
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nucleic acid constructs for the interrogation of multivalent protein interactions
Sean B.
Yeldell
and
Oliver
Seitz
 *
*
Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: oliver.seitz@chemie.hu-berlin.de
First published on 1st September 2020
Abstract
Multivalency is nature's way to establish firm and specific interactions when the binding sites of a protein receptor have only low affinity for monovalent ligands. Recently, researchers are increasingly using nucleic acid architectures for multivalent ligand presentation to unravel the mechanisms of multivalency-enhanced interactions and create high affinity binding agents. In contrast to other polymers, nucleic acid materials are capable of accessing a wide variety of rigid three-dimensional structures through the sequence-programed self-assembly of component strands. By controlling the number of ligands and their distances, researchers can construct tailor-made probes for interrogating multivalent interactions with Ångstrom precision. Nucleic acid assemblies have been used to address fundamental questions of multivalency in order to unravel how monovalent interaction strength, scaffold flexibility, distances between interacting sites and spatial arrangement influence the achievable affinity gains. In a slightly different approach, nucleic acid constructs have been applied as chemical dimerizers of protein receptors, to investigate the importance of receptor proximity or construct tools that provide control over biological signal transduction processes. In this review, we discuss multivalent nucleic acid–ligand conjugates in the context of the biological protein receptors they interrogate. We recount pioneering work and seminal studies performed within the last 10 years describing the in vitro interrogation of proteins recognizing carbohydrate ligands, small molecules, peptides and nucleic acid aptamers and we portray work performed with viruses, cell models, and whole organisms.
 Sean B. Yeldell | Sean Yeldell received his BSc in Biochemistry from Case Western Reserve University before pursuing his PhD in Chemistry under the guidance of Dr Ivan Dmochowski at the University of Pennsylvania. There, his thesis work centered on the development of oligonucleotide probes for the capture of transcriptomes from spatially-resolved, living cells. After graduating, he followed his passion for oligonucleotide-based technologies by accepting a postdoctoral research position in the laboratory of Prof. Dr Oliver Seitz at the Humboldt-Universität zu Berlin. His recent work has focused on new ways to apply multivalent nucleic acid constructs to cell surface receptors. |
 Oliver Seitz | Oliver Seitz obtained his PhD from the University of Mainz in 1995. After postdoctoral research at the Scripps Research Institute in La Jolla he moved to the University of Karlsruhe, Germany. In 2000, he became group leader at the Max-Planck Institute of Molecular Physiology in Dortmund, Germany, and in 2003 he was appointed Full Professor at the Humboldt-Universität zu Berlin. Oliver Seitz has a keen interest on developing chemistry that allows the interrogation and perturbation of cellular and biochemical processes. Recently, he is focusing on protein synthesis, peptide- and nucleic acid-templated chemistry and RNA and protein imaging. He is a recipient of an ERC Advanced Grant and the Max Bergmann Medal 2018. |
Key learning points• Multivalent ligand–receptor systems are employed by nature to enhance interaction affinity and specificity.• Oligonucleotide scaffolds presenting multivalent ligands can be used to probe and manipulate biological systems with unrivaled precision and ease of preparation. • Multivalent nucleic acid constructs are compatible with a wide range of ligand types and receptor classes, and can be utilized to create high affinity binding agents for studies in solution and on surfaces. • Nucleic acid hybridization and strand displacement allows reversible switching of bioactivity. |
1. Introduction
The binding of ligands to cell-surface receptors is a cornerstone of life, initiating processes such as cellular adhesion, communication, metabolism, and more. The execution of these processes is highly dependent on how tightly and specifically a given ligand binds to its target receptor. Notably, there are many ligands which exhibit poor binding characteristics individually, but – when linked by a scaffold – are capable of producing a high collective affinity to a corresponding receptor with multiple binding sites.The binding enhancements observed for bi- or multivalent interactions can arise from a combination of mechanisms, including chelation, cross-linking, statistical rebinding and steric shielding.1,2 Chelation refers to bridging of adjacent binding sites (Fig. 1A, left). When a multivalent ligand cluster initially binds its first ligand to a receptor site, the framework connecting the ligands arranges a second ligand in the vicinity of a second receptor site. The better the distance between receptor sites matches the ligand–ligand distance at a given scaffold flexibility the higher the increase in the effective molarity and the stronger the multivalency-induced binding enhancement. However, an entropic cost may need to be paid when a flexible scaffold is confined in an arrangement that allows bridging of binding sites. As a result, rigid scaffolds, if available, typically provide higher binding enhancements. Other types of chelation can also occur when a multivalent ligand connects a primary binding site with a secondary binding site (Fig. 1A, right). Binding enhancements may be the result of interactions involving a multivalent ligand and binding sites on two surface-bound receptors (crosslinking, Fig. 1B). The balance between chelation, subsite binding, and cross-linking is intricate, and depends on scaffold properties, individual ligand affinities, concentration, and more. Constructs which present multiples of ligands in close proximity – clusters of glycans, for example – also utilize the statistical rebinding effect, in which the abundance of ligands around a single receptor site allows for rapid re-filling of the site as soon as a ligand is released (Fig. 1C). Fast rebinding produces slower off-rates, which further enhances overall binding affinities. Steric shielding contributes to multivalency-enhanced interaction when the polyvalent ligand is large enough to hinder the approach of competing non-scaffolded ligands (Fig. 1D).
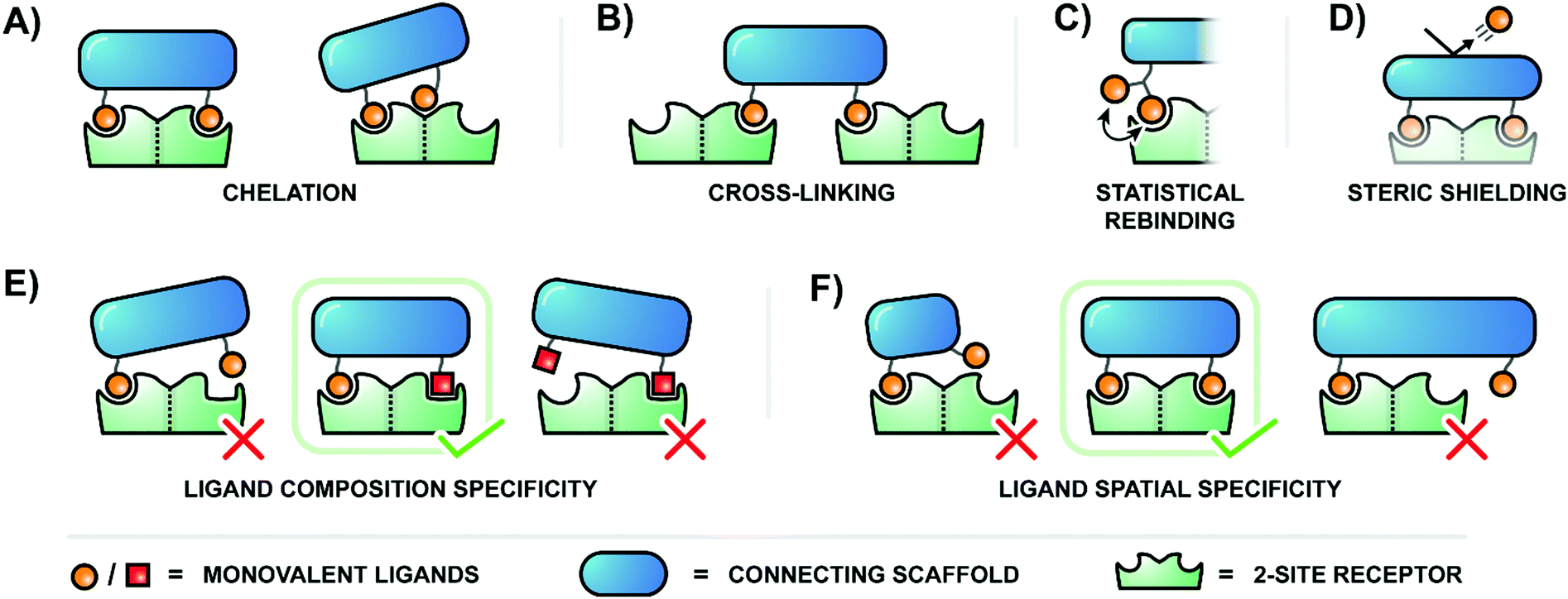 | ||
| Fig. 1 Bi- or multivalent ligand–receptor interactions can produce significant affinity (A–D) or specificity (E and F) enhancements relative to monovalent interactions through several contributing mechanisms. (A) Chelation bridges either two primary binding sites (left) or a primary and a secondary binding site of a single receptor (right). (B) Cross-linking spans binding sites on two different receptor molecules. (C) Statistical rebinding involves the rapid exchange of locally clustered ligands. (D) Steric shielding from a large scaffold can hinder the approach of competing monovalent ligands. Multivalent binding agents must present ligands of the correct composition (E) and spatial context (F) to be recognized by their target receptor. | ||
Multivalent systems can also enhance specificity of binding, mainly through two avenues. The first is to create a more unique binding agent by combining several relatively common ligands (Fig. 1E). For example, this could be as simple as connecting two different sugar residues, or become more complex by incorporating a greater diversity of sugars linked through a variety of branching points. Additionally, the geometric layout of a multivalent receptor also dictates binding specificity (Fig. 1F). In the simplest scenario a receptor presenting two binding sites 100 Å apart would require a bivalent agent to similarly display the two correct ligands at 100 Å apart to bind tightly; a 50 Å separation would fail to reach the second binding site, and a 150 Å separation would overshoot it. A more complex receptor may present three or more binding sites along its curved surface, and the binding pockets of the receptors may require ligands to be presented with a particular orientation. Therefore, the geometry and rigidity of the scaffold connecting the ligands is crucial to ensure that the ligands are correctly displayed in a three-dimensional pattern matching the target receptor protein.
The importance of multivalent ligand–protein interactions, both in fundamental biological processes and when applied to therapeutic agents targeting viral infection and cancerous cells, has motivated the ongoing development of methods to study and modulate these systems.3 While many efforts have focused on displaying multivalent ligands on traditional scaffolds such as polymers, dextrans, or dendrimers,1–3 these materials often struggle to reproduce complex three-dimensional ligand geometries owing to their non-uniformity, their flexibility, or the difficulty in conjugating ligands to specific sites of the polymer.
In contrast to traditional polymers, oligonucleotides are capable of accessing a wide variety of rigid three-dimensional structures through the programed self-assembly of component strands.4 The most common oligonucleotide structure is a rigid double helix (or “duplex”), formed from two complementary, hydrogen-bonded strands (Fig. 2A). A typical B-form double helix has a diameter of 20 Å and completes its right-handed helical rotation every 34 Å, or 10.5 bases. In addition to a two-stranded duplex, three strands can form a triple helix (or “triplex”, Fig. 2B), four strands can create a G-quadruplex (Fig. 2C), and multiple strands can also be joined together via 3-way (Fig. 2D) or 4-way (Fig. 2E) junctions. By carefully combining structural motifs like rigid double helices, flexible single stranded regions, and connecting junctions it is possible to design a precisely-defined scaffold with nearly any geometry imaginable. Furthermore, these complex scaffolds are programmed to self-assemble from component strands through predictable Watson–Crick and Hoogsteen base-pairing rules. Ligands can be site-specifically incorporated into these component strands during solid-phase synthesis, either directly using ligand-modified monomers or through post-synthetic conjugations (Fig. 2F). Lastly, the structure of an oligonucleotide scaffold can be easily tuned by swapping out any number of component strands for alternatives of different lengths or ligand configurations. In this manner, a small handful of component strands can be combined in different permutations to produce a library of hundreds or even thousands of unique arrangements.
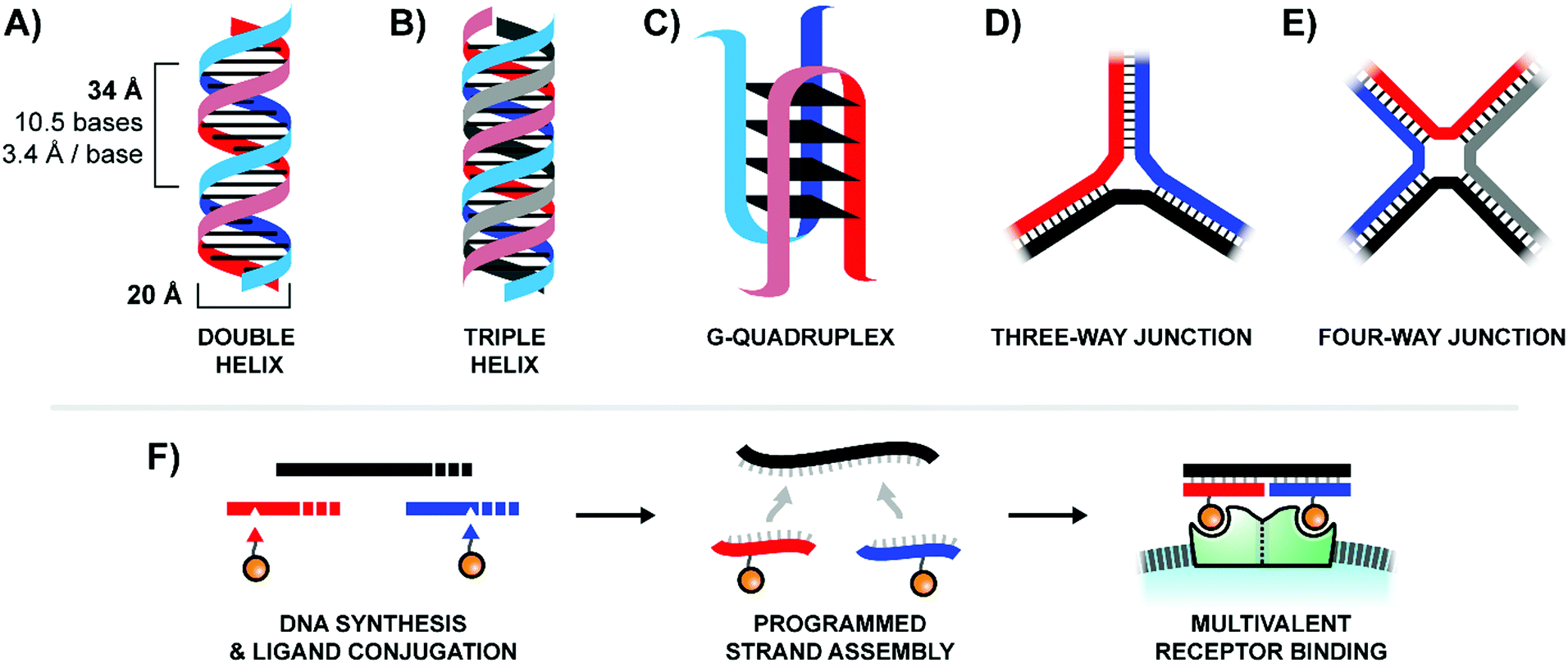 | ||
| Fig. 2 Common architectures of nucleic acids (A–E) and a simple example of the production and application process for a bivalent binding agent based around a double helix design (F). | ||
Within the last 10 years, an increasing number of researchers have recognized the advantages provided by nucleic acid-programmed multivalency. Various excellent reviews have surveyed the diverse architectures of multivalent nucleic acid constructs.5,6 In this review, we contextualize these designs within the biological protein receptors they interrogate. We first begin by introducing the multivalent constructs for the in vitro interrogation of proteins recognizing carbohydrate ligands and then advance to recognition of small molecules, peptides and nucleic acid aptamers before progressing to work performed with viruses, cell models, and whole organisms. We then close with a discussion of trends observed in the progression of the field as well as general guidelines to consider when designing multivalent nucleic acid constructs.
2. Multivalent protein–carbohydrate interactions
Carbohydrates are key components of biological recognition events governing the interactions between two cells or between cells and bacteria or viruses. The protein receptors that bind specific carbohydrates are known as lectins. Typically, lectins have low affinity for a single sugar residue and multivalent presentation of both carbohydrate recognition domains (CRDs) and carbohydrate ligands is required to establish firm interactions at low concentration. As a result, multivalent ligand presentation has been the key approach in studies aiming for inhibitors of lectin–carbohydrate interactions.7 Highest potencies are usually achieved with high-valent sugar displays based on multivalent scaffolds such as dendrimers, polymers or nanoparticles. The Kazukiyo Kobayashi group was first to recognize the potential provided by the use of DNA scaffolds. In their initial 2001 publication, they devised a system based around a DNA duplex featuring a 10mer double-stranded core flanked by unpaired 10mer overhangs on either side (Fig. 3A).8 Because these sticky ends were complementary, a large number of these “half-sliding” complexes link together into a nanometer-sized rigid scaffold. Galactose was aligned along a single helical face by attachment at 34 or 68 Å intervals. Binding measurements with the well-characterized plant lectin Ricinus communis agglutinin (RCA120), which binds clusters of up to four sugars, showed cooperative binding suggesting that the lectin recognized two or more galactose units on the DNA complex. Of note, reduced RCA120 affinity was observed when the 20 nucleotide register was shortened or elongated by two nucleotides.9 These results highlight a core principle: the spatial arrangement of ligands presented to multivalent receptor systems such as RCA120 can be more important than the raw ligand density.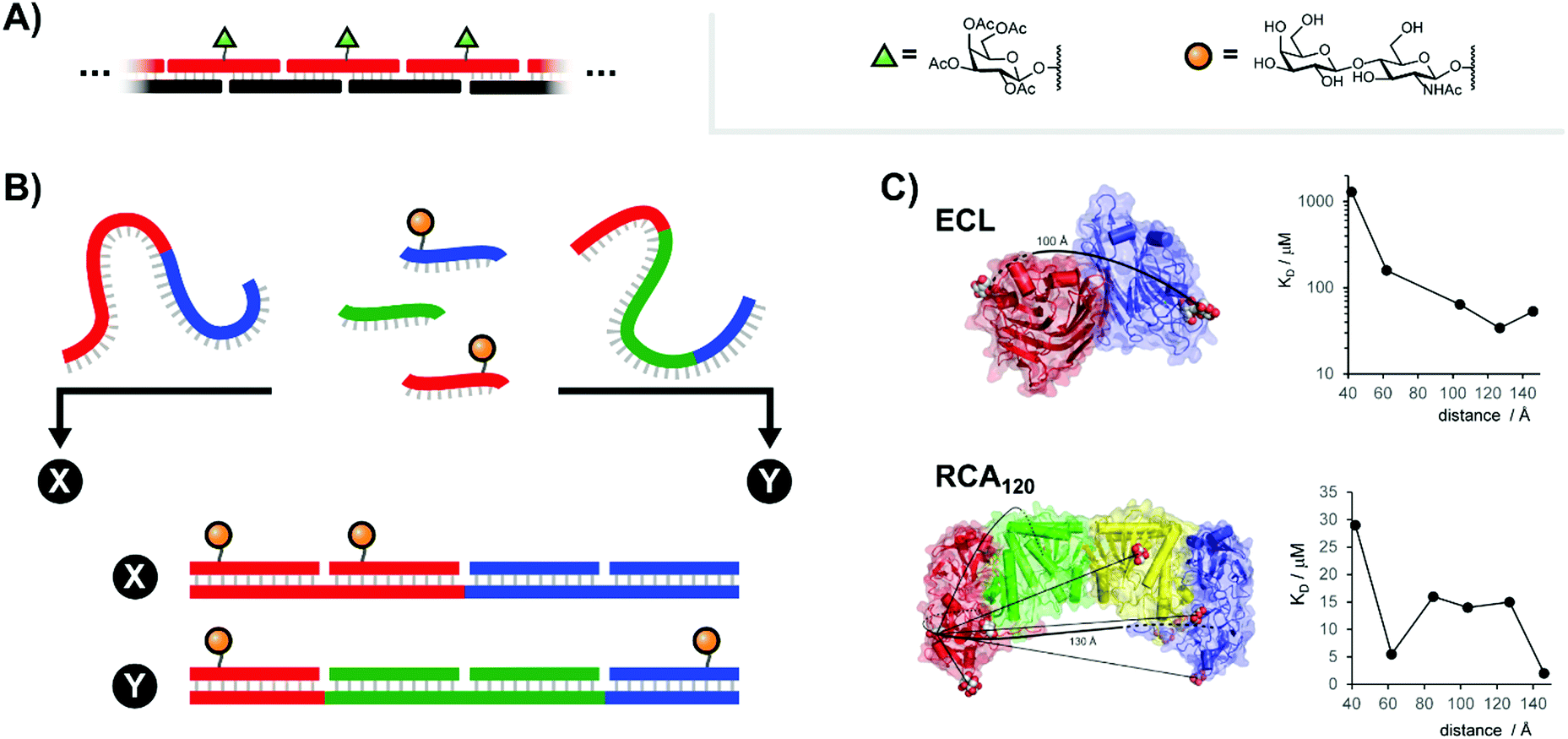 | ||
| Fig. 3 Architectures of multivalent nucleic acid probes presenting carbohydrate ligands. (A) Repeating “half-slide” formation for binding to Ricinus communis agglutinin (RCA120) (ref. 8). (B) Tunable “molecular ruler” duplex design for targeting Erythrina cristagalli lectin (ECL) (ref. 10) or RCA120 (ref. 12). By using different combinations of scaffold strands and ligand strands, constructs could be assembled with unique ligand arrangements (example: “X” vs. “Y”). (C) Crystal structures for ECL and RCA120 depicting distances between binding sites as well as the distance-affinity curves obtained from the corresponding multivalent screening experiments (reproduced from ref. 13 with permission from John Wiley & Sons Inc., copyright 2019). | ||
In highly polyvalent polymer systems, only a few out of the many glyco ligands are needed for binding interactions and the majority of the expensive sugar units will be redundant. A higher ligand economy is offered by approaches that allow the presentation of glyco ligands in a distance matching the arrangement of CRDs on the lectin. We were interested to find out whether DNA scaffolds would enable bivalent sugar recognition if the lectin's CRDs are separated by distances larger than the 68 Å assessed by Kobayashi.10 As a test system, we selected the Erythrina cristagalli lectin (ECL) which exhibits specific binding to N-acetyl-lactosamine (LacNAc) groups. ECL's receptor sites are situated on opposite sides of a dimer at 65 Å Eucledian distance (Fig. 3C, top left). Bridging of the two biding sites, however, would require a roughly 100 Å long spacer to bend over the convex protein surface. In order to reduce the length of scaffold strands and enable a more fine-grained alignment of ligands, we used peptide nucleic acid (PNA) strands, that have higher affinity for complementary DNA than DNA itself. The helical twist of DNA–PNA duplexes comprises 13 base pairs with 42 Å pitch.11 The design involved three different PNA-13mer strands and two different LacNAc–PNA-13mer conjugates. Hybridization of four PNA-13mers to 52 nt long DNA templates could give rise to 324 combinations, differing by the number of LacNAc ligands and their spatial arrangement (Fig. 3B). Binding was assessed by surface plasmon resonance (SPR). The study showed that care has to be taken in assays with immobilized lectins, as distances between LacNAc units did not matter at high ECL surface loads. However, a distinct distance-affinity profile (Fig. 3C, top right) became apparent at low ECL density. Amongst the bivalent complexes tested, highest affinity (33-fold enhancement over affinity of the free ligand) was obtained when the LacNAc ligands were spaced in 104 Å distance. Molecular dynamics (MD) simulations showed that nick sites and single stranded template regions provide sufficient flexibility to allow bending over a curved protein surface. In subsequent work, the approach was applied to a spatial screening of RCA120.12 This tetrameric 120 kDa lectin features four carbohydrate-binding subunits arranged in a BAAB pattern, with each B subunit binding to galactose or LacNAc. SPR affinity screening of a library of oligonucleotide scaffolds presenting LacNAc groups at increasing distances (42–146 Å) revealed a 70-fold binding enhancement for the 140 Å-separated bivalent probe relative to the monovalent control, in approximate agreement with the known ≥130 Å length required to connect the RCA120's B subunit binding sites along the protein surface (Fig. 3C, bottom).13 Additionally, a 62 Å-separated probe also exhibited significant binding enhancements. This spacing had also been identified by Kobayashi.9 Because this distance is too short to span the B subunits of a single RCA120 protein and because our experiments were conducted at concentrations dilute enough to minimize cross-linking, this result implies the engagement of a different LacNAc binding site.
A recurring question in the design of scaffolds for multivalent binding is: how important is scaffold rigidity? We compared two entirely different scaffold types based on self-assembled rigid nucleic acid complexes and flexible polyethylene glycol (PEG).14 The study centered on hemagglutinin (HA) of an Influenza A virus (IAV). In the HA trimer from IAVs, binding sites for the recognition of sialylα2-6LacNAc are positioned in 42 Å distance. We conjugated sialyl-LacNAc ligands to PNA and polyethylene glycol comprised of 3–144 monomer units. Microscale thermophoresis measurements with a soluble HA trimer exposed the limitations of the flexible PEG tethers as the bivalent PEG conjugates failed to show enhanced binding. Computational modeling was used to assess the effective molarity (ceff) of the second ligand at the second binding site after the first receptor–ligand interaction had taken place. The best fitting PEG scaffold provided an effective molarity of 45 μM, which is too low to establish the second interaction with a ligand that has only millimolar affinity. By contrast, a PNA–DNA scaffold arranging the sugar residues in 52 Å distance provides a three orders of magnitude higher effective molarity, resulting in a 107-fold enhanced affinity (KD = 29 μM). This comparison points to the power of DNA scaffolds which provide high effective concentration of ligands, thereby facilitating multivalency enhanced binding even for those cases in which the strength of the monovalent receptor–ligand interaction would be too weak to elicit avidity improvements with conventional, less rigid linkers.
Extending the repertoire of nucleic acid scaffolds applied to lectin studies, the Ebara group explored a Y-shaped, DNA 3-way junction (3WJ) to produce a high-affinity binder for the model lectin concanavalin A (ConA) (Fig. 4A).15 ConA arranges three of its four sugar-binding subunits in a triangular pattern, a layout shared by both mannose binding protein (MBP-A) and influenza hemagglutinin (HA). A small panel of constructs was generated with varying placements of maltose and lactose units, unpaired junction sizes, and the scaffold rigidity was tuned by leaving the arms single- or double-stranded. Fluorescence titrations against ConA revealed that several 3WJ DNA-carbohydrate conjugates featured relatively high affinities, and this high affinity could be abolished by reducing the number of decorated arms or enforcing an overly rigid design through fully-double-stranded arms. With 18 maltose ligands, the optimal binder had a binding affinity more than 700-fold higher than monovalent maltose. Follow-up experiments with lactose-modified 3WJs targeting RCA120 yielded similar trends.
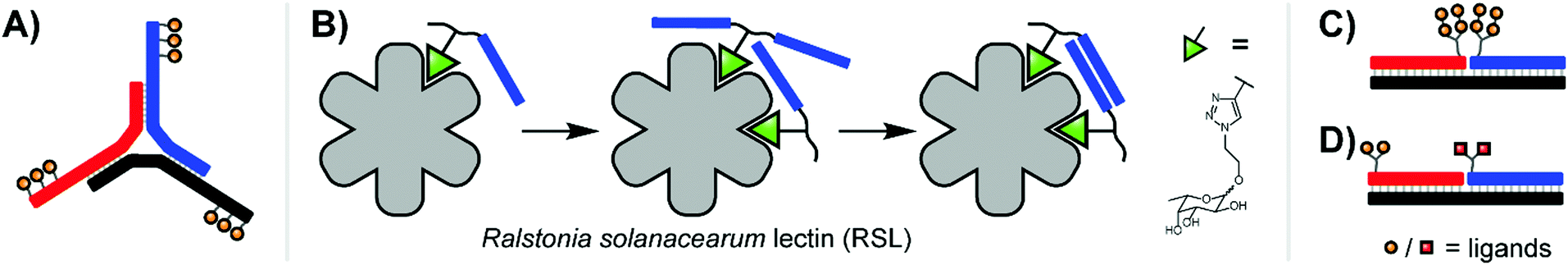 | ||
| Fig. 4 (A) 3-Way junction construct for interrogating concanavalin A (ConA) (ref. 15). (B) PNA–fucose conjugates dynamically assemble on the surface of Ralstonia solanacearum lectin, establishing a bivalent interaction with a binding affinity more than 700-fold higher than that of the monovalent ligand (ref. 16). (C) Duplex design for emulating HIV gp120 epitope (ref. 20). (D) Duplex design targeting DC-SIGN (ref. 17). | ||
Recent work from Machida et al. explored binding of fucose-PNA conjugates to a trimeric Burkholderia ambifaria lectin (BambL).16 Bacteria of the Burkholderia species can cause severe lung infections in Cystic Fibrosisi patients. BambL arranges 6 glycane binding sites on a propeller fold, which recognizes fucosylated epitopes and is used by the bacterium for attachment, suggesting that multivalency-enhanced binding is achievable with PNA assemblies. To comply with the 20 Å distance between binding sites, fucose ligands were incorporated via highly flexible junctions that connected 8 nt long PNA–PNA duplex segments. Constructs presenting 1–6 fucose residues were screened for binding of BambL by SPR. The highest affinity gain (from KD = 362 nM to 3.3 nM) was obtained for a construct displaying two fucose ligands (Fig. 4B). Subsequently, the authors found that affinity enhancements could also be achieved by using a dynamic assembly of fucose–PNA-4mer conjugates. This is a remarkable observation. Duplexes formed by PNA-4mers are thermodynamically unstable and at room temperature the majority of conjugates will exist in single-stranded form. Of note, protein binding and PNA–PNA hybridization cooperate and reinforce each other and, as a result, chelate binding can be induced despite low thermodynamic stability of the assemblies. Further studies proved that dynamic fucose–PNA assemblies inhibit binding of BambL to H1299 lung epithelial cells 723-fold more potently than unconjugated fucose. Dynamic DNA assembly has also been used for the construction of DNA-encoded dynamic combinatorial libraries of small molecules (see next section).17–19
Antibodies are another important class of carbohydrate recognizing proteins. In 2009, the Winssinger group reported an ingenious approach to emulate the architecture of HIV's gp120 epitope, a trimer of mannose-rich nonasaccharides presented on the viral surface which had garnered significant attention as a potential route to a HIV vaccine.20 However, not only are these nonasaccharides difficult to synthesize, but it's believed that only an outer subset of their glycans are actually involved in receptor interactions. The Winssinger lab therefore broke the nonasaccharide into smaller linear or branched sugar clusters and arranged mixtures of these components at 30–90 Å spacings on DNA templates to create a library of ∼30 architectures (Fig. 4C). To assess the accuracy of their emulation, binding of the glycan–DNA complexes to an antibody with established broad-spectrum activity against HIV (“2G12 Antibody”) was measured by SPR. An α-1,2-mannose disaccharide unit with 11 atom branch separation proved critical for binding, and placing two branched disaccharides as close together as possible produced the optimal binder, with a KD of 4.2 μM. In subsequent work, the Winssinger group screened a DNA-templated library of PNA–sugar conjugates against DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin), which is normally involved in modulating the immune response of immature dendritic cells, but its typical preference for mannose-rich glycans can be hijacked by HIV to infect the dendritic cells.17 The PNA-encoded glycans were used for the assembly of a bivalent library (Fig. 4D). A heterobivalent mannose(6N3)-α1,2-mannose assembly emerged as a highly selected glycan providing submicromolar affinity (KD = 0.13 μM) for DC-SIGN.
Studies of multivalent interactions on DNA-type scaffolds typically rely on well-defined 3D structures of nucleic acid complexes. To test a different approach Krauss and co-workers used directed evolution to screen for DNA scaffolds that have high affinity for the HIV neutralizing antibody 2G12.21 Their approach centered on a cyclical selection process known as SELMA (selection of modified aptamers) to refine the structure of the DNA scaffold that presents azido groups used afterwards for attachment of tetramannosides via cupper click chemistry (Fig. 5). After 7 rounds of selection, mutagenesis, and truncation studies, the optimal sequence produced a submicromolar binder for 2G12 (KD = 220 nM). Based on the number of alkinyl nucleotides in the emerging sequence, the best binder displayed 9–10 tetramannose units.
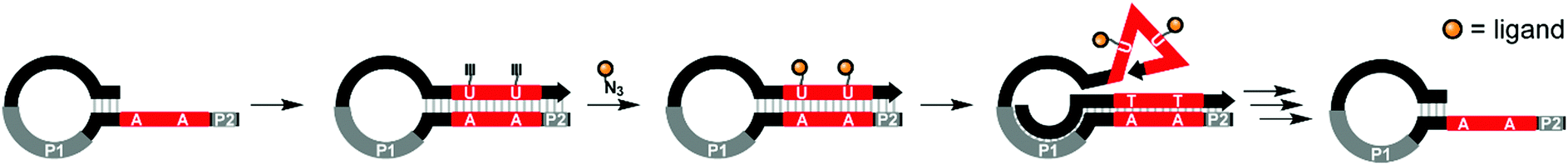 | ||
| Fig. 5 SELMA (selection of modified aptamers) process: a library of hairpins is extended by polymerase which incorporates alkyne-modified uridines. Azide-modified ligands are conjugated through Cu-mediated click chemistry. Extension from primer site P1 ejects the ligand-decorated strand from the hairpin, allowing it to fold into a three-dimensional shape, which is screened against a target compound. Through multiple subsequent steps the starting hairpin can be regenerated for further selection rounds (ref. 21). | ||
3. Multivalent protein interactions with small molecules
The effect of affinity increase by multivalence is not restricted to protein–carbohydrate interactions but rather a generic principle. Antibodies are a prominent example. In seminal work, Baird interrogated the initiating step of the allergic response pathway, in which a multivalent antigen cross-links receptor-bound IgE antibodies on the surface of mast cells or basophils and triggers degranulation of histamine.22 Two complementary DNA strands were labeled at the 5′-end with the model antigen dinitrophenol (DNP) (Fig. 6A). Hybridization provided DNA duplexes presenting two ligands for DNP-reactive IgE antibodies in 13–30 nt distance. The shortest bivalent complexes triggered the highest rates of degranulation pointing to the need for communication between adjacent IgE antibodies and immune cell receptors. A similar trend was observed with a trimeric design, arranging three 5′ DNP-antigens on a DNA 3-way junction in 50–150 Å distance (Fig. 6B).23 Shorter complex lengths once again produced the strongest degranulation response, nearly matching the degree of degranulation afforded by the multivalent DNP-labeled BSA control.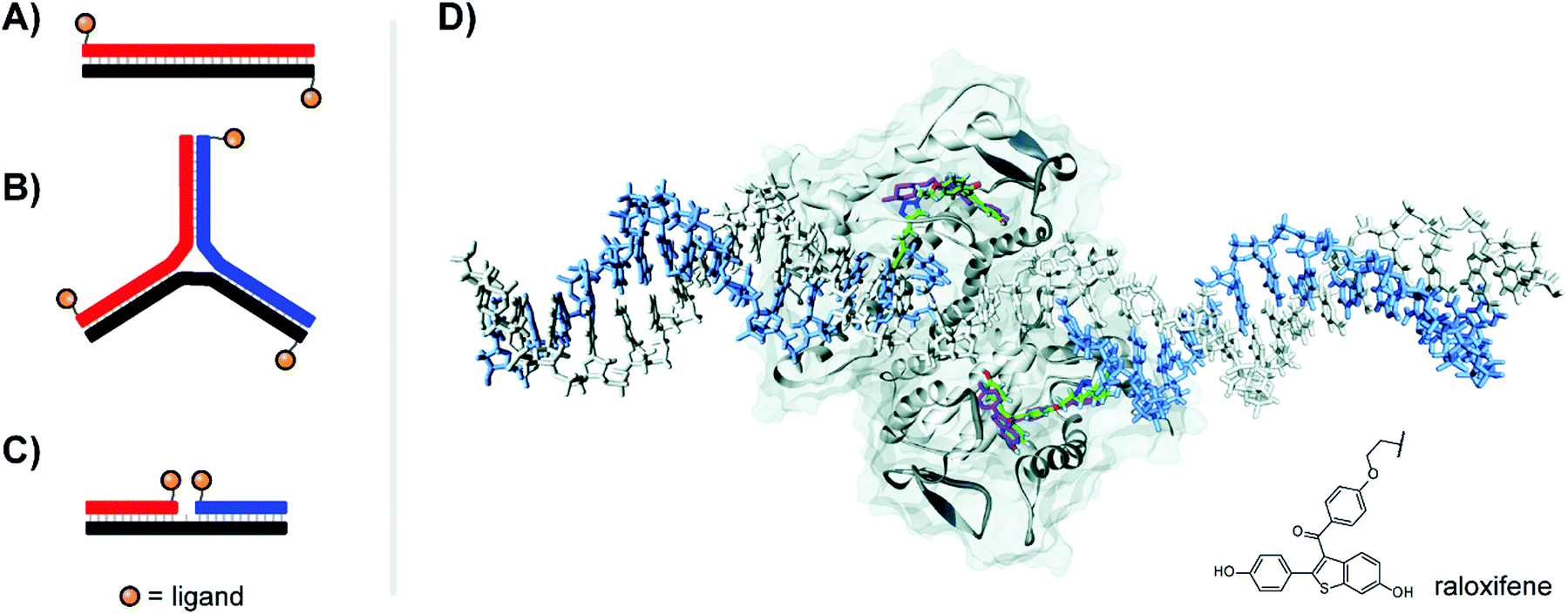 | ||
| Fig. 6 Architectures of multivalent nucleic acid probes presenting small molecule ligands. Duplex with dinitrophenyl (DNP) ligand attached to the 5′ end of each strand (A, ref. 22) or 3-way junction configuration (B, ref. 23), both for targeting the FcεRI receptor of immune cells. (C) Ternary complex design for presentation of selective estrogen receptor modulators (SERMs) to the estrogen receptor (ER-α) (ref. 24). (D) Docking of ternary DNA complex presenting two raloxifene ligands (green) to the ligand-binding domain of ER-α (ref. 24). The position of unconjugated raloxifene is shown in magenta, and the structure of conjugated raloxifene is shown in the lower right. Reproduced from ref. 24 with permission from John Wiley & Sons Inc., copyright 2011. | ||
Our lab applied DNA-programmed spatial screening to the estrogen receptor (ER).24 At nanomolar concentrations the ER is a dimer. Binding of a hormone such as estradiol triggers binding to hormone response elements on DNA to influence transcription. It has been a longstanding idea to present selective estrogen receptor modulators (SERMs) such as hexestrol, raloxifene or 4-hydroxytamoxifene as dimers to the ER. We found that flexibly linked SERMs suffer from intramolecular hydrophobic interactions that penalize bivalent recognition.25 Such interactions cannot occur when the SERMs are arranged on rigid DNA scaffolds. Indeed, for the first time a 270% or 400% binding enhancement relative to the monovalent ligand was observed when two raloxifene units were displayed in 3 nt or 6 nt distance from ternary complexes (Fig. 6C). The display of tamoxifene revealed a similar distance-affinity profile. While the 6–7 nt (38–40 Å including tethers between DNA and SERM) distance is in agreement with the spacing of the two estradiol binding sites (Fig. 6D), a 3 nt (23 Å) distance is too short for bridging. Computational studies predicted the existence of a hydrophobic patch nearby the canonical binding pocket, which was further supported by experiments replacing SERMs with non-specific hydrophobic groups capable of also binding to this region.24 The discovery of secondary binding sites adjacent to the primary binding site was a recurring phenomenon both in our work in others, and will be discussed further in Section 6.
In a burgeoning field, small molecules are attached to DNA, which serves as a code to identify binders selected by affinity purification.26 The technique also allows the combinatorial display of pharmacophores for fragment-based drug discovery. Recently, small molecule–DNA conjugates have been designed in a way that allows the conversion of monovalent binders into high-affinity bidentate binders via formation of dynamic duplex assemblies.18,19 The approach relies on the synergistic interactions between (i) the protein and the small molecule ligand and (ii) two complementary DNA strands. In a noteworthy example, DNA-encoded dynamic combinatorial libraries enabled the selection of inhibitors of the sirtuin SIRT3.19
4. Multivalent protein–peptide interactions
Antibodies are indispensable tools in molecular and cell biology as well as analytical sciences. Traditional antibodies can engage two or up to 10 (in IgM) binding sites for enhanced interaction with antigens. With the aim to create synthetic high affinity binding agents Chaput and co-workers designed a nucleic acid conjugate architecture which combines two different peptides on a single DNA scaffold to produce a bispecific complex with high binding affinity for a protein of interest (Fig. 7A).27 In a proof of concept study, ∼4000 unique 12-mer peptide ligands were screened for binding of the yeast regulatory protein Gal80 (Fig. 7B). Binding peptides targeting different binding sites were attached to oligonucleotides and paired by hybridization resulting in bispecific dsDNA complexes with 30–90 Å distance between peptides. In total, ∼400 bispecific complex variants were simultaneously tested for interaction to Gal80 by SPR analysis, and the optimal configuration produced a nearly 1000-fold improvement in binding (KD = 4.2 nM) relative to the monovalent, monospecific probes on their own dsDNA duplexes.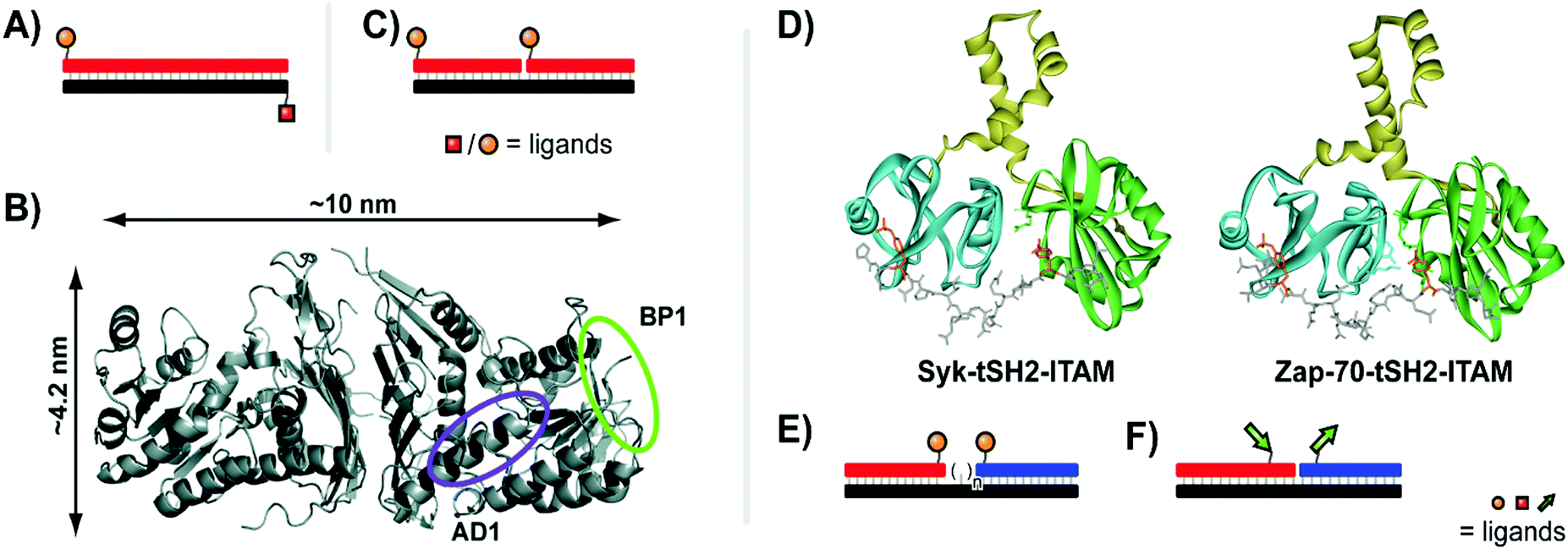 | ||
| Fig. 7 Architectures of multivalent nucleic acid probes presenting peptide ligands. Duplex design for targeting Gal80 (A, ref. 27), and crystal structure of Gal80 labeled with binding sites for two different peptides (B, reproduced from ref. 27 with permission from American Chemical Society, copyright 2009). (C) Bivalent design for binding to Death Receptor 5 (ref. 28). (D) Crystal structures of spleen tyrosine kinase (Syk) and ζ-chain-associated protein kinase (Zap-70) in complex with diphosphorylated immunoreceptor tyrosine-based interaction motifs (ITAMs) (from protein database entry 1A81 and 2OQ1, reproduced from ref. 30 with permission from American Chemical Society, copyright 2017). (E) A bivalent design for targeting Syk featuring a variable number of central unpaired bases to improve ligand orientation (ref. 29). (F) Recognition of bipartite ITAM motifs on an oligonucleotide scaffold by the Zap-70 tSH2 domain requires a specific strand orientation (ref. 30). | ||
In the pursuit of high affinity binders for the Death Receptor 5 (DR5), Winssinger et al. created bivalent PNA–peptide constructs.28 DR5 belongs to a family of receptors which transmit an apoptogenic signal upon binding of the trimeric TRAIL cytokine. Seeking to improve binding, a cyclic peptide ligand of DR5 was dimerized via conjugation with PNA and hybridization of two ligand–PNA conjugates with a DNA template strand (Fig. 7C). SPR screening of the constructs against immobilized DR5 identified close positioning of the two peptide ligands (10 base pairs) and an inter-oligo-peptide PEG spacer as critical components of high affinity binders. The best binder bound DR5 on the SPR chip with one order of magnitude higher affinity than the non-conjugated cyclopeptide.
Many recognition modules that orchestrate intracellular protein–protein interactions are arranged in tandem. For example, the tandem Src homology 2 (tSH2) domains, which cooperate in the spleen tyrosine kinase (Syk), recognize diphosphorylated immunoreceptor tyrosine-based interaction motifs (ITAMs) (Fig. 7D, left). Syk is a critical component in activation of B cells and IgE receptor signaling. Recognition of ITAMs created upon engagement of the B cell receptor complex activates Syk and triggers downstream signaling. We explored the recognition repertoire of the Syk tSH2 domain by DNA-programmed spatial screening.29 A library of bivalent DNA–phosphotetrapeptide conjugates presenting the ligands at a range from 1–20 nuclotide distance was assessed in a solution phase binding assay involving the tSH2 domain and a fluorescence-labelled reference binder. Bivalent probes with single-stranded DNA backbones produced low IC50 values but with little correlation to distance, likely attributed to the high flexibility of single-stranded DNA. By adding a second DNA strand to the duplex, the more rigid dsDNA enforced structures with IC50 minima at 11 nt (37 Å) and 2 nt (7 Å). These binding trends correspond with alignment of the two ligands along the same helical face, favoring intervals of 10.4 nt corresponding to a complete helical turn. The analysis of complexes presenting the phosphopeptide ligands from the same helix side revealed that the interdomain linker connecting the two SH2 domains in Syk tSH2 is flexible and able to arrange the SH2 domains in distances of 7 Å and 37 Å but not to the 71 Å required to simultaneously bind ligand peptide arranged in 21 nt distance. Regions of unpaired nucleotides were introduced to remedy the orientation problem (Fig. 7E). As the flexible nature of this unpaired region undermined the predictability of the molecular ruler approach, experimental distance measurements were conducted using a FRET reporter pair in place of ligands. The FRET-corrected distance measurements exposed a high flexibility of the tSH2 interdomain domain which supports bivalent binding until a distance of 50 Å between the two phosphotyrosine motifs.
In follow-up work, Marczynke et al. expanded the interrogation and compared the tSH2 domains of Syk with the closely related tSH2 domain of the ζ-chain-associated protein kinase (ZAP-70) (Fig. 7D, right).30 Both kinases bind to similar ITAM peptides which poses the question as to how differential binding (and activation) can occur when both kinases are co-expressed. Binding characteristics of the two tSH2 domains were probed with a panel of bivalent oligonucleotide–phosphopeptide constructs that varied ITAM separation distance, rigidity, as well as the orientation at which ITAM residues were attached (Fig. 7F). The Syk tSH2 domain accepted a variety of substrates regardless of orientation and distance between the phosphopeptide motifs. By contrast, the Zap-70–tSH2 domain showed a remarkably different behavior and required a proximal arrangement of the motifs at a defined orientation. The results suggest that ITAM folding within a protein could contribute to achieving specificity for activation of one of the tSH2-containing kinases, Syk or Zap-70, and provided a guideline for the design of “DNA-free” ligands, which discriminate between Zap-70 and Syk tSH2.
Concurrent work within our lab centered on the endocytic Adapter Complex 2 (AP-2), which serves as a central interaction hub for clathrin-mediated endocytosis.31 AP-2 features a core domain and two appendage domains known as α- and β2-appendage domains (Fig. 8A). Both of the appendage domains utilize peptide-binding grooves to mediate protein–protein interactions controlling the formation and maturation of clathrin- or AP-2-coated pits. By applying DNA-programmed heterobivalent probes, we found that the two binding sites within the α-appendage domain cooperate in a synergistic manner and provide for affinity enhancements upon bivalent recognition. We also addressed the question of whether a bivalency-enhancement is obtainable when both ear domains interact simultaneously with two peptide motifs found in a single protein. Given that the two ear domains are connected to the AP-2 core region via a 71 and 113 amino acid long, unstructured peptide tether, the two peptide motifs were presented in 10–140 nt (34–476 Å) distance. However, none of the assemblies were able to bind AP-2 with a higher affinity than monovalent peptide–DNA conjugates. We inferred that AP-2 was probably evolutionarily optimized for cross-linking of two protein molecules and we hypothesized that the distance between the two ear domains was too large to allow bivalency-enhanced interactions. To test this hypothesis, we constructed a model system comprised of synthetic receptor–ligand pairs.32 Two adamantane or two cucurbit[7]uril (CB[7]) units were attached to DNA scaffolds and the distances varied from 21–105 nt (Fig. 8B). Adamantane and CB[7] form host–guest complexes. Distance matched bivalent displays of adamantane and CB[7] were allowed to interact (Fig. 8C). We found that the affinity gain provided by bivalency is dependent on (i) the strength of the monovalent interaction, (ii) the distance between the host–guest complexes and (iii) the flexibility of the scaffold. In other words, strength of monovalent interactions and scaffold flexibility critically control the reach of bivalency-enhanced interactions. Of note, cross-linking became competitive as the bivalency-enhanced affinity gain decreased with increases of scaffold length/stability. This suggests that binding systems destined to act as cross-linkers must connect low affinity binding sites via long flexible linkers as is the case in AP-2.
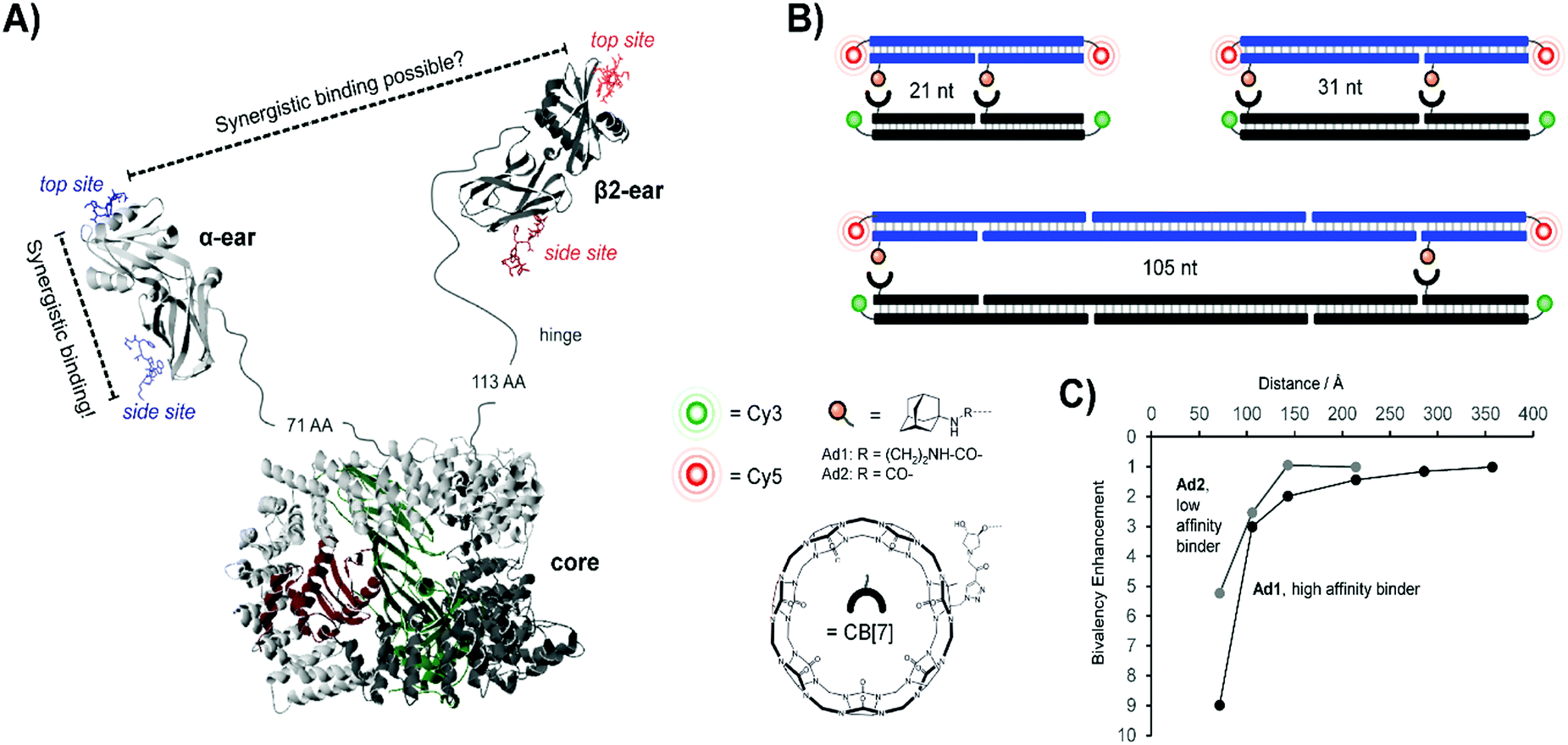 | ||
| Fig. 8 Bivalent binding studies interrogating Adapter Complex 2 (AP-2) (reproduced from ref. 13 with permission from John Wiley & Sons Inc., copyright 2019 and adapted from ref. 32 with permission from John Wiley & Sons Inc., copyright 2019). (A) Crystal structure of AP-2, including core domain as well as α- and β2-ear appendages (model based on PDB IDs 2VGL, 2VJO, 2G30, and 3HS9). Each appendage features a top and side binding site – bivalent binding between two sites of a single ear fell within the range of previous works, but the feasibility of linking the binding sites of two different ear appendages was uncertain. (B) To probe the limits of bivalency-based binding enhancement, a bivalent library was generated which separated cucurbit[7]uril (CB[7])-adamantane host–guest pairs at distances from 21 nt to 105 nt. A Cy3/Cy5 FRET pair was utilized to assess binding interactions. (C) Distance dependency of the affinity gain provided by interactions between bivalent [CB7] displays and distance-matched bivalent displays of either high-affinity ligands (Ad1) or low-affinity ligands (Ad2). Reproduced from ref. 13 with permission from John Wiley & Sons Inc., copyright 2019. | ||
An antibody's capacity for high-affinity binding interactions makes it attractive as a bivalent targeting agent. However, once an antibody-targeted payload is introduced into an organism, the effect is typically achieved systemically, with minimal control over the exact region and timing of dosing. Merkx and colleagues therefore explored innovative approaches to create activatable antibodies. Their first tactic relied on a bivalent activity-blocking group that non-covalently bound each of the antigen binding sites of an anti-HIV1-p17 antibody. Given that the two antigen binding sites of an antibody are 120 Å apart (though the hinge region in IgG antibodies induces flexibility), a rigid scaffold is required to enable bivalency-enhanced binding (see also ref. 15 and 38). Janssen et al. tethered two peptide antigens by a 20mer dsDNA helix, with each ligand connected via a peptide recognition sequence cleavable by matrix metalloprotease 2 (MMP2).33 Upon addition of MMP2 the recognition sequences were severed, resulting in a 500-fold loss of affinity for the individual monovalent ligands relative to formerly intact bivalent complex (Fig. 9A). SPR analyses demonstrated that roughly 87% of the blocked antibody's binding activity was inhibited prior to MMP2 treatment, and that the full binding potential of the blocked antibody could be restored after 2 h of MMP2 addition. The Merkx lab recently debuted an approach that puts antibody activity under the control of pH.34 Through clever design of overlapping duplex and triplex structures, the lab created bivalent assemblies which spontaneously dissociate when moved from acidic to neutral pH (7.3) (Fig. 9B), as well as a strategy for dissociation at low pH ranges (pH 5.0–6.5). The latter is clinically relevant in cancer treatment, where the acidified microenvironment of a tumor could potentially trigger local dissociation of the caging complex and activation of the therapeutic antibody. The Merkx group provided preliminary evidence supporting this approach through specific labeling of human carcinoma cells, both in bulk as well as in lipid droplet microenvironments. Both the light- and pH-triggered bivalent peptide–DNA caging complexes could theoretically be applied to any antibody given a suitable low-affinity antigen.
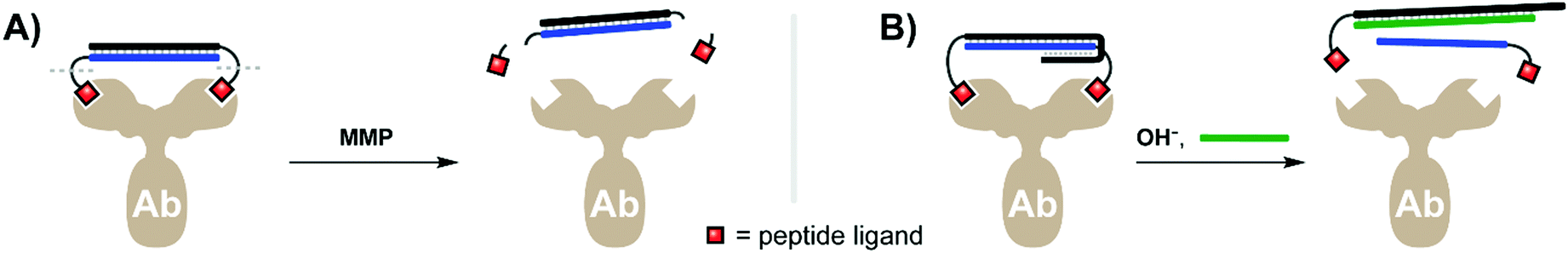 | ||
| Fig. 9 Bivalent duplex constructs can be combined with low-affinity peptide antigens to transiently block antibody activity until activated by a proteolytic enzyme such as MMP (A, ref. 33), or until a triplex structure is disrupted by a pH change (B, ref. 34). | ||
5. Multivalent interactions with receptors on the surface of cells and viruses
The surfaces of viruses assemble an array of receptor molecules and their combined action allows firm adhesion to host cells. Likewise, receptors on the surface of cells constitute a multivalent system that should be amenable to multivalency-enhanced binding via cross-linking (Fig. 1B). In addition, interaction of cell surface receptors with multivalent ligand systems can change the proximity relationship of receptors and, thereby, alter receptor activation. Nucleic acid complexes provide an ideal tool to create high affinity agents for targeting of cells and probing of receptor proximity.5.1 Multivalent interactions with carbohydrate receptors on cells and viruses
Early work by the Seeberger lab explored targeting of bone-marrow derived dendritic cells, which are known to express receptors for Lewis-X (Galβ1,4(Fucα1,3)GlcNAc) and Tn (Ser/Thr(αGalNAc)) sugar ligands.35 They arranged Lewis X trisaccharide, α-GalNAc, and combinations thereof on DNA duplexes. Unfortunately, the unmodified duplex exhibited high non-specific uptake during flow cytometry, but the cells incubated with duplexes bearing both sugars still demonstrated a ∼50% increase in uptake.In a study aiming to identify the requirements for bivalency enhanced interactions with weak monovalent binders, we submitted hemagglutinin on Influenza A virus particles to a spatial screening.14 The Influenza A virus is decorated with hundreds of hemagglutinin (HA) trimers, with which it recognizes patterns of host sialic acid–galactose linkages to mediate attachment and subsequent infection. On the IAV particles, binding sites for sialyl-LacNAc are arranged in 42 Å distance (Fig. 10A). Additionally, neighboring HA trimers allow for intermolecular bivalent binding, although the vertical rotation of HA trimers within the membrane means that the distance between intermolecular receptor sites ranges from 49–154 Å. With this uncertain receptor geometry in mind, we explored whether bivalent recognition favors “intratrimeric” chelation within one HA trimer or “intertrimeric” cross-linking across two adjacent HA trimers. We attached two sialyl-LacNAc ligands to two different scaffold types (Fig. 10B). For reasons outlined in Section 2, PEG tethers failed to provide affinity enhancements. Spatial screening of HA on IAV particles with sialyl-LacNAc on PNA–DNA duplexes revealed a bimodal distance-affinity relationship, with a local optimum at 26 Å ligand separation and an absolute optimum at 52–59 Å (Fig. 10C). The shorter distance likely corresponds to binding between the primary binding site and a secondary site, while the longer distance aligns with values predicted by statistical mechanics simulations for intratrimeric binding between two primary sites. A hemagglutination inhibition assay showed that the optimal arrangement exhibited an inhibition constant Ki(HAI) = 44 μM, which corresponds to 1360-fold binding enhancement relative to the free sugar ligand and a relative potency of 680-fold per ligand.
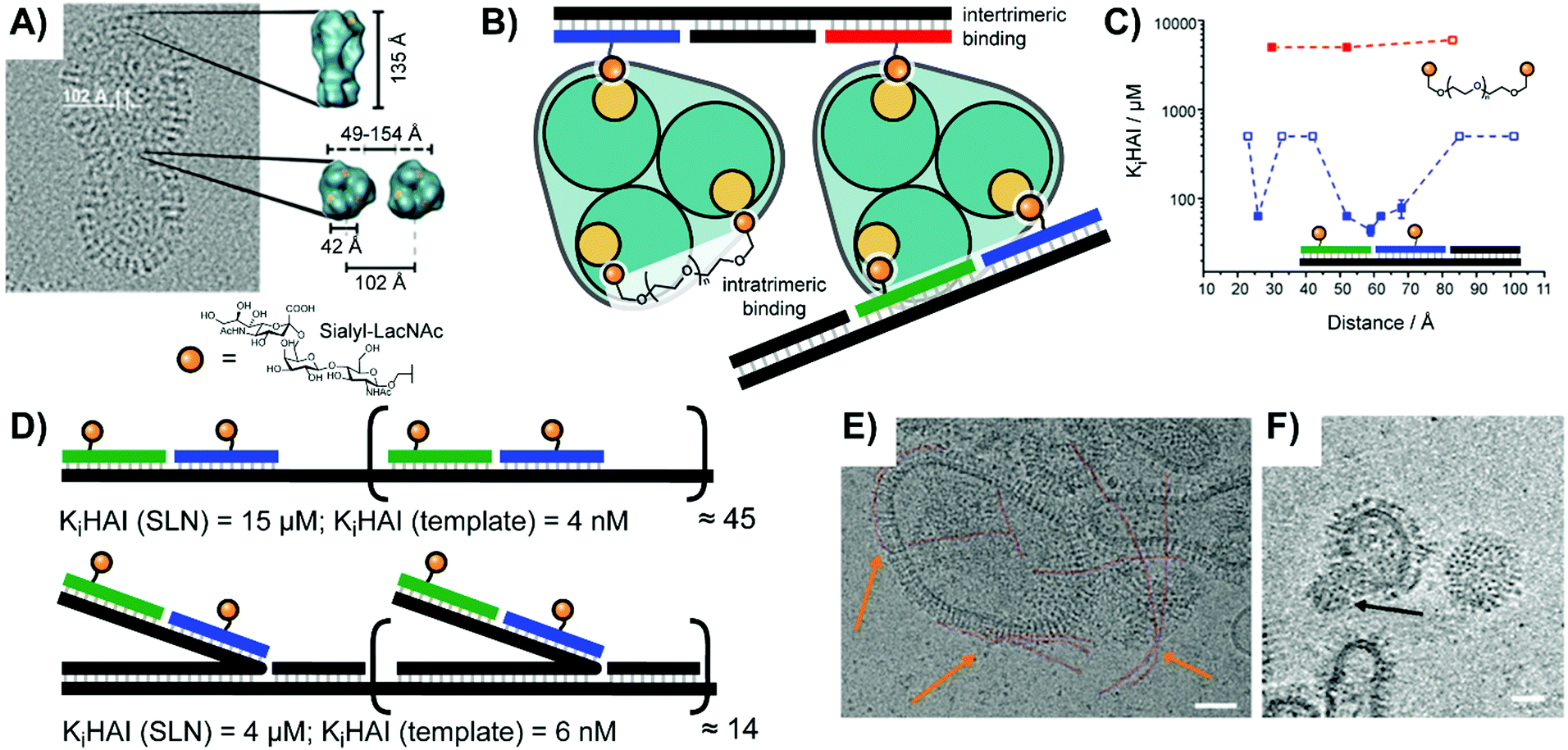 | ||
| Fig. 10 Multivalent studies probing Influenza A virus's hemagglutinin (HA) protein (adapted from ref. 14 and 36). (A) Cryo-electron micrograph of HA structure. (B) Design of oligonucleotide- or polyethylene glycol (PEG)-based bivalent agents for intratrimeric binding of one HA trimer or intertrimeric binding of two HA trimers. (C) Distance–affinity curve comparison of oligonucleotide- and PEG-based bivalent binding agents. (D) Oligomerization of optimized intratrimeric binders via concatenation on templates produced by rolling circle amplification (RCA). Linear version shown above, and bottlebrush version shown below. (E and F) Cryo-electron micrograph of DNA-concatenated multivalent constructs binding to Influenza A virus X31. Linear spaghetti-type structures are highlighted in red and orange arrows. (F) Slice of the reconstructed 3D volume of tilt series showing a virusbound wool-type sialyl-LacNAc concatemer. Scale bars: 50 nm. (A, E and F) Reproduced from ref. 36 with permission from John Wiley & Sons Inc., copyright 2019. | ||
Seeking to boost the affinity for IAV particles, we used rolling circle amplification (RCA) to oligomerize the optimized bivalent sialyl-LacNAc assemblies.36 After hybridization of sugar-containing strands with the long repeating template strand afforded by RCA, the resulting complex could present bivalent LacNAc groups at extremely high effective concentrations. The tandem repeats encoded during RCA provided a binding platform, which we utilized for two different attachment strategies (Fig. 10D): a “flat” design aligning the ligand strands directly against the scaffold, as well as a “bottlebrush” approach with the ligand-containing strands extending off into solution. Depending upon whether the structural repeat region was hybridized to a simple complementary strand or was left as single-stranded DNA the resulting complex could also be made more or less rigid. The best performing complexes fully blocked IAV binding at 10−9 M, a 107-fold enhancement over monovalent sialyl-LacNAc sugars. Cryo TEM analysis showed that the assembly adopted spaghetti-type linear forms and cottonball-like clusters, which both were able to bridge adjacent HA trimers in the IAV surface (Fig. 10E and F).
In their quest for binders of Influenza virus particles, Ebara and co-workers used a DNA 3-way junction to arrange 2,3-sialyllactose (2,3-SL) groups in distances that match the position of binding sites on the trimeric hemagglutinin (HA) structure (Fig. 11A).37 Optimal binding was obtained by 3WJ complexes presenting three 2,3-SL groups on each arm, yielding a roughly 8 × 104-fold improvement over unconjugated 2,3-SL. The arrangement provided each ligand with a 890-fold binding enhancement. These binding improvements are likely attributable to its ability to induce both statistical rebinding on a single HA site and a chelate effect between multiple HA binding sites. Recently, the Ebara lab engineered a new trigonal DNA scaffold structure (Fig. 11B), which provides increased resistance to nucleases.38
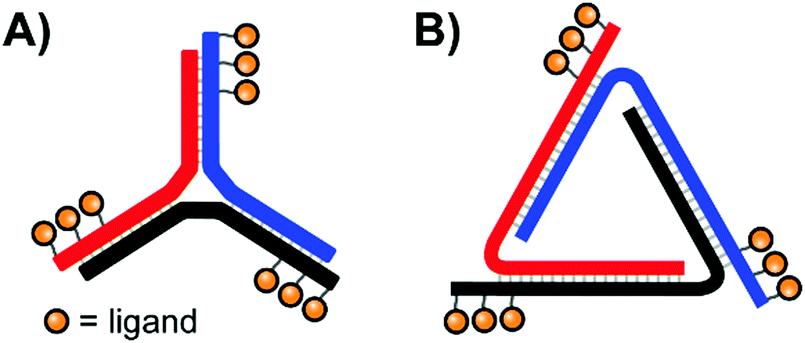 | ||
| Fig. 11 Architectures of trimeric nucleic acid probes targeting Influenza A hemagglutinin. (A) 3-Way junction design (ref. 37) and (B) trigonal design (ref. 38). | ||
5.2 Cell receptor interactions with multivalent proteins, peptides and small molecules
Pioneering work by the Appella lab in 2012 established that a nucleic acid-assembled multivalent oligo-peptide could successfully make the leap from benchtop cellular assays to whole-organism in vivo applications.39 Their research focused on integrins, cell-surface receptors that mediate adhesion through multivalent interactions with the extracellular matrix. Overexpression of integrins can promote metastasis of tumors which has motivated the development of competitive integrin inhibitors such as the derivatives based around a cyclic Arg-Gly-Asp (cRGD) motif. To increase their potency, c(RGDfK) ligands were displayed in multiple copies from PNA–DNA complexes (Fig. 12A). By using PNA strands containing 1–3 c(RGDfK) ligands and DNA templates offering up to 15 sequence repeats, the group generated arrays of 1–45 ligands. The ability to inhibit attachment of C32 human melanoma cells to a vitronectin-coated surface scaled with the number of ligands up to a plateau at 9–15 copies, peaking at an improvement of 2 orders of magnitude relative to the free monovalent c(RGDfK) peptide. Subsequent in vivo follow-up experiments assessed the ability of a DNA:PNA hybrid with 15 c(RGDfK) ligands to inhibit the growth of lung carcinomas in a murine xenograft model. Relative to an untreated control group, the mice given 0.1 mg of DNA:PNA-c(RGDfK) construct exhibited a ∼50% reduction in tumor mass, compared to only ∼30% tumor reduction for the group given 2.0 mg of free c(RGDfK) peptide. This is an impressive demonstration of the power of nucleic acid scaffolding considering that the ligand load carried by the nucleic acid system amounted to 1% of the amount introduced in the unconjugated form.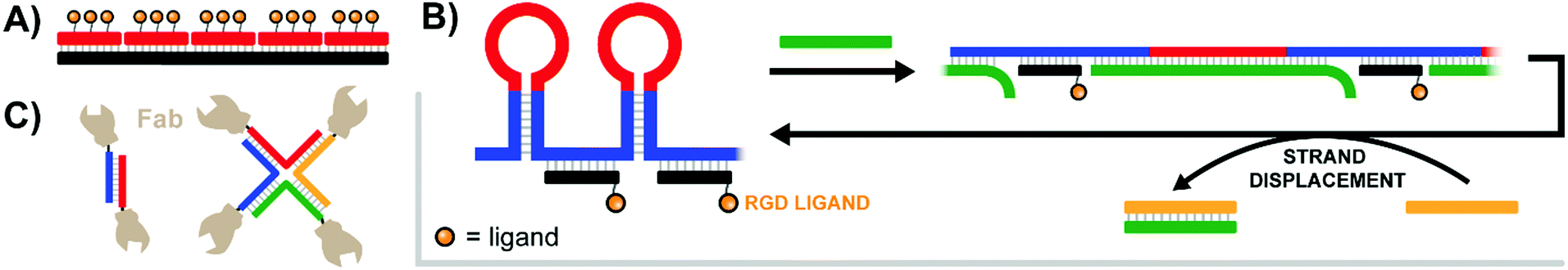 | ||
| Fig. 12 Architectures of multivalent nucleic acid probes presenting protein, peptide, and small molecule ligands to cells. (A) Tetravalent PNA–DNA duplex construct targeting multiple trimeric integrin receptors (ref. 39). (B) DNA-spring RCA product features alternating hairpins and linear regions hybridized to ligand-functionalized strands. Addition of an initiating strand (green) results in binding to the hairpin, which linearizes and extends the distance between ligands. Addition of second control strand (yellow) triggers strand displacement of green control strand, returning the construct to its original contracted state (ref. 40). (C) Antibody variable regions conjugated to a duplex or 4-way junction structure to unite αHER2- and aCD3-expressing cells (ref. 41). | ||
Li and colleagues mounted DNA-conjugated RGD peptides on a so-called DNA-spring to alter cell shape.40 A circular RCA template encoded a series of repeats comprising a sequence for annealing of RGD-modified oligonucleotides followed by a hairpin structure (Fig. 12B). In the starting conformation the RGD ligands were positioned in spacings of ∼13.2 nm. However, upon addition of a DNA strand complementary to the repeating hairpin segments the resulting hybridization forces the hairpins to unravel into longer and more rigid double stranded duplexes. Consequently, the spacing between each RGD ligand was extended to ∼22.5 nm, and any bound integrin receptors should also be pushed into the corresponding spacing. By mechanically manipulating bound receptors in this way it was possible to induce the formation of filopodia protrusions extending out from the central mass of HeLa cells, while also modulating the expression of integrin-related mRNAs. A strand displacement reaction was used to return to the original 13.2 nm spacing.
The labs of Schultz, Winssinger and Smider collaborated in an effort to explore PNA-antibody assemblies as an alternative approach to producing bispecific antibodies.41 In a rapidly emerging field, bispecific antibodies are used to recognize tumor cells with enhanced specificity or to guide T cells to tumor cells by bridging one surface molecule on one cell with another surface molecules on the other cell. The antibody's variable targeting region (sFv or Fab) was connected to a DNA or PNA strand. For this purpose, a ketone group was introduced into the antibodies by means of an unnatural amino acid. The conjugates were formed in reactions with alkoxyamine-modified DNA or PNA. Dimerization of the Fab fragment of an anti αHer2 antibody via DNA hybridization provided a bivalent assembly that had a lower ability to inhibit Her2 phosphorylation in SK-BR-3 breast cancer cells than the parent antibody trastuzumab. Of note, the activity loss was not observed when dimerization was induced by PNA rather than DNA. A PNA-assembled Fab tetramer (Fig. 12C) showed even higher activity. The authors speculated that affinity losses may be due to repulsion between the negatively charged phospholipid membrane and the polyanionic DNA scaffold. Further efforts yielded αHER2-aCD3 and αCD20-aCD3 heterodimers, which could recruit cytotoxic T lymphocytes to HER2- or CD20-expressing cancer cells.
The Teixeira lab aimed to probe distance effects of receptor communication networks, specifically examining ephrin receptor activation.42 Ephrin receptors are receptor tyrosine kinases (RTKs) that regulate cellular migration and proliferation by recognizing ephrin on adjacent cells. To probe the influence of ephrin distribution on signaling from ephrin receptors, Teixeira built multivalent rafts called “nanocalipers” around a base of 18 parallel double helices, which projected up to two single-stranded DNA threads off into solution (Fig. 13A). Ephrin ligands were coupled with short DNA strands complementary to the dangling strands of the origami base. The attachment of ephrin ligands was varied from a single ligand (NC0) to bivalent ligands at 100 nm (NC100) or 43 nm spacings (NC40), or 8 ligands at roughly 14.3 nm spacings (NC-Sat). Perhaps unsurprisingly, the more closely-spaced bivalent NC40 construct induced the highest activation of ephrin receptors on MDA-MB-231 cells and also the greatest reduction in cell invasion rates. The results point to the importance of receptor distribution upon binding.
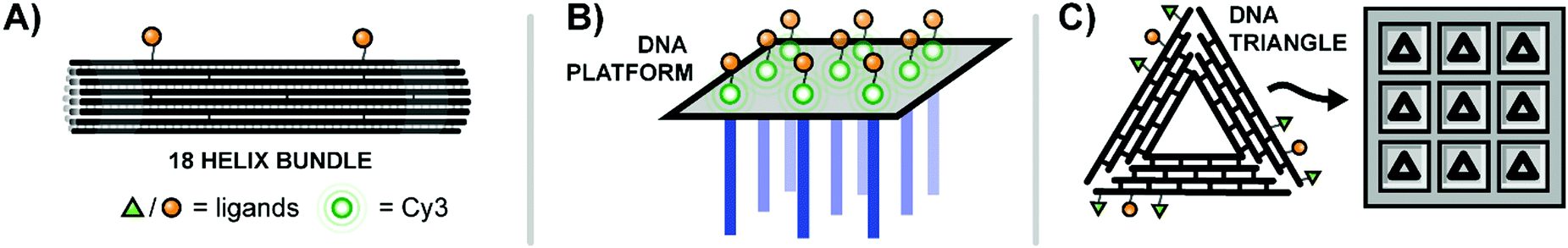 | ||
| Fig. 13 Multivalent binding agents constructed using DNA origami. (A) 18-Bundle helix origami provides a rigid platform for bivalent ligands targeting Ephrin receptors (ref. 42). (B) Rectangular DNA origami platform with dangling strands for hybridization to nanoarray (blue) and Cy3–EGF ligands for receptor binding (green) (ref. 43). (C) Triangular DNA origami platforms decorated with ligands for EGFR and β6 integrin are loaded into etched wells to create a surface with finely-tunable ligand patterning (ref. 44). | ||
Niemeyer investigated the response of MCF7 cells to the spatial organization of EGF.43 Rectangular (91 nm2) DNA origami constructs offered on one face 9 DNA single strands for immobilization on a DNA nanoarray platform. The other face was functionalized with 4, 5, 8 or 12 biotin residues, which arranged Cy3-labeled streptavidin molecules in an evenly distributed fashion or in a clustered mode. The origami constructs were then loaded with biotinylated EGF and immobilized to a DNA nanoarray, leading to a 5 μm spot pattern. MCF7 cells were allowed to adhere and, after fixation, activated EGFR was stained with Cy5-labeled specific anti-phosphoEGFR antibodies. The number of Cy5-spots under each immobilized cell was used as an indicator of EGFR activation. It was found that origami platforms with evenly distributed EGF induced stronger activation than with clustered EGF.
Recent work by the Palma lab has established a method for studying the communication between different cell receptors with nanoscale control.44 Their approach begins by constructing large triangular DNA origami platforms decorated with carefully spaced receptor ligands (Fig. 13C, left). Next, a type of lithography – focused ion beam patterning – was used to etch repeating wells into a glass surface, and the exposed regions were functionalized with carboxylic-terminating silane. The multivalent ligand-decorated DNA origami were then bound to the wells in the glass surface, roughly one DNA origami per well, using amino-terminating ends on the origami sequences Fig. 13C, right). To establish the method the Palma lab studied how cells expressing β6 integrin and epidermal growth factor (EGF) receptors spread over artificial surfaces presenting the corresponding ligands, modeling the movement of cells through an extracellular matrix. A peptide ligand for β6 integrin (A20FMDV2) and EGF were conjugated to the DNA origami triangles in ∼60 nm spacings previously identified as preferential for cell spreading. Then, wells were etched in ∼300 nm intervals and DNA origami fixed to the wells. The group identified a cooperative effect between the two ligand–receptor pairs which enhanced cell spreading. At least three A20FMDV2 peptides per DNA origami were necessary for optimal cellular adhesion to the surface, and a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio of A20FMDV2 peptide to EGF ligands was found to produce optimal cell spreading.
:6 ratio of A20FMDV2 peptide to EGF ligands was found to produce optimal cell spreading.
5.3 Cell receptor interactions with multivalent aptamers
According to the original definition aptamers are oligonucleotides that bind specific target molecules. Nowadays this definition has been broadened to include peptides. Unlike binders based on carbohydrates and small molecules, aptamers can be identified and optimized by in vitro evolution processes. A unique advantage offered by nucleic acid aptamers is their facile integration into nucleic acid structures offering opportunities also for those labs less experienced in conjugation chemistry.An early contribution to DNA-programmed assembly of multivalent aptamers came from Yan Liu and Yung Chang. Their aim was to develop oligonucleotide-based analogs of bispecific antibodies. Aptamers with specificity for Ramos cells (B cells) and aptamers that bind L-selectin on Jurkat cells (T cells) were used as recognition molecules.45 Their optimization efforts progressed through increasingly higher valency structures, ultimately arriving at a DNA origami structure featuring a central bridge connecting a pair of four-fingered hands (Fig. 14A). The left hand's fingers were each tipped with a T cell-specific aptamer, and the right hand was similarly decorated with B cell-specific aptamers. Though the affinity increase provided by the tetravalent presentation was rather low, flow cytometry studies demonstrated that a rigid bi-tetravalent complex was able to enforce connections between ∼37% of the observed T cells and B cells. This methodology could be used in similar applications to bispecific antibodies to generate a T cell-dependent B cell response, or with modified aptamers it might be used to bring tumor cells into contact with immune cells for targeted killing.
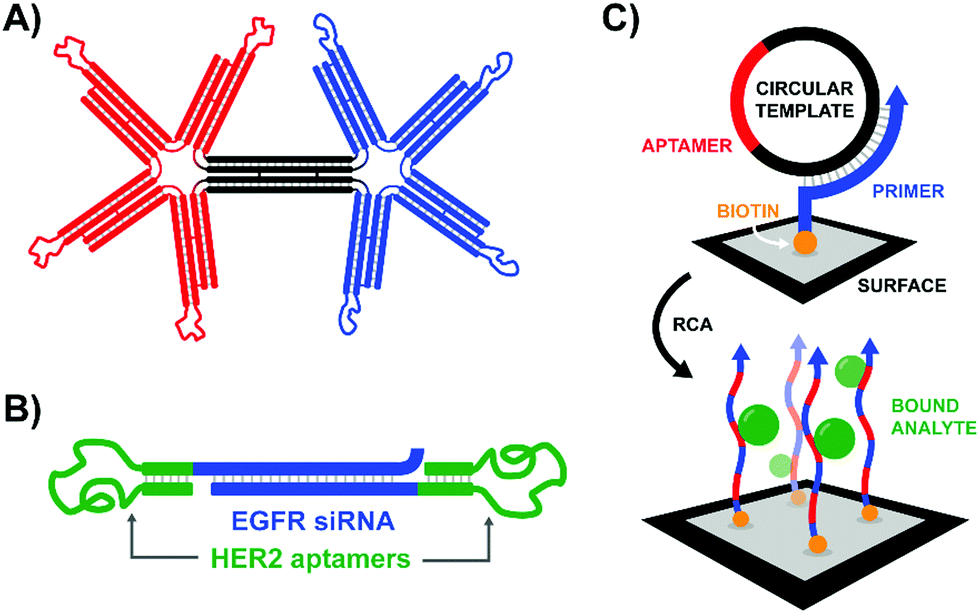 | ||
| Fig. 14 Architectures of multivalent nucleic acid probes presenting aptamer ligands to live cells. (A) Bi-tetravelent “hands” with aptamer-tipped “fingers” to bridge B cells and T cells (ref. 45). (B) EGFR siRNA duplex flanked by HER2-targeting aptamers. After aptamer-mediated delivery of construct to cells, siRNA knockdown can provide a secondary therapeutic effect (ref. 46). (C) Rolling circle amplification of an aptamer-containing circular template off of a surface-bound primer functionalizes the surface with a network of aptamers capable of trapping target-expressing analytes during flow experiments (ref. 47). | ||
Work in our lab addressed the interaction between L-selectin on leucocytes and the sialyl-Lewis-X (sLex) tetrasaccharide.12 In order to infiltrate inflamed tissue, leukocytes passing through the bloodstream utilize selectins to bind to clusters of sLex on endothelial cell surfaces and trigger vascular extravasation at the site of inflammation. Excessive leukocyte infiltration can lead to inflammatory disorders such as rheumatoid arthritis, asthma, or psoriasis. Seeking to design more efficient L-selectin binders, we constructed high precision PNA–DNA scaffolds for bivalent presentation of the sLex tetrasaccharide or an L-selectin-targeting DNA aptamer. Monovalent sLex has orders of magnitude lower affinity than the aptamer. It is thus not surprising that bivalent aptamer complexes outperformed the bivalent sLex complexes. When tested against L-selectin-expressing K562 cells, scaffolds presenting the aptamers in 62 or 104 Å distance showed 25-fold improved binding over monovalent conjugates. The binding enhancement dropped to 7-fold when the aptamers were arranged 146 Å apart. In this case, and all other cases discussed in this review, affinities for cell-based assemblies of receptors often increase with decreasing distance. This could be indicative for clustered receptors or, perhaps more trivially, simply reflect the distance-dependence of the bivalency enhancement discussed in Section 4.
The strategy of bivalent aptamer agents was recently elaborated further by the Hong Yan Liu lab, adding functionality by building the scaffold from siRNA sequences (Fig. 14B).46 The resulting Aptamer-siRNA Chimera (AsiC) successfully targeted HER2-expressing breast cancer cells using its twin aptamers, which triggered uptake of the complex and release of EGFR-targeting siRNA within the cell's cytosol, ultimately interfering with signaling activity of both receptors. Treatment was also capable of inducing low levels of apoptosis in cell line models, and a fluorescently tagged chimera localized to tumors within a murine xenograft model with a half-life of roughly 12 hours.
Seminal work from Karnik and Karp introduced rolling circle amplification (RCA) as a method for increasing the effective concentration of receptor-targeting ligands.47 By encoding an aptamer against protein tyrosine kinase-7 (PTK7) into a circular template and attaching the primer strand to the surface of a microfluidic device, RCA produced a dense network of polymeric PTK7 aptamers. The resulting three-dimensional web was capable of capturing PKT7-expressing human leukocytes as they flowed through a microfluidic chamber with higher specificities and a 3- to 5-fold binding enhancement relative to similar chambers coated with two-dimensional monovalent aptamers or anti-PKT7 antibodies (Fig. 14C). The aptamer-bound leukocytes could be gently released with DNase I treatment. In follow-up work the Zhao lab loaded the polymeric aptamers with a cytotoxic payload of doxorubicin.48 Confocal fluorescence microscopy and flow cytometry utilizing the intrinsic fluorescence of doxorubicin confirmed that polymeric aptamers generated from RCA exhibited faster and more thorough uptake in PTK7-expressing cells, with minimal binding to control cell lines. Apoptosis rates were similarly increased for the Dox-loaded polymeric aptamers.
Sando and colleagues used aptamers as a mimic of the Hepatocyte Growth Factor (HGF), which upon binding and promoting the dimerization of the Met receptor bolsters cellular proliferation, differentiation, and migration (Fig. 15A).49 Two copies of the HGF mimetic aptamer were connected via a single stranded or double stranded DNA linker with lengths ranging from 0 to 60 nt. In theory, each aptamer would bind to a Met receptor, and the linker would encourage the two receptors to dimerize, autophosphorylate, and induce proliferative signaling. Expectedly, bivalent mimetic constructs with the shortest and most flexible linkers exhibited the highest Met phosphorylation rates in trials with Met-expressing A549 cells, likely due to increased Met dimerization at closer proximities. Mimetics coupled by short, flexible linkers were also the most successful in cell scattering and cell migration assays. Follow-up work further developed the ligand dimerization strategy by replacing the fixed central linker with a aptamer-based bridge that could be selectively connected upon binding a bridge molecule (Fig. 15B).50 Once the trigger molecule was added, the two bridging aptamers induce dimerization of the HGF mimetic aptamers facilitating Met binding and downstream signaling. In this way, the ligand that typically triggered receptor activation could be functionally replaced by whatever molecule was chosen to link the bridge aptamers. For a proof-of-principle case study thrombin protein was used as an external trigger to bridge two thrombin-specific aptamers and dimerize the Met-specific aptamers. Met phosphorylation was inducible in a thrombin-concentration-dependent manner, and could only be triggered when all components were present. The use of bridge aptamers that recognize the platelet-derived growth factor (PDGF) rendered Met-signaling responsive to PDGF. The method should be applicable to a variety of trigger molecules so long as they provide two binding sites for bridge aptamers.
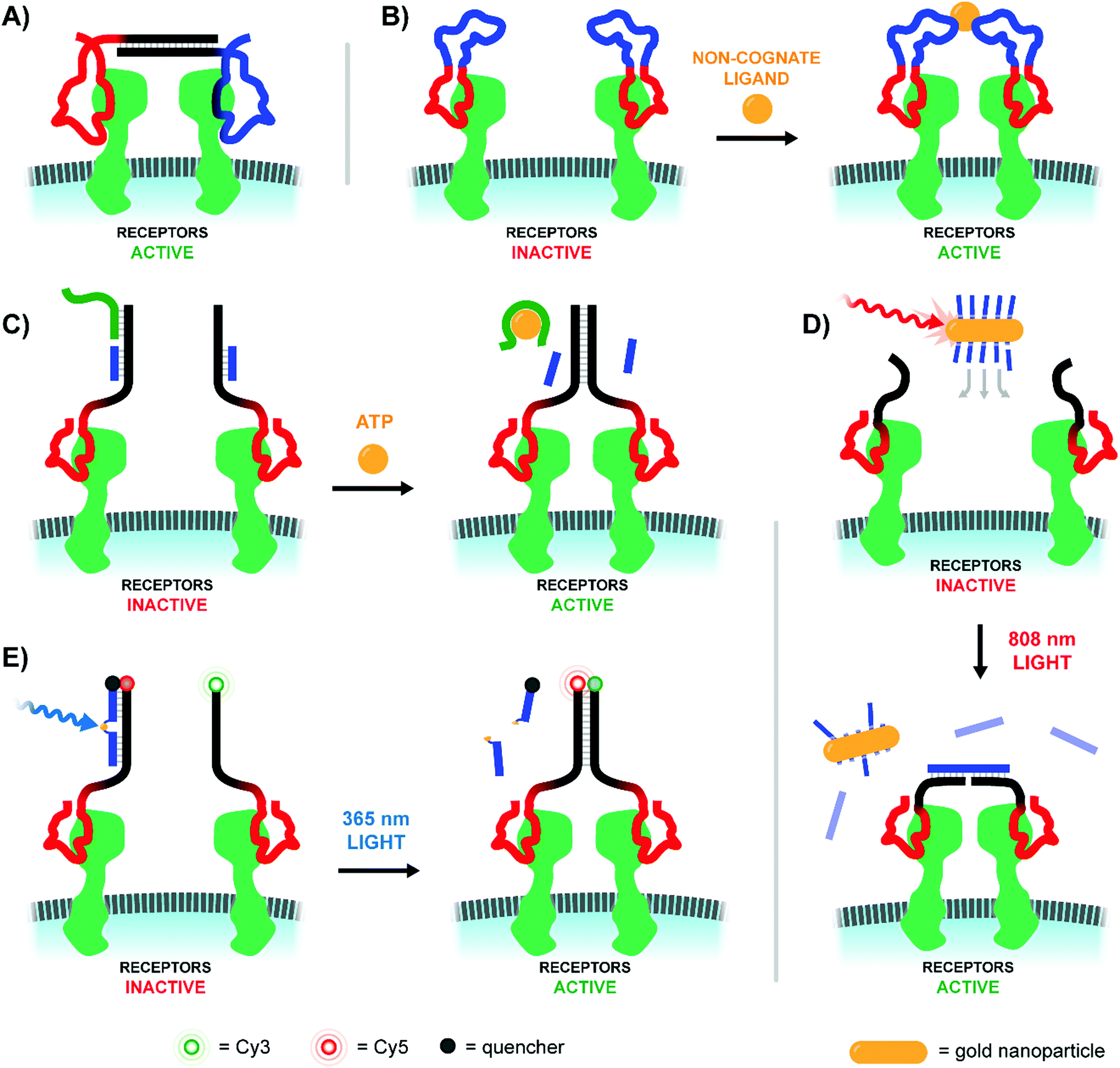 | ||
| Fig. 15 Architectures of multivalent nucleic acid probes utilizing aptamers to modulate cell receptor spacing and activity. (A) Two aptamers targeting hepatocyte growth factor receptors are modified with a duplex bridge region (black) capable of uniting and subsequently activating the two aptamer-bound receptors (ref. 49). (B) A variant of (A) which replaces the static bridge duplex with another pair of aptamers that can be united by binding to different sites of a non-cognate protein ligand (ref. 50). (C) Another aptamer approach which replaces the need for a two-site activating ligand by incorporating a ligand-sensitive aptamer. Binding of the sensing strand (green) to the ligand initiates a strand displacement reaction uniting the two aptamer-bound receptors (ref. 51). (D) When a gold nanoparticle decorated with bridge strands is irradiated at 808 nm, it releases those strands to connect two aptamer-bound receptors (ref. 52). (E) Light activation can be accomplished by utilizing a blocking strand with a nitrobenzyl-based photocleavable linker. Upon 365 nm irradiation the blocking strand is fragmented into two shorter sequences, which can be displaced by a nearby receptor-bound aptamer strand (ref. 53). | ||
The Nie lab soon introduced a method that circumvented the need for a trigger molecule with multiple binding sites.51 Their technique similarly leveraged a pair of receptor-specific aptamers (Fig. 15C). The system was based on a “sensor” DNA strand and an “actuator” DNA strand, each hybridized to a single strand extending off of the receptor-specific aptamer. The sensor strand featured a linear toehold aptamer which selectively folded around a small molecule trigger. Upon folding the sensor strand disassociated, allowing the freed single strand to bind the actuator single strand and form a new dimerizing DNA bridge. To prove feasibility, an ATP-responsive sensor was hybridized with the single strand extension of a Met-specific aptamer. In the presence of ATP the sensor strand dissociated, and dimerization with the single strand extension of a second anti-Met aptamer could occur. In this way, ATP served as a trigger for Met activation. The method was expanded to DNAzyme sensors which fold upon addition of histidine or Zn2+. In recent work, the Nie lab further expanded the toolkit of activating agents to include near-IR light using a DNA-decorated gold nanorods (Fig. 15D).52 As the gold nanorods (∼60 nm long, ∼14 nm wide) absorbed near-IR light and converted the energy into heat, the attached DNA agonists were released into solution where they could bridge two receptor-bound DNA aptamers. Roughly half of the ∼200 nanorod-bound DNA agonists could be deployed within 4 minutes of irradiation using an 808 nm laser. After a proof of concept trial demonstrated photo-controlled dimerization of Met receptors and basic spatiotemporal control of cultured cells, the group next explored deep-tissue activation of their complexes to stimulate skeletal muscle regeneration in mice. Following a liquid-nitrogen induced leg wound, mice treated with the complexes and near-IR light exhibited 300% higher Met activation at the injury site, coupled with increased differentiation and cellular proliferation rates of muscle cells.
A rather straightforward method to achieve optical control of receptor activation was introduced by the Yang lab (Fig. 15E).53 The method, again, relied on a pair of Met-specific aptamers. Met-specific aptamer 1 was functionalized with a DNA duplex terminating in either a Cy5 fluorophore or BHQ-2 quencher, with the quencher-bearing strand incorporating a nitrobenzyl-based photocleavable linker into its backbone. Meanwhile, Met-specific aptamer 2 was functionalized with a Cy3-labeled, single stranded sequence complementary to Aptamer 1's Cy5-labeled sequence. Upon photoactivation with 200 nm light, the quencher-labeled strand's photocleavable linker was broken, separating the strand into two strands short enough to be displaced by the Cy3-labeled strand of Aptamer 2. The formation of a new duplex connecting the two aptamer-bound Met receptors also displaced the quenching group and united the Cy3- and Cy5-labeled DNA strands for a robust Cy3/Cy5 FRET turn-on signal. This technique was successfully employed to dimerize receptors of Met-expressing DU145 cells, resulting in increased cellular proliferation and migration rates comparable to addition of the native Met-receptor ligand, HGF.
In addition to ways of modulating receptor dimerization, there is also a need for techniques which visualize these interactions, even in low abundances. The Juan Li lab visualized receptor-dimerization using multivalent oligonucleotide interactions, including signal amplification via hybridization chain reaction (HCR) (Fig. 16A).54 The Yang lab's approach relied on receptor-specific aptamers – again targeting Met dimerization – with each aptamer's single-stranded DNA tails extending into solution. When the receptor-bound aptamer complexes were in close proximity, they offered a bipartite recognition sequence for a separate DNA hairpin strand. Binding opened the hairpin and liberated an initiator strand for HCR which involved two additional hairpin species. Because one of the two hairpins included a FITC fluorophore, the ensuing HCR product chain emitted an intense fluorescence signal at the site of Met receptor dimerization. Not only was the method useful for imaging Met dimerization events in plated cells, but it also proved effective for imaging cell-receptor dimerization events in live zebrafish embryos under confocal scanning laser microscopy (Fig. 16B).
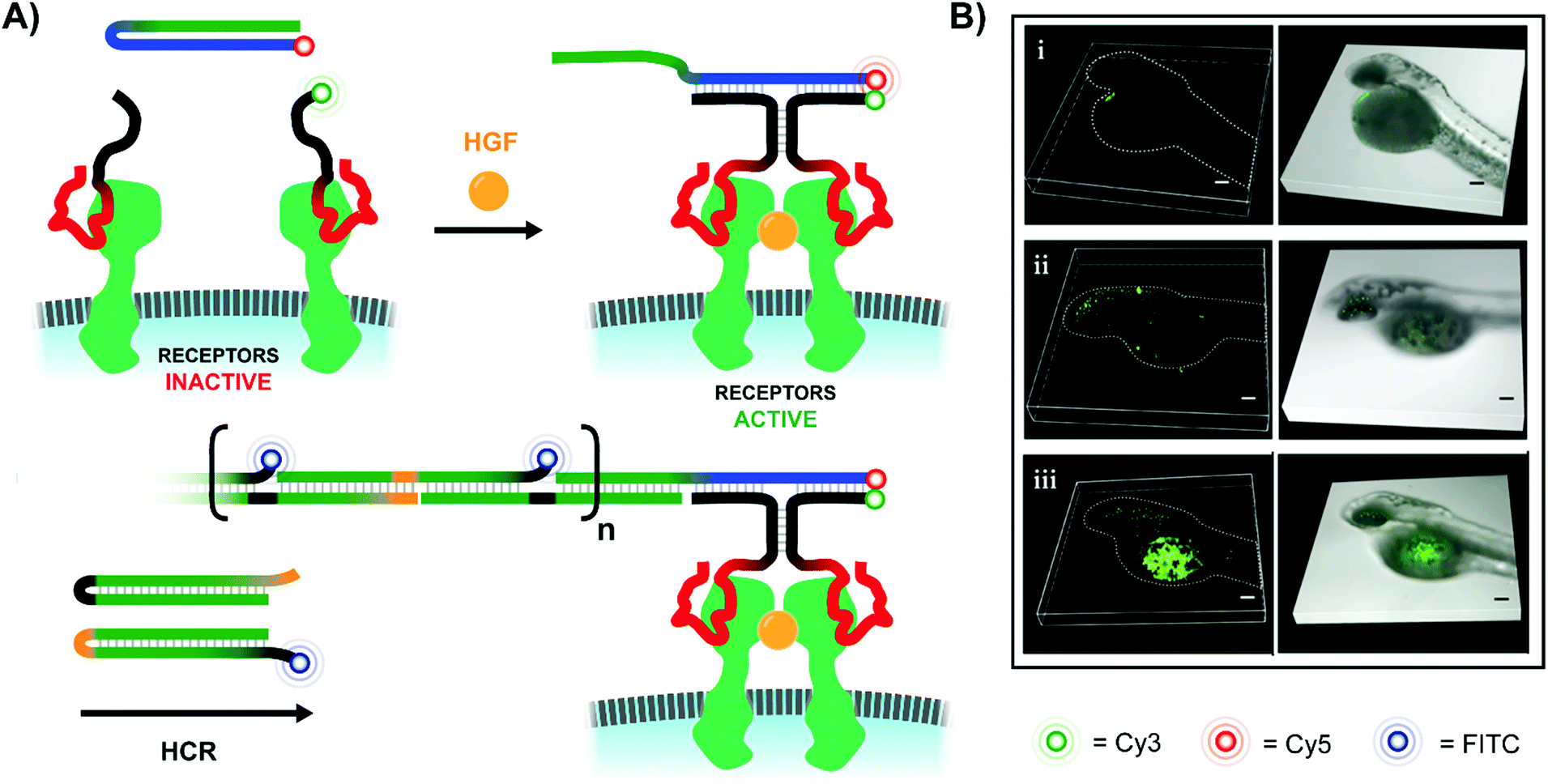 | ||
| Fig. 16 Amplified fluorescence detection of HGF-activated Met receptors (ref. 54). (A) When two aptamer-bound receptors are united by their native ligand in the presence of an additional Cy5-labeled hairpin (blue-green), a 3-way junction is formed, exposing an initiator strand for hybridization chain reaction (HCR). If one of the two hairpins used for HCR is labeled with a FITC fluorophore, the resulting HCR product amplifies the receptor-detection signal. (B) 3D-stacked confocal laser scanning microscopy images of zebrafish embryos from the Juan Li lab (reproduced from ref. 54 with permission from American Chemical Society, copyright 2018). Embryos were injected with Met-expressing MKN-45 cells at 48 h post-fertilization, and then Met was imaged using the hybridization chain reaction-based imaging technique. Embryos were incubated with either (i) no DNA probes, (ii) all DNA probes except trigger hairpin, or (iii) all DNA probes present. | ||
6. Overall trends and outlook
There are few hard rules for multivalent construct design. However, work performed in the last 10 years offers guidelines that may facilitate the design process. For many applications, it is key to maximize the enhancement of affinity that can be obtained through multivalent presentation.(A) It has become clear that the strength of the monovalent interactions is of prime importance. The higher the affinity of a monovalent ligand for a single receptor site, the higher the fold increase of affinity achievable by multivalency. It therefore is worthwhile to invest efforts in the selection or synthesis of suitable ligands.
(B) The affinity gain provided by chelate binding depends on the distance between receptor–ligand pairs. Even when the distance between two ligands seems to perfectly match the distance between binding sites on the multivalent receptor, no linker is perfectly rigid and at large distances the conformational entropy of the linker will decrease the effective molarity of ligands. As a result, while flexible linkers may work when distances are in the 5–30 Å range, rigid linkers are required at larger distances. The higher the linker rigidity, the greater the distance multivalent effects can reach. If chelation requires bridging over >200 Å it will be difficult to obtain affinity gains.
(C) As a combination of the prior two points, the distance reach of multivalency is a function of the strength of the monovalent interaction. While millimolar affinity may be sufficient to provide significant affinity gains when distances between binding sites are in the 40 Å range, higher affinities are required to enable multivalency-enhanced interactions when distances are large.
(D) The nucleic acid scaffold is not innocent. As the scaffold may engage in attractive or repulsive interactions with the receptor surface, a nucleic acid-conjugated ligand may have higher or lower target affinity than the unconjugated ligand. A positive influence has frequently been observed in studies of carbohydrate–lectin interactions.10,14 Negative influences of DNA scaffolds have been reported for studies of cell surface receptors, which has been attributed to the repulsion between the negatively charged outer layer of the plasmamembrane and the polyanionic DNA.
(E) DNA-type linkers alter the solubility properties. It is, thus, feasible to study hydrophobic ligands in aqueous solution.
Beyond these criteria, potential nucleic acid architectures must be carefully considered in relation to the receptor system of interest. A variety of scaffold architectures are possible, ranging from linear duplexes; to three-way junctions and trigonal structures; long, repeating RCA products; complex aptamer structures; and even large DNA origami superstructures. Intuitively, a more rigid scaffold would be more energetically favorable by lowering the entropic cost of binding and enhancing multivalent rebinding rates. While the display of multivalent ligands on flexible polymeric scaffolds may fail to generate the necessary binding enhancements,14,24 the distance-dependent relationships for multivalent binding were not revealed until the scaffold was made sufficiently rigid.29,31 If a dsDNA scaffold still isn’t rigid enough, a DNA origami structure made from helical bundles can provide a platform for even larger, and more rigid constructs.42,45 However, the higher the rigidity of the scaffold, the more closely it must conform to the geometry of the receptor sites to produce a binding enhancement. In the cases of receptor sites with highly complex, curved, or unknown binding geometries, it may be advantageous to reduce the rigidity of the construct, either by introducing nick sites in a dsDNA duplex,10 leaving regions of unhybridized ssDNA,36 and/or emphasizing flexible linkers between the oligonucleotide scaffold and the presented ligands.28
The most commonly employed backbones for multivalent nucleic acid constructs, DNA and PNA, both present unique challenges and opportunities. PNA duplexes can obtain the same thermal stability as DNA duplexes at significantly shorter strand lengths, allowing for more compact scaffolds.10,55 Furthermore, the peptide linkages in PNA backbones make it resistant to degradation by nucleases, which is a very attractive quality as the field progresses from cellular assays to applications in whole organisms. The neutral PNA backbone can enhance ligand binding interactions,28 but can also create significant challenges for solubilizing the probe in biological buffers. Conversely, DNA-based probes may be larger and more susceptible to degradation, but are cheaper to synthesize at larger scales. DNA is also compatible with many important enzymatic techniques such as PCR, RCA, HCR and SELMA,21 and utilizing polymerases with ligand- or handle-modified uridines remains a viable approach for site-specifically incorporating ligands into DNA scaffolds.15 We’ve also seen that non-traditional secondary structures such as the trigonal design employed by Ebara and colleagues can offer their own degree of protection from nucleases,38 potentially lessening the need for PNA and other modified backbones in whole organism trials. Finally, the type of ligand employed may guide selection of backbone chemistry: pairing a peptide ligand with a PNA scaffold or an aptamer with a DNA scaffold might allow for a more convenient, all-in-one approach during solid phase synthesis, without the need for downstream conjugation.
When choosing the range of interligand distances to probe there are several factors to consider. If studying a previously-characterized protein system, the upper range limit can be informed by knowledge of the receptor site geometry. Additionally, close interligand spacings – even those too short to bridge the predicted distance between binding sites – are still worthy to be explored given the possibility of rebinding and the presence of secondary binding sites fortuitously discovered adjacent to known binding sites.12,14,24 While the resulting construct may bridge a primary and secondary binding site on a single protein instead of bivalently spanning two proteins, the goal of obtaining a binding agent with high affinity and specificity can still be achieved.
After nearly two decades of work in the field, the core promise of utilizing multivalent nucleic acid scaffolds to potentially enhance ligand–receptor affinities by orders of magnitude remains as alluring as ever. Furthermore, the recent surge in multivalent oligonucleotide publications highlights the community's growing interest in an approach capable of probing protein interactions with incredible precision, tunability, and ease of preparation. Because the strategy is compatible with a wide variety of ligand chemistries and receptor types, it can be adapted for almost any multivalent biological system. Given the progression of the field from in vitro assays, to cellular experiments, and now finally to the study of live viruses and whole organisms, we may soon see the strategy make the leap from fundamental biological research and into clinic applications.
Conflicts of interest
There are no conflicts to declare.References
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS.
- F. Diezmann and O. Seitz, Chem. Soc. Rev., 2011, 40, 5789–5801 RSC.
- Y. J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- M. Xiao, W. Lai, T. Man, B. Chang, L. Li, A. R. Chandrasekaran and H. Pei, Chem. Rev., 2019, 119, 11631–11717 CrossRef CAS PubMed.
- V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492–4503 RSC.
- K. Matsuura, M. Hibino, Y. Yamada and K. Kobayashi, J. Am. Chem. Soc., 2001, 123, 357–358 CrossRef CAS PubMed.
- K. Matsuura, M. Hibino, T. Ikeda, Y. Yamada and K. Kobayashi, Chem. – Eur. J., 2004, 10, 352–359 CrossRef CAS PubMed.
- C. Scheibe, A. Bujotzek, J. Dernedde, M. Weber and O. Seitz, Chem. Sci., 2011, 2, 770–775 RSC.
- M. Eriksson and P. E. Nielsen, Nat. Struct. Biol., 1996, 3, 410–413 CrossRef CAS PubMed.
- C. Scheibe, S. Wedepohl, S. B. Riese, J. Dernedde and O. Seitz, ChemBioChem, 2013, 14, 236–250 CrossRef CAS PubMed.
- O. Seitz, J. Pept. Sci., 2019, 25, e3198 CrossRef PubMed.
- V. Bandlow, S. Liese, D. Lauster, K. Ludwig, R. R. Netz, A. Herrmann and O. Seitz, J. Am. Chem. Soc., 2017, 139, 16389–16397 CrossRef CAS PubMed.
- M. Matsui and Y. Ebara, Bioorg. Med. Chem. Lett., 2012, 22, 6139–6143 CrossRef CAS PubMed.
- T. Machida, A. Novoa, E. Gillon, S. Zheng, J. Claudinon, T. Eierhoff, A. Imberty, W. Romer and N. Winssinger, Angew. Chem., Int. Ed., 2017, 56, 6762–6766 CrossRef CAS PubMed.
- M. Ciobanu, K.-T. Huang, J.-P. Daguer, S. Barluenga, O. Chaloin, E. Schaeffer, C. G. Mueller, D. A. Mitchell and N. Winssinger, Chem. Commun., 2011, 47, 9321–9323 RSC.
- F. V. Reddavide, W. Lin, S. Lehnert and Y. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7924–7928 CrossRef CAS PubMed.
- Y. Zhou, C. Li, J. Peng, L. Xie, L. Meng, Q. Li, J. Zhang, X. D. Li, X. Li, X. Huang and X. Li, J. Am. Chem. Soc., 2018, 140, 15859–15867 CrossRef CAS PubMed.
- K. Gorska, K.-T. Huang, O. Chaloin and N. Winssinger, Angew. Chem., Int. Ed., 2009, 48, 7695–7700 CrossRef CAS PubMed.
- I. S. MacPherson, J. S. Temme, S. Habeshian, K. Felczak, K. Pankiewicz, L. Hedstrom and I. J. Krauss, Angew. Chem., Int. Ed., 2011, 50, 11238–11242 CrossRef CAS PubMed.
- J. M. Paar, N. T. Harris, D. Holowka and B. Baird, J. Immunol., 2002, 169, 856–864 CrossRef CAS PubMed.
- D. Sil, J. B. Lee, D. Luo, D. Holowka and B. Baird, ACS Chem. Biol., 2007, 2, 674–684 CrossRef CAS PubMed.
- F. Abendroth, A. Bujotzek, M. Shan, R. Haag, M. Weber and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 8592–8596 CrossRef CAS PubMed.
- M. Shan, A. Bujotzek, F. Abendroth, A. Wellner, R. Gust, O. Seitz, M. Weber and R. Haag, ChemBioChem, 2011, 12, 2587–2598 CrossRef CAS PubMed.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- B. A. R. Williams, C. W. Diehnelt, P. Belcher, M. Greving, N. W. Woodbury, S. A. Johnston and J. C. Chaput, J. Am. Chem. Soc., 2009, 131, 17233–17241 CrossRef CAS PubMed.
- K. Gorska, J. Beyrath, S. Fournel, G. Guichard and N. Winssinger, Chem. Commun., 2010, 46, 7742–7744 RSC.
- H. Eberhard, F. Diezmann and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 4146–4150 CrossRef CAS PubMed.
- M. Marczynke, K. Groger and O. Seitz, Bioconjugate Chem., 2017, 28, 2384–2392 CrossRef CAS PubMed.
- F. Diezmann, L. von Kleist, V. Haucke and O. Seitz, Org. Biomol. Chem., 2015, 13, 8008–8015 RSC.
- N. Dubel, S. Liese, F. Scherz and O. Seitz, Angew. Chem., Int. Ed., 2019, 58, 907–911 CrossRef CAS PubMed.
- B. M. G. Janssen, E. H. M. Lempens, L. L. C. Olijve, I. K. Voets, J. L. J. van Dongen, T. F. A. de Greef and M. Merkx, Chem. Sci., 2013, 4, 1442–1450 RSC.
- W. Engelen, K. Zhu, N. Subedi, A. Idili, F. Ricci, J. Tel and M. Merkx, ACS Cent. Sci., 2020, 6, 22–31 CrossRef CAS PubMed.
- M. K. Schlegel, J. Hütter, M. Eriksson, B. Lepenies and P. H. Seeberger, ChemBioChem, 2011, 12, 2791–2800 CrossRef CAS PubMed.
- V. Bandlow, D. Lauster, K. Ludwig, M. Hilsch, V. Reiter-Scherer, J. P. Rabe, C. Böttcher, A. Herrmann and O. Seitz, ChemBioChem, 2019, 20, 159–165 CAS.
- M. Yamabe, K. Kaihatsu and Y. Ebara, Bioconjugate Chem., 2018, 29, 1490–1494 CrossRef CAS PubMed.
- M. Yamabe, K. Kaihatsu and Y. Ebara, Bioorg. Med. Chem. Lett., 2019, 29, 744–748 CrossRef CAS PubMed.
- E. A. Englund, D. Wang, H. Fujigaki, H. Sakai, C. M. Micklitsch, R. Ghirlando, G. Martin-Manso, M. L. Pendrak, D. D. Roberts, S. R. Durell and D. H. Appella, Nat. Commun., 2012, 3, 614 CrossRef PubMed.
- K. Zhang, R. Deng, Y. Sun, L. Zhang and J. Li, Chem. Sci., 2017, 8, 7098–7105 RSC.
- S. A. Kazane, J. Y. Axup, C. H. Kim, M. Ciobanu, E. D. Wold, S. Barluenga, B. A. Hutchins, P. G. Schultz, N. Winssinger and V. V. Smider, J. Am. Chem. Soc., 2013, 135, 340–346 CrossRef CAS PubMed.
- A. Shaw, V. Lundin, E. Petrova, F. Fordos, E. Benson, A. Al-Amin, A. Herland, A. Blokzijl, B. Hogberg and A. I. Teixeira, Nat. Methods, 2014, 11, 841–846 CrossRef CAS PubMed.
- A. Angelin, S. Weigel, R. Garrecht, R. Meyer, J. Bauer, R. K. Kumar, M. Hirtz and C. M. Niemeyer, Angew. Chem., Int. Ed., 2015, 54, 15813–15817 CrossRef CAS PubMed.
- D. Huang, K. Patel, S. Perez-Garrido, J. F. Marshall and M. Palma, ACS Nano, 2019, 13, 728–736 CrossRef CAS PubMed.
- X. Liu, H. Yan, Y. Liu and Y. Chang, Small, 2011, 7, 1673–1682 CrossRef CAS PubMed.
- L. Xue, N. J. Maihle, X. Yu, S. C. Tang and H. Y. Liu, Mol. Pharmaceutics, 2018, 15, 4801–4813 CrossRef CAS PubMed.
- W. Zhao, C. H. Cui, S. Bose, D. Guo, C. Shen, W. P. Wong, K. Halvorsen, O. C. Farokhzad, G. S. Teo, J. A. Phillips, D. M. Dorfman, R. Karnik and J. M. Karp, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19626–19631 CrossRef CAS PubMed.
- Z. Zhang, M. M. Ali, M. A. Eckert, D. K. Kang, Y. Y. Chen, L. S. Sender, D. A. Fruman and W. Zhao, Biomaterials, 2013, 34, 9728–9735 CrossRef CAS PubMed.
- R. Ueki, A. Ueki, N. Kanda and S. Sando, Angew. Chem., Int. Ed., 2016, 55, 579–582 CrossRef CAS PubMed.
- R. Ueki, S. Atsuta, A. Ueki and S. Sando, J. Am. Chem. Soc., 2017, 139, 6554–6557 CrossRef CAS PubMed.
- H. Li, M. Wang, T. Shi, S. Yang, J. Zhang, H. H. Wang and Z. Nie, Angew. Chem., Int. Ed., 2018, 57, 10226–10230 CrossRef CAS PubMed.
- M. Wang, F. He, H. Li, S. Yang, J. Zhang, P. Ghosh, H. H. Wang and Z. Nie, Nano Lett., 2019, 19, 2603–2613 CrossRef CAS PubMed.
- S. Chen, J. Li, H. Liang, X. H. Lin, J. Li and H. H. Yang, Chem. – Eur. J., 2018, 24, 15988–15992 CrossRef CAS PubMed.
- L. Wang, W. Li, J. Sun, S. Y. Zhang, S. Yang, J. Li, J. Li and H. H. Yang, Anal. Chem., 2018, 90, 14433–14438 CrossRef CAS PubMed.
- S. Barluenga and N. Winssinger, Acc. Chem. Res., 2015, 48, 1319–1331 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |