
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent labeling of nucleic acids
Nils
Klöcker
a,
Florian P.
Weissenboeck
a and
Andrea
Rentmeister
 *ab
*ab
aInstitute of Biochemistry, University of Muenster, Corrensstraße 36, D-48149 Münster, Germany. E-mail: a.rentmeister@uni-muenster.de
bCells in Motion Interfaculty Center, University of Muenster, Waldeyerstrasse 15, D-48149 Münster, Germany
First published on 21st October 2020
Abstract
Labeling of nucleic acids is required for many studies aiming to elucidate their functions and dynamics in vitro and in cells. Out of the numerous labeling concepts that have been devised, covalent labeling provides the most stable linkage, an unrivaled choice of small and highly fluorescent labels and – thanks to recent advances in click chemistry – an incredible versatility. Depending on the approach, site-, sequence- and cell-specificity can be achieved. DNA and RNA labeling are rapidly developing fields that bring together multiple areas of research: on the one hand, synthetic and biophysical chemists develop new fluorescent labels and isomorphic nucleobases as well as faster and more selective bioorthogonal reactions. On the other hand, the number of enzymes that can be harnessed for post-synthetic and site-specific labeling of nucleic acids has increased significantly. Together with protein engineering and genetic manipulation of cells, intracellular and cell-specific labeling has become possible. In this review, we provide a structured overview of covalent labeling approaches for nucleic acids and highlight notable developments, in particular recent examples. The majority of this review will focus on fluorescent labeling; however, the principles can often be readily applied to other labels. We will start with entirely chemical approaches, followed by chemo-enzymatic strategies and ribozymes, and finish with metabolic labeling of nucleic acids. Each section is subdivided into direct (or one-step) and two-step labeling approaches and will start with DNA before treating RNA.
 Nils Klöcker | Nils Klöcker carried out his Chemistry studies at the University of Münster and obtained a BSc degree under the supervision of Prof. W. Uhl. During his master studies, he joined the group of Prof. A. Rentmeister at the same University to work on enzymatic synthesis and investigation of photocaged bioactive compounds. In 2019, he obtained the MSc degree under the supervision of Prof. A. Rentmeister. Since then he started his doctoral studies in the same group focusing on the development of photo-caged mRNA for optochemical control of translation. |
 Florian P. Weissenboeck | Born in 1993 in Bad Aibling (Germany), Florian P. Weissenboeck studied Chemistry at the University of Münster, Germany. In 2019, he investigated aptamer-based FRET systems in the research group of Prof. P. Unrau at the Simon Fraser University in Burnaby, Canada. During his master studies, he joined the group of Prof. A. Rentmeister to work on photocaging groups and their application on biomolecules. Since then he started his PhD studies in the same group, focusing on the investigation of optochemical tools and their utilization to control biological processes. |
 Andrea Rentmeister | Andrea Rentmeister received her PhD in Chemistry (2007) from the University of Bonn under the guidance of Prof. Michael Famulok. She then moved to the California Institute of Technology, where she was a postdoctoral fellow in the group of Prof. Frances H. Arnold. Afterwards, she joined the Department of Chemistry at the University of Hamburg as a Junior Professor and Emmy Noether group leader. In 2013, she was appointed as Associate Professsor for Biomolecular Label Chemistry at the University of Münster and promoted to Full Professor in 2020. Research in her lab focuses on RNA at the interface of chemistry and biochemistry and aims to understand and ultimately control the processes affecting mRNA expression and turnover. |
1 Introduction
Labeling is a prerequisite to investigate and understand all aspects of nucleic acid function. The underlying labeling concepts form the basis for discoveries in basic research, biotechnology and medical applications. Some techniques, like on-chip labeling and detection in Next Generation Sequencing (NGS),1 are so well established and commercialized that users are not aware of labeled nucleic acids being involved. Scheme 1 provides an overview of areas in research and technology, which rely on nucleic acid labeling. The selection illustrates how diverse applications are; a complete list of applications would require a review on its own. | ||
| Scheme 1 Overview of select applications of labeled nucleic acids in different areas. | ||
In basic research, fluorescently labeled nucleic acids are widely used in bulk and single-molecule experiments to elucidate fundamental processes, such as folding,2 replication, transcription and translation.3–6 In cells, fluorescent labeling is needed to visualize the subcellular localization of nucleic acids, most importantly mRNAs.5 The spatial information obtained in fluorescence microscopy cannot be obtained after harvesting and breaking the cells and has become very precise thanks to superresolution techniques.7 Fluorescently labeled nucleic acids already form the basis of commercially available methods used in fixed cells, such as Fluorescence In Situ Hybridization (FISH) and variations thereof.7–9 Challenges in the field of covalent labeling are methods for live cell labeling and imaging, aiming to track the molecule of interest.10 As a complement to fluorescent labeling, other reporters and analytic modalities, such as spin-labeling of nucleic acids together with Electron Paramagnetic Resonance (EPR)-based techniques, have proven particularly suitable for probing conformational changes in long range interactions in vitro and in cells.11–16
In biotechnology, the rise of NGS since its discovery in 2005 would not have been possible without fluorescently labeled nucleotides, their enzymatic incorporation and localized detection. Nucleic acid samples are amplified in situ (sequencing by synthesis) followed by the sequencing-reaction chemistry used in the commercially available platform (e.g. 454, Illumina, Ion Torrent and Gene Reader).1 NGS and emerging sequencing techniques like nanopore and single-molecule sequencing benefit enormously from the modification tolerance of polymerases17 and developments in covalent labeling.18
As the throughput of NGS increased and the costs decreased, transcriptome-wide studies have now become a common practice. Combined with chemical, chemo-enzymatic or metabolic labeling, NGS can shed light on subsets of nucleic acids, their modifications or interactions—in principle even for individual cells.19,20 Hence, modifications to isolate nucleic acids of interest or metabolic labeling, including cell-type specific labeling bear high potential for broad applications and commercialization.
The analysis of nucleic acids from cell samples and tissues by NGS is tightly linked to diagnostic questions and provides a route to personalized medicine.21,22 Some studies also point out applications of nucleic acid labeling in molecular imaging of tumors and infection.23,24 The existing and potential medical applications are in turn a driving force for biotechnology.
This review article provides an in-depth view on cutting edge covalent labeling approaches for DNA and RNA, including chemical and chemo-enzymatic as well as direct and two-step labeling strategies. The key advantage of covalent labeling compared to methods based on binding (hybridization of RNA-binding proteins) is the permanent linkage. This is particularly important in cells, where otherwise an equilibrium between bound and unbound reporter would have to be considered. Nevertheless, non-covalent methods are very important and popular, notably for mRNA labeling and tracking in living cells and we would like to refer the reader to excellent review articles in this field.25,26 Alternative methods for non-covalent labeling are also covered in other chapters of this special issue (fluorogenic aptamers) and recent reviews.27
We will start with fluorescent labeling approaches as these are widely applied and offer a great variety of applications (Fig. 1). Sections 2.1 and 2.2 will highlight the chemical direct and two-step introduction of fluorescent labels via solid-phase synthesis. Sections 2.3 and 2.4 will focus on chemo-enzymatic fluorescent labeling of nucleic acids in either one or two steps, involving methyltransferases (MTases), polymerases or other enzymes. Additionally, ribozymes and other reporters applied in various contexts will be treated in Sections 2.5 and 3, respectively. Finally, Section 4 will give a detailed view on the field of metabolic labeling, which provides access to dynamic aspects of replication or transcription in cells. This review will not cover radioactive or isotope labeling. For the following closely related topics, we would like to refer readers to other reviews within this themed issue: non-covalent labeling via aptamers (Unrau), labeling with photocages (Deiters), novel DNA base pairs (Hirao), fundamental photophysics of nucleic acids (Seidel) and fundamental studies of ribozymes/DNAzymes.28 For the detection of natural modifications of DNA and RNA, we would like to refer to an excellent recent review.29
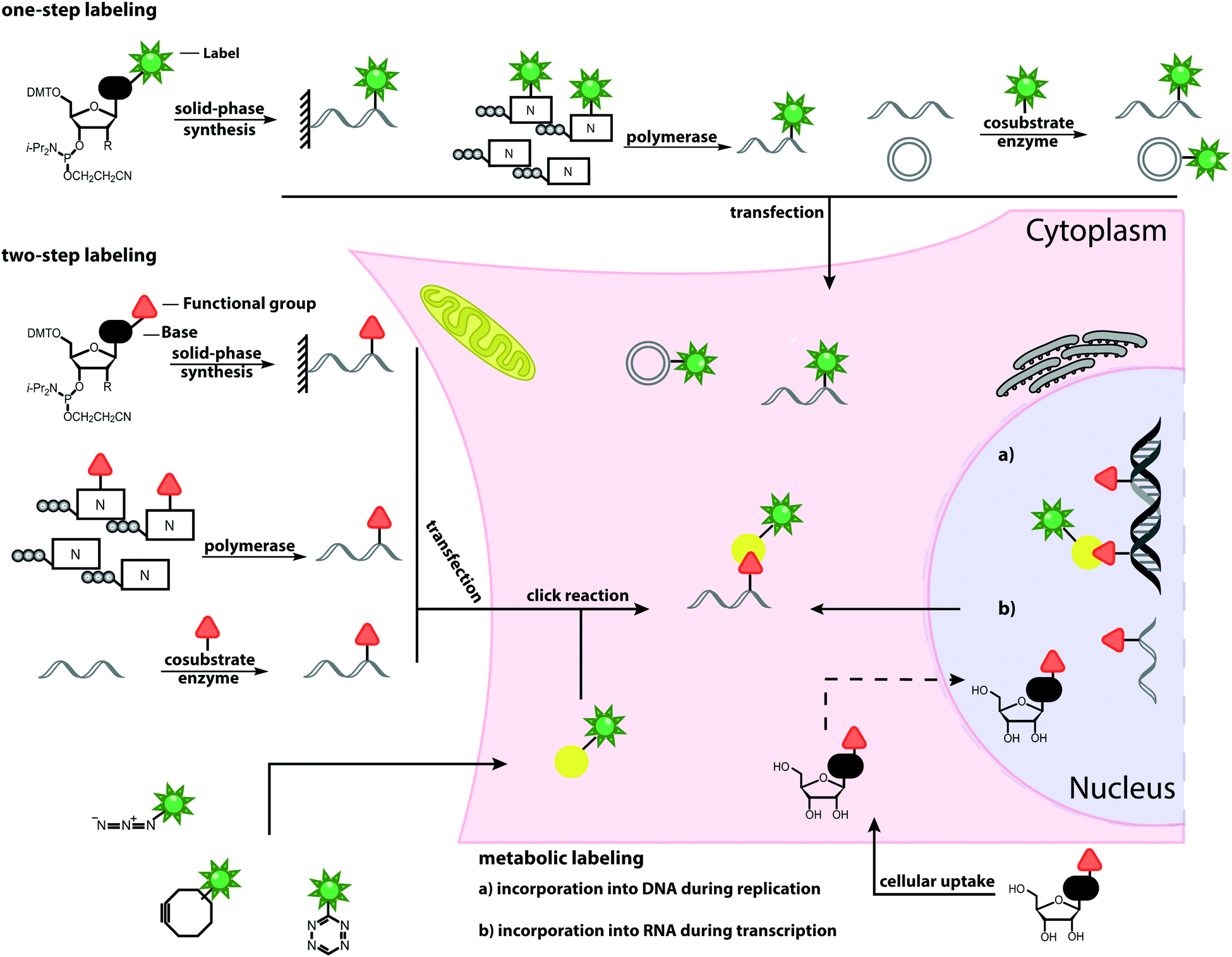 | ||
| Fig. 1 Schematic overview of labeling approaches and their application in cells. The illustrated methods include chemical and chemo-enzymatic one-step labeling, chemical and chemo-enzymatic two-step labeling and metabolic labeling. | ||
1.1 Click chemistry
Numerous labeling methods – especially the two-step approaches mentioned in this review – rely on click chemistry. In this brief introduction, we will therefore cover the basics of click chemistry and provide an overview of the benefits and drawbacks of the most relevant click reactions. For more comprehensive information about this topic, we refer the reader to dedicated reviews on click reactions.30–34Click reactions have become highly attractive for biological applications because they are by definition fast, stereospecific (not necessarily enantioselective) and can be performed in aqueous solutions with high yields.35 These characteristics make click reactions suitable for the conjugation of biomolecules in vitro, or – if the reaction is non-toxic – even in cells or in vivo, where the concentration of targeted biomolecules is usually low.36–38 The Copper(I)-catalyzed Azide–Alkyne Cycloaddition (CuAAC) between a terminal alkyne and an azide is widely used and has been applied to label nucleic acids, proteins, lipids and glycans39 (Fig. 2A). The reaction works fast (rate constants around 200 M−1 s−1)40 and the required functional groups are small. However, the CuAAC reaction is not bioorthogonal as the copper ions are cytotoxic at the required concentrations (μM range) and can generate reactive oxygen species. These impair the functionality of biomolecules, making the application of CuAAC in living cells challenging.41,42 Nevertheless, CuAAC reactions in living cells have been reported—enabled by stabilizing ligands.43
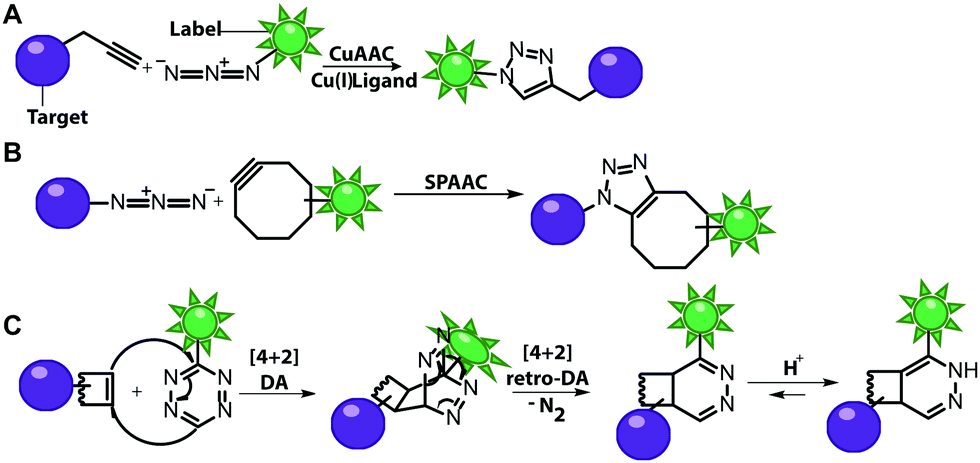 | ||
| Fig. 2 Schematic illustration of selected click reactions commonly used for nucleic acid labeling. (A) Copper(I)-catalyzed Azide–Alkyne Cycloaddition (CuAAC). (B) Reaction scheme of the Strain-Promoted Azide–Alkyne Cycloaddition (SPAAC). (C) Reaction scheme of the Inverse-Electron Demand Diels–Alder cycloaddition (IEDDA). | ||
Copper-free click reactions, like the Strain-Promoted Azide–Alkyne Cycloaddition (SPAAC) and the Inverse Electron-Demand Diels–Alder cycloaddition (IEDDA), represent valuable alternatives as they do not require addition of a catalyst. In case of the SPAAC, a strained alkyne, such as cyclooctyne, is reacted with an azide (Fig. 2B). However, this reaction shows relatively slow kinetics (rate constants around 0.12 M−1 s−1) when dibenzocyclooctyne derivatives are used and faces restrictions when performed in cells.44 The higher reactivity of strained alkynes can lead to unwanted side-reactions with cellular thiols.45 In contrast to the small terminal alkynes, the cyclooctyne derivates are bulky and usually not transferred by enzymes, excluding applications in metabolic labeling up to now. Azides, on the other hand, are easily reduced and should thus not be exposed to the intracellular environment for long periods of time.46
The most promising reaction for nucleic acid labeling in cells is currently the tetrazine ligation—an IEDDA reaction between a tetrazine and an alkene (Fig. 2C), because it is bioorthogonal and fast – at least with strained alkenes (rate constants 103 M−1 s−1 up to 105 M−1 s−1) for trans-cyclooctene (TCO)-derivatives.47,48 The fluorescence of the reaction product can be markedly increased compared to the starting material, by choosing suitable tetrazine-conjugates. In these cases, the tetrazine acts as an internal quencher of the dye, whose quenching abilities are lost upon bioconjugation.34 Various turn-on fluorophores allowing wash-free imaging have already been reported.49–51 Furthermore, for live-cell labeling, cell permeability of the reagent is important.
Both SPAAC and IEDDA reactions have also been used for covalent labeling of nucleic acids as many examples in this review will illustrate.
2 Fluorescent labeling of nucleic acids
Nucleic acids can be fluorescently labeled via binding, intercalation or covalent modification. This review will cover exclusively the latter. For fluorescent labeling by non-covalent interactions, we refer the reader to recent reviews in this field.7,25,52,53 Covalent labeling of nucleic acids has become the gold standard for numerous biophysical studies, such as elucidation of structure and dynamics of different functional nucleic acids,54,55 probing of local viscosity,56 optical genome mapping,57 or electro-optical nanopore sensing.58In general, a covalent fluorescent label can either be introduced directly into the nucleic acid of interest, or in a two-step approach, meaning that first a reactive handle is installed that allows for subsequent post-synthetic functionalization using the above mentioned click reactions. In the following sections, we will give a comprehensive overview of current chemical and enzymatic approaches for covalent fluorescent labeling. Furthermore, we would like to point out recent reviews about post-synthetic labeling of DNA and RNA.29,33,59–63
2.1 Direct chemical labeling via solid-phase synthesis
Solid-phase synthesis is the method of choice to make labeled RNA and DNA of up to 150–200 nucleotides.64 This approach uses nucleoside phosphoramidites as key building blocks and is compatible with almost any natural or non-natural modification as long as undesired side reactions during synthesis are avoided and reactive hydroxyl and amino groups can be protected. As the coupling efficiency is not directly affected by the steric demand of the modification, fluorescent nucleobase analogs can be directly incorporated (Fig. 3A). Alternatively, labeling can be achieved in a two-step approach (Fig. 3B).65–67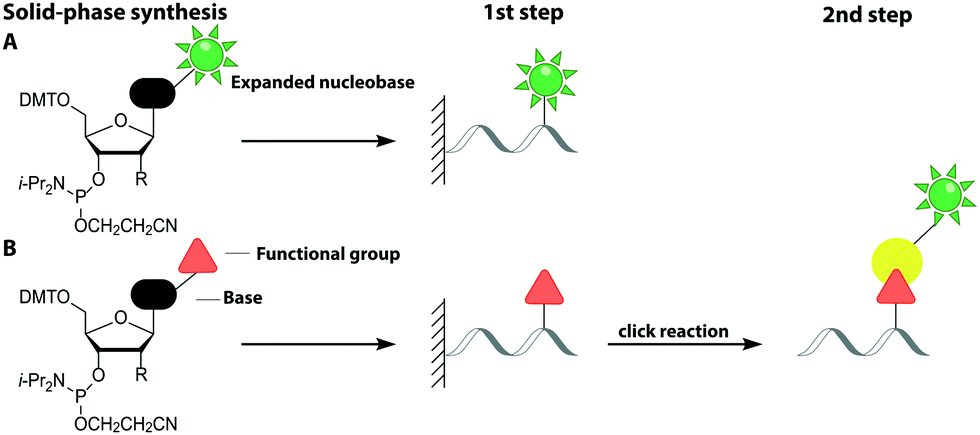 | ||
| Fig. 3 Strategies for labeling nucleic acids based on solid-phase synthesis. (A) Co-synthetic incorporation of expanded nucleobases. (B) Post-synthetic fluorescent labeling, achieved by a two-step approach: co-synthetic incorporation of a functional group and subsequent labeling via click chemistry. Similar approaches are usually used for DNA (R = H) and RNA (R = O-TOM) labeling. | ||
This sub-chapter will cover approaches that introduce the fluorescent label directly during solid-phase synthesis. The entire nucleoside can be replaced by a fluorophore, which is connected to a terminal phosphate group of the nucleic acid. These non-nucleoside-based fluorescent labels cannot form a duplex and are therefore preferably used for 5′- or 3′-end labeling. Already in 1992, the 5-carboxyfluorescein (FAM) phosphoramidite was synthesized and incorporated into DNA.68 In recent years, several non-nucleoside-based phosphoramidites have been developed, like the xanthene 4′,5′-dichloro-2′,7′-dimethoxy-fluorescein (JOE) and tetramethylrhodamine (TAMRA).69 For the investigation of the unwinding mechanism of dsDNA induced by the hepatitis C virus nonstructural protein 3 helicase (NS3h) a DNA was labeled at the 5′-end with Cy3 via solid-phase synthesis. In combination with a Cy5-labeled protein Förster Resonance Energy Transfer (FRET) was measured to elucidate the dynamics of enzyme translocation during unwinding.70
Another category is denoted chromophoric base analogs. Here, the entire nucleobase is exchanged for a fluorophore like pyrene, coumarin or stilbene. These fluorophores provide high fluorescence quantum yields and extinction coefficients, but lack a hydrogen bonding face for Watson–Crick or Hoogsteen interactions, limiting their general applicability.
More versatile are expanded nucleobase analogs, i.e. fluorescent nucleotides, which can be incorporated into a nucleic acid chain without perturbing the secondary structure and pertaining intra- and intermolecular hydrogen bond interactions. The latter can be achieved by fusing aromatic rings to the pyrimidine or purine nucleobases (Fig. 4B).71 These are also termed isomorphic, in that they closely resemble the corresponding natural nucleobases with respect to their overall dimensions, hydrogen bonding patterns and ability to form isostructural Watson–Crick base pairs. The design of such nucleobase analogs is challenging, but qAN1, qAN4 and qAnitro represent recent and notable examples (Fig. 4B).71 The respective phosphoramidites were successfully used for DNA synthesis.72 Moreover, qAN1 or qAN4 were suitable as a FRET pair with qAnitro and thus used to determine the FRET efficiency when installed at different positions of a DNA helix.73 Another example of an expanded nucleobase analog is pyrrolo-(d)C, a fluorescent analog of (deoxy)cytidine (Fig. 4B).74
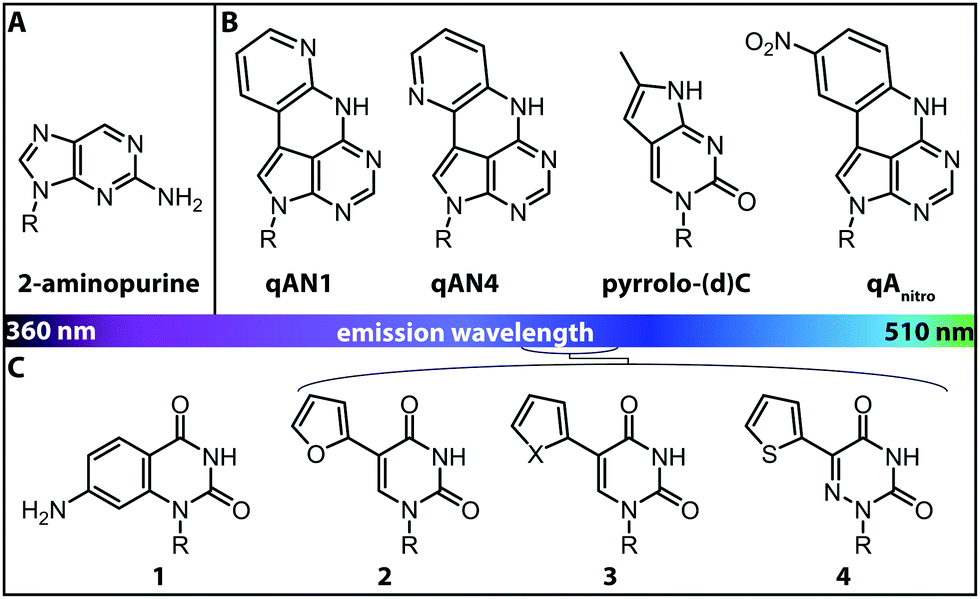 | ||
| Fig. 4 Structures of fluorescent nucleobase analogs sorted according to their emission wavelengths. (A) Structure of 2-aminopurine. (B) Examples of expanded nucleobase analogs that are fluorescent. (C) Examples of expanded nucleobase analogs that are fluorescent and two-photon excitable. 1: 7-amino-1-ribose-quinazoline-2,4(1H,3H)-dione, 2: 5-(furan-2-yl)-2′-deoxyuridine, 3: X = S: 5-(thiophene-2-yl)-2′-deoxyuridine, X = Se: 5-(selenophene-2-yl)-2′-deoxyuridine, 4: 5-(thiophene-2-yl)-6-aza-uridine. R denotes the (deoxy)ribose.76 | ||
Interestingly, the incorporation of qAN4 even stabilized the B-form of DNA. However, other expanded nucleobase analogs (although they retain their Watson–Crick face) rather perturb the resulting oligonucleotide structures due to their large surface area. Moreover, compared to typical commercially available fluorophores, such as Cy3, they are less fluorescent, as they exhibit lower extinction coefficients and fluorescence quantum yields and are also more sensitive to photo-bleaching.75
Isomorphic nucleobases do not necessarily have to be expanded. The most commonly used fluorescent ribonucleoside is 2-aminopurine (Fig. 4A), which usually pairs with thymine as an adenine-analog, but can also pair with cytosine as a guanine-analog.77 Excitation of 2-aminopurine is selective, due to a red-shifted absorption from 260 nm to 303 nm in an aqueous solution at neutral pH.78 The 2-aminopurine nucleoside was extensively used for elucidation of structure and dynamics of different functional nucleic acids, like the SAM-IV riboswitch54 or the hammerhead ribozyme.55 However, several studies revealed that the fluorescence of 2-aminopurine is highly susceptible to several parameters: the pH,78 the polarity of the solvent, the concentration of nucleotides in solution77 and the stacking of 2-aminopurine with purines and thymidines79—these affected the excitation and emission wavelengths, the fluorescence lifetimes and the quantum yield. Therefore, conclusions drawn from experimental results with 2-aminopurine have to be assessed with great care.
More recently, uridine-mimicking nucleobase analogs were developed (Fig. 4C, 2–4). These expanded and isomorphic nucleobase analogs proved suitable for two-photon excitation at 690 nm and 740 nm and the corresponding phosphoramidites were used in solid-phase synthesis of labeled RNA.76 A new addition to this set of uridine analogs is extended by the addition of a selenophene group at the C5-position. This SeU (Fig. 4C, 3) was incorporated into the bacterial ribosomal decoding site (A-site) via solid-phase synthesis and used to study RNA–drug interactions by fluorescence titration and X-ray crystallography.80
Considering the scope of fluorescent probes that can be introduced into nucleic acids, solid-phase synthesis is the most versatile methodology. Plenty of fluorophores can be introduced at every given position of an oligonucleotide, even if they are sterically demanding. Its biggest drawback is that the yield of the synthesis drops with the length of the oligonucleotide, limiting synthesis to around 150–200 nt.64 Therefore, although many interesting short RNAs – such as siRNAs and microRNAs or test substrates – can be made directly, large nucleic acids, such as mRNAs and lncRNAs (typically >1000 nt) are not directly accessible. In these cases, splint-ligation of chemically synthesized short and enzymatically produced long fragments provides a solution, as exemplified in labeling a SAM-I riboswitch for single-molecule FRET analysis to investigate ligand-induced structural changes.81
An interesting and conceptually different approach – named functionality transfer – for sequence- and site-specific labeling of RNA that – at least in theory – is not limited by the length of the target RNA was developed by the Sasaki group (Fig. 5).82 This method relies on the transfer of an α,β-unsaturated carbonyl unit of a 6-thioguanosine derivative onto the N4-position of a desired cytosine within the target RNA via enamine formation. Sequence-specificity is achieved by embedding this S-functionalized 6-thioguanosine within a complementary guide-DNA. With this approach, it was possible to label a desired cytidine within a specific sequence of the target RNA either with a reactive handle for subsequent reactions or directly with a fluorophore, in this case pyrene. By adjusting the pH, the functionality-transfer could be used to target the amino function of either cytidine (pH 7) or of a mismatched guanosine (pH 9.6).83
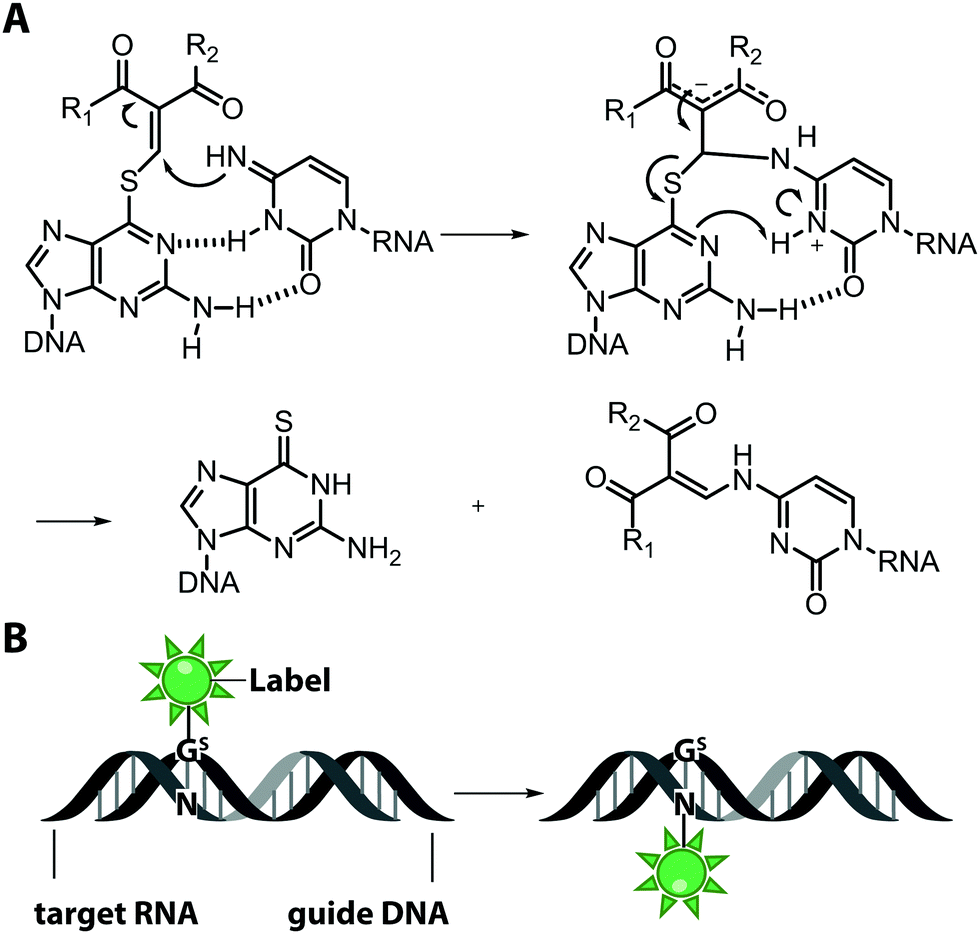 | ||
| Fig. 5 Functionality transfer for RNA labeling. (A) Proposed reaction mechanism. The 1-aryl-2-methylydene-1,3-diketone building block from the S-functionalized 6-thio-9-methyl-guanine is transferred onto the exocyclic amino function of cytidine or guanosine depending on the pH in the area where the guide-DNA hybridizes with the target RNA. R1 is usually a methyl or phenyl group, R2 is the fluorophore (pyrene, ATTO550, Cy3). (B) Scheme illustrating RNA labeling guided by a DNA oligonucleotide containing modified 6-thioguanosine (Gs). | ||
2.2 Two-step chemical labeling via solid-phase synthesis
This sub-chapter will show how solid-phase synthesis has been used to incorporate functional handles for subsequent post-synthetic labeling of nucleic acids (Fig. 3B).The attachment of fluorescent labels may influence both structure and function of nucleic acids depending on whether it involves the Watson–Crick, the Hoogsteen or the sugar edge of a given nucleoside. As already shown, isomorphic nucleobase analogs aim to preserve those interactions, but sensitive detection is hampered by their low extinction coefficients, poor fluorescence quantum yields and potential photo-bleaching.75 To circumvent these issues, two-step approaches first introduce a small reactive handle and then use selective chemistry to install a fluorescent label with better properties. In this chapter, we focus on click chemistry for labeling, which has become very common in the field of nucleic acids. However, we would like to mention that numerous typical conjugation chemistries, using thiols, amines84 and maleimides85 have also been successfully combined with solid-phase synthesis. For a more detailed overview, we recommend the reviews from Madsen et al. and Ivancová et al.29,86
Various functional handles like terminal alkynes,87,88 cyclooctynes89,90 and norbornenes91 were successfully incorporated during solid-phase synthesis without the necessity of additional protection chemistry. Fluorescent labeling was then achieved in a second step by click chemistry. Alkynes were introduced at the C5-position of thymidine analogs, converted to the corresponding phosphoramidites, used for DNA synthesis and subsequently labeled with coumarin and fluorescein azides.88 Other examples are the alkyne-modifications at the phosphate backbone,92 the exocyclic O4- or N4 position of pyrimidines and the N3-position of thymine.93
Schoch et al. achieved incorporation of an alkyne and a trans-cyclooctene group into the same oligonucleotide by standard phosphoramidite chemistry on the solid-phase. The resulting modified oligonucleotides were suitable for dual labeling via CuAAC and IEDDA reactions with different fluorophores like Cy5 azide and TAMRA tetrazine, and then applicable for FRET studies.90
The IEDDA reaction has also been performed on synthetic RNA in mammalian cells. Kath-Schorr and coworkers synthesized a norbornene-modified uridine phosphoramidite and incorporated it into a 21 nt long RNA. The RNA was labeled with Oregon Green 488-tetrazine conjugates in transfected NCI-H460 cells.91 Internally quenched tetrazines were developed to make the click reaction fluorogenic. Combined with proper antisense probes for the target oligonucleotide, it was possible to fluorescently label DNA and RNA in HeLa and SKBR3 cells.94 With a similar approach miRNA was detected in SKBR3, MCF-7 and HeLa cells.95
The Wagenknecht group synthesized a 2′-deoxyuridine DNA building block with a cyclooctyne at the C5-position attached via a flexible or rigid linker. The building block was incorporated into oligonucleotides using solid-phase synthesis. Subsequent copper-free click reaction with azide-conjugated fluorophores was performed.96 In a follow-up study, HeLa cells were transfected with DNA containing the cyclooctyne moiety. The DNA was click labeled with a set of new water-soluble and membrane-permeable benzothiazolium-based fluorescent dyes.97
While cyclooctynes are compatible with solid-phase synthesis, the introduction of azido-groups poses a challenge. The preparation of a classical azido-modified phosphoramidite is difficult, due to an unwanted Staudinger reaction, which occurs between the azido and phosphoramidite group, forming an iminophosphorane.98,99 To circumvent this issue, an azido-modified H-phosphonate was synthesized and used in common solid-phase synthesis. Here, an important observation was that the Staudinger reaction was not disturbing the azido group in the support-bound growing nucleotide chain upon further assembly using phosphoramidites.100 Later, Fomich et al. presented an interesting azido-bearing phosphoramidite based on 4-azaribose, which was stable (i.e. did not undergo the unwanted Staudinger reaction) and successfully incorporated into a DNA oligonucleotide during solid-phase synthesis.101
To circumvent the difficulties associated with azides in phosphoramidite chemistry, Winz et al. developed an alternative route and used it for site-specific one-pot triple click labeling of DNA and RNA. In this approach, different bioorthogonal groups are combined: first, a norbornene and an alkyne moiety were introduced during solid-phase synthesis. Then, an azido-modified nucleotide was added enzymatically, using the terminal deoxynucleotidyl transferase (TdT) or the poly(A)-polymerase (PAP). A one-pot click reaction was performed to label all three different reactive handles with three different fluorophores via CuAAC, SPAAC and IEDDA reaction. This triple labeling can be used to study the dynamics of nucleic acid structures during folding and the population of multiple folding states using FRET.102
In this subchapter, we tried to illustrate the broad scope of phosphoramidites and solid-phase synthesis for position-specific incorporation of a plethora of modified nucleobases into oligonucleotides and subsequent labeling via click chemistry. Most of the two-step approaches mentioned above are suitable for both DNA and RNA labeling. Nevertheless, all approaches based on solid-phase synthesis, irrespective of one-step or two-step strategies, bear one great disadvantage, which is the length limit of the oligonucleotides.
2.3 Direct chemo-enzymatic labeling
Enzymes can also be used to label nucleic acids co- or post-synthetically in one- or two-step procedures if they accept suitable substrate analogs. We will present non-natural nucleoside triphosphates that are converted by DNA- and RNA-polymerases as well as substrate analogs of nucleic acid-modifying enzymes, most notably MTases.Compared to solid-phase synthesis, the scope of enzymatic modifications is certainly limited, although protein-engineering bears the potential to fundamentally broaden it. On the positive side, the length of the nucleic acid does usually not limit the enzymatic approaches.
In this sub-chapter, we will elaborate on polymerase-based labeling strategies to directly incorporate fluorescent reporters into nucleic acids – a co-synthetic approach (Fig. 6A). In 2001, for the first time, the replacement of the complete set of dNTPs by analogs labeled with different fluorophores was reported. This primer extension synthesized DNA of up to 40 basepairs. Modified dUTPs bearing a coumarin or a fluorescein residue at the C5-position were successfully incorporated by three different DNA polymerases and full-length PCR products were obtained. Polymerases belonging to family B, such as Vent exo− and Tgo exo−, were more efficient at incorporating labeled dNTPs compared to the commonly used Taq polymerase, which belongs to family A.106
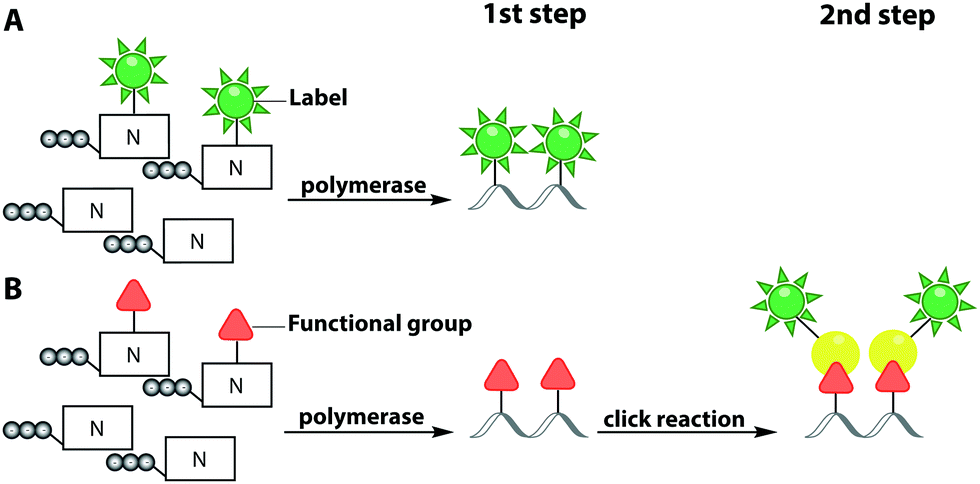 | ||
| Fig. 6 Polymerase-based fluorescent labeling strategies for nucleic acids. (A) Direct enzymatic incorporation of fluorescent nucleotides. (B) Two-step labeling approach. Enzymatic incorporation of a functional group for subsequent fluorescent labeling via click chemistry. Templates for polymerases are omitted. | ||
While the fluorophores are typically connected via alkyl,107 amide106 and polyethyleneglycol87 linkers, a recent study revealed that rigid phenylethyne linkers (Fig. 7A) improve the fluorescence properties of the labeled nucleic acid significantly. Specifically, fluorescently labeled dTTP and ddTTP analogs (i.e., modified with fluorescein at the C5-postion via different linkers) were successfully incorporated into DNA during primer extension using TaqFS polymerase. It turned out that longer, fully conjugated linkers shifted the absorption maximum of the label to longer wavelengths and increased the extinction coefficient of the modified nucleotide. The authors hypothesized that the phenylethyne linker acts like a donor that channels more energy into fluorescein during absorption, which enhances the fluorescence intensity. Moreover, it was shown that shorter linkers (like in 5 and 6) were not well tolerated by TaqFS, whereas a longer linker (like in 7) placing the dye further away from the nucleobase is efficiently incorporated.108
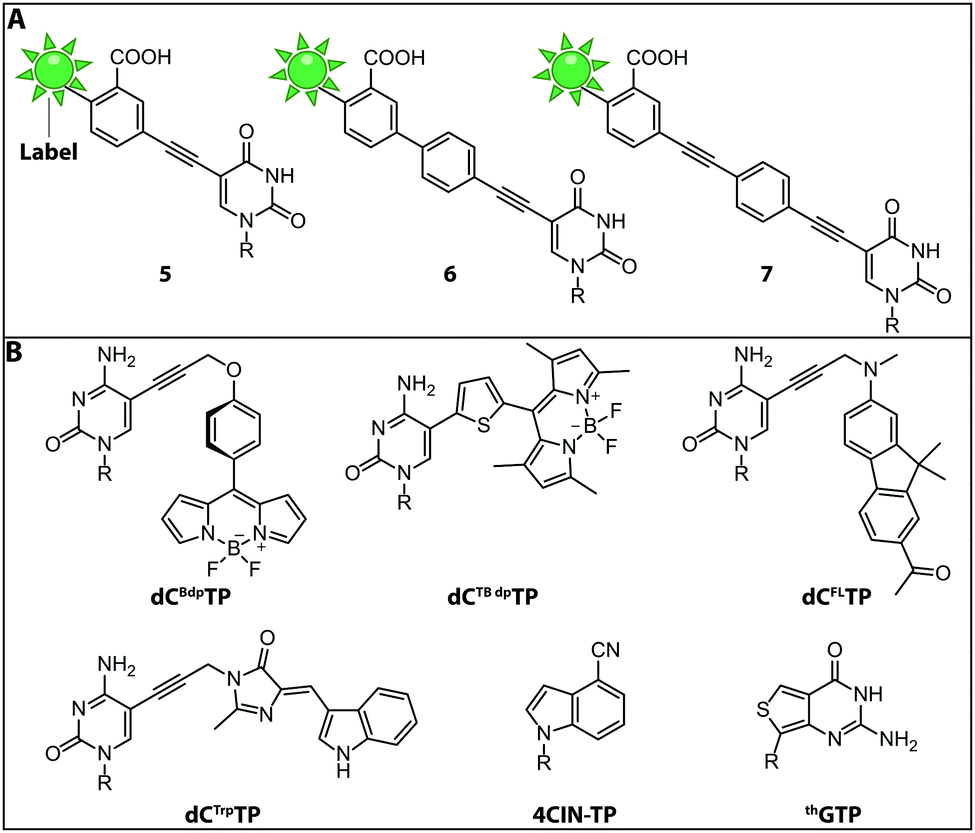 | ||
| Fig. 7 Structures of fluorescent (d)NTPs. (A) Structures of rigid phenylethyne linkers of increasing lengths lead to a bathochromic shift increasing with the length of the linker. (B) Structures of the fluorescent dNTP/NTP analogs dCBdpTP (BODIPY), dCTBdpTP (thiophene-linked tetramethylbodipy), dCFLTP (fluorene), dCTrpTP (tryptophan), 4CIN-TP (4-cyanoindole-2′-deoxyribonucleoside-5′-triphosphate) and thGTP (thieno). R denotes 5′-triphosphorylated (deoxy)ribose. | ||
More recently, the rat DNA polymerase β was found to be suitable for the consecutive incorporation of multiple fluorescently labeled nucleotides. Interestingly, the incorporation kinetics for polymerase β with different commercially available dNTPs (fluorescein-12-dNTP) revealed similar Km values for unlabeled and fluorescently labeled dNTPs. This means that polymerase β can tolerate sterically demanding modifications (such as fluorescein and its linker) at the C7 of deazapurine and the C5 of pyrimidine bases without affecting the incorporation efficiency.109
Fluorescent Molecular Rotors (FMRs) are tools to probe local viscosity. The Hocek group developed a novel fluorescent environment-sensitive nucleoside analog dCBdpTP (BODIPY) (Fig. 7B) for fluorescence lifetime-based biophysical assays and successfully incorporated it into nascent DNA using KOD XL DNA polymerase. This approach allowed the investigation of complexes between transfection agents (here DOTAP) and nucleic acids, rendering this approach suitable for the screening of new transfection reagents. Furthermore, transfection of HeLa cells with DNABdpvia DOTAP-lipoplexes revealed that the local environment of exogenous DNA undergoes significant changes during the process of transfection. The phenyl-substituted BODIPY fluorophores have proven adequate sensors for measuring local viscosity in cells. Furthermore, protein–DNA interactions can be observed due to changes in the fluorescence lifetime after the BODIPY rotor is bound to a hydrophobic pocket of the protein.56
Along these lines, the Hocek group synthesized a novel dCTP analog whose fluorescence lifetime depends on the viscosity of the environment—the thiophene-linked tetramethylbodipy dCTBdpTP (Fig. 7B). This new fluorescent probe, which was also accepted by DNA polymerases, provides longer fluorescence lifetimes especially in viscous media than the previous phenyl BODIPY construct.110
Another approach to observe interactions between DNA and DNA-binding proteins or lipids, uses a solvatochromic push–pull fluorophore. This fluorene based fluorophore was attached to the C5-position of dCTP analogs (dCFLTP) (Fig. 7B). The KOD XL and Bst DNA polymerases were able to use dCFLTP as a substrate for DNA synthesis. A 50 nt long dsDNA containing two dCFL nucleotides was synthesized to determine DNA–protein and DNA–lipid interactions by observing the shift of the fluorescence emission maxima.111
Furthermore, a dCTP analog bearing a tryptophan-based fluorophore from cyan fluorescent protein (dCTrpTP) (Fig. 7B) was synthesized, incorporated into DNA, and used to sense protein–DNA interactions. To this end, a single stranded 19-mer DNA bearing one dCTrp nucleotide was synthesized and the fluorescence emission was detected in absence and presence of single-strand binding protein from E. coli (SSB) revealing a 2-fold increase of fluorescence in presence of SSB.112 Importantly, dCTBdpTP, dCFLTP and dCTrpTP were not only successfully used for primer extension but also in DNA amplification by PCR.
Minimal perturbation of Watson–Crick and Hoogsteen interactions can be achieved by the use of isomorphic nucleotides. Recently, the fluorescent 4-cyanoindole-2′-deoxyribonucleoside-5′-triphosphate (4CIN-TP) (Fig. 7B) was synthesized and successfully incorporated into DNA by DNA polymerase I Klenow exo− fragment.113
Similar to DNA polymerases, RNA polymerases were also exploited to introduce fluorescently labeled nucleotides into nascent RNA. Thieno[3,4-d]pyrimidine represents an isomorphic GTP analog termed thGTP (Fig. 7B) which was incorporated into nascent RNA by T7 RNA polymerase. This thGTP could be used instead of GTP in transcription reactions, yielding a highly emissive RNA transcript in which all guanosine residues had been replaced. However, this approach is not generally applicable as the T7 polymerase accepts only a limited set of modified NTPs. Also, termini-modifying enzymes such as alkaline phosphatase and T4 polynucleotide kinase tolerated thGTP and were used to modify RNA at the 5′-end. Such modified transcripts contain a pppthG at their 5′-end.114
The examples above illustrate that fluorescent reporters can be directly incorporated by various polymerases to produce labeled nucleic acids. However, due to the size of the fluorescent reporter molecules and the limited, not completely predictable promiscuity of the polymerases, the compatibility of each new (d)NTP analog needs to be carefully evaluated. Long linkers between the base and the fluorophores have proven useful to prevent abrogation of elongation in several cases. Furthermore, it is important to keep in mind that polymerases incorporate the non-natural nucleotides randomly unless a nucleoside triphosphate was completely replaced. To overcome the size limitations associated with direct incorporation of fluorescent nucleotides, the two-step polymerase-based approach represent attractive alternatives (Section 2.4.1).
The Weinhold group developed a DNA labeling strategy, based on an engineered fluorescent cofactor for the DNA MTase from Thermus aquaticus (M.TaqI). Normally, M.TaqI transfers the methyl group from the cofactor S-adenosyl-L-methionine (AdoMet or SAM) to the exocyclic amino group of adenosine within the double-stranded 5′-TCGA-3′ DNA sequence. For direct labeling, a non-natural cofactor containing an aziridinyl residue instead of a methionine sidechain was synthesized (Fig. 8A). This cofactor led to the transfer of the entire nucleoside onto the exocyclic amino function of adenosine. Importantly, the C8-position of the adenosine moiety of the non-natural cofactor could be modified with a fluorophore (in this case dansyl-based), because this position points out of the enzyme and does not impair binding.115
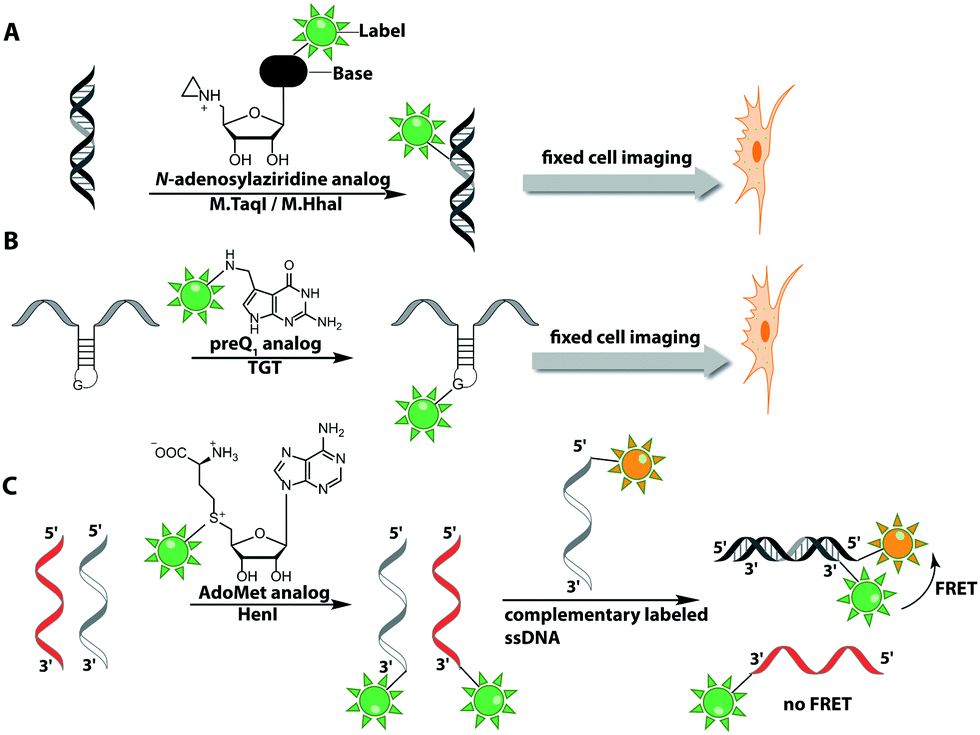 | ||
| Fig. 8 Direct post-synthetic enzymatic labeling strategies for nucleic acids. (A) M.TaqI was used to transfer a Cy3 bearing aziridine cofactor onto the exocyclic amino function of adenine in the 5′-TCGA-3′ sequence of the two plasmids, pUC19 and pBR322. Mammalian CHO-K1 cells were transfected with these labeled plasmids, which could then be detected by fluorescence microscopy. (B) TGT from E. coli can transfer fluorescent PreQ1 analogs onto a small 17 nt hairpin motif and label RNA in fixed cells. (C) Hen1 2′-O-methyltransferase utilizing a Cy3-bearing AdoMet analog for labeling ssRNA at the 3′-end. Together with a Cy5-labeled complementary ssDNA for the labeled ssRNA was detected by FRET. | ||
In 2008, M.TaqI was used, together with a newly developed Cy3 bearing aziridine cofactor, to label the two DNA plasmids, pUC19 and pBR322. After transfection of mammalian CHO-K1 cells, the labeled plasmids were visualized by fluorescence microscopy.116 Using the DNA cytosine-C5 MTase from Haemophilus haemolyticus (M.HhaI), Weinhold and coworkers presented a similar approach for direct labeling of DNA (Fig. 8A). In this case, the fluorophore was attached to the C7-position of the cosubstrate, yielding a stable 7-deazaadenosylaziridine moiety, which was transferred to the C5-position of cytosine within the 5′-GCGC-3′ motif in short duplex oligonucleotides and plasmid DNA.117 By using M.TaqI and a TAMRA-modified AdoMet analog (AdoYnTAMRA), it was possible to fluorescently label the genomes of λ and T7 bacteriophages in a single step. This optical mapping strategy provides an excellent method for accurate genotyping.118
Recently, a novel approach was developed to circumvent tedious synthesis of non-natural AdoMet analogs for MTase-based labeling strategies of nucleic acids. Usually, the reaction of S-adenosyl-L-homocysteine (SAH) with allyl bromides suffers from low yields and requires a high excess of the bromide. Hofkens and coworkers presented the synthesis of cysteine (instead of homocysteine)-based AdoMet analogs – i.e. one methylene group shorter. The respective unnatural cofactors were successfully used to transfer rhodamine fluorophores onto the plasmid DNA, pUC19 by M.TaqI. Despite reduced enzymatic transfer efficiency compared to homocysteine-based cofactor analogs, various fluorophores like Cy3, Cy5 and cascade blue were successfully linked to DNA.119
Direct post-synthetic labeling was also successful with several RNA-modifying enzymes. To label RNA in fixed mammalian cells (CHO cells), an approach using bacterial (E. coli) tRNA guanine transglycosylase (TGT) was developed. Prokaryotic TGT naturally performs a transglycosylation reaction, in which the guanine is exchanged with an amine-containing queuine pre-cursor, PreQ1 (Fig. 8B). The remarkable promiscuity of TGT allowed the direct transfer of fluorophores like Thiazol Orange, Cy7 and BODIPY onto its 17 nt hairpin recognition motif.120
Recently, 3′-end labeling of ssRNA was achieved by the animal Hen1 2′-O-methyltransferase utilizing AdoMet analogs bearing biotin and Cy3 moieties (Fig. 8C). Selective detection of a specific 3′-labeled ssRNA was then accomplished using a Cy5-labeled ssDNA counter strand and FRET measurements. Interestingly, the human Hen1 enzyme, piRNA MTase HsHEN1, requires cobalt ions (Co2+ or Co3+) for enzymatic activity, whereas Drosophila DmHen1 is also active in the presence of other divalent cations, like Mn2+, Mg2+ and Ca2+.121 However, cobalt ions are cytotoxic, limiting the application of this enzyme to in vitro RNA labeling.122
Although a one-step procedure appears to be most straightforward to label nucleic acids, there are a few disadvantages that should not be overlooked. Most importantly, many modifying enzymes show a limited promiscuity regarding the rather big fluorescent reporter molecules.
2.4 Two-step chemo-enzymatic labeling
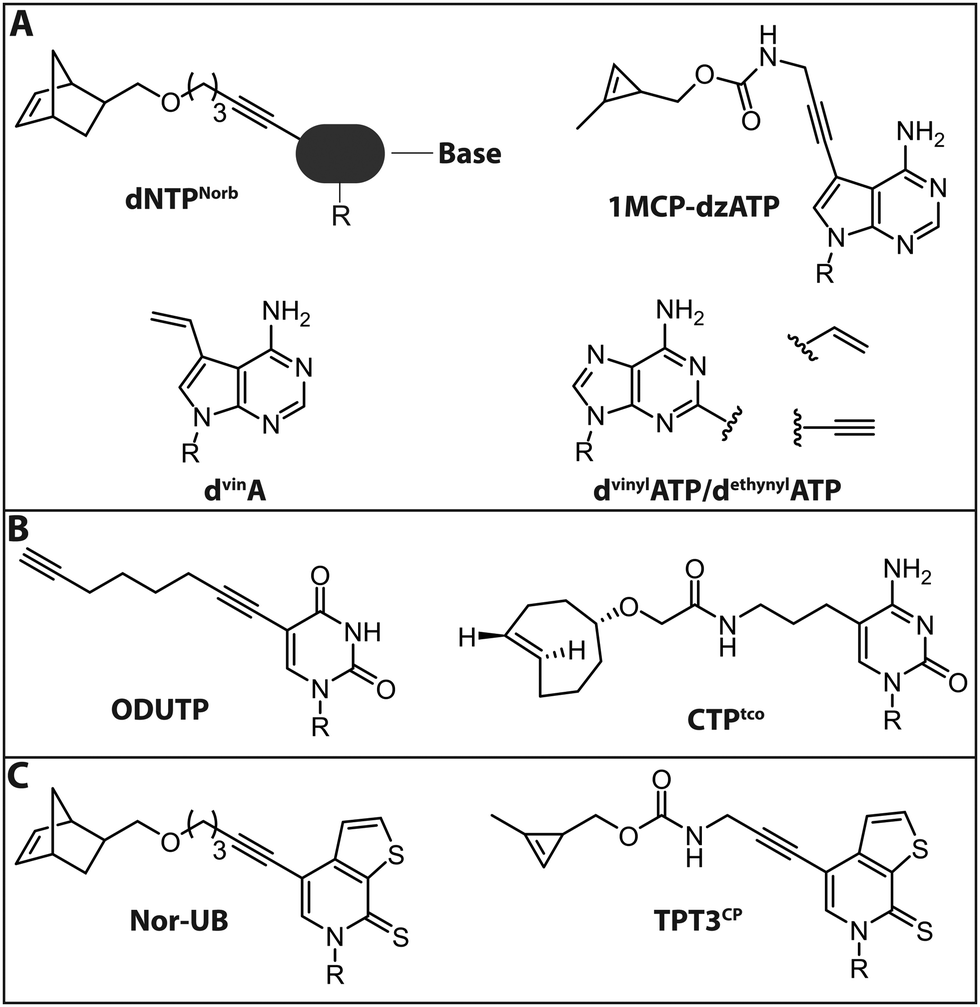 | ||
| Fig. 9 Modified (d)NTPs for chemo-enzymatic two-step labeling of nucleic acids. (A) dNTPs for chemo-enzymatic two-step labeling of DNA. (B) NTPs for chemo-enzymatic two-step labeling of RNA. (C) NTPs with unnatural base-pairs for chemo-enzymatic labeling of RNA using the dTPT3–dNaM system. R denotes 5′-triphosphorylated (deoxy)ribose. | ||
Jäschke and coworkers investigated the polymerase-based incorporation of norbornene-modified pyrimidine- (dUNorb, dCNorb) and deaza-purine-nucleotides (dANorb) into nascent DNA. Building on the known acceptance of C5-modified pyrimidines and C7-modified 7-deazapurines by DNA polymerases, they synthesized the respective norbornene-modified nucleoside triphosphates (Fig. 9A). The Taq DNA polymerase accepted dUTPNorb as a substrate and 38 nt long full-length products were obtained. The PEX Phusion polymerase was able to incorporate all three modified nucleotides but failed in the PCR experiment. The amplification of DNA is more challenging because the enzyme must additionally tolerate the modifications in the template strand. However, the KOD-XL DNA polymerase, an engineered DNA polymerase from the archaeon Thermococcus kodakarensis, successfully amplified a 429 nt long dsDNA containing around 600 norbornene moieties.124 In this study, no fluorescent reporter was attached, although the click reaction with tetrazine-bearing fluorophores should be straightforward (see Introduction: Click chemistry).
In a related strategy, 7-vinyl-7-deaza-2′-deoxyadenosines (dvinA) were incorporated into dsDNA using KlenTaq DNA polymerase (Fig. 9A). In this case, the modified dATP substituted its natural counterpart completely and the modification was reacted with fluorescently labeled tetrazines in a subsequent IEDDA reaction.125 In addition to the different cycloadditions, other reactions were used for the attachment of fluorophores. In 2018, the group of Hocek reported the introduction of a fluorophore in a two-step manner by first incorporating an aldehyde modified UTP that could be reacted in a second step via reductive amination.126 The same group further reported the introduction of a coumarine via thiol–ene click chemistry, only utilizing a single modification in the minor groove of DNA.127
For proteins and ligands binding to the DNA minor groove, labeling at the respective positions could provide new insights into their molecular interactions. However, these positions of the nucleobases are difficult to modify since they are crucial both for Watson–Crick base pairing and for key minor groove interactions with DNA polymerase that are vital for the extension of the chain. Nevertheless, several 2-substituted dATP derivatives were synthesized and incorporated into DNA by PEX, Vent exo− or Bst DNA polymerase. Vinyl- and ethynyl-modified dATPs (dvinylATP and dethynylATP) allowed post-synthetic labeling of DNA via thiol–ene or CuAAC reactions with thiols or azides, respectively (Fig. 9A).128 Wagenknecht and coworkers recently presented a DNA polymerase-based approach to incorporate the 1-methylcyclopropene (1MCP) group at the 7-position of 7-deaza-adenine (dzA) nucleotides into oligonucleotides by primer extension. Subsequently, a tetrazine-bearing fluorescent dye was attached via click chemistry to obtain fluorescently labeled DNA (Fig. 9A). Additionally, HeLa cells were transfected with the modified DNA oligonucleotides, a click reaction was performed inside living cells and the DNA was visualized by confocal microscopy.129
In 2018, the group of Jäschke reported site-specific one-pot labeling of DNA and RNA via click chemistry. They combined three different click reactions (CuAAC, SPAAC and IEDDA) utilizing a DNA or RNA equipped with three different functional moieties (azide, alkyne and norbornene; see also Section 2.2). With this approach, it was possible to triple label, for instance, the preQ1 riboswitch RNA, rendering this strategy applicable for folding and dynamic studies.102
For RNA labeling, the polymerase-based approaches are, in principle, similar to the DNA-labeling approaches, but based on in vitro transcription with a DNA-dependent RNA polymerase, such as T7. In 2014, a cytidine triphosphate analog bearing a trans-cyclooctene at the 5-position (CTPtco) was synthesized and incorporated into RNA by T7 RNA polymerase (Fig. 9B). A fluorescein with a tetrazine functionality was then used to label the RNA via IEDDA.130 Recently, the Srivatsan group developed an alkyne-modified UTP analog, 5-(1,7-octadiynyl)uridine triphosphate (ODUTP), with a disubstituted alkyne as Raman scattering label (Fig. 9B).131
Slightly different is the development of a two-step labeling approach for RNA at its 3′-terminus that uses poly(A) polymerases (PAPs). Winz et al. presented this template-independent strategy in 2012. Several PAPs were tested to incorporate different azido-modified NTPs. The PAP from yeast turned out to exhibit the highest incorporation activity for all modified NTPs. The NTPs were modified with azido groups at various positions (8C-N3-ATP, 2′-N3-2′-dNTP, 3′-N3-2′,3′-ddATP/ddTTP). Except for 2′-N3-2′-dATP, where multiple successive incorporations were observed, the yeast PAP was able to incorporate up to two azido-modified nucleotides. In a second step, a CuAAC or SPAAC reaction was performed to fluorescently label the RNA. This robust approach was successfully used to label RNA that was isolated from E. coli.132
We used the latter approach to 3′-end label the poly(A) tail of mRNAs (eGFP-, Gaussia luciferase (GLuc) or Cypridina luciferase (CLuc) mRNA) with yeast PAP and 2′-N3-2′-dATP or 3′-N3-2′,3′-ddATP. After introduction of the azido-groups, a SPAAC reaction for fluorescent labeling was performed in vitro and in HeLa cells. Interestingly, it turned out that the modified poly(A) tail, before and after click reaction with a rhodamine fluorophore, increased the translation efficiency of the mRNA both in vitro and in HeLa cells. Further experiments showed that the increase in the translation efficiency is not caused by transfection efficiency or stabilization at the mRNA level, rendering this a potential strategy in the field of therapeutic mRNAs, where the change of pharmacokinetic properties of mRNAs is an important point of research.133
Recently, together with the Raz group, we expanded the applicability of the 3′ poly(A) tail labeling strategy. By using yeast PAP and 2′-N3-2′-dATP as cosubstrate, reporter mRNAs bearing localization-specifying 3′-UTRs were successfully azido-modified. Subsequently, SPAAC reactions were performed to label the mRNAs with different fluorophores. Afterwards, the in vitro-labeled mRNAs were injected into 1-cell stage zebrafish embryos. Further experiments revealed that the label neither negatively affected the translational activity nor altered the subcellular localization of the injected mRNAs.233 This strategy was used for live imaging of labeled mRNA in vivo, which is important for understanding the dynamic aspects underlying its function.
The seminal recent development of unnatural base pairs (UBPs) that are treated in detail in a dedicated review in this themed collection by Hirao, provided also the basis for site-specific RNA labeling. First, a DNA containing UBPs is synthesized on the solid phase and then amplified by PCR, including the dNTP analogs.134 Subsequently, the DNA with UBPs is used as template for transcription with modified ribonucleoside triphosphates, including the NTP analog. In a final step, the RNA is labeled via click chemistry.135–137 Recently, Feldman et al. were successful in delivering the necessary (d)NTPs for the UBP-approach into E. coli by overexpression of a nucleoside triphosphate transporter from Phaeodactylum tricornutum (ptNTT2).138 The application of this method to eukaryotic cells remains a challenge, due to low delivery efficiency of (d)NTPs.
The Kath–Schorr group presented an approach to incorporate an unnatural ribonucleoside triphosphate (Nor-UB) containing a norbornene moiety for post-transcriptional functionalization by IEDDA with Oregon Green 488 tetrazine dyes (Fig. 9C). The unnatural NTP was accepted by T7 RNA polymerase and the subsequent click reaction with the dye-tetrazine conjugate labeled the UBP-containing RNA efficiently. Along these lines, an unnatural base of the dTPT3–dNaM system developed by Romesberg and Li was modified with a cyclopropene residue (TPT3CP, Fig. 9C).139 The cyclopropene group bears an additional methyl group to ensure prolonged stability in solution allowing IEDDA reactions on biomolecules (Fig. 9C). With this approach the highly structured 77 nt Methynococcus jannaschii tRNA was fluorescently labeled in the anticodon loop.137
One significant advantage of the two-step labeling approach is the higher tolerance of several polymerases for small functional groups, compared to sterically demanding reporter moieties required in one-step approaches. Current work in the directed evolution of polymerases suggests that the scope of compatible functional groups can be significantly expanded in the future.140 However, this approach – with exception of the UBPs – still leads to random incorporation which results in a lack of specificity.
Two main strategies have been established to label DNA using MTases and fluorophore bearing cofactors. The Sequence-specific Methyltransferase-Induced Labeling (SMILing) approach relies on N-mustard analogs of AdoMet that form aziridine AdoMet analogs carrying an aziridine ring at the 5′-sulfonium in an in situ reaction. A nucleophilic attack by the target compound (e.g. an amino group of a DNA) induces a ring opening and transfers the entire cofactor following an SN2 reaction mechanism similar to the direct labeling approach described in 2008 with M.TaqI mentioned in Section 2.3.2.149
Du et al. used this method to transfer azido- and alkyne-functionalized AdoMet analogs to DNA by the sequence-specific MTases, M.TaqI and M.HhaI. The subsequent labeling with fluorophores was achieved in a CuAAC reaction. Since DNA is labeled sequence-specifically this approach shows promise to further investigate biological methylation sites.150 This strategy can also be used in one-step labeling experiments, as mentioned above (Section 2.3.2).116,151
The other widely used method in the field of two-step MTase labeling is called Methyltransferase-directed Transfer of Activated Groups (mTAG). Like the SMILing method, it relies on MTases and AdoMet analogs. However, in mTAG, the AdoMet analogs are not completely transferred but act as cosubstrates for transfer of an extended alkyl group. This approach was implemented for numerous DNA and RNA MTases, but also for other biomolecules.152 The AdoMet analogs often carry a sulfonium-bound extended side chain with a terminal functional group. If accepted by MTases, the target nucleic acid will be covalently modified with functional groups that can be labeled with a reporter group in a second step (Fig. 10B, 11).153
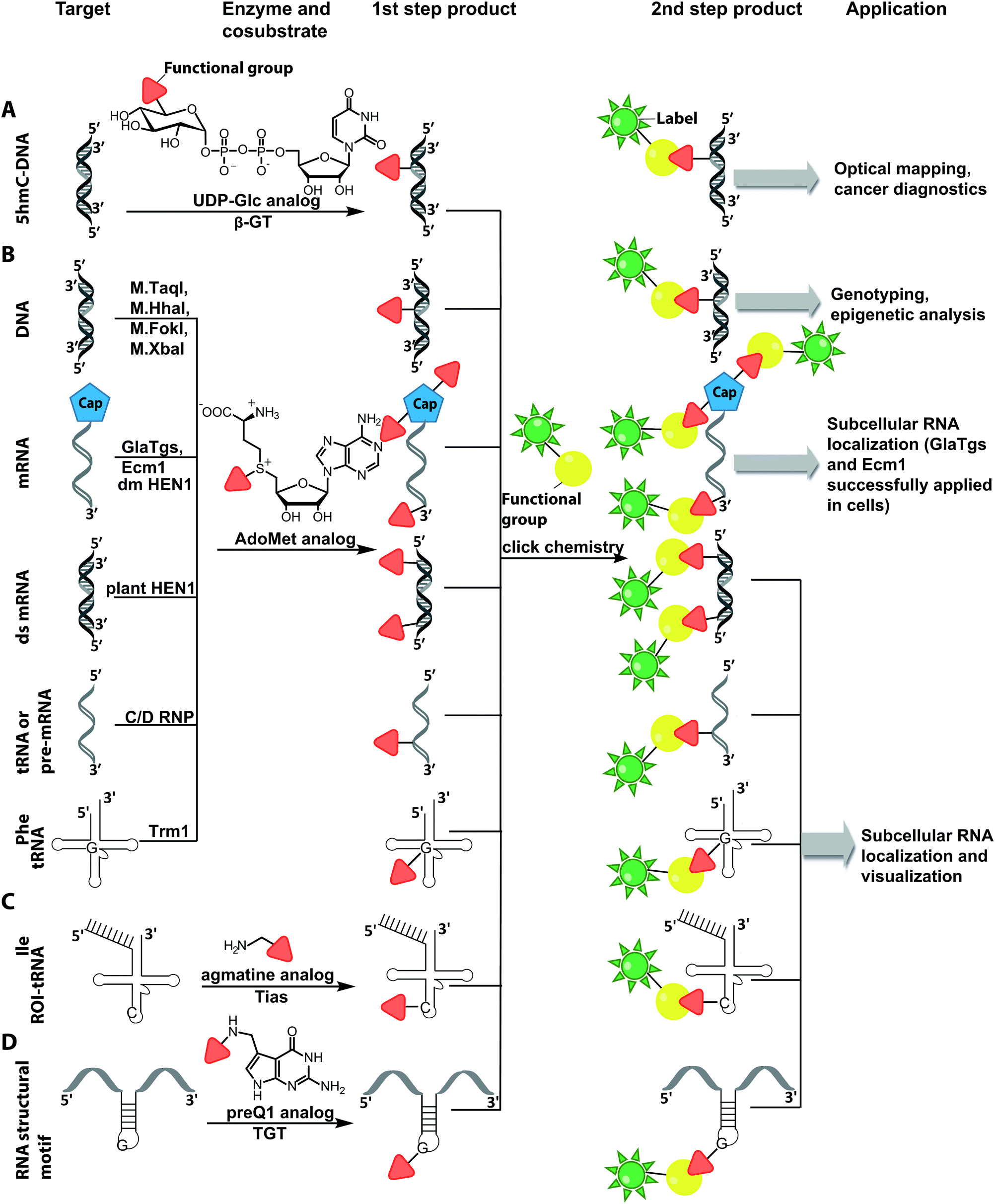 | ||
| Fig. 10 Schematic overview of post-synthetic enzyme-mediated two-step covalent labeling of nucleic acids and its applications. (A) Beta-glucosyltransferase (β-GT)-directed tagging of 5-hydroxymethylcytosine (5-hmC) in DNA. (B) Labeling of DNA and different RNA species using AdoMet analogs and various MTases. (C) tRNAIle-agmatine synthetase (Tias)-directed tagging of RNA. (D) tRNA guanine transglycosylase (TGT)-mediated labeling. | ||
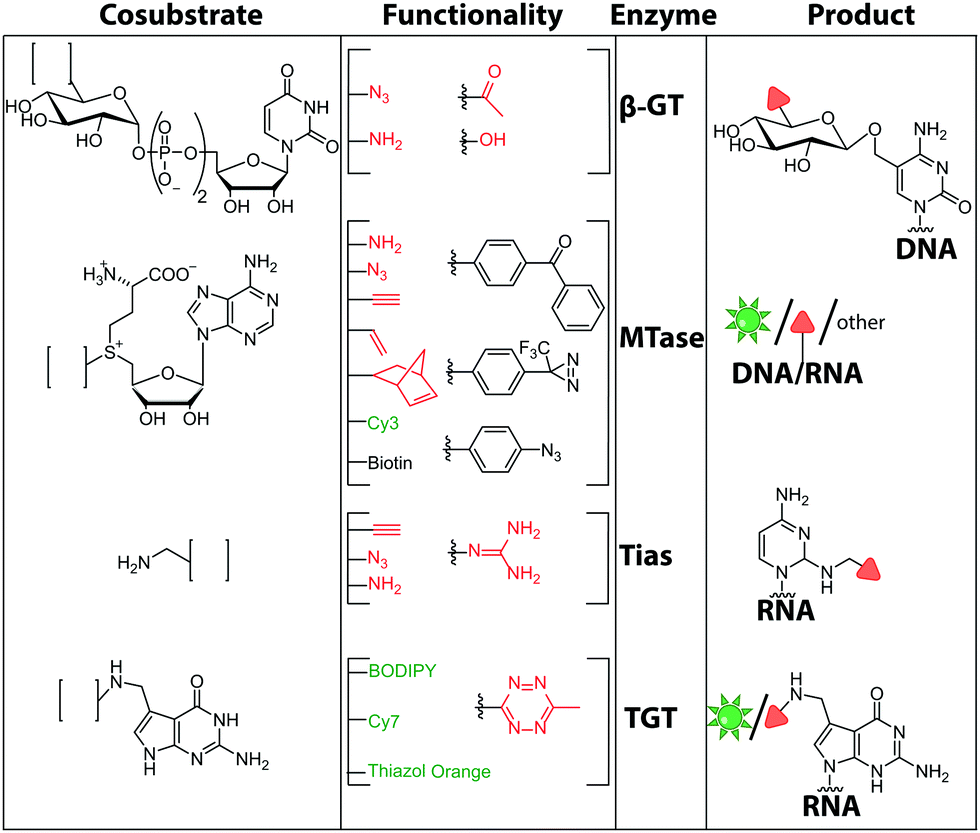 | ||
| Fig. 11 Chemical structures of functionalities transferred to nucleic acids using β-GT, MTase, Tias or TGT and the corresponding cofactors. | ||
In 2007, the groups of Klimašauskas and Weinhold demonstrated sequence-specific labeling of oligonucleotides by functionalizing methylation sites of DNA with an amino group. Plasmid DNA was modified using an AdoMet analog with an amino-alkyl group within the side chain using the enzymes M.TaqI or M.HhaI. A subsequent NHS-esterification resulted in the desired fluorescence or biotin-labeled plasmid.154 More recent studies expanded the scope of transferred side chain functionalities (amino-, azido-, alkyne-groups) leading to a variety of modified DNAs amenable for labeling with click reactions in vitro and in cell lysate.155
These advances in cosubstrate chemistry together with the identification of suitable MTases156 have contributed to the development of techniques like optical genome mapping57 or electro-optical nanopore sensing.58
There are only few other classes of DNA-modifying enzymes that have been harnessed to label DNA. Of note is the use of β-glucosyltransferase (β-GT) from T4 bacteriophage for labeling 5-hydroxymethylcytosine in DNA (Fig. 10A, 11). Its cosubstrate uridine 5′-diphosphate glucose (UDP-glucose) can be modified at the 6-position of the D-glucose residue with a functional group. Similar to the above-mentioned procedures, the functionalized DNA can be labeled in a second step using click chemistry.157 This method was used to map the epigenetic modification that leads to the formation of 5-hydroxymethylcytosine on genomic DNA.158 In clinical diagnostics, the single-molecule detection of 5-hydroxymethylcytosine shows potential to diagnose several types of blood and colon cancers by distinguishing between malignant and healthy tissue.159
Also for RNA, a number of post-synthetic two-step labeling strategies have been reported (Fig. 10B, 11). Similar to the DNA labeling strategies mentioned before, a suitable MTase can be used to transfer a functional group site-specifically from an AdoMet analog to the RNA of interest for subsequent chemical conjugation to a fluorophore. A privileged labeling position for mRNA is the 5′ cap, which is involved in several processes like nuclear export, translation and stabilization of mRNA.160 Furthermore, the cap provides various positions for enzymatic modification. In 2013, our group presented a two-step labeling approach based on a trimethylguanosinesynthase (Tgs) variant, which recognizes the N7 methylguanosine triphosphate of the cap and enables the transfer of functional groups to the N2 position of this guanosine. A variant of the Tgs from Giardia lamblia (GlaTgs) was able to transfer alkene and alkyne groups from an AdoMet analog to the 5′ cap. The alkyne-functionalized mRNA was then fluorescently labeled via CuAAC.143 The same enzyme also proved suitable to transfer an azido and an alkene group to label RNA using the SPAAC, IEDDA or photo-click reaction.145,161 To provide a way for intracellular functionalization and click labeling reactions, the two-step approach was implemented in a biocatalytic cascade starting from stable methionine analogs, which – in contrast to AdoMet analogs – are cell permeable.162
More recently, the N7 position of the guanosine of the cap has been modified using the highly promiscuous MTase from Encephalitozoon cuniculi (Ecm1). This enzyme efficiently transfers bulky side chains and thus expands the scope of enzymatic modifications. In 2016, we reported a method to label target mRNA in cells. The N7 position of the 5′ cap was enzymatically modified with Ecm1 using various AdoMet analogs. The functionalized mRNA was used for transfection of HeLa cells. Subsequently, the cells were incubated with a clickable fluorophore conjugate. This intracellular bioorthogonal RNA labeling may prove useful for investigating the subcellular RNA localization during dynamic processes.163
Due to its cosubstrate promiscuity, wild-type Ecm1 was suitable for the transfer of norbornene moieties that were tethered to SAH via benzylic linkers, stabilizing the corresponding AdoMet analog and promoting rapid transfer. The norbornene was subsequently reacted in a tetrazine ligation to label mRNA in cell lysate. These results are promising for future application in cells.147 The GlaTgs and Ecm1 based strategies have also been combined, resulting in a double labeled 5′ cap.164
Although mRNA plays a key role in gene expression and is extensively studied, the majority of RNAs are non-coding, necessitating approaches to label them specifically for biophysical and cellular studies. In particular, miRNAs and siRNAs have attracted a lot of attention as crucial regulators of gene expression in organisms ranging from plants and insects to mammals.165 The 2′-O-methyltransferase from Arabidopsis thaliana (HEN1) was utilized to label miRNA and siRNA duplexes. Similar to Ecm1, GlaTgs or M.TaqI it uses AdoMet as a cosubstrate and is sufficiently promiscuous to transfer extended side chains. HEN1 was used to functionalize small double stranded RNAs, like miRNA or siRNA, by transferring a functional group (amine, thiol, alkyne, azide) with an extended linker onto the 2′-O position of a 3′-terminal nucleotide of a small RNA duplex or heteroduplex (Fig. 10). Subsequent esterification or click chemistry completed the labeling.166,167 While the HEN1 MTase from plants only modifies dsRNA, the HEN1 enzyme from Drosophila turned out to modify the 3′-end of all RNAs, further expanding the scope. The labeling approach with HEN1 has not been tested in cells yet. However, due to the recently optimized conditions for the enzyme, resulting in improved efficiency, this strategy can form the basis for in vitro and in vivo labeling of specific RNAs in single stranded RNA pools, which could become highly relevant for RNA sequencing.121
Comparable strategies can be applied for tRNA labeling. The MTase Trm1 from Pyrococcus furiosus was used to fluorescently label the N2-position of guanosine-26 in tRNAPhe. Trm1 requires an RNA substrate forming a stem loop structure.168 However, it is promiscuous regarding the cosubstrate and can transfer an azide bearing extended side chain from an AdoMet analog to tRNA. Subsequent click chemistry enabled fluorescent labeling of RNA.169
The only enzyme reported to date that can be used for sequence-specific modification of RNA is a box C/D small ribonucleoprotein RNA 2′-O methyltransferase (C/D RNP) from the thermophilic archaeon, Pyrococcus abyssi. It has been used to functionalize tRNA, rRNA and spliceosomal RNA with alkyne moieties at specific target sequences corresponding to a programmable guide RNA.170 The required functionalization of the alkyne group based on the CuAAC reaction renders these methods disadvantageous for applications in cells.
Although the methods of using nucleic-acid modifying enzymes are widely used, chemo-enzymatic RNA labeling is not restricted to AdoMet analogs as cosubstrates. For example, the tRNAIle2-agmatidine synthetase (Tias) with its unique RNA sequence-specificity and cosubstrate promiscuity has been applied to site-specifically transfer azido and alkyne groups to an RNA target in vitro and in mammalian cells.171 The RNA was subsequently labeled via click chemistry. For the modification, Tias requires an agmatine analog instead of the AdoMet analogs used by MTases (Fig. 10C, 11). Agmatine analogs with suitable functional groups were synthesized and accepted by Tias. A related approach that also provides site-specific labeling of RNA in mammalian cells is based on tRNA guanine transglycosylase (TGT) from Escherichia coli.120 This enzyme is a key element of the recently established RNA-TAG (transglycosylation at guanosine) strategy, which allows genetically encoded site-specific labeling of large transcripts by employing modified mRNA (mRNA bearing 5-methylcitidine and pseudouridine substitutions). The TGT enzyme together with a 7-deazaguanine derivative (preQ1), which served as cosubstrate, were used in an RNA-TAG experiment that permitted the enzymatic transfer of a tetrazine to mRNA (Fig. 10D, 11). In a subsequent step a fluorophore was attached to the mRNA by tetrazine ligation.172
2.5 Ribozymes
In recent years, ribozymes and deoxyribozymes have become versatile tools to label RNA with fluorescent tags, either directly or in two steps.Vauléon et al. engineered a small twin ribozyme that mediates specific exchange of RNA patches. If the patch is functionalized, e.g. by an amino group, the entire RNA will be functionalized and prepared for subsequent chemical conjugation (Fig. 12A). This patch needs to be provided and chemically modified, if an application in cells is envisioned.173 Recently, a 42 nt long catalytic RNA from the thermophilic archaeon, Aeropyrum pernix was discovered, which catalyzes a reaction between a guanosine and a disubstituted epoxide (Fig. 12B). To spot this activity, the assay comprised epoxides with clickable/functional groups, which specifically react with RNA containing functional groups with enhanced nucleophilicity. The newly formed C–N bond resulted in covalently functionalized RNA that was subsequently labeled via click chemistry.174
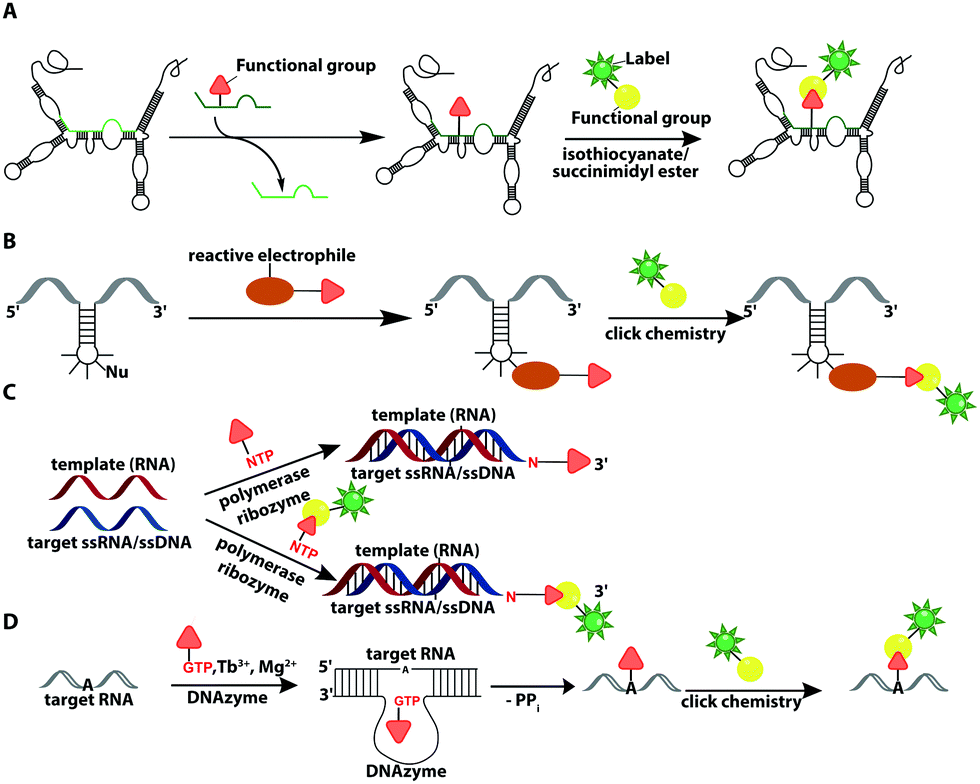 | ||
| Fig. 12 Ribozyme-assisted labeling of nucleic acids. (A) Twin ribozyme-mediated insertion of short-labeled RNA fragment by strand exchange. (B) Ribozyme-directed reaction between a labeled electrophile and nucleophilic groups in RNA. (C) Polymerase ribozyme-based strategy to attach functionalized or labeled nucleotides at the 3′-end. (D) DNAzyme-assisted labeling of 2′-OH of internal adenosine residues using functionalized GTP. | ||
In 2018, a polymerase ribozyme was reported, which attaches one of various possible functionalized nucleotide analogs at the 3′-end (Fig. 12C). This ribozyme successfully added nucleotides, which already carried a fluorophore or a clickable functional group for fluorescent labeling and successfully labeled 5S-rRNA in total RNA.175
An alternative way to post-transcriptionally label RNA is based on deoxyribozymes (DNAzymes) (Fig. 12D). Studies report successful labeling by modifying an internal 2′-hydroxy group of an adenosine with either a GMP that carries a fluorophore (or other reporters) or a functionalized tagging-RNA, which can be further modified by NHS-esterification. DNAzymes catalyze a sequence-specific nucleophilic attack at the desired position16,176 and attach a guanosine or adenosine equipped with a fluorescent dye or a reactive handle under formation of a 2′,5′-linked phosphodiester with the RNA. The group of Höbartner also developed another approach to transfer dyes or functional groups to RNA with ribozymes, utilizing orthogonal substrates that are based on the antiviral-drug tenofovir. Labeling of 16S and 23S rRNAs with fluorescent dyes and attachment of functional groups was successful in total cellular RNA.177
Furthermore, they developed a labeling strategy called DEoxyribozyme-CAtalyzed Labeling (DECAL). To this end, a tagging RNA with a 5-aminoallylcytidine was first labeled with either a fluorescein or TAMRA fluorophore via an NHS esterification. Subsequently, the 10DM24 deoxyribozyme, a catalytic DNA, was used to covalently connect the 2′-OH of a specific adenosine of the target RNA with the 5′-triphosphate of the labeled tagging RNA. With this approach the folding of the P4–P6 domain of Tetrahymena was observed via FRET.176 Based on DECAL, Höbartner et al. developed, in 2018, the FH series ribozymes, FH14 and FH31. With these ribozymes, the 5S rRNA from E. coli was covalently labeled with fluorescein, ATTO550 and Cy3. The FH ribozymes, similar to the 10DM24 deoxyribozyme, catalyze the phosphodiester formation by connecting the 2′-OH of the target RNA with the α-phosphate of the 5′-triphosphate of the labeled RNA.178 The DECAL strategy allows labeling, even multiple color labeling, of long RNAs, which is difficult to achieve by solid-phase synthesis. For every new target site the binding arms of the deoxyribozymes have to be adapted.176
Ribozyme- and deoxyribozyme-assisted strategies are highly versatile for in vitro labeling, but not yet suited for in-cell experiments. Even if problems like NTP delivery in cells are solved, obstacles like competition with endogenous nucleotides or background fluorescence must be overcome before experiments in cells can succeed.
3 Labeling nucleic acids with other reporters
Fluorescent labels are the most widely applied reporters attached to nucleic acids and undoubtedly enable numerous biophysical and intracellular studies. In the previous chapters, we elaborated on methods for their random or site-specific incorporation and pointed out which ones are compatible with applications in cells. This chapter aims to introduce different reporters than fluorescent labels for nucleic acids including a glimpse of their current and future applications. The methods to incorporate these reporters rely on the strategies outlined previously (Section 2) and thus, again, can be divided into chemical and chemo-enzymatic approaches.3.1 Chemical non-fluorescent labeling of nucleic acids
One important factor for the investigation of nucleic acids is to use biologically relevant conditions. Therefore, the determination of long-range distances is critical to probe conformational changes in folding and function in complex systems.11,15,16 Two strategies, which fulfill these demands, are FRET (based on fluorescent labeling) and electron paramagnetic resonance (EPR) spectroscopy.179,180 EPR spectroscopy requires an unpaired electron, which can be introduced by a spin label, usually as a nitroxyl radical (N–O) that is incorporated into a heterocyclic ring (Fig. 13A). Spin labeling of nucleic acids can be performed during chemical oligonucleotide synthesis (Section 2.1)181,182 or post-synthetically via click chemistry.183 A current limitation for the application of spin labels in cells is the stability of the unpaired electron, which is quickly reduced under cellular conditions. The broadly applied gem-dimethyl nitroxide spin label is therefore not suited for cell experiments. For that reason, other spin labels were recently investigated and showed that gem-diethyl nitroxide labels, which were introduced via click chemistry, provide higher stability against reduction in HeLa cell lysate.184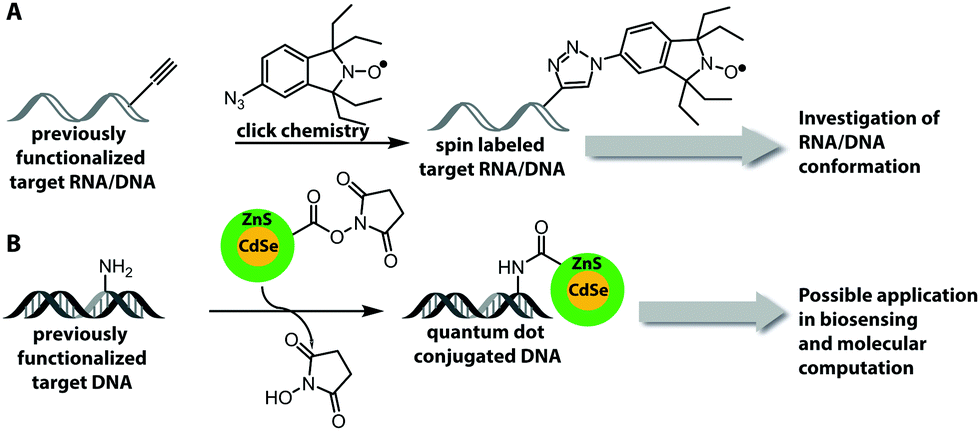 | ||
| Fig. 13 Chemically introduced reporters (besides fluorophores) and their application. (A) Introduction of spin labels in RNA and DNA using click chemistry. (B) Attachment of quantum dots to functionalized DNA via NHS-esterification. | ||
Fluorophores are suitable for a wide range of biophysical investigations but face limitations with respect to photo-bleaching, chemical stability and quantum yields. Quantum dots are luminescent semiconductor nanocrystals and show promising optical properties like broad absorption and narrow photoluminescence, allowing for a bioorthogonal setup without influencing chromophores present in biological systems, high resistance to photo-bleaching and high quantum yield.185 These quantum dots can be covalently attached to DNA by NHS-esterification between amine-functionalized DNA and carboxylic groups that are present on the quantum dot surface (Fig. 13B).186 However, the quantum dot strategy requires an excess of reagent, several purification steps and the surface of the quantum dots could react with other molecules within cells or cell lysate.186
3.2 Chemo-enzymatic non-fluorescent labeling of nucleic acids
To study weak or transient interactions between nucleic acids and proteins, photo-cross-linking is widely applied.187,188 However, strategies to site-specifically introduce photo-cross-linking moieties using enzymes are rare. Recently, MTases in combination with AdoMet analogs were used to install photo-cross-linking moieties in RNA (Fig. 14A), based on the procedure described in Section 2 of this review. AdoMet analogs that contain a side chain with photo-cross-linking moieties were enzymatically transferred with Ecm1 to the 5′ cap of mRNA. The resulting product was successfully linked to the cap-binding protein eIF4E enabling the study of RNA–protein interactions.189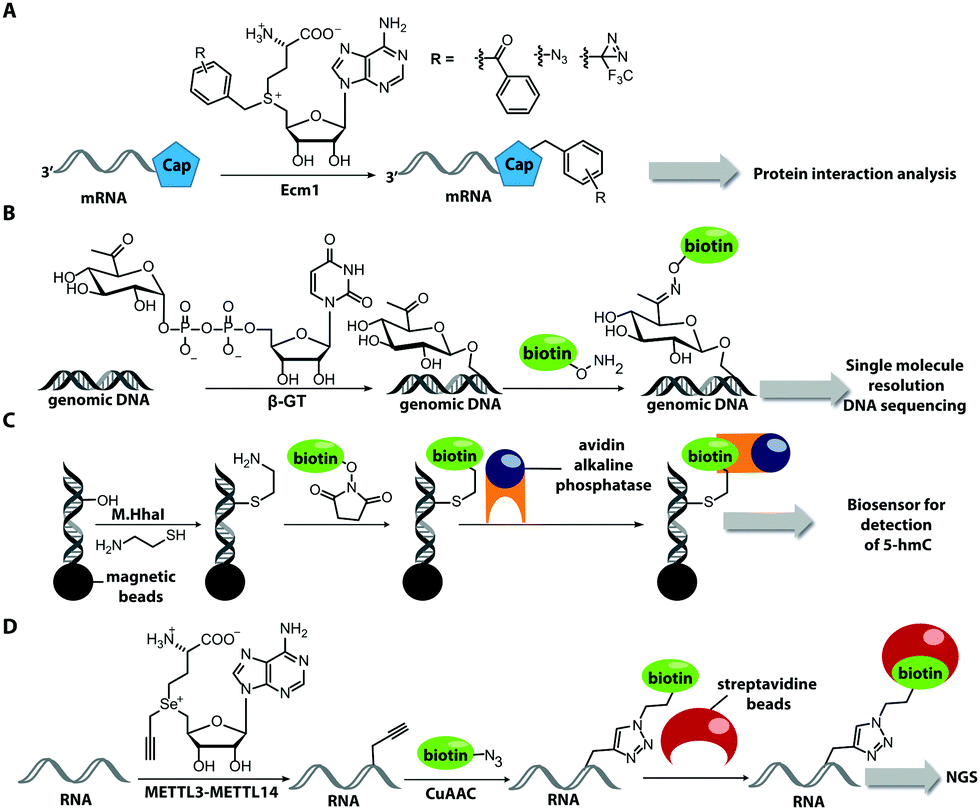 | ||
| Fig. 14 Chemo-enzymatically introduced reporters (besides fluorophores) and their applications. (A) Incorporation of photocrosslinker via MTase. (B) DNA biotinylation based on β-GT modification. (C) Biosensor for detection of 5-hmC by modifying immobilized DNA (magnetic beads are illustrated as black circle). (D) RNA MTase-directed modification and subsequent clicking with biotin. | ||
Biotin is another widely applied label for nucleic acids as it can be used as reliable interface for enrichment and detection. It can be introduced in the same way as fluorescent labels but its enzymatic introduction is particularly relevant to make certain nucleic acids, motifs or modifications in biological samples accessible to further analysis, in particular by next generation sequencing.
Song et al. biotinylated DNA at sites containing 5-hydroxymethylcytosine. They utilized the β-GT MTase and an analog of the cofactor uridine 5′-diphosphate glucose, with a ketogroup to functionalize DNA at these modification sites. Subsequent chemical conversion with biotin enabled enrichment of DNA and detection of the modification sites at single base resolution in DNA sequencing (Fig. 14B).190 A different approach to detect 5-hydroxymethylcytosine in DNA via biotinylation was presented in 2016. The DNA was functionalized with an amino group at 5-hydroxymethylcytosine with the help of the MTase, M.HhaI and cysteamine. Biotinylation was then achieved with a biotin-NHS-ester. Binding of an avidin–alkaline phosphatase fusion protein forms a photoelectrochemical biosensor that enables highly sensitive detection of 5-hydroxymethylcytosine (Fig. 14C).191,192
Biotin labeling was also used in the Rentmeister group in cooperation with the Leidel group to detect MTase target sites in RNA. The RNA was modified with alkyne groups at target sites of wild-type METTL3–METTL14, which is an MTase complex that usually transfers methyl groups to the N6 position of adenosine but is also able to transfer propargyl groups (Fig. 14D). The RNA was further conjugated to a biotin using click chemistry. The biotin-labeled RNA in combination with next generation sequencing allowed detection of the METTL3–METTL14 target sites.193
Aside from biotin labels or optical detection, redox labeling offers a viable alternative that is applied in sequencing and diagnostics.194,195 A common strategy is to attach a redox active group to a dNTP, which can be incorporated into DNA during the polymerase based synthesis.196 In a recent report it was shown that differently substituted ferrocene labels could be incorporated into DNA using the above mentioned strategy.197 Due to the orthogonality of the oxidizable labels it was possible to directly measure the relative abundance of two different nucleotides in a target sequence.197 With future work, orthogonal labels for all four nuclebase may become available and provide a valuabe strategy for sequencing.
4 Metabolic labeling
4.1 Feeding nucleoside pre-cursors
Metabolic labeling typically makes use of cell-permeable nucleotide pre-cursors, i.e. nucleosides or nucleobases. These need to enter the salvage pathway and be processed to (d)NTPs that can be used by polymerases (see Sections 2.3.1. and 2.4.1). Hence, small modifications that do not interfere with metabolism are preferred. Here, we will focus on modifications that can be reacted with reporters via click reactions, typically to achieve fluorescent labeling, but also other labels will be mentioned where appropriate. For thiol-reactive chemistry, which has proven particularly useful for studying transcriptional dynamics, we refer to recent review articles.63,198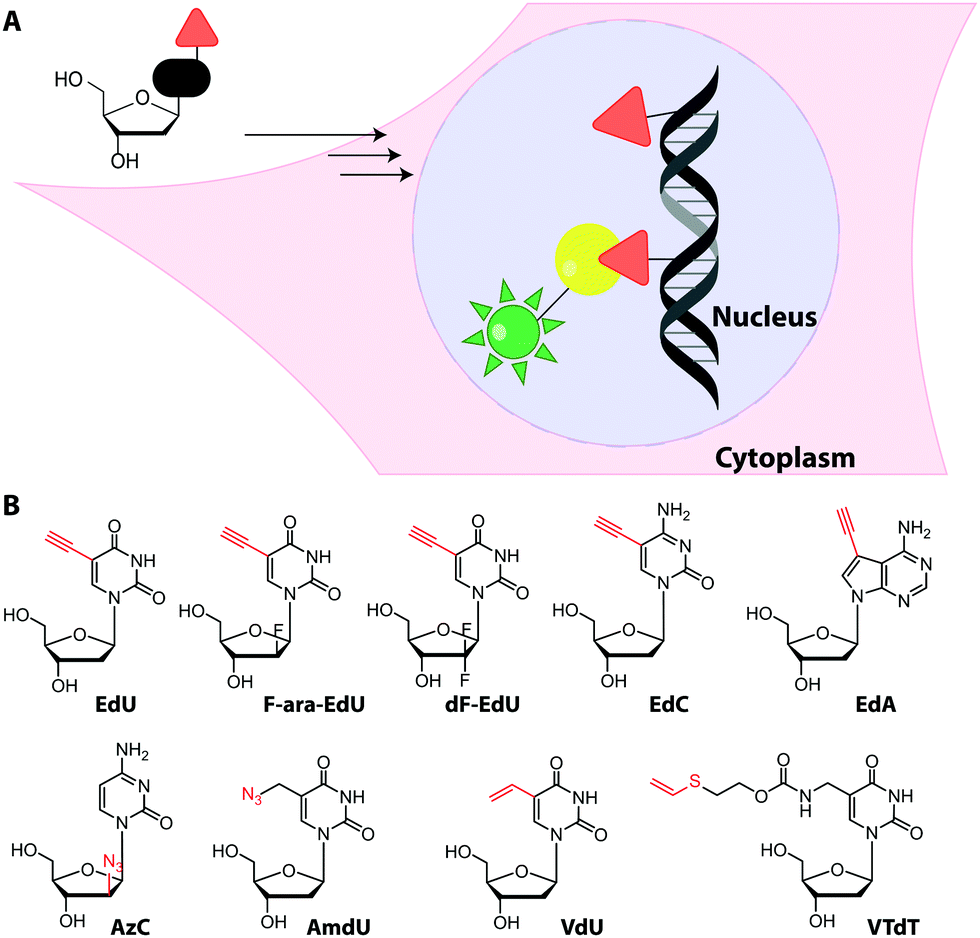 | ||
| Fig. 15 (A) Concept of metabolic DNA labeling. Deoxynucleosides with small modifications (red triangle) are taken up by cells and enter the salvage pathway to dNTPs. After polymerase-based incorporation into DNA, click chemistry is used for (fluorescent) labeling. (B) Deoxynucleosides successfully used for metabolic labeling. Abbreviations: EdU: 5-ethynyl-2′-deoxyuridine, F-ara-EdU: 2′-deoxy-2′-fluoro-5-ethynyluridine, dF-EdU: 2′-deoxy-2′,2′-difluoro-5-ethinyluridine, EdC: 2′-deoxy-5-ethynylcytidine, EdA: 7-deaza-7-ethynyl-2′-deoxyadenosine; AzC: 1-(2-azido-2′-deoxy-β-D-arabinofuranosyl)cytosine, AmdU: 5-(azidomethyl)-2′-deoxyuridine; VdU: 5-vinyl-2′-deoxyuridine; VTdT: 5-(vinylthio-ethoxycarbamoyl-methyl)-2′-deoxythymidine. | ||
Recently, EdU labeling was used in vivo in a mouse xenograft model. EdU was injected intraperitoneally, the mice were sacrificed and the DNA was labeled with a fluorescent azide. Importantly, EdU labeling indicated proliferating areas of the tumors, as validated by immunostaining.23
An interesting application for metabolic labeling of DNA was to mark newly synthesized DNA in different viruses (adenovirus, herpes virus, vaccinia virus). During a significant part of their replication cycles, viruses do not exist in the form of morphologically distinct virus particles, but either as subviral nucleoprotein complexes, as genetic information only, or as newly-synthesized virion components.24 Detecting viral nucleic acids is therefore a central objective for scientists who want to image virus–cell interactions. After metabolic labeling using various ethynyl-containing nucleosides at 1–2.5 μM (and fluorescent labeling via CuAAC), DNA trafficking could be visualized at single-molecule resolution within mammalian cells using super-resolution microscopy. Interestingly, different ethynyl-modified nucleosides were incorporated with variable efficiencies into DNA virus replication sites, indicating that screening is valuable to optimize virus genome labeling. The procedure did not affect viral infectivity.201
Despite the widespread use of EdU for metabolic labeling, it should be noted that this compound is toxic, and perturbs DNA function and stability at concentrations in the micromolar range.202–204 The arabinofuranosyl derivative, F-ara-EdU, was found to be an alternative for metabolic incorporation into DNA. F-ara-EdU (Fig. 15) proved less toxic than EdU and was successfully used in cells and zebrafish. It proved superior in experiments where long-term cell survival and/or deep-tissue imaging was desired.205
Similar to EdU, the cytidine analog, 2′-deoxy-5-ethynylcytidine (EdC, Fig. 15), was shown to work for DNA labeling in vivo with comparable efficiency.206 Ethynyl-modified purine analogs were also reported. Here, 7-deaza-7-ethynyl-2′-deoxyadenosine (EdA, Fig. 15) exhibited selective metabolic labeling in cells and zebrafish embryos, whereas 7-deaza-7-ethynyl-2′-deoxyadenosine (EdG) was not suitable.207
An azido group was also installed at the 2′-position of arabinose-analogs of cytosine (AzC, Fig. 15), albeit not with labeling as the primary purpose.210 AzC is not only a clickable analog but also a known anticancer prodrug. Surprisingly, the compound – in combination with click chemistry – led to efficient labeling of DNA in drug-resistant cells, whereas drug-sensitive cells were less efficiently labeled. Detailed evaluation and super-resolution imaging revealed stalled replication forks as reason for high incorporation and – later – resumption of DNA synthesis as reason for drug-resistance of these cancer cells—a prime example of how nucleoside analogs can contribute to understanding a fundamental mechanism of toxicity.210
The potential alternative approach to realize SPAAC via metabolic incorporation of strained alkynes has not been realized to the best of our knowledge and is unlikely to be compatible with the cellular machinery.
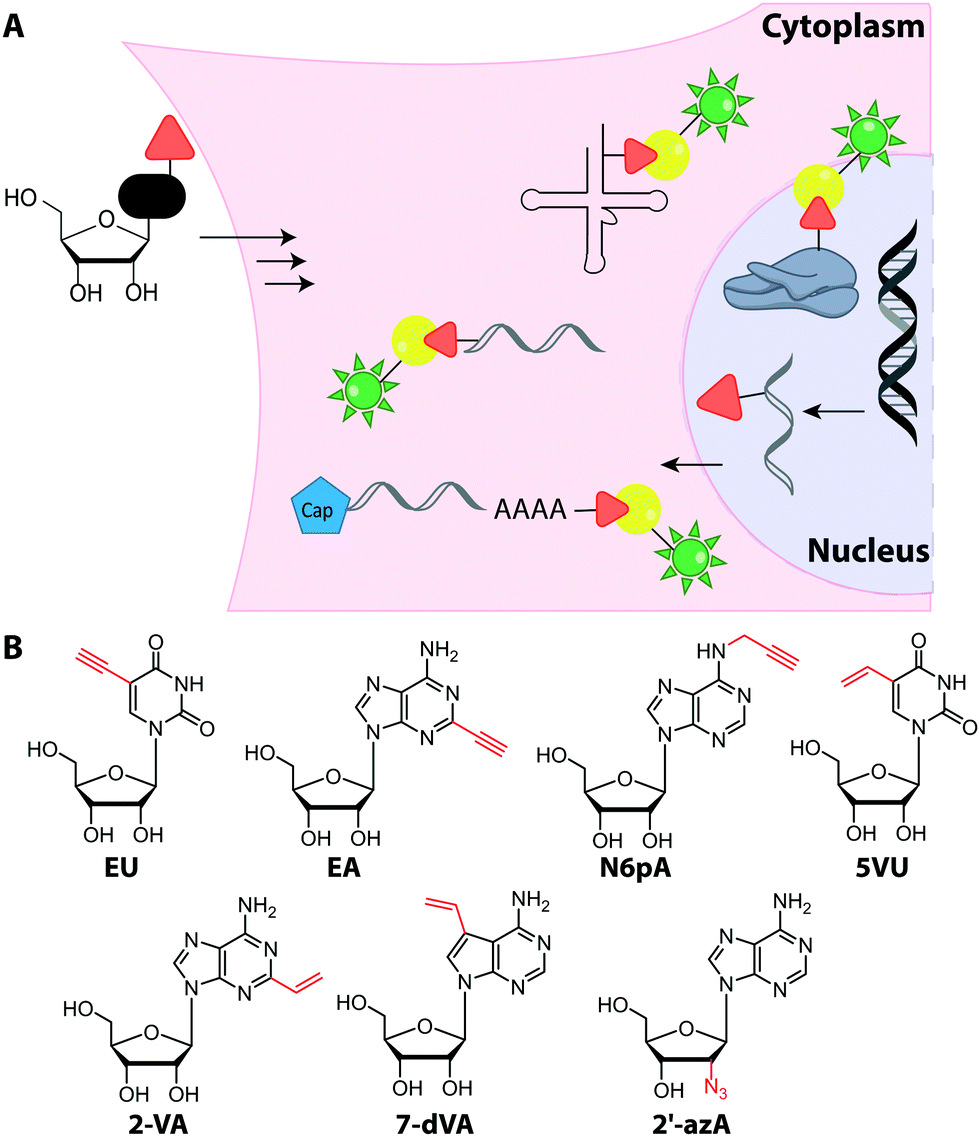
Metabolic labeling of RNA with EU provides a way to label all newly transcribed RNAs using the CuAAC in fixed cells but cannot distinguish between different types of RNA.214 Although widely used, it does impair cell proliferation, especially at long incubations times (48 h).215 EU was injected into mice intraperitoneally and click chemistry was performed in the harvested organs 5 h later.214 It should be mentioned that CuAAC can also be used to enrich nascent RNAs after metabolic labeling.216 Together with NGS, this approach has recently emerged as a powerful method to get insights into transcriptional dynamics – also with other, notably thiol-sensitive, chemistries as reviewed by Yamada et al.198
In addition to EU, the adenosine analogs N6-propargyladenosine (N6pA) and 2-ethynyladenosine (EA) have been used for metabolic labeling and subsequent click reactions in eukaryotic cells (Fig. 16).217,218 In addition to RNA polymerase I, II and III, the modified adenosines are also used by PAPs, providing access to monitoring poly(A) tail dynamics. To achieve selective poly(A) tail labeling in eukaryotic cells using EA, transcription was blocked by actinomycin. EA was also used in frog oocytes, where polyadenylation of stored mRNAs happens during development and can be induced by addition of progesterone.218 This experiment was not used to label but to isolate RNA for subsequent NGS analysis.
 | ||
| Fig. 16 (A) Concept of metabolic RNA labeling. Nucleosides with small modifications are taken up by cells and enter the salvage pathway to NTPs. After incorporation into different types of RNA during transcription or by PAPS, click chemistry is used for (fluorescent) labeling. (B). Nucleosides successfully used for RNA metabolic labeling. Abbreviations: EU: 5-ethynyluridine, EA: 2-ethynyladenosine, N6pA: N6-propargyl-adenosine, 5VU: 5-vinyluridine, 2VA: 2-vinyladenosine, 7-dVA: 7-deaza-7-vinyl-adenosine, 2′-azA: 2′-azido-2′-deoxy-adenosine. Arrows indicate uptake of nucleosides and export of RNA. | ||
Only few azido-modified nucleosides – most notably 2′-azido-2′-deoxyadenosine (2′-azA, Fig. 16) – were successfully incorporated in amounts relevant for RNA imaging in HeLa cells.221 In most cases, the formation of the respective NTP is inefficient, because the first kinase of the salvage pathway does not accept the nucleoside analog. To circumvent this limitation, a kinase of the salvage pathway responsible for the first phosphorylation step but expressed only in few tissues, was targeted. The overexpression and engineering of this uridine-cytidine kinase 2 (UCK 2) enabled efficient metabolic labeling with azidomethyl-uridine (5AmU, Fig. 17) in HeLa cells—an important step towards cell-selective labeling.222
 | ||
| Fig. 17 Approaches for cell-specific RNA labeling. (A) Cells overexpressing UPRT, which is lacking in mammals can convert 5EC to EU-monophosphate. WT cells can use 5EU to a limited extent, causing background. (B and C) Cells overexpressing UCK2 or an improved variant can process 5AmU and 2′AzUd to the respective monophosphates. The monophosphates are processed to the respective triphosphates and incorporated into RNA, which can be labeled. In mammalian wildtype cells, low (A and B) or no (C) background labeling is detected. | ||
4.2 Cell-selective metabolic labeling
Building on the concept of overexpression, it is also conceivable to achieve metabolic labeling of nucleic acids exclusively in a subset of cells—namely the ones expressing the particular enzyme. However, this requires absence of metabolic labeling in wildtype cells.Cell-selective metabolic labeling of DNA was achieved for cells infected with Herpes Simplex Virus-1 (HSV-1) and could thus be used to visualize pathogen-infected cells.223 Here, a gemcitabine metabolite analog, 2′-deoxy-2′,2′-difluoro-5-ethynyluridine (dF-EdU, Fig. 15) was used. The analog's phosphorylation depends on the herpes virus thymidine kinase and is not catalyzed by the human enzyme.
In the field of RNA labeling, an interesting strategy for cell-specific metabolic labeling is based on feeding the modified nucleobase 5-ethynylcytosine (5EC, Fig. 17). 5EC is readily taken up by cells and can be processed to 5-ethynyluridine-monophosphate (EU-monophosphate) in two steps. First, cytosine deaminase converts 5EC to 5EU, then uracil phosphoribosyltransferase (UPRT) catalyzes formation of EU-monophosphate (Fig. 17). Importantly, UPRT is lacking in mammals but expressed in protozoan parasites, enabling tissue-specific metabolic RNA labeling upon heterologous expression, as originally demonstrated with 4-thiouracil and termed “TU-tagging”.224 The ethynyl group was used to detect the RNAs and allowed cell-type specific gene expression data to be gathered from Drosophila larvae.225 Cell-type specific RNA labeling is an important step forward as it overcomes the inherent non-specificity of metabolic RNA labeling approaches.
However, recent work showed that off-target cells can use 5EC to a limited extent, which causes background. Instead, the combination of feeding 2′-azidouridine (2′AzUd, Fig. 17) and urine-cytidine kinase 2 (UCK2) overexpression proved superior to achieve cell-selective metabolic labeling.226 The above mentioned 5AmU also led to more efficient labeling in combination with overexpression of suitable UCK2 mutants. However, in cell viability and proliferation tests, cells fed with 2′AzUd performed better than cells fed with 5AmU.226 Subsequent CuAAC was used to visualize nascent RNAs and confirmed cell-specificity for 2′AzUd, whereas 2′-azidocytidine resulted in RNA labeling also in the absence of UCK2 expression. As a proof of concept, the method was used to enrich RNA from a mixture of cells, as confirmed by RNA sequencing.226 This combination of cell-selective metabolic RNA labeling with isolation and sequencing bears huge potential for studying dynamic aspects of the transcriptome. For labeling subtypes of cells, the metabolic RNA labeling still has to compete with the plethora of fluorescent proteins that can also be routinely expressed in specific cells and tissues.
4.3 Feeding methionine analogs
Both DNA and RNA can be naturally modified after their polymerase-based synthesis and the most prevalent modification is methylation. Although simple and small, the effects of methylation are remarkably relevant, with restriction/methylation systems in prokaryotes and epigenetic silencing by methylation of CpG sites being the most remarkable examples. The methyl group in the AdoMet originates from methionine. Therefore, metabolic labeling of methyl-derived modifications can be accomplished by feeding clickable analogs of methionine, as performed with propargyl-selenohomocysteine (PSH) for total RNA.193Like in all metabolic labeling approaches, the specificity is a major bottleneck for this approach as the resulting AdoMet analog can be used by all sufficiently promiscuous MTases. Therefore, this approach has not been used for labeling but for isolation of the respective species after pre-enrichment of the biomolecule class. RNA sequencing then gave insights into the MTase target sites.193
4.4 Approaches to increase uptake or production of nucleotides
Feeding cells with cell-permeable nucleosides or nucleobases is convenient. However, for generation of the corresponding NTPs, the first phosphorylation is often the critical step as the kinase is highly specific and thus susceptible to non-natural modifications—more than the subsequent kinases. Thus, the first kinase is in many cases the limiting factor for the metabolic incorporation of certain modified nucleotides. There are two strategies to address this issue and they are detailed in the following sections.EdU was chemically monophosphorylated and esterified to improve its function and maintain uptake,228 resulting in PEdU (Fig. 18). Similar to F-ara-EdU, PEdU was less toxic than EdU and suitable for DNA labeling in cells and mice that were injected with PEdU.228 A cyclosal-phosphortriester of EdU was also a suitable pro-label for feeding cells and labeling nuclei.229 Also azido groups were incorporated into DNA in cells and in zebrafish by using membrane-permeable nucleotide triesters, such as POM-AdmdU (Fig. 18).230
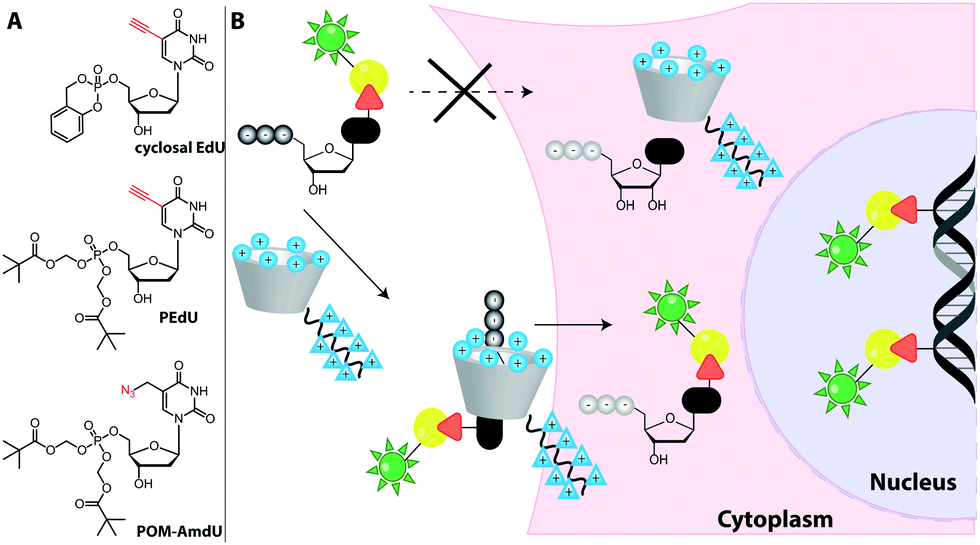 | ||
| Fig. 18 Approaches to increase uptake of deoxynucleoside phosphates. (A) Membrane-permeable phosphotriester derivatives successfully used as pro-labels for DNA metabolic labeling. (B) Schematic illustration of the synthetic nucleoside-triphosphate transporter (SNTT).231 A labeled dNTP binds to the transporter that enters the cell via a cell-penetrating peptide. Cellular ATP binds to the transporter, facilitating release of the labeled dNTP and subsequent DNA metabolic labeling. | ||
Recently, a synthetic nucleoside triphosphate transporter (SNTT) was developed to address this issue. The SNTT is based on a cyclodextrin scaffold equipped with a receptor moiety for the triphosphate and a cell-penetrating agent (Fig. 18).231 This SNTT managed to transport fluorescently labeled NTPs into cells and was used for DNA labeling in live cells.232
5 Conclusions
In summary, many approaches exist to label nucleic acids for a specific purpose but there is no general solution that fits all.Site-specific labeling of short nucleic acids for biophysical studies can best be achieved by solid-phase synthesis. Isomorphic nucleobases that do not interfere with the secondary structures are particularly useful, and we presented well-known examples like 2-aminopurine as well as recent additions to this toolbox, that are red-shifted or compatible with two-photon excitation (qAN1–4, extended uridines). However, if fluorophores with high photostability and quantum yield are needed or different labels have to be tested (e.g. for cellular applications or as efficient FRET pairs), two-step-approaches provide more flexibility regarding the label. Most of the bioorthogonal groups presented in Section 1 – namely alkynes (both terminal and strained cyclic ones), tetrazines, and several strained alkenes – are compatible with solid-phase synthesis. Azides have proven to be difficult in solid-phase synthesis, although approaches are being developed to solve this limitation.
For longer nucleic acids, ligation of a short chemically modified fragment and a long enzymatically produced fragment is a widely used strategy – in particular for RNA labeling. However, the additional step makes the overall procedure tedious and reduces the yield. Thus, straightforward efficient, site-specific labeling of long nucleic acids is still highly sought after.
The chemo-enzymatic post-synthetic modification of nucleic acids represents the other side of the spectrum and nicely complements the possibilities of solid-phase synthesis. It proves useful for sequence- and site-specific modification of nucleic acids and is – in principle – independent of the length. MTases, TGT and Tias have proven highly valuable to introduce numerous labels – either directly or in combination with click chemistry. The TGT or Tias recognition motif can be appended to the nucleic acid sequence, partially solving the above mentioned goal of labeling long nucleic acids site-specifically, albeit at the cost of having changed the nucleic acid sequence.
The mild reaction conditions typically required for enzymatic conversions are compatible with introduction of azides, although attention has to be paid to reducing conditions in buffers or the cellular milieu. On the other hand, in contrast to solid-phase synthesis, enzymes have not been used to directly attach bulky strained alkenes, such as DBCO or TCO to nucleosides. If such moieties were enzymatically introduced (both by polymerases or MTases), they were usually attached via an extended linker, such as a rigid alkyne-based linker (Fig. 7) or a benzylic linker (Fig. 11).
Polymerases have been thoroughly tested for their ability to accept modified (d)NTPs. The resulting nucleic acids are partially or completely labeled at specific nucleotides, however, the labeling is usually not position-specific. To achieve site-specific labeling of nucleic acids with polymerases, the UBPs are a great recent addition to the toolbox. UBPs are treated in a separate article in this special edition. Future developments in this field will show, whether this approach – that currently requires significant synthetic efforts – will become straightforward and widely used in life sciences, e.g. by commercialization of primers containing UBPs for site-specific introduction of modification sites by PCR. The same is true for recent advances in (deoxy)ribozymes and their application as a general labeling strategy. If the design rules for a specific sites are straightforward, and both the (deoxy)ribozyme and the fluorescent substrates are commercially available, this approach bears potential to become a standard technique in molecular biology for labeling long nucleic acids.
The last part of this review introduced methods for metabolic labeling of nucleic acids. For this approach, three aspects are of key importance: (i) cell permeability, (ii) compatibility with the salvage pathway, where the critical step is usually the first phosphorylation of nucleosides and (iii) compatibility with polymerases. Similar to the in vitro labeling with polymerases, metabolic labeling lacks position-specificity. In the case of RNA, the different types of RNA (rRNA, tRNA, mRNA and more) cannot be specifically addressed. Nevertheless, metabolic labeling brings a new twist to studying nucleic acids in cells, as dynamic aspects can be followed in combination with NGS. Thus, labeling of nucleic acids can be combined with click chemistry for affinity enrichment rather than fluorescent labeling and imaging. The combination of metabolic labeling and NGS is an emerging field enabling studies of transcriptional dynamics and potentially modification dynamics in the future. Therefore, the recent development of cell-specific metabolic labeling approaches bears exceptional potential, especially in the light of single-cell sequencing to understand the function and development of individual cells in complex organisms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. thanks the ERC (772280) and the DFG (RE2796/6-1, RE2796/7-1 and SFB858) for funding. We would like to thank all members of the Rentmeister group for their input and critically reading the manuscript.Notes and references
- E. R. Mardis, Nat. Protoc., 2017, 12, 213–218 CrossRef CAS.
- F. D. Steffen, M. Khier, D. Kowerko, R. A. Cunha, R. Borner and R. K. O. Sigel, Nat. Commun., 2020, 11, 104 CrossRef CAS.
- J. Chen, A. Tsai, S. E. O'Leary, A. Petrov and J. D. Puglisi, Curr. Opin. Struct. Biol., 2012, 22, 804–814 CrossRef CAS.
- S. Ray, J. R. Widom and N. G. Walter, Chem. Rev., 2018, 118, 4120–4155 CrossRef CAS.
- M. Vera, J. Biswas, A. Senecal, R. H. Singer and H. Y. Park, Annu. Rev. Genet., 2016, 50, 267–291 CrossRef CAS.
- H. M. Chen and D. R. Larson, Genes Dev., 2016, 30, 1796–1810 CrossRef CAS.
- K. Rau and A. Rentmeister, ACS Cent. Sci., 2017, 3, 701–707 CrossRef CAS.
- Y. Xia, R. Zhang, Z. Wang, J. Tian and X. Chen, Chem. Soc. Rev., 2017, 46, 2824–2843 RSC.
- X. Long, J. Colonell, A. M. Wong, R. H. Singer and T. Lionnet, Nat. Methods, 2017, 14, 703–706 CrossRef CAS.
- S. H. Kim, M. Vieira, J. Y. Shim, H. Choi and H. Y. Park, RNA Biol., 2019, 16, 1108–1118 CrossRef.
- K. Halbmair, J. Seikowski, I. Tkach, C. Hobartner, D. Sezer and M. Bennati, Chem. Sci., 2016, 7, 3172–3180 RSC.
- K. Wawrzyniak-Turek and C. Hobartner, Methods Enzymol., 2014, 549, 85–104 CAS.
- L. Buttner, J. Seikowski, K. Wawrzyniak, A. Ochmann and C. Hobartner, Bioorg. Med. Chem., 2013, 21, 6171–6180 CrossRef.
- C. Hobartner, G. Sicoli, F. Wachowius, D. B. Gophane and S. T. Sigurdsson, J. Org. Chem., 2012, 77, 7749–7754 CrossRef.
- A. Collauto, S. von Bulow, D. Gophane, S. Saha, L. Stelzl, G. Hummer, S. T. Sigurdsson and T. Prisner, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202009800.
- L. Buttner, F. Javadi-Zarnaghi and C. Hobartner, J. Am. Chem. Soc., 2014, 136, 8131–8137 CrossRef.
- A. Hottin and A. Marx, Acc. Chem. Res., 2016, 49, 418–427 CrossRef CAS.
- M. Hocek, Acc. Chem. Res., 2019, 52, 1730–1737 CrossRef CAS.
- F. Imdahl, E. Vafadarnejad, C. Homberger, A. E. Saliba and J. Vogel, Nat. Microbiol., 2020, 5, 1202–1206 CrossRef.
- A. Kulkarni, A. G. Anderson, D. P. Merullo and G. Konopka, Curr. Opin. Biotechnol., 2019, 58, 129–136 CrossRef CAS.
- A. A. Friedman, A. Letai, D. E. Fisher and K. T. Flaherty, Nat. Rev. Cancer, 2015, 15, 747–756 CrossRef CAS.
- B. Rabbani, H. Nakaoka, S. Akhondzadeh, M. Tekin and N. Mahdieh, Mol. Omics, 2016, 12, 1818–1830 RSC.
- Y. J. He, J. Wen, Q. H. Cui, F. F. Lai, D. L. Yin and H. Q. Cui, Front. Pharmacol., 2018, 9, 812 CrossRef.
- T. G. Mueller, V. Sakin and B. Mueller, Molecules, 2019, 24, 481 CrossRef.
- A. K. Rath and A. Rentmeister, Curr. Opin. Biotechnol., 2015, 31, 42–49 CrossRef CAS.
- A. R. Buxbaum, G. Haimovich and R. H. Singer, Nat. Rev. Mol. Cell Biol., 2015, 16, 513 CrossRef CAS.
- F. Bouhedda, A. Autour and M. Ryckelynck, Int. J. Mol. Sci., 2018, 19, 44 CrossRef.
- R. Micura and C. Hobartner, Chem. Soc. Rev., 2020 10.1039/d0cs00617c.
- I. Ivancova, D. L. Leone and M. Hocek, Curr. Opin. Chem. Biol., 2019, 52, 136–144 CrossRef CAS.
- Y. Tian and Q. Lin, ACS Chem. Biol., 2019, 14, 2489–2496 CrossRef CAS.
- P. Shieh and C. R. Bertozzi, Org. Biomol. Chem., 2014, 12, 9307–9320 RSC.
- K. Krell, D. Harijan, D. Ganz, L. Doll and H. A. Wagenknecht, Bioconjugate Chem., 2020, 31, 990–1011 CrossRef CAS.
- D. Ganz, D. Harijan and H.-A. Wagenknecht, RSC Chem. Biol., 2020, 1, 86–97 RSC.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS.
- W. J. Tipping, M. Lee, V. G. Brunton, G. C. Lloyd-Jones and A. N. Hulme, Faraday Discuss., 2019, 220, 71–85 RSC.
- K. S. Chandrasekaran and A. Rentmeister, Biochemistry, 2019, 58, 24–30 CrossRef CAS.
- L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef.
- T. Machida and N. Winssinger, ChemBioChem, 2016, 17, 811–815 CrossRef CAS.
- D. C. Kennedy, C. S. McKay, M. C. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS.
- B. Cao, Y. Zheng, T. Xi, C. Zhang, W. Song, K. Burugapalli, H. Yang and Y. Ma, Biomed. Microdevices, 2012, 14, 709–720 CrossRef CAS.
- V. Bevilacqua, M. King, M. Chaumontet, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., Int. Ed., 2014, 53, 5872–5876 CrossRef CAS.
- J. Dommerholt, F. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113–4119 CrossRef CAS.
- I. L. Cartwright, D. W. Hutchinson and V. W. Armstrong, Nucleic Acids Res., 1976, 3, 2331–2339 CrossRef CAS.
- I. Nikic, T. Plass, O. Schraidt, J. Szymanski, J. A. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245–2249 CrossRef CAS.
- R. A. Carboni and R. V. Lindsey, J. Am. Chem. Soc., 1959, 81, 4342–4346 CrossRef CAS.
- M. E. Jun, B. Roy and K. H. Ahn, Chem. Commun., 2011, 47, 7583–7601 RSC.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- F. Eggert, K. Kulikov, C. Domnick, P. Leifels and S. Kath-Schorr, Methods, 2017, 120, 17–27 CrossRef CAS.
- E. A. Specht, E. Braselmann and A. E. Palmer, in Annual Review of Physiology, ed. D. Julius, 2017, vol. 79, pp. 93–117 Search PubMed.
- L. V. Mannack, S. Eising and A. Rentmeister, F1000Research, 2016, 5(F1000 Faculty Rev), 775, DOI:10.12688/f1000research.8151.1.
- A. A. Sun, C. Gasser, F. D. Li, H. Chen, S. Mair, O. Krasheninina, R. Micura and A. M. Ren, Nat. Commun., 2019, 10, 1–13 CrossRef.
- S. R. Kirk, N. W. Luedtke and Y. Tor, Bioorg. Med. Chem., 2001, 9, 2295–2301 CrossRef CAS.
- D. Dziuba, P. Jurkiewicz, M. Cebecauer, M. Hof and M. Hocek, Angew. Chem., Int. Ed., 2016, 55, 174–178 CrossRef CAS.
- J. Jeffet, A. Kobo, T. Su, A. Grunwald, O. Green, A. N. Nilsson, E. Eisenberg, T. Ambjornsson, F. Westerlund, E. Weinhold, D. Shabat, P. K. Purohit and Y. Ebenstein, ACS Nano, 2016, 10, 9823–9830 CrossRef CAS.
- T. Gilboa, C. Torfstein, M. Juhasz, A. Grunwald, Y. Ebenstein, E. Weinhold and A. Meller, ACS Nano, 2016, 10, 8861–8870 CrossRef CAS.
- F. Muttach, N. Muthmann and A. Rentmeister, Org. Biomol. Chem., 2017, 15, 278–284 RSC.
- L. Anhauser and A. Rentmeister, Curr. Opin. Biotechnol., 2017, 48, 69–76 CrossRef.
- J. M. Holstein and A. Rentmeister, Methods, 2016, 98, 18–25 CrossRef CAS.
- D. Schulz and A. Rentmeister, ChemBioChem, 2014, 15, 2342–2347 CrossRef CAS.
- N. Muthmann, K. Hartstock and A. Rentmeister, Wiley Interdiscip. Rev.: RNA, 2020, 11, e1561 CAS.
- S. Nagata, T. Hamasaki, K. Uetake, H. Masuda, K. Takagaki, N. Oka, T. Wada, T. Ohgi and J. Yano, Nucleic Acids Res., 2010, 38, 7845–7857 CrossRef CAS.
- M. Goldeck, T. Tuschl, G. Hartmann and J. Ludwig, Angew. Chem., Int. Ed., 2014, 53, 4694–4698 CrossRef CAS.
- J. Schoch, M. Wiessler and A. Jaschke, J. Am. Chem. Soc., 2010, 132, 8846–8847 CrossRef CAS.
- S. Pitsch, P. A. Weiss, L. Jenny, A. Stutz and X. Wu, Helv. Chim. Acta, 2001, 84, 3773–3795 CrossRef CAS.
- P. Theisen, C. McCollum, K. Upadhya, K. Jacobson, H. N. Vu and A. Andrus, Tetrahedron Lett., 1992, 33, 5033–5036 CrossRef CAS.
- M. V. Kvach, D. A. Tsybulsky, V. V. Shmanai, I. A. Prokhorenko, I. A. Stepanova and V. A. Korshun, Curr. Protoc. Nucleic Acid Chem., 2013, 52(1), 4.55.1–4.55.33, DOI:10.1002/0471142700.nc0455s52.
- C. J. Ablenas, Y. Gidi, M. H. Powdrill, N. Ahmed, T. A. Shaw, M. Mesko, M. Götte, G. Cosa and J. P. Pezacki, ACS Infect. Dis., 2019, 5, 2118–2126 CrossRef CAS.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS.
- M. S. Wranne, A. F. Fuchtbauer, B. Dumat, M. Bood, A. H. El-Sagheer, T. Brown, H. Graden, M. Grotli and L. M. Wilhelmsson, J. Am. Chem. Soc., 2017, 139, 9271–9280 CrossRef CAS.
- A. F. Fuchtbauer, M. S. Wranne, S. Sarangamath, M. Bood, A. H. El-Sagheer, T. Brown, H. Graden, M. Grotli and L. M. Wilhelmsson, ChemPlusChem, 2020, 85, 319–326 CrossRef.
- D. A. Berry, K.-Y. Jung, D. S. Wise, A. D. Sercel, W. H. Pearson, H. Mackie, J. B. Randolph and R. L. Somers, Tetrahedron Lett., 2004, 45, 2457–2461 CrossRef CAS.
- J. E. Sanabia, L. S. Goldner, P. A. Lacaze and M. E. Hawkins, J. Phys. Chem. B, 2004, 108, 15293–15300 CrossRef CAS.
- R. S. K. Lane, R. Jones, R. W. Sinkeldam, Y. Tor and S. W. Magennis, ChemPhysChem, 2014, 15, 867–871 CrossRef CAS.
- E. L. Rachofsky, R. Osman and J. B. A. Ross, Biochemistry, 2001, 40, 946–956 CrossRef CAS.
- D. C. Ward, E. Reich and L. Stryer, J. Biol. Chem., 1969, 244, 1228–1237 CAS.
- J. M. Jean and K. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 37–41 CrossRef CAS.
- A. Nuthanakanti, M. A. Boerneke, T. Hermann and S. G. Srivatsan, Angew. Chem., Int. Ed., 2017, 56, 2640–2644 CrossRef CAS.
- C. Manz, A. Y. Kobitski, A. Samanta, B. G. Keller, A. Jaschke and G. U. Nienhaus, Nat. Chem. Biol., 2017, 13, 1172–1178 CrossRef CAS.
- K. Onizuka, Y. Taniguchi and S. Sasaki, Bioconjugate Chem., 2009, 20, 799–803 CrossRef CAS.
- K. Onizuka, Y. Taniguchi and S. Sasaki, Nucleic Acids Res., 2010, 38, 1760–1766 CrossRef CAS.
- S. Jin, C. V. Miduturu, D. C. McKinney and S. K. Silverman, J. Org. Chem., 2005, 70, 4284–4299 CrossRef CAS.
- A. Sanchez, E. Pedroso and A. Grandas, Org. Biomol. Chem., 2012, 10, 8478–8483 RSC.
- M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384–6458 CAS.
- A. Panattoni, R. Pohl and M. Hocek, Org. Lett., 2018, 20, 3962–3965 CrossRef CAS.
- J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639–3642 CrossRef CAS.
- A. M. Jawalekar, S. Malik, J. M. Verkade, B. Gibson, N. S. Barta, J. C. Hodges, A. Rowan and F. L. van Delft, Molecules, 2013, 18, 7346–7363 CrossRef CAS.
- J. Schoch, M. Staudt, A. Samanta, M. Wiessler and A. Jaeschke, Bioconjugate Chem., 2012, 23, 1382–1386 CrossRef CAS.
- A. M. Pyka, C. Domnick, F. Braun and S. Kath-Schorr, Bioconjugate Chem., 2014, 25, 1438–1443 CrossRef CAS.
- J. Lietard, A. Meyer, J.-J. Vasseur and F. Morvan, J. Org. Chem., 2008, 73, 191–200 CrossRef CAS.
- S. Lingala, L. U. Nordstrom and P. R. Mallikaratchy, Tetrahedron Lett., 2019, 60, 211–213 CrossRef CAS.
- J. Seckute, J. Yang and N. K. Devaraj, Nucleic Acids Res., 2013, 41, e148 CrossRef CAS.
- H. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, J. Am. Chem. Soc., 2014, 136, 17942–17945 CrossRef CAS.
- C. Stubinitzky, G. B. Cserep, E. Batzner, P. Kele and H. A. Wagenknecht, Chem. Commun., 2014, 50, 11218–11221 RSC.
- A. Eordogh, J. Steinmeyer, K. Peewasan, U. Schepers, H. A. Wagenknecht and P. Kele, Bioconjugate Chem., 2016, 27, 457–464 CrossRef CAS.
- T. Wada, A. Mochizuki, S. Higashiya, H. Tsuruoka, S. Kawahara, M. Ishikawa and M. Sekine, Tetrahedron Lett., 2001, 42, 9215–9219 CrossRef CAS.
- A. M. Jawalekar, N. Meeuwenoord, J. S. Cremers, H. S. Overkleeft, G. A. van der Marel, F. P. Rutjes and F. L. van Delft, J. Org. Chem., 2008, 73, 287–290 CrossRef CAS.
- A. Kiviniemi, P. Virta and H. Lonnberg, Bioconjugate Chem., 2008, 19, 1726–1734 CrossRef CAS.
- M. A. Fomich, M. V. Kvach, M. J. Navakouski, C. Weise, A. V. Baranovsky, V. A. Korshun and V. V. Shmanai, Org. Lett., 2014, 16, 4590–4593 CrossRef CAS.
- M. L. Winz, E. C. Linder, J. Becker and A. Jaschke, Chem. Commun., 2018, 54, 11781–11784 RSC.
- P. Kielkowski, J. Fanfrlik and M. Hocek, Angew. Chem., Int. Ed., 2014, 53, 7552–7555 CrossRef CAS.
- H. Cahova, A. Panattoni, P. Kielkowski, J. Fanfrlik and M. Hocek, ACS Chem. Biol., 2016, 11, 3165–3171 CrossRef CAS.
- A. Hottin, K. Betz, K. Diederichs and A. Marx, Chem. – Eur. J., 2017, 23, 2109–2118 CrossRef CAS.
- M. A. Augustin, W. Ankenbauer and B. Angerer, J. Biotechnol., 2001, 86, 289–301 CrossRef CAS.
- K. Tram, D. Twohig and H. B. Yan, Nucleosides, Nucleotides Nucleic Acids, 2011, 30, 1–11 CrossRef CAS.
- L. H. Thoresen, G. S. Jiao, W. C. Haaland, M. L. Metzker and K. Burgess, Chem. – Eur. J., 2003, 9, 4603–4610 CrossRef CAS.
- K. Hirano, Y. Yoshida, T. Ishido, Y. Wada, N. Moriya, N. Yamazaki, Y. Mizushina, Y. Baba and M. Ishikawa, Anal. Biochem., 2010, 405, 160–167 CrossRef CAS.
- P. Guixens-Gallardo, J. Humpolickova, S. P. Miclea, R. Pohl, T. Kraus, P. Jurkiewicz, M. Hof and M. Hocek, Org. Biomol. Chem., 2020, 18, 912–919 RSC.
- D. Dziuba, P. Pospíšil, J. Matyašovský, J. Brynda, D. Nachtigallová, L. Rulíšek, R. Pohl, M. Hof and M. Hocek, Chem. Sci., 2016, 7, 5775–5785 RSC.
- M. Kuba, R. Pohl and M. Hocek, Tetrahedron, 2018, 74, 6621–6629 CrossRef CAS.
- K. T. Passow, N. M. Antczak, S. J. Sturla and D. A. Harki, Curr. Protoc. Nucleic Acid Chem., 2020, 80, e101 CAS.
- L. S. McCoy, D. Shin and Y. Tor, J. Am. Chem. Soc., 2014, 136, 15176–15184 CrossRef CAS.
- G. Pljevaljcic, M. Pignot and E. Weinhold, J. Am. Chem. Soc., 2003, 125, 3486–3492 CrossRef CAS.
- F. H. Schmidt, M. Huben, B. Gider, F. Renault, M. P. Teulade-Fichou and E. Weinhold, Bioorg. Med. Chem., 2008, 16, 40–48 CrossRef CAS.
- F. Kunkel, R. Lurz and E. Weinhold, Molecules, 2015, 20, 20805–20822 CrossRef CAS.
- A. Grunwald, M. Dahan, A. Giesbertz, A. Nilsson, L. K. Nyberg, E. Weinhold, T. Ambjornsson, F. Westerlund and Y. Ebenstein, Nucleic Acids Res., 2015, 43, e117 CrossRef.
- V. Goyvaerts, S. Van Snick, L. D'Huys, R. Vitale, M. Helmer Lauer, S. Wang, V. Leen, W. Dehaen and J. Hofkens, Chem. Commun., 2020, 56, 3317–3320, 10.1039/c9cc08938a.
- S. C. Alexander, K. N. Busby, C. M. Cole, C. Y. Zhou and N. K. Devaraj, J. Am. Chem. Soc., 2015, 137, 12756–12759 CrossRef CAS.
- M. Mickute, M. Nainyte, L. Vasiliauskaite, A. Plotnikova, V. Masevicius, S. Klimasauskas and G. Vilkaitis, Nucleic Acids Res., 2018, 46, e104 CrossRef.
- D. D. Agranoff and S. Krishna, Mol. Microbiol., 1998, 28, 403–412 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS.
- J. Schoch and A. Jaeschke, RSC Adv., 2013, 3, 4181–4183 RSC.
- H. Busskamp, E. Batroff, A. Niederwieser, O. S. Abdel-Rahman, R. F. Winter, V. Wittmann and A. Marx, Chem. Commun., 2014, 50, 10827–10829 RSC.
- M. Kromer, K. Bartova, V. Raindlova and M. Hocek, Chem. – Eur. J., 2018, 24, 11890–11894 CrossRef.
- J. Matyasovsky, R. Pohl and M. Hocek, Chem. – Eur. J., 2018, 24, 14938–14941 CrossRef CAS.
- J. Matyasovsky, P. Perlikova, V. Malnuit, R. Pohl and M. Hocek, Angew. Chem., Int. Ed., 2016, 55, 15856–15859 CrossRef CAS.
- D. Ploschik, F. Roenicke, H. Beike, R. Strasser and H.-A. Wagenknecht, ChemBioChem, 2018, 19, 1949–1953 CrossRef CAS.
- P. N. Asare-Okai, E. Agustin, D. Fabris and M. Royzen, Chem. Commun., 2014, 50, 7844–7847 RSC.
- A. A. Sawant, A. A. Tanpure, P. P. Mukherjee, S. Athavale, A. Kelkar, S. Galande and S. G. Srivatsan, Nucleic Acids Res., 2016, 44, e16 CrossRef.
- M. L. Winz, A. Samanta, D. Benzinger and A. Jaschke, Nucleic Acids Res., 2012, 40, e78 CrossRef CAS.
- L. Anhaeuser, S. Huwel, T. Zobel and A. Rentmeister, Nucleic Acids Res., 2019, 47, e42 CrossRef.
- A. M. Leconte, G. T. Hwang, S. Matsuda, P. Capek, Y. Hari and F. E. Romesberg, J. Am. Chem. Soc., 2008, 130, 2336–2343 CrossRef CAS.
- T. Ishizuka, M. Kimoto, A. Sato and I. Hirao, Chem. Commun., 2012, 48, 10835–10837 RSC.
- C. Domnick, F. Eggert and S. Kath-Schorr, Chem. Commun., 2015, 51, 8253–8256 RSC.
- F. Eggert and S. Kath-Schorr, Chem. Commun., 2016, 52, 7284–7287 RSC.
- A. W. Feldman, E. C. Fischer, M. P. Ledbetter, J. Y. Liao, J. C. Chaput and F. E. Romesberg, J. Am. Chem. Soc., 2018, 140, 1447–1454 CrossRef CAS.
- L. Li, M. Degardin, T. Lavergne, D. A. Malyshev, K. Dhami, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2014, 136, 826–829 CrossRef CAS.
- G. Houlihan, S. Arangundy-Franklin, B. T. Porebski, N. Subramanian, A. I. Taylor and P. Holliger, Nat. Chem., 2020, 12, 683–690 CrossRef CAS.
- S. Klimasauskas and E. Weinhold, Trends Biotechnol., 2007, 25, 99–104 CrossRef CAS.
- M. Tomkuvienė, E. Kriukienė and S. Klimašauskas, in DNA Methyltransferases – Role and Function, ed. A. Jeltsch and R. Z. Jurkowska, Springer International Publishing, Cham, 2016, pp. 511–535 DOI:10.1007/978-3-319-43624-1_19.
- D. Schulz, J. M. Holstein and A. Rentmeister, Angew. Chem., Int. Ed., 2013, 52, 7874–7878 CrossRef CAS.
- D. Stummer, C. Herrmann and A. Rentmeister, ChemistryOpen, 2015, 4, 295–301 CrossRef CAS.
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362–1369 RSC.
- M. H. Lauer, C. Vranken, J. Deen, W. Frederickx, W. Vanderlinden, N. Wand, V. Leen, M. H. Gehlen, J. Hofkens and R. K. Neely, Chem. Sci., 2017, 8, 3804–3811 RSC.
- F. Muttach, N. Muthmann, D. Reichert, L. Anhaeuser and A. Rentmeister, Chem. Sci., 2017, 8, 7947–7953 RSC.
- L. Anhaeuser, F. Muttach and A. Rentmeister, Chem. Commun., 2018, 54, 449–451 RSC.
- J. Zhang and Y. G. Zheng, ACS Chem. Biol., 2016, 11, 583–597 CAS.
- Y. Du, C. E. Hendrick, K. S. Frye and L. R. Comstock, ChemBioChem, 2012, 13, 2225–2233 CAS.
- J. Deen, S. Wang, S. Van Snick, V. Leen, K. Janssen, J. Hofkens and R. K. Neely, Nucleic Acids Res., 2018, 46, e64 CrossRef.
- W. Peters, S. Willnow, M. Duisken, H. Kleine, T. Macherey, K. E. Duncan, D. W. Litchfield, B. Luscher and E. Weinhold, Angew. Chem., Int. Ed., 2010, 49, 5170–5173 CrossRef CAS.
- G. M. Hanz, B. Jung, A. Giesbertz, M. Juhasz and E. Weinhold, J. Visualized Exp., 2014, e52014, DOI:10.3791/52014.
- G. Lukinavicius, V. Lapiene, Z. Stasevskij, C. Dalhoff, E. Weinhold and S. Klimasauskas, J. Am. Chem. Soc., 2007, 129, 2758–2759 CrossRef CAS.
- G. Lukinavicius, M. Tomkuviene, V. Masevicius and S. Klimasauskas, ACS Chem. Biol., 2013, 8, 1134–1139 CrossRef CAS.
- C. Vranken, J. Deen, L. Dirix, T. Stakenborg, W. Dehaen, V. Leen, J. Hofkens and R. K. Neely, Nucleic Acids Res., 2014, 42, e50 CrossRef CAS.
- N. Dai, J. Bitinaite, H. G. Chin, S. Pradhan and I. R. Correa, Jr., ChemBioChem, 2013, 14, 2144–2152 CrossRef CAS.
- G. Nifker, M. Levy-Sakin, Y. Berkov-Zrihen, T. Shahal, T. Gabrieli, M. Fridman and Y. Ebenstein, ChemBioChem, 2015, 16, 1857–1860 CrossRef CAS.
- N. Gilat, T. Tabachnik, A. Shwartz, T. Shahal, D. Torchinsky, Y. Michaeli, G. Nifker, S. Zirkin and Y. Ebenstein, Clin. Epigenet., 2017, 9, 70 CrossRef.
- M. Gao, D. T. Fritz, L. P. Ford and J. Wilusz, Mol. Cell, 2000, 5, 479–488 CrossRef CAS.
- J. M. Holstein, D. Schulz and A. Rentmeister, Chem. Commun., 2014, 50, 4478–4481 RSC.
- F. Muttach and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 1917–1920 CrossRef CAS.
- J. M. Holstein, L. Anhaeuser and A. Rentmeister, Angew. Chem., Int. Ed., 2016, 55, 10899–10903 CrossRef CAS.
- J. M. Holstein, F. Muttach, S. H. H. Schiefelbein and A. Rentmeister, Chem. – Eur. J., 2017, 23, 6165–6173 CrossRef CAS.
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS.
- A. Plotnikova, A. Osipenko, V. Masevicius, G. Vilkaitis and S. Klimasauskas, J. Am. Chem. Soc., 2014, 136, 13550–13553 CrossRef CAS.
- A. Osipenko, A. Plotnikova, M. Nainyte, V. Masevicius, S. Klimasauskas and G. Vilkaitis, Angew. Chem., Int. Ed., 2017, 56, 6507–6510 CrossRef CAS.
- T. Awai, A. Ochi, I. Ihsanawati, T. Sengoku, A. Hirata, Y. Bessho, S. Yokoyama and H. Hori, J. Biol. Chem., 2011, 286, 35236–35246 CrossRef CAS.
- Y. Motorin, J. Burhenne, R. Teimer, K. Koynov, S. Willnow, E. Weinhold and M. Helm, Nucleic Acids Res., 2011, 39, 1943–1952 CrossRef CAS.
- M. Tomkuviene, B. Clouet-d'Orval, I. Cerniauskas, E. Weinhold and S. Klimasauskas, Nucleic Acids Res., 2012, 40, 6765–6773 CrossRef CAS.
- F. Li, J. Dong, X. Hu, W. Gong, J. Li, J. Shen, H. Tian and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 4597–4602 CrossRef CAS.
- F. Ehret, C. Y. Zhou, S. C. Alexander, D. Zhang and N. K. Devaraj, Mol. Pharmaceutics, 2018, 15, 737–742 CrossRef CAS.
- S. Vauleon, S. A. Ivanov, S. Gwiazda and S. Muller, ChemBioChem, 2005, 6, 2158–2162 CrossRef CAS.
- R. I. McDonald, J. P. Guilinger, S. Mukherji, E. A. Curtis, W. I. Lee and D. R. Liu, Nat. Chem. Biol., 2014, 10, 1049–1054 CrossRef CAS.
- B. Samanta, D. P. Horning and G. F. Joyce, Nucleic Acids Res., 2018, 46, e103 CrossRef.
- D. A. Baum and S. K. Silverman, Angew. Chem., Int. Ed., 2007, 46, 3502–3504 CrossRef CAS.
- M. G. Maghami, S. Dey, A. K. Lenz and C. Hobartner, Angew. Chem., Int. Ed., 2020, 9421–9425, DOI:10.1002/anie.202001300.
- M. G. Maghami, C. P. M. Scheitl and C. Hobartner, J. Am. Chem. Soc., 2019, 141, 19546–19549 CrossRef.
- N. K. Kim, A. Murali and V. J. DeRose, Chem. Biol., 2004, 11, 939–948 CrossRef CAS.
- M. F. Juette, D. S. Terry, M. R. Wasserman, Z. Zhou, R. B. Altman, Q. Zheng and S. C. Blanchard, Curr. Opin. Chem. Biol., 2014, 20, 103–111 CrossRef CAS.
- N. Piton, Y. G. Mu, G. Stock, T. F. Prisner, O. Schiemann and J. W. Engels, Nucleic Acids Res., 2007, 35, 3128–3143 CrossRef CAS.
- T. Weinrich, E. A. Jaumann, U. Scheffer, T. F. Prisner and M. W. Gobel, Chem. – Eur. J., 2018, 24, 6202–6207 CrossRef CAS.
- U. Jakobsen, S. A. Shelke, S. Vogel and S. T. Sigurdsson, J. Am. Chem. Soc., 2010, 132, 10424–10428 CrossRef CAS.
- C. Wuebben, S. Blume, D. Abdullin, D. Brajtenbach, F. Haege, S. Kath-Schorr and O. Schiemann, Molecules, 2019, 24, 4482 CAS.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Diaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS.
- A. Samanta, S. Buckhout-White, E. Oh, K. Susumu and I. L. Medintz, Mol. Syst. Des. Eng., 2018, 3, 314–327 RSC.
- Z. H. Qiu, L. H. Lu, X. Jian and C. He, J. Am. Chem. Soc., 2008, 130, 14398–14399 CrossRef CAS.
- U. K. Shigdel, J. Zhang and C. He, Angew. Chem., Int. Ed., 2008, 47, 90–93 CrossRef CAS.
- F. Muttach, F. Masing, A. Studer and A. Rentmeister, Chem. – Eur. J., 2017, 23, 5988–5993 CrossRef CAS.
- C. X. Song, Y. Sun, Q. Dai, X. Y. Lu, M. Yu, C. G. Yang and C. He, ChemBioChem, 2011, 12, 1682–1685 CrossRef CAS.
- Z. Yang, Y. Shi, W. Liao, H. Yin and S. Ai, Sens. Actuators, B, 2016, 223, 621–625 CrossRef CAS.
- Z. Liutkeviciute, E. Kriukiene, I. Grigaityte, V. Masevicius and S. Klimasauskas, Angew. Chem., Int. Ed., 2011, 50, 2090–2093 CrossRef CAS.
- K. Hartstock, B. S. Nilges, A. Ovcharenko, N. V. Cornelissen, N. Püllen, A. M. Lawrence-Dörner, S. A. Leidel and A. Rentmeister, Angew. Chem., Int. Ed., 2018, 57, 6342–6346 CrossRef CAS.
- M. Hocek and M. Fojta, Chem. Soc. Rev., 2011, 40, 5802–5814 RSC.
- E. Palecek and M. Bartosik, Chem. Rev., 2012, 112, 3427–3481 CrossRef CAS.
- P. Havranova-Vidlakova, M. Kromer, V. Sykorova, M. Trefulka, M. Fojta, L. Havran and M. Hocek, ChemBioChem, 2020, 21, 171–180 CAS.
- A. Simonova, I. Magrina, V. Sykorova, R. Pohl, M. Ortiz, L. Havran, M. Fojta, C. K. O'Sullivan and M. Hocek, Chem. – Eur. J., 2020, 26, 1286–1291 CAS.
- T. Yamada and N. Akimitsu, Wiley Interdiscip. Rev.: RNA, 2019, 10, e1508 Search PubMed.
- S. L. Hong, T. Chen, Y. T. Zhu, A. Li, Y. Y. Huang and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 5827–5831 CrossRef CAS.
- T. Ishizuka, H. S. Liu, K. Ito and Y. Xu, Sci. Rep., 2016, 6, 1–10 CrossRef.
- I. H. Wang, M. Suomalainen, V. Andriasyan, S. Kilcher, J. Mercer, A. Neef, N. W. Luedtke and U. F. Greber, Cell Host Microbe, 2013, 14, 468–480 CrossRef CAS.
- P. V. Danenberg, R. S. Bhatt, N. G. Kundu, K. Danenberg and C. Heidelberger, J. Med. Chem., 1981, 24, 1537–1540 CrossRef CAS.
- H. H. Ross, M. Rahman, L. H. Levkoff, S. Millette, T. Martin-Carreras, E. M. Dunbar, B. A. Reynolds and E. D. Laywell, J. Neuro-Oncol., 2011, 105, 485–498 CrossRef CAS.
- S. Diermeier-Daucher, S. T. Clarke, D. Hill, A. Vollmann-Zwerenz, J. A. Bradford and G. Brockhoff, Cytometry, Part A, 2009, 75a, 535–546 CrossRef CAS.
- A. B. Neef and N. W. Luedtke, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20404–20409 CrossRef CAS.
- D. Qu, G. Wang, Z. Wang, L. Zhou, W. Chi, S. Cong, X. Ren, P. Liang and B. Zhang, Anal. Biochem., 2011, 417, 112–121 CrossRef CAS.
- A. B. Neef, F. Samain and N. W. Luedtke, ChemBioChem, 2012, 13, 1750–1753 CrossRef CAS.
- G. L'Abbe, Chem. Rev., 1969, 69, 345–363 CrossRef.
- A. B. Neef and N. W. Luedtke, ChemBioChem, 2014, 15, 789–793 CrossRef CAS.
- T. Triemer, A. Messikommer, S. M. K. Glasauer, J. Alzeer, M. H. Paulisch and N. W. Luedtke, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E1366–E1373 CrossRef CAS.
- U. Rieder and N. W. Luedtke, Angew. Chem., Int. Ed., 2014, 53, 9168–9172 CrossRef CAS.
- Y. X. Wu, G. L. Guo, J. D. Zheng, D. Xing and T. Zhang, ACS Sens., 2019, 4, 44–51 CrossRef CAS.
- A. Gubu, L. Li, Y. Ning, X. Y. Zhang, S. Lee, M. K. Feng, Q. Li, X. G. Lei, K. B. Jo and X. J. Tang, Chem. – Eur. J., 2018, 24, 5895–5900 CrossRef CAS.
- C. Y. Jao and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15779–15784 CrossRef CAS.
- M. Kubota, S. Nainar, S. M. Parker, W. England, F. Furche and R. C. Spitale, ACS Chem. Biol., 2019, 14, 1698–1707 CrossRef CAS.
- K. Abe, T. Ishigami, A. B. Shyu, S. Ohno, S. Umemura and A. Yamashita, Biochem. Biophys. Res. Commun., 2012, 428, 44–49 CrossRef CAS.
- M. Grammel, H. Hang and N. K. Conrad, ChemBioChem, 2012, 13, 1112–1115 CrossRef CAS.
- D. Curanovic, M. Cohen, I. Singh, C. E. Slagle, C. S. Leslie and S. R. Jaffrey, Nat. Chem. Biol., 2013, 9, 671 CrossRef CAS.
- H. S. Liu, T. Ishizuka, M. Kawaguchi, R. Nishii, H. Kataoka and Y. Xu, Bioconjugate Chem., 2019, 30, 2958–2966 CrossRef CAS.
- Y. Wang, W. J. Hu, W. Song, R. K. Lim and Q. Lin, Org. Lett., 2008, 10, 3725–3728 CrossRef CAS.
- S. Nainar, S. Beasley, M. Fazio, M. Kubota, N. Dai, I. R. Corrêa, Jr. and R. C. Spitale, ChemBioChem, 2016, 17, 2149–2152 CrossRef CAS.
- Y. Zhang and R. E. Kleiner, J. Am. Chem. Soc., 2019, 141, 3347–3351 CrossRef CAS.
- A. B. Neef, L. Pernot, V. N. Schreier, L. Scapozza and N. W. Luedtke, Angew. Chem., Int. Ed., 2015, 54, 7911–7914 CrossRef CAS.
- M. R. Miller, K. J. Robinson, M. D. Cleary and C. Q. Doe, Nat. Methods, 2009, 6, 439–457 CrossRef CAS.
- N. Hida, M. Y. Aboukilila, D. A. Burow, R. Paul, M. M. Greenberg, M. Fazio, S. Beasley, R. C. Spitale and M. D. Cleary, Nucleic Acids Res., 2017, 45, e138 CrossRef.
- S. Nainar, B. J. Cuthbert, N. M. Lim, W. E. England, K. Ke, K. Sophal, R. Quechol, D. L. Mobley, C. W. Goulding and R. C. Spitale, Nat. Methods, 2020, 17, 311–318 CrossRef CAS.
- H. J. Jessen, T. Schulz, J. Balzarini and C. Meier, Angew. Chem., Int. Ed., 2008, 47, 8719–8722 CrossRef CAS.
- S. Seo, K. Onizuka, C. Nishioka, E. Takahashi, S. Tsuneda, H. Abe and Y. Ito, Org. Biomol. Chem., 2015, 13, 4589–4595 RSC.
- N. Huynh, C. Dickson, D. Zencak, D. H. Hilko, A. Mackay-Sim and S. A. Poulsen, Chem. Biol. Drug Des., 2015, 86, 400–409 CrossRef CAS.
- M. Tera, S. M. K. Glasauer and N. W. Luedtke, ChemBioChem, 2018, 19, 1939–1943 CrossRef CAS.
- Z. Zawada, A. Tatar, P. Mocilac, M. Budesinsky and T. Kraus, Angew. Chem., Int. Ed., 2018, 57, 9891–9895 CrossRef CAS.
- P. Guixens-Gallardo, Z. Zawada, J. Matyasovsky, D. Dziuba, R. Pohl, T. Kraus and M. Hocek, Bioconjugate Chem., 2018, 29, 3906–3912 CrossRef CAS.
- K. J. Westerich, K. S. Chandrasekaran, T. Gross-Thebing, N. Kueck, E. Raz and A. Rentmeister, Chem. Sci., 2020, 11, 3089–3095 RSC.
| This journal is © The Royal Society of Chemistry 2020 |