave, ChemSusChem, 2020, 13, 173–179 CrossRef CAS PubMed.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS PubMed.
- D. Gianolo, X-Ray Absorption and X-Ray Emission Spectroscopy, Wiley, 2016, ch. 5, pp. 99–124 DOI:10.1002/9781118844243.ch5.
- T. Masuda, Top. Catal., 2018, 61, 2103–2113 CrossRef CAS.
- B. Lassalle-Kaiser, S. Gul, J. Kern, V. K. Yachandra and J. Yano, J. Electron Spectrosc. Relat. Phenom., 2017, 221, 18–27 CrossRef CAS PubMed.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- K. Cheaib, B. Maurice, T. Mateo, Z. Halime and B. Lassalle-Kaiser, J. Synchrotron Radiat., 2019, 26, 1980–1985 CrossRef CAS PubMed.
- S. P. Best, A. Levina, C. Glover, B. Johannessen, P. Kappen and P. A. Lay, J. Synchrotron Radiat., 2016, 23, 743–750 CrossRef CAS PubMed.
- O. Wolter and J. Heitbaum, Ber. Bunsenges. Phys. Chem., 1984, 88, 2–6 CrossRef CAS.
- P. Dubé and G. M. Brisard, J. Electroanal. Chem., 2005, 582, 230–240 CrossRef.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS PubMed.
- Y. Lai, R. J. R. Jones, Y. Wang, L. Zhou and J. M. Gregoire, ACS Comb. Sci., 2019, 21, 692–704 CrossRef CAS PubMed.
- R. Kortlever, C. Balemans, Y. Kwon and M. T. M. Koper, Catal. Today, 2015, 244, 58–62 CrossRef CAS.
- A. H. Wonders, T. H. M. Housmans, V. Rosca and M. T. M. Koper, J. Appl. Electrochem., 2006, 36, 1215–1221 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- R. Kortlever, K. H. Tan, Y. Kwon and M. T. M. Koper, J. Solid State Electrochem., 2013, 17, 1843–1849 CrossRef CAS.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- H. Baltruschat, J. Am. Soc. Mass Spectrom., 2004, 15, 1693–1706 CrossRef CAS PubMed.
- T. Hartung and H. Baltruschat, Langmuir, 1990, 6, 953–957 CrossRef CAS.
- Y. Gao, H. Tsuji, H. Hattori and H. Kita, J. Electroanal. Chem., 1994, 372, 195–200 CrossRef CAS.
- K. Jambunathan, S. Jayaraman and A. C. Hillier, Langmuir, 2004, 20, 1856–1863 CrossRef CAS.
- Y. Hirata, K. Suga and M. Fujihira, Chem. Lett., 1990, 1155–1158 CrossRef CAS.
- H. Kazuya, T. Katsuhiro, S. Hideo and T. Shinobu, Chem. Lett., 1977, 1137–1140 Search PubMed.
- H. Kazuya, T. Katsuhiro, S. Hideo and T. Shinobu, Chem. Lett., 1979, 305–308 Search PubMed.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- L. Chen, Z. Guo, X.-G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K.-C. Lau, T.-C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed.
- K.-W. Fung and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1988, 2153–2159 Search PubMed.
- D. C. Lacy, C. C. L. McCrory and J. C. Peters, Inorg. Chem., 2014, 53, 4980–4988 CrossRef CAS PubMed.
- M. Zhang, M. El-Roz, H. Frei, J. L. Mendoza-Cortes, M. Head-Gordon, D. C. Lacy and J. C. Peters, J. Phys. Chem. C, 2015, 119, 4645–4654 CrossRef CAS.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS PubMed.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 RSC.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- J. P. Collin, A. Jouaiti and J. P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1992, 322, 325–335 CrossRef CAS.
- K. Bujno, R. Bilewicz, L. Siegfries and T. A. Kaden, J. Electroanal. Chem., 1998, 445, 47–53 CrossRef CAS.
- S. Sakaki, J. Am. Chem. Soc., 1992, 114, 2055–2062 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- Y. Wu, B. Rudshteyn, A. Zhanaidarova, J. D. Froehlich, W. Ding, C. P. Kubiak and V. S. Batista, ACS Catal., 2017, 7, 5282–5288 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1994, 116, 5015–5016 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Phys. Chem., 1996, 100, 19981–19985 CrossRef CAS.
- C. Costentin, M. Robert, J. M. Saveant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef CAS PubMed.
- C. Costentin, G. Passard, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- C. G. Margarit, N. G. Asimow, C. Costentin and D. G. Nocera, ACS Energy Lett., 2020, 5, 72–78 CrossRef CAS.
- M. N. Mahmood, D. Massheder and C. J. Harty, J. Appl. Electrochem., 1987, 17, 1223–1227 CrossRef CAS.
- N. Furuya and K. Matsui, J. Electroanal. Chem., 1989, 271, 181–190 CrossRef CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulie, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Isci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- K. Torbensen, C. Han, B. Boudy, N. von Wolff, C. Bertail, W. Braun and M. Robert, Chem. – Eur. J., 2020, 26, 3034–3038 CrossRef CAS PubMed.
- A. Tatin, C. m. Comminges, B. Kokoh, C. Costentin, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5526–5529 CrossRef CAS PubMed.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y. F. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- H. Wang, D. Y. C. Leung and J. Xuan, Appl. Energy, 2013, 102, 1057–1062 CrossRef CAS.
- M. Bevilacqua, J. Filippi, A. Lavacchi, A. Marchionni, H. A. Miller, W. Oberhauser, E. Vesselli and F. Vizza, Energy Technol., 2014, 2, 522–525 CrossRef CAS.
- I. Merino-Garcia, E. Alvarez-Guerra, J. Albo and A. Irabien, Chem. Eng. J., 2016, 305, 104–120 CrossRef CAS.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- N. Furuya and S. Koide, Electrochim. Acta, 1991, 36, 1309–1313 CrossRef CAS.
- N. Sonoyama, M. Kirii and T. Sakata, Electrochem. Commun., 1999, 1, 213–216 CrossRef CAS.
- X. Lu, Y. Wu, X. Yuan, L. Huang, Z. Wu, J. Xuan, Y. Wang and H. Wang, ACS Energy Lett., 2018, 3, 2527–2532 CrossRef CAS.
- C. Jiang, A. W. Nichols, J. F. Walzer and C. W. Machan, Inorg. Chem., 2020, 59, 1883–1892 CrossRef CAS PubMed.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- M. Schreier, P. Gao, M. T. Mayer, J. Luo, T. Moehl, M. K. Nazeeruddin, S. D. Tilley and M. Gratzel, Energy Environ. Sci., 2015, 8, 855–861 RSC.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- M. G. Bradley, T. Tysak, D. J. Graves and N. A. Viachiopoulos, J. Chem. Soc., Chem. Commun., 1983, 349–350 RSC.
- Y. Matsubara, ACS Energy Lett., 2017, 2, 1886–1891 CrossRef CAS.
- M. Beley, J.-P. Collin, J.-P. Sauvage, J.-P. Petit and P. Chartier, J. Electroanal. Chem. Interfacial Electrochem., 1986, 206, 333–339 CrossRef CAS.
- J. P. Petit, P. Chartier, M. Beley and J. P. Sauvage, New J. Chem., 1987, 11, 751 CAS.
- J.-P. Petit, P. Chartier, M. Beley and J.-P. Deville, J. Electroanal. Chem., 1989, 269, 267–281 CrossRef CAS.
- J. O. M. Bockris and J. C. Wass, Mater. Chem. Phys., 1989, 22, 249–280 CrossRef CAS.
- C. R. Cabrera and H. D. Abruña, J. Electroanal. Chem. Interfacial Electrochem., 1986, 209, 101–107 CrossRef CAS.
- J. P. Collin and J. P. Sauvage, Coord. Chem. Rev., 1989, 93, 245–268 CrossRef CAS.
- B. Kumar, J. M. Smieja and C. P. Kubiak, J. Phys. Chem. C, 2010, 114, 14220–14223 CrossRef CAS.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- T. Arai, S. Tajima, S. Sato, K. Uemura, T. Morikawa and T. Kajino, Chem. Commun., 2011, 47, 12664–12666 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- B. Kumar, J. M. Smieja, A. F. Sasayama and C. P. Kubiak, Chem. Commun., 2012, 48, 272–274 RSC.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 RSC.
- E. Torralba-Peñalver, Y. Luo, J.-D. Compain, S. Chardon-Noblat and B. Fabre, ACS Catal., 2015, 5, 6138–6147 CrossRef.
- K. Alenezi, S. K. Ibrahim, P. Li and C. J. Pickett, Chem. – Eur. J., 2013, 19, 13522–13527 CrossRef CAS PubMed.
- D. He, T. Jin, W. Li, S. Pantovich, D. Wang and G. Li, Chem. – Eur. J., 2016, 22, 13064–13067 CrossRef CAS PubMed.
- H. Gerischer, J. Electroanal. Chem., 1977, 82, 133–143 CrossRef CAS.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- A. H. A. Tinnemans, T. P. M. Koster, D. H. M. W. Thewissen and A. Mackor, Recl. Trav. Chim. Pays-Bas, 1984, 103, 288–295 CrossRef CAS.
- M. Schreier, J. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Grätzel, J. Am. Chem. Soc., 2016, 136, 1938–1946 CrossRef PubMed.
- K. Sekizawa, S. Sato, T. Arai and T. Morikawa, ACS Catal., 2018, 8, 1405–1416 CrossRef CAS.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354–365 CrossRef CAS.
- Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef CAS.
- T.-T. Li, B. Shan and T. J. Meyer, ACS Energy Lett., 2019, 4, 629–636 CrossRef CAS.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef CAS PubMed.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- B. Shan, S. Vanka, T.-T. Li, L. Troian-Gautier, M. K. Brennaman, Z. Mi and T. J. Meyer, Nat. Energy, 2019, 4, 290–299 CrossRef CAS.
- A. J. Bard, A. B. Bocarsly, F. R. F. Fan, E. G. Walton and M. S. Wrighton, J. Am. Chem. Soc., 1980, 102, 3671–3677 CrossRef CAS.
- J. J. H. Pijpers, M. T. Winkler, Y. Surendranath, T. Buonassisi and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10056–10061 CrossRef CAS PubMed.
- T. E. Rosser, C. D. Windle and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 7388–7392 CrossRef CAS PubMed.
- Y.-S. Kim, S. Kriegel, K. D. Harris, C. Costentin, B. Limoges and V. Balland, J. Phys. Chem. C, 2017, 121, 10325–10335 CrossRef CAS.
- N. E. Mendieta-Reyes, W. Cheuquepán, A. Rodes and R. Gómez, ACS Catal., 2020, 10, 103–113 CrossRef CAS.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- D. Bae, T. Pedersen, B. Seger, M. Malizia, A. Kuznetsov, O. Hansen, I. Chorkendorff and P. C. K. Vesborg, Energy Environ. Sci., 2015, 8, 650–660 RSC.
- M.-K. Son, L. Steier, M. Schreier, M. T. Mayer, J. Luo and M. Gratzel, Energy Environ. Sci., 2017, 10, 912–918 RSC.
- K. Mochizuki, S. Manaka, I. Takeda and T. Kondo, Inorg. Chem., 1996, 35, 5132–5136 CrossRef CAS.
- M. Shinjiro, Y. Kiichi, P. Chyongjin and Y. Shozo, Chem. Lett., 1991, 2099–2100 Search PubMed.
- S. Matsuoka, K. Yamamoto, T. Ogata, M. Kusaba, N. Nakashima, E. Fujita and S. Yanagida, J. Am. Chem. Soc., 1993, 115, 601–609 CrossRef CAS.
- T. Ogata, Y. Yamamoto, Y. Wada, K. Murakoshi, M. Kusaba, N. Nakashima, A. Ishida, S. Takamuku and S. Yanagida, J. Phys. Chem., 1995, 99, 11916–11922 CrossRef CAS.
- E. Fujita, D. J. Szalda, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1988, 110, 4870–4871 CrossRef CAS.
- E. Fujita, C. Creutz, N. Sutin and B. S. Brunschwig, Inorg. Chem., 1993, 32, 2657–2662 CrossRef CAS.
- T. Ogata, S. Yanagida, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 1995, 117, 6708–6716 CrossRef CAS.
- T. Ogata, S. Yanagida, B. S. Brunschwig and E. Fujita, Energy Convers. Manage., 1995, 36, 669–672 CrossRef CAS.
- Y. Qin, L. Chen, G. Chen, Z. Guo, L. Wang, H. Fan, M. Robert and T.-C. Lau, Chem. Commun., 2020, 56, 6249–6252 RSC.
- T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and E. Fujita, J. Phys. Chem. A, 1999, 103, 7742–7748 CrossRef CAS.
- J. Grodkowski and P. Neta, J. Phys. Chem. A, 2000, 104, 4475–4479 CrossRef CAS.
- J. Grodkowski, D. Behar, P. Neta and P. Hambright, J. Phys. Chem. A, 1997, 101, 248–254 CrossRef CAS.
- J. Bonin, M. Chaussemier, M. Robert and M. Routier, ChemCatChem, 2014, 6, 3200–3207 CrossRef CAS.
- J. Grodkowski, T. Dhanasekaran, P. Neta, P. Hambright, B. S. Brunschwig, K. Shinozaki and E. Fujita, J. Phys. Chem. A, 2000, 104, 11332–11339 CrossRef CAS.
- J. Grodkowski, P. Neta, E. Fujita, A. Mahammed, L. Simkhovich and Z. Gross, J. Phys. Chem. A, 2002, 106, 4772–4778 CrossRef CAS.
- J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed.
- H. Rao, J. Bonin and M. Robert, Chem. Commun., 2017, 53, 2830–2833 RSC.
- H. Rao, J. Bonin and M. Robert, ChemSusChem, 2017, 10, 4447–4450 CrossRef CAS PubMed.
- H. Rao, J. Bonin and M. Robert, J. Phys. Chem. C, 2018, 122, 13834–13839 CrossRef CAS.
- P. G. Alsabeh, A. Rosas-Hernández, E. Barsch, H. Junge, R. Ludwig and M. Beller, Catal. Sci. Technol., 2016, 6, 3623–3630 RSC.
- A. Rosas-Hernandez, P. G. Alsabeh, E. Barsch, H. Junge, R. Ludwig and M. Beller, Chem. Commun., 2016, 52, 8393–8396 RSC.
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC.
- H. Takeda, K. Ohashi, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 4354–4357 CrossRef CAS PubMed.
- E. Kimura, X. Bu, M. Shionoya, S. Wada and S. Maruyama, Inorg. Chem., 1992, 31, 4542–4546 CrossRef CAS.
- C. Herrero, A. Quaranta, S. El Ghachtouli, B. Vauzeilles, W. Leibl and A. Aukauloo, Phys. Chem. Chem. Phys., 2014, 16, 12067–12072 RSC.
- K. Nobuko, H. Yuichiro, H. Takuji, S. Hideki and K. Kazuyuki, Bull. Chem. Soc. Jpn., 1999, 72, 725–731 CrossRef.
- X. Wang, V. Goudy, G. Genesio, J. Maynadié, D. Meyer and M. Fontecave, Chem. Commun., 2017, 53, 5040–5043 RSC.
- A. D. Handoko, K. Li and J. Tang, Curr. Opin. Chem. Eng., 2013, 2, 200–206 CrossRef.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- T. Jin, C. Liu and G. Li, J. Coord. Chem., 2016, 69, 1748–1758 CrossRef CAS.
- T. Fenton, S. Gillingham, T. Jin and G. Li, Dalton Trans., 2017, 46, 10721–10726 RSC.
- X. Wang, I. Thiel, A. Fedorov, C. Copéret, V. Mougel and M. Fontecave, Chem. Sci., 2017, 8, 8204–8213 RSC.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- T. Atoguchi, A. Aramata, A. Kazusaka and M. Enyo, J. Chem. Soc., Chem. Commun., 1991, 156–157 RSC.
- W. W. Kramer and C. C. L. McCrory, Chem. Sci., 2016, 7, 2506–2515 RSC.
- N. Morlanés, K. Takanabe and V. Rodionov, ACS Catal., 2016, 6, 3092–3095 CrossRef.
- S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 12180–12184 CrossRef CAS PubMed.
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y.-F. Han, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- S. Lian, M. S. Kodaimati and E. A. Weiss, ACS Nano, 2018, 12, 568–575 CrossRef CAS PubMed.
- T. Jin, C. Liu and G. Li, Chem. Commun., 2014, 50, 6221–6224 RSC.
- C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Chem. – Eur. J., 2015, 21, 3746–3754 CrossRef CAS PubMed.
- C. Liu, T. Jin, M. E. Louis, S. A. Pantovich, S. L. Skraba-Joiner, T. Rajh and G. Li, J. Mol. Catal. A: Chem., 2016, 423, 293–299 CrossRef CAS.
- G. Neri, M. Forster, J. J. Walsh, C. M. Robertson, T. J. Whittles, P. Farras and A. J. Cowan, Chem. Commun., 2016, 52, 14200–14203 RSC.
- T. Wu, L. Zou, D. Han, F. Li, Q. Zhang and L. Niu, Green Chem., 2014, 16, 2142–2146 RSC.
- L. Lin, C. Hou, X. Zhang, Y. Wang, Y. Chen and T. He, Appl. Catal., B, 2018, 221, 312–319 CrossRef CAS.
- C. Cometto, R. Kuriki, L. Chen, K. Maeda, T.-C. Lau, O. Ishitani and M. Robert, J. Am. Chem. Soc., 2018, 140, 7437–7440 CrossRef CAS PubMed.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T.-C. Lau, J. Bonin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef CAS PubMed.
- M. F. Kuehnel, C. D. Sahm, G. Neri, J. R. Lee, K. L. Orchard, A. J. Cowan and E. Reisner, Chem. Sci., 2018, 9, 2501–2509 RSC.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- S. Lian, M. S. Kodaimati, D. S. Dolzhnikov, R. Calzada and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 8931–8938 CrossRef CAS PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- H. Rao, C.-H. Lim, J. Bonin, G. M. Miyake and M. Robert, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed.
- M. D. Rail and L. A. Berben, J. Am. Chem. Soc., 2011, 133, 18577–18579 CrossRef CAS PubMed.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313 CrossRef CAS PubMed.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- A. Taheri, E. J. Thompson, J. C. Fettinger and L. A. Berben, ACS Catal., 2015, 5, 7140–7151 CrossRef CAS.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281–1285 CrossRef CAS PubMed.
- C.-Y. Zhu, Y.-Q. Zhang, R.-Z. Liao, W. Xia, J.-C. Hu, J. Wu, H. Liu and F. Wang, Dalton Trans., 2018, 47, 13142–13150 RSC.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed.
- U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Nat. Commun., 2014, 5, 5883 CrossRef CAS PubMed.
- N. D. Loewen, E. J. Thompson, M. Kagan, C. L. Banales, T. W. Myers, J. C. Fettinger and L. A. Berben, Chem. Sci., 2016, 7, 2728–2735 RSC.
- K. Kusuda, R. Ishihara, H. Yamaguchi and I. Izumi, Electrochim. Acta, 1986, 31, 657–663 CrossRef CAS.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X.-a. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- S. Kapusta and N. Hackerman, J. Electrochem. Soc., 1984, 131, 1511–1514 CrossRef CAS.
- N. Furuya and K. Matsui, J. Electroanal. Chem. Interfacial Electrochem., 1989, 271, 181–191 CrossRef CAS.
- T. V. Magdesieva, T. Yamamoto, D. A. Tryk and A. Fujishima, J. Electrochem. Soc., 2002, 149, D89–D95 CrossRef CAS.
- V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed.
- J. D. Cope, N. P. Liyanage, P. J. Kelley, J. A. Denny, E. J. Valente, C. E. Webster, J. H. Delcamp and T. K. Hollis, Chem. Commun., 2017, 53, 9442–9445 RSC.
- S. Gonglach, S. Paul, M. Haas, F. Pillwein, S. S. Sreejith, S. Barman, R. De, S. Müllegger, P. Gerschel, U.-P. Apfel, H. Coskun, A. Aljabour, P. Stadler, W. Schöfberger and S. Roy, Nat. Commun., 2019, 10, 3864 CrossRef PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- K. Ogura and S. Yamasaki, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 267–271 RSC.
- K. Ogura and K. Takamagari, J. Chem. Soc., Dalton Trans., 1986, 1519–1523 RSC.
- K. Ogura and I. Yoshida, J. Mol. Catal., 1988, 47, 51–57 CrossRef CAS.
- C. Cometto, L. Chen, D. Mendoza, B. Lassalle-Kaiser, T.-C. Lau and M. Robert, ChemSusChem, 2019, 12, 4500–4505 CrossRef CAS PubMed.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S.-M. Ng, C.-C. Ko, G. Liu, L. Chen, M. Robert and T.-C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |