
Low-loading Pt nanoparticles embedded on Ni, N-doped carbon as superior electrocatalysts for oxygen reduction†
Xinliang
Wang
ab,
Shaoxuan
Yang
ab,
Yihuan
Yu
ab,
Meiling
Dou
ab,
Zhengping
Zhang
*ab and
Feng
Wang
 *ab
*ab
aState Key Laboratory of Chemical Resource Engineering, Beijing Key Laboratory of Electrochemical Process and Technology for Materials, Beijing University of Chemical Technology, Beijing 100029, P. R. China
bBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 27th November 2019
Abstract
As one of the efficient and classic nanoscale catalysts, Pt nanoparticles play predominant roles in multiple energy conversion systems, especially for electrochemical devices involving the oxygen reduction reaction (ORR). It is important to develop a scalable method for synthesis of more efficient Pt-based electrocatalysts with higher activity and stability. In this work, a low-loading Pt-based electrocatalyst (8.0 wt%) is fabricated by the galvanic replacement reaction, presenting well-dispersed Pt nanoparticles adjacent to atomic Ni–N–C complexes (Pt@NiNC). Due to the synergetic effect associated with the nanoscale/atomic scale joint active sites and the strong metal–support interaction, the resulting Pt@NiNC hybrid exhibits better ORR performance and higher mass activity than the benchmark Pt/C, as well as enhanced electrochemical stability. This research not only opens a new route to develop heterogeneous catalysts with multi-scale joint sites but also provides bright prospects for high-performance and low-cost energy conversion and storage.
Introduction
Proton exchange membrane fuel cells (PEMFCs) have attracted much attention due to their high efficiency and zero emission characteristics, making them promising energy conversion devices. As the cathodic reaction of PEMFCs, the oxygen reduction reaction (ORR) is one kind of multi-step electrochemical reaction with sluggish kinetics.1 Although platinum has been considered as the state-of-the-art and indispensable ORR electrocatalyst in practical applications, the high usage and high cost of Pt still severely hamper the large-scale applications of PEMFCs.2 To break these obstacles, many efforts have been devoted to developing Pt-based electrocatalysts with lower Pt loading and higher Pt mass activity (MA) while maintaining long-term stability in harsh oxygen reduction environments.3 However, for the low Pt loading limitation, there will not be enough nanoparticle-involved active sites over the electrode surface to expose to the reaction interface; meanwhile, Pt nanoparticles (NPs) tend to suffer from severe degradation due to the dissolution, migration, and/or Ostwald ripening/coalescence by highly reactive oxygen species (ROS, such as –OH, –OOH, and H2O2).4,5 On the other hand, greatly downsizing Pt NPs, and even dispersing Pt to the atomic level, seems to be in favor of enlarging the electrochemically active surface area (ECSA), but resulting in a poor catalytic specific activity (SA) due to the excess ROS adsorption.6Besides optimizing Pt NPs themselves, the opposite approach, developing support materials (e.g., carbon, metal oxides), is one of the effective ways to improve the strong metal–support interaction (SMSI) due to the possible synergistic effects.7,8 Of these, significant progress has been made with respect to carbon supports, especially for doped carbons.3,9,10 The SMSI between Pt NPs and doped carbons not only works on the Pt nucleation and growth, but also ameliorates the catalyst behavior. As one kind of platinum-group-metal-free (PGM-free) catalyst, carbon materials with atomic metal–nitrogen–carbon (M–N–C) complexes have been demonstrated as promising ORR electrocatalysts and have shown promise to associate with Pt NPs to form joint catalysis systems.3,11,12 In addition, M–N–C supports can also provide Pt NPs with a stable ORR-favorable chemical environment, where oxophilic M–N–C complexes could facilitate the ROS cleavage. In this case, improving the SMSI of the synergistically coupled sites of Pt NPs and atomic M–N–C complexes is a promising strategy for low-loading Pt electrocatalysts to maintain or exceed the excellent ORR performance.
In this work, we developed a scalable method for preparation of low-loading Pt NPs (8.0 wt%) embedded on Ni, N-doped carbon (Pt@NiNC) electrocatalysts (Scheme S1, Table S1†). The Pt@NiNC electrocatalysts were derived from the galvanic replacement reaction between H2PtCl6 and Ni, N-doped carbon (NiNC)-supported Ni NPs (Ni@NiNC, which was the pyrolyzed product of nickel polyphthalocyanine13) in ethylene glycol solution. Several solutions (water, ethanol and ethylene glycol) were used for the galvanic replacement reaction, and it was found that the resulting Pt@NiNC hybrid synthesized in the ethylene glycol solution showed the best ORR activity (Fig. S1–S3, and see the ESI† for details), and hence only the Pt@NiNC hybrid synthesized in ethylene glycol solution was used for subsequent analysis.
Results and discussion
Transmission electron microscopy (TEM) and high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) were firstly performed to observe Ni@NiNC and Pt@NiNC. As shown in Fig. 1a and S4 and S5,† the Ni NPs in Ni@NiNC and the Pt NPs in Pt@NiNC showed different sizes of ca. 24 and 2.4 nm, respectively, but both had uniform dispersion on the supports. The lattice fringes of metal NPs in Pt@NiNC were measured as the close d-spacing of 0.14 and 0.23 nm as shown in Fig. 1b, which were indexed to the Pt(220) and Pt(111) planes. A highly jagged surface with low-coordinate atomic steps (i.e., the {311} step) was also observed around the surface of a Pt nanoparticle. The presence of a high density of low-coordinate atomic steps could facilitate the bonding of oxygen molecules to enhance the ORR activity.14,15 Notably, the corresponding HAADF-STEM image in Fig. 1c showed that the Pt nanoparticle was adjacent to numerous atomic metals (Ni or Pt) marked by yellow circles. To provide further information on the chemical components of Pt@NiNC, electron energy loss spectroscopy (EELS) elemental mapping of Pt@NiNC on an Enfina spectrometer was employed (Fig. 1d). It was clearly shown that metal NPs presented the distribution of the element Pt but without Ni, whilst there were strong signals of elements Ni, N and C with good dispersion on the support. According to these observations, we designed the resulting hybrid as Pt NPs embedded on nickel, nitrogen-doped carbon (Pt@NiNC). | ||
| Fig. 1 (a) TEM image of Ni@NiNC and the particle diameter distribution histogram (inset). (b) STEM and (c) the corresponding HAADF-STEM images of Pt@NiNC. (d) Elemental mapping images of Pt, C, N and Ni for Pt@NiNC. (e) XRD patterns, (f) N2 adsorption–desorption isotherms (left) and the corresponding DFT pore size distributions (right) of polyphthalocyanine, Ni@NiNC and Pt@NiNC. | ||
To further analyse the Pt@NiNC hybrid, X-ray diffraction (XRD) analyses were conducted for the polyphthalocyanine, Ni@NiNC and Pt@NiNC samples. As shown in Fig. 1e, the characteristic diffraction peaks of polyphthalocyanine demonstrated a good correlation with our previous work.16 The Pt@NiNC sample exhibited several diffraction peaks centered at 26.3°, 39.8°, 46.2° and 67.5°, which were indexed to the C(002), Pt(111), Pt(200), and Pt(220) planes (JCPDS: no. 41-1487 and no. 04-0802), respectively.17 Compared with the sharp peaks for metallic Ni (44.5° for Ni(111), 51.8° for Ni(200), and 76.4° for Ni(220)) in the Ni@NiNC sample, the relatively broad diffraction peaks of Pt NPs demonstrated their smaller particle size and lower crystallinity.18 The non-shifting characteristic diffraction peaks of Pt implied the generated Pt NPs without Pt–Ni alloy, which was consistent with the observation in the STEM and EELS results. The texture coefficients of Pt NPs on Pt@NiNC calculated from the XRD patterns were 1.28, 0.95, and 0.77 for Pt(111), Pt(200) and Pt(220) peaks, respectively. The dominant Pt(111) peaks suggested that the Pt@NiNC sample had a preferred orientation along the Pt(111) planes.19 The surface-exposed Pt(111) plane could participate in constituting the low-coordinate atomic steps (such as {211} and {311} steps), which was consistent with the STEM results.20 In addition, we also carried out measurements of N2 adsorption–desorption isotherms and the corresponding pore size distributions to investigate the porous structure of Pt@NiNC and Ni@NiNC (Fig. 1f and Table S3†). The isotherms for Ni@NiNC and Pt@NiNC both exhibited type-IV characteristics for mesoporous materials. Compared with Ni@NiNC (micropore volume: 0.005 cm3 g−1; mesopore volume: 0.296 cm3 g−1), the Pt@NiNC sample showed a lower micropore volume of 0.002 cm3 g−1 with a rapid decrease in nitrogen uptake (p/p0 < 0.1), but a similar mesopore volume of 0.297 cm3 g−1. The doping-enriched micropores should be the anchoring sites for Pt growth, whilst the maintained mesopores could facilitate mass transfer.
To probe the detailed chemical structure of the resultant Pt@NiNC sample, we performed X-ray analytical techniques. Fig. 2a shows the normalized X-ray absorption near-edge structure (XANES) spectra of the Pt@NiNC sample and Pt foil. The XANES spectra showed that the threshold energy and the maximum energy of Pt@NiNC were similar to those of Pt foil in the Pt L3-edge, demonstrating that most Pt atoms formed into metallic Pt NPs. The higher white-line intensity in Pt@NiNC than that in the Pt foil (inset in Fig. 2a) suggested that the Pt NPs in Pt@NiNC carried more positive charge.21 The extended X-ray absorption fine structure (EXAFS) analysis of Pt@NiNC (Fig. 2b) also showed the presence of the first, second, third and fourth Pt–Pt bonds (at distances of 2.58, 3.68, 4.60 and 5.12 Å, respectively) and collinear Pt–Pt–Pt linkages, which were the distinct features of a face-centered cubic structure.22 The interfacial interaction of Pt–N and Pt–Ni contributions (at 1.60 and 2.20 Å) could enhance the SMSI effect between Pt NPs and the NiNC support. Additionally, Fig. 2c shows that the Pt@NiNC hybrid presented a clear pre-edge peak at 8340 eV in the Ni K-edge XANES spectrum, attributed to the fingerprint of square-planar Ni–N4 complexes (inset of Fig. 2c).23 Ni–Pt bonds were also observed in the Pt@NiNC sample (inset of Fig. 2d), consistent with the Pt–Ni bonds in the Pt EXAFS spectra.
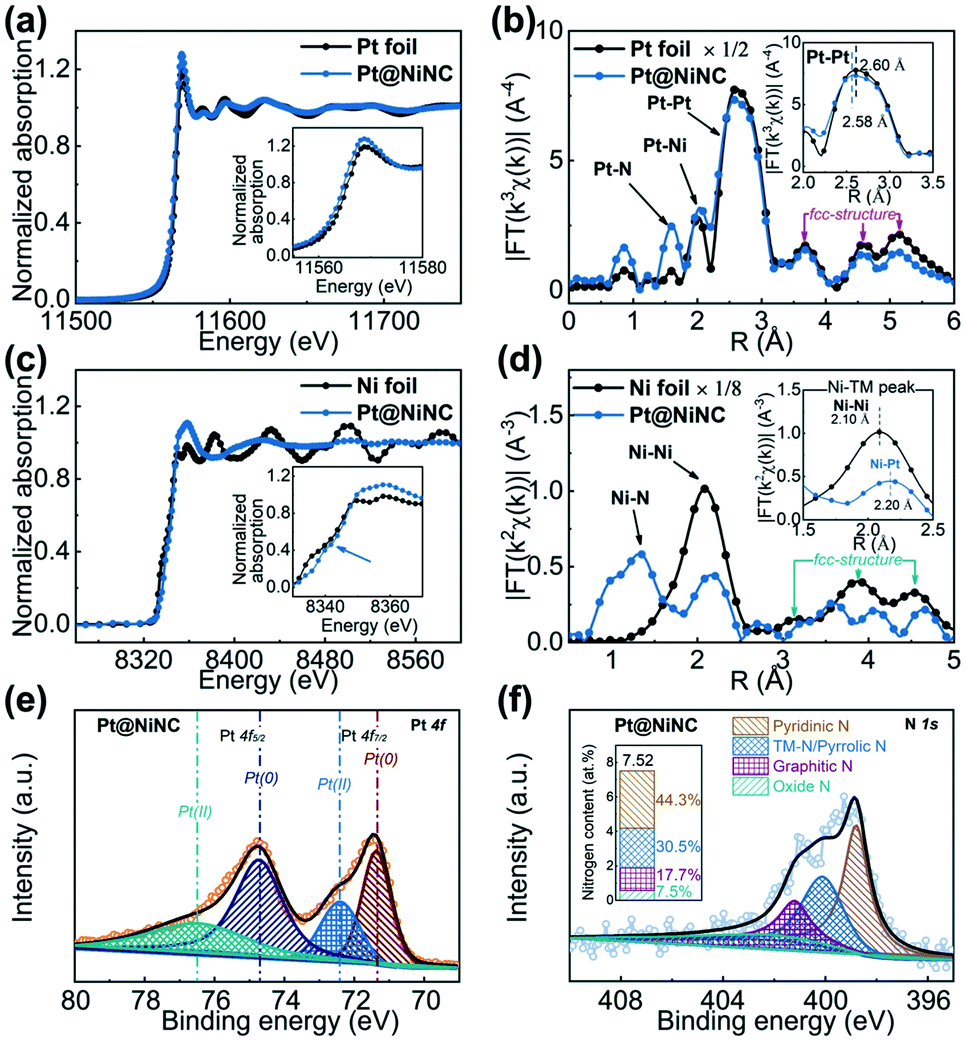 | ||
| Fig. 2 (a) Pt L3-edge XANES and (b) EXAFS spectra of Pt@NiNC and Pt foil. (c) Ni K-edge XANES and (d) EXAFS spectra of Pt@NiNC and Ni foil. (e) Pt 4f and (f) N 1s XPS spectra of Pt@NiNC. The inset in (f) shows the nitrogen content of Pt@NiNC. | ||
As shown in Fig. 2e, the low-coordinated electronic state of Pt NPs in Pt@NiNC was reconfirmed by X-ray photoelectron spectroscopy (XPS), which showed a high-binding-energy-shift Pt 4f peak (71.37 eV) compared with the standard Pt sample (71.20 eV).24 The chemical structure of Ni was only illustrated as ionic Ni in the Pt@NiNC sample (Fig. S6c†). In addition, the chemical structure of element N was further investigated with the deconvolution of four well-resolved peaks, which were related to pyridinic N (398.8 eV), TM-N/pyrrolic N (399.8 eV), graphitic N (401.2 eV) and oxide N (402.4 eV, Fig. 2f). The high content of pyridinic N and pyrrolic N could provide numerous anchoring sites for the Ni–N–C, Pt–N–C or Pt–Ni–N–C configuration and further strengthened the SMSI between Pt NPs and the NiNC support.25
To evaluate the ORR performance of the resultant Pt@NiNC hybrid, we also prepared chemically-reduced nitrogen-doped carbon supported Pt (Pt/NC) and chemically-reduced NiNC supported Pt NPs (Pt/NiNC, see the ESI† for details). Fig. 3a shows the linear sweep voltammetry (LSV) curves of Pt@NiNC, Pt/NiNC, Pt/NC, NiNC, and the benchmark 20% Pt/C on a rotating-disk electrode in O2-saturated 0.5 M H2SO4 electrolyte. The half-wave potential (E1/2) of Pt@NiNC was measured to be 0.85 V (versus reversible hydrogen electrode, vs. RHE, the same below). This indicated that Pt@NiNC performed a higher ORR activity than the other homemade Pt-based electrocatalysts (Pt/NiNC: 0.81 V; Pt/NC: 0.78), and even better than Pt/C (0.82 V). In addition, the kinetic current density (Jk) and Tafel slope were calculated with the mass-transport correction of the corresponding LSV curves. The Pt@NiNC sample exhibited the highest transfer coefficient for ORR kinetics (54 mV per decade) with the largest Jk (0.87 mA cm−2) at 0.90 V among all the Pt-based electrodes (Fig. S8a, Table S4†). The MA and SA of Pt@NiNC, Pt/NiNC, Pt/NC and commercial Pt/C are summarized in Fig. 3b for the detailed comparison. As expected, the Pt@NiNC sample exhibited the highest MA and SA among these four samples. Notably, although Pt/NiNC and Pt@NiNC possessed the same NiNC support, as well as similar particle size and Pt loading (Fig. S9 and Table S1†), the improved ORR activity of Pt@NiNC demonstrated that the galvanic replacement reaction was in favor of the Pt growth adjacent to the Ni–N–C complexes, further generating more active joint sites. As shown in Fig. 3c, the Jk of Pt/NiNC was much larger than the sum of those of Ni–N–C and Pt/NC, demonstrating that a synergistic effect existed between Pt NPs and Ni–N–C sites. The advanced ORR performance of Pt@NiNC was attributed to the enhanced interaction between Pt and adjacent Ni–N–C complexes, which was confirmed by the XPS measurement (Fig. S6, see the detailed discussion in the ESI†).
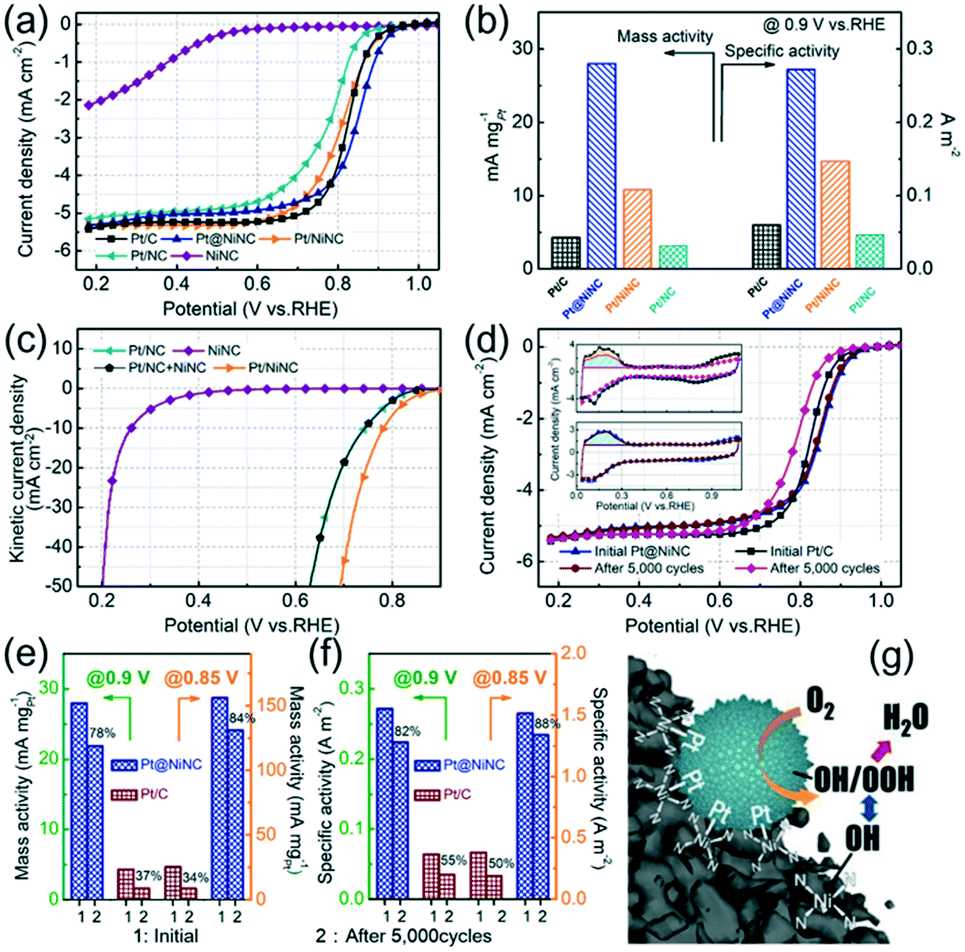 | ||
| Fig. 3 (a) LSV curves of Pt/C, Pt@NiNC, Pt/NiNC, Pt/NC and NiNC at a sweep rate of 5 mV s−1 with 1600 rpm. (b) Comparison of MA and SA at 0.90 V for the above electrodes. (c) The corrected LSV curves of Pt/NiNC, Pt/NC, NiNC and Pt/NC + NiNC. (d) LSV curves of Pt@NiNC and Pt/C for the ORR before and after 5000 potential cycles and the corresponding CV curves at a sweep rate of 50 mV s−1 (inset). Comparison of (e) MA and (f) SA for the Pt@NiNC and Pt/C electrodes. (g) Schematic mechanism of the enhanced catalytic performance for the ORR. | ||
Furthermore, the ORR LSV curves of Pt@NiNC before and after 5000 potential cycles were also measured to evaluate its catalytic stability toward the ORR. The commercial Pt/C electrode was also tested as reference. Fig. 3d shows that the Pt/C electrode exhibited obvious degradation with a 34 mV negative shift of its E1/2, whereas the E1/2 of the Pt@NiNC electrode remained nearly unchanged (ca. 3 mV negative shift) under the same measurement conditions. To assess the ORR performance in detail, the electrochemically active surface areas (ECSAs) were calculated from the hydrogen underpotential deposition (HUPD) area. As shown in the HUPD measurements (inset of Fig. 3d), the initial ECSA of Pt@NiNC was calculated to be 102.9 m2 gPt−1, about 1.5-fold enhancement compared with that of the Pt/C catalyst (66.8 m2 gPt−1). The CO-stripping CV curves (Fig. S10†) reconfirmed this result (Pt@NiNC: 109.8 m2 gPt−1, about 1.5-fold enhancement compared with that of the Pt/C catalyst). The larger ECSA of Pt for Pt@NiNC was most likely due to the high dispersion of Pt NPs with small size and jagged surficial morphology.15 After 5000 potential cycles, the Pt@NiNC sample showed much better ECSA retention with 95% ECSA remaining, but the ECSA of Pt/C decreased by almost 31%. To better elucidate the enhanced catalytic activity and stability, we calculated the MA (Fig. 3e) and SA (Fig. 3f) of Pt@NiNC and Pt/C before and after long-term operations. The Pt@NiNC exhibited a large initial MA of 28.0/156.0 mA mgPt−1 at 0.90/0.85 V (6.5-/6.2-fold enhancements compared with Pt/C, respectively), and the MA of Pt@NiNC was still maintained at 21.9/131.0 mA mgPt−1 after 5000 potential cycles. However, the MA of Pt/C decreased from 4.3/25.3 to 1.6/8.7 mA mgPt−1 at 0.90/0.85 V. Similarly, the Pt@NiNC hybrid also exhibited much higher SA before and after the long-term operation (initial: 0.272/1.516 A m−2 at 0.90/0.85 V; after 5000 cycles: 0.224/1.340 A m−2) compared with Pt/C (initial: 0.064/0.379 A m−2; after 5000 cycles: 0.035/0.189 A m−2). The TEM images before and after the long-term operation reconfirmed the enhanced electrochemical stability of Pt@NiNC (Fig. S11†). The enhanced catalytic stability for the ORR can be attributed to the fact that the oxophilic Ni–N–C complexes could produce strong lateral repulsion between the oxygen-containing groups and Pt-adsorbed ROS, which effectively facilitate the ROS cleavage.26 On the other hand, the generated intimate contact between Pt NPs and the NiNC support exhibits the enhanced SMIS effect to impede the segregation of Pt NPs from the NiNC support, and further improve the electrochemical stability (Fig. 3g).
Conclusions
In summary, a multi-scale hybrid with low-loading Pt NPs and a NiNC support has been elaborately constructed and performed as an excellent ORR electrocatalyst. The synergistically coupled sites of nanoscale Pt particles adjacent to atomic Ni–N–C complexes can be prepared by the galvanic replacement reaction, which enables the maximum accessibility of low-loading Pt with high ORR activity. Multiple characterization and electrochemical testing methods reveal that the superior ORR activity and stability are ascribed to the multi-scale joint active sites and the strong SMSI effect. Compared with the benchmark 20% Pt/C, the resultant 8 wt% Pt@NiNC hybrid exhibits a higher MA and SA, along with a low activity decrease, promising to serve as efficient ORR electrocatalysts for further applications. This work opens a new window to develop more efficient heterogeneous Pt-based electrocatalysts for high-performance and low-cost energy conversion and storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFB0105500), the National Natural Science Foundation of China (51432003, 51802011 and 51125007), the Start-Up Fund for Talent Introduction of Beijing University of Chemical Technology (buctrc201806), the Fund of High-Performance Carbon-Based Electrodes for Energy Storage, and the Fundamental Research Funds for the Central Universities (JD1906). The authors acknowledge the beamline 1W1B station of the Beijing Synchrotron Radiation Facility (BSRF) for the XAFS tests.Notes and references
- C. Wang, X. Sang, J. T. L. Gamler, D. P. Chen, R. R. Unocic and S. E. Skrabalak, Nano Lett., 2017, 17, 5526–5532 CrossRef CAS.
- C. Li, H. Tan, J. Lin, X. Luo, S. Wang, J. You, Y. M. Kang, Y. Bando, Y. Yamauchi and J. Kim, Nano Today, 2018, 21, 91–105 CrossRef CAS.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D. J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- W. Gao, Z. Zhang, M. Dou and F. Wang, ACS Catal., 2019, 9, 3278–3288 CrossRef CAS.
- A. Kumar and V. Ramani, ACS Catal., 2014, 4, 1516–1525 CrossRef CAS.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS.
- C. Su, T. Yang, W. Zhou, W. Wang, X. Xu and Z. Shao, J. Mater. Chem. A, 2016, 4, 4516–4524 RSC.
- M. C. Tsai, T. T. Nguyen, N. G. Akalework, C. J. Pan, J. Rick, Y. F. Liao, W. N. Su and B. J. Hwang, ACS Catal., 2016, 6, 6551–6559 CrossRef CAS.
- Y. Qin, L. Chao, J. J. He, Y. Liu, F. Chu, J. Cao, Y. Kong and Y. Tao, Power Sources, 2016, 335, 31–37 CrossRef CAS.
- X. X. Wang, S. Hwang, Y. T. Pan, K. Chen, Y. He, S. Karakalos, H. Zhang, J. S. Spendelow, D. Su and G. Wu, Nano Lett., 2018, 18, 4163–4171 CrossRef CAS.
- B. C. Hu, Z. Y. Wu, S. Q. Chu, H. W. Zhu, H. W. Liang, J. Zhang and S. H. Yu, Energy Environ. Sci., 2018, 11, 2208–2215 RSC.
- Q. Lai, L. Zheng, Y. Liang, J. He, J. Zhao and J. Chen, ACS Catal., 2017, 7, 1655–1663 CrossRef CAS.
- Z. Zhang, Y. Qin, M. Dou, J. Ji and F. Wang, Nano Energy, 2016, 30, 426–433 CrossRef CAS.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C. j. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard III, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS.
- Z. Zhang, M. Dou, H. Liu, L. Dai and F. Wang, Small, 2016, 12, 4193–4199 CrossRef CAS.
- Z. Wen, S. Ci and J. Li, J. Phys. Chem. C, 2009, 113, 13482–13487 CrossRef CAS.
- J. Zeng, C. Francia, C. Gerbaldi, V. Baglio, S. Specchia, A. S. Aricò and P. Spinelli, Electrochim. Acta, 2013, 94, 80–91 CrossRef CAS.
- T. L. Hsieh, H. W. Chen, C. W. Kung, C. C. Wang, R. Vittal and K. C. Ho, J. Mater. Chem., 2012, 22, 5550 RSC.
- S. Bai, S. Qi, Y. Feng, L. Bu and X. Huang, Small, 2017, 13, 1604311 CrossRef.
- X. Cheng, Y. Li, L. Zheng, Y. Yan, Y. Zhang, G. Chen, S. Sun and J. Zhang, Energy Environ. Sci., 2017, 10, 2450–2458 RSC.
- H. Mistry, F. Behafarid, E. Zhou, L. K. Ono, L. Zhang and B. R. Cuenya, ACS Catal., 2014, 4, 109–115 CrossRef CAS.
- C. D. Douglas, A. V. Dias and D. B. Zamble, Dalton Trans., 2012, 41, 7876–7878 RSC.
- P. Marcus and C. Hinnen, Surf. Sci., 1997, 392, 134–142 CrossRef CAS.
- M. Nadeem, G. Yasin, M. H. Bhatti, M. Mehmood, M. Arif and L. Dai, J. Power Sources, 2018, 402, 34–42 CrossRef CAS.
- Y. Wu, Y. Zhao, J. Liu and F. Wang, J. Mater. Chem. A, 2018, 6, 10700–10709 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supporting tables and figures. See DOI: 10.1039/c9cy01654f |
| This journal is © The Royal Society of Chemistry 2020 |