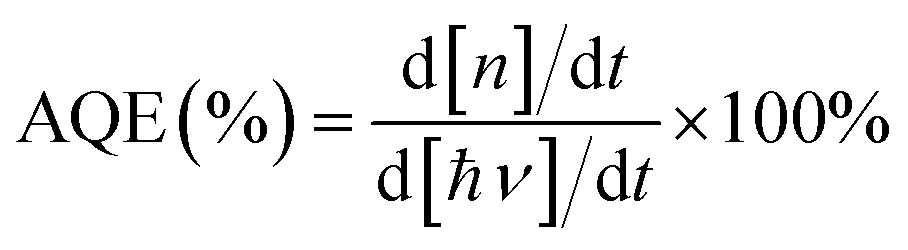Ultra-small subnano TiOx clusters as excellent cocatalysts for the photocatalytic degradation of tetracycline on plasmonic Ag/AgCl†
Received
17th September 2019
, Accepted 29th October 2019
First published on 30th October 2019
Abstract
The separation efficiency and recombination rate of photogenerated electron–hole pairs play a vital role in photocatalytic activity. We report the incorporation of ultra-small subnano TiOx clusters as outstanding cocatalysts on the surface of plasmonic Ag/AgCl to boost the simulated sunlight photoreactivity. The formed structure is in favour of an efficient photoinduced electron–hole transfer process and suppressing the recombination rate, yielding an enhanced photocatalytic activity. The experimental results show that TiOx@Ag/AgCl exhibits an extraordinarily high activity in the photodegradation of tetracycline (TC) under simulated sunlight irradiation, which is about 21.7 times higher than that of bare Ag/AgCl. Their corresponding apparent quantum efficiency (AQE) is up to 0.917% at 435 nm and 17.459% at 350 nm during the photodegradation of TC, respectively. Moreover, TiOx@Ag/AgCl shows an outstanding long-term stability without obvious performance loss even after ten cycles of photodegradation. The possible enhancement mechanism of the reaction rate is proposed based on the compositional, structural, and optical property analysis. We found that h+ was the major reactive species in the photodegradation process of TC. Our findings on ultra-small TiOx cocatalysts would shed light on the design and development of highly efficient oxide cluster cocatalysts.
Introduction
In the field of photocatalysis, a tremendous number of catalysts have been investigated to solve the energy crisis and environmental contamination by CO2 reduction,1,2 H2 evolution,3,4 de-NOx and degradation of organic pollutants.5,6 However, some catalysts have limited applications owing to their low reactive efficiency and vulnerability. Recently, Ag/AgX (X = Cl, Br, I) has attracted enormous attention in the design of composite photocatalysts for their efficient absorption of visible light by the surface plasmon resonance (SPR) of Ag NPs. The surface plasmon resonance of Ag can be combined with the high photosensitivity of Ag halides.7,8 Wang et al. synthesized Ag/AgCl@chiral TiO2 nanofibers and an enhanced surface plasmon resonance (SPR) effect and improved photocatalytic activity were observed.9 Yu et al. synthesized hollow octahedral AgCl/TiO2 composites and the excellent photodegradation performance for MO was identified due to their unique hollow octahedral structure.10 Unfortunately, halide and sulfide based photocatalysts usually have weak stability and suffer from photodecomposition under light irradiation.11,12 There is still a lack of an effective strategy to achieve efficient and stable halide based photocatalysts in an ambient environment.
Cocatalysts play crucial roles in semiconductor photocatalysts, as suitable cocatalyst loading could promote the photocatalytic process. In general, the reasons for the promotion of catalytic reactions via cocatalyst loading mainly involve the following three aspects: (i) the cocatalyst is capable of facilitating the separation of electron–hole pairs at the interface between the cocatalyst and semiconductor; (ii) the presence of cocatalysts on semiconductors suppresses photo-corrosion and improves the photostability of the catalyst by consuming photogenerated charge carriers in time; (iii) the cocatalyst can reduce the activation energy of the reaction. Currently, most of the developed photocatalytic systems mainly improve the photocatalytic activity through noble metal-based and noble metal oxide catalysts. It has been reported that highly efficient cocatalysts, such as Au, Pd, Pt, RuO2, PtOx, and IrO2, are used to catalyse photocatalytic water splitting, photodegradation of pollutants and CO2 reduction.13–18 However, the above noble metal based cocatalysts cannot be industrially applied on a large scale due to their high prices and scarce reserves. Therefore, it is necessary to develop earth-abundant and low-cost element-based cocatalysts with high efficiency and anti-photocorrosion ability. Recently, transition metal, metal oxide and hydroxide based novel cocatalyst systems with high performance photocatalytic activity have been extensively reported.19–24 For example, quantum Cu(II) nanodot loaded TiO2 nanosheets exhibited an H2 evolution rate up to 25-fold higher than that of bare TiO2; the CuO cocatalyst enhanced the photoactivity of Ag/AgCl/TiO2 for the photodegradation performance for methyl orange and phenol under visible light. Amorphous transition metal borides were used as cocatalysts for nanostructured NiCoB/CdS composites for hydrogen evolution, and the H2 production rate was 36 times greater than that of bare CdS alone. Typical transition metal sulfide cocatalysts were employed with g-C3N4 for photocatalytic hydrogen evolution, and the maximum photocatalytic hydrogen production rate was as high as 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 μmol g−1 h−1, which was about 2500 times higher than that of bare g-C3N4. Cu(OH)2 and Ni(OH)2 clusters for the first time were utilized as highly efficient cocatalysts for improving the photocatalytic activity of various semiconductors, e.g. TiO2, CdS and g-C3N4. However, as far as we know, there is no report on ultra-small subnano titanium based metal oxide co-catalysts.
400 μmol g−1 h−1, which was about 2500 times higher than that of bare g-C3N4. Cu(OH)2 and Ni(OH)2 clusters for the first time were utilized as highly efficient cocatalysts for improving the photocatalytic activity of various semiconductors, e.g. TiO2, CdS and g-C3N4. However, as far as we know, there is no report on ultra-small subnano titanium based metal oxide co-catalysts.
Herein, ultra-small subnano titanium oxide (TiOx) clusters are utilized for the first time as novel non-noble metal cocatalysts to decorate plasmonic Ag/AgCl. The TiOx@Ag/AgCl composites exhibit an extraordinarily high activity in the degradation of tetracycline under simulated sunlight irradiation with outstanding long-term stability. We find that the ultra-small subnano TiOx clusters play key roles in the photocatalytic system and we propose a plausible mechanism for this enhancement. Our findings on ultra-small TiOx cocatalysts may have potential applications in environmental purification and sustainable energy development.
Experimental
Synthesis
General procedure for the synthesis of compound Ag/AgCl.
For the synthesis of Ag/AgCl, 776 μL HCl was added into 10 ml deionized water, in which 0.9 g AgNO3 was dissolved with stirring for half an hour. The precipitates were washed with ethanol three times and finally dried in a vacuum oven at 60 °C for 8 h.
General procedure for the synthesis of compound TiOx@Ag/AgCl.
For the synthesis of TiOx solution, we followed our previous procedure.25 236 μL TiCl4 was added dropwise into 30 ml EG under stirring until the initial solution changed to clear and transparent (solution A). After that, 26 mL of solution A (0.3 g) and 0.9 g AgNO3 were added into 54 ml EG with stirring for half an hour. The precipitates were washed with ethanol three times and finally dried in a vacuum oven at 60 °C for 8 h.
General procedure for the synthesis of compound X-TiOx@Ag/AgCl.
For the synthesis of various concentrations of TiOx@Ag/AgCl, 26 ml TiOx solution (0.3 g) and 0.9 g AgNO3 were dissolved separately into 774 ml, 134 ml, and 0.4 ml EG with stirring for half an hour to synthesize 0.1TiOx@Ag/AgCl, 0.5TiOx@Ag/AgCl, and 3TiOx@Ag/AgCl. 1.18 ml TiCl4 was added dropwise in 30 ml EG under stirring until the initial solution change to clear and transparent. Then, 5.25 ml 5TiOx solution (0.3 g) and 0.9 g AgNO3 were dissolved into 11 ml EG to synthesize 5TiOx@Ag/AgCl. All the samples were washed with ethanol three times and finally dried in a vacuum oven at 60 °C for 8 h.
Material characterization.
Powder X-ray diffraction (XRD) analysis was carried out on a PANalytical X'pert apparatus at 40 kV and 40 mA with monochromatic Cu Kα radiation. The particle sizes and surface morphology were observed with a field-emission scanning electron microscope (FEI Quanta 650) and spherical aberration corrected electron microscopy (FEI, Titan Themis). UV-vis diffuse reflectance spectra were collected using a UV-vis spectrophotometer (UV3600, Shimadzu) (the absorption spectra were referenced to BaSO4); photoluminescence (PL) measurements were carried out on a RF-6000 Shimadzu instrument; FTIR spectroscopy was conducted on a Thermo Nicolet IS-50 spectrophotometer. X-ray absorption fine structure spectra (Ti K-edge) were collected at the 1W1B station in Beijing Synchrotron Radiation Facility (BSRF, operated at 2.5 GeV with a maximum current of 250 mA); total organic carbon (TOC) was measured on a TOC-4200 (Shimadzu). Electron spin resonance (ESR) spectra were obtained on a JES FA200 spectrometer (JEOL, Japan). LC-MS was performed by using an LCMS-8060.
Photocatalytic experiment.
The photocatalytic performance of the catalysts was evaluated by the photocatalytic degradation of tetracycline (TC) under simulated sunlight irradiation with an intensity of 100 mW cm−2. Simulated sunlight irradiation was achieved using a 300 W xenon lamp with an AM 1.5G filter. Typically, 0.11 g of the photocatalyst was added into 20 mL of 40 mg L−1 TC aqueous solution. The suspension was magnetically stirred in the dark for 30 min to establish adsorption equilibrium. For cycling performance tests, the samples were collected by centrifugation after the latest cycle and re-utilized on the next degradation cycle test.
Method of calculating AQE.
The apparent quantum efficiency of photodegradation was calculated using the following equation:
where d[n]/dt is the number of photodegraded molecules per unit time; d[ℏν]/dt is the number of the incident photons per unit time.
Results and discussion
Materials characterization
The XRD patterns of the as-prepared TiOx@Ag/AgCl and TiOx@Ag/AgCl across ten cycling runs are shown in Fig. S1.† The as-prepared TiOx@Ag/AgCl exhibits four main peaks at 28°, 32.4°, 46.2° and 57.6°, which can be indexed to the cubic AgCl (111), (200), (220) and (222) crystal planes, respectively (JCPDF 31-1238). Compared with fresh TiOx@Ag/AgCl, the peaks located at 38.2°, 44.4° and 64.5° of TiOx@Ag/AgCl after ten consecutive cyclic degradations are assigned to metallic Ag (JCPDF 01-1167), indicating that Ag NPs were produced via the photoreduction of Ag+ ions on the surface of AgCl. All phase structures of the used samples are similar to that of the fresh sample, suggesting that AgCl is relatively stable during the degradation of organic pollutants. As for TiOx@Ag/AgCl loaded with different concentrations of TiOx and pristine Ag/AgCl (Fig. S1a†), there are no obvious peak shifts and other impurity phases (e.g. anatase TiO2), and the peak positions of all the samples are consistent without deviation.
The SEM and TEM images reveal the morphology and crystalline properties of the as-prepared samples (Fig. 1). Fig. 1a shows the typical low magnification SEM image of fresh TiOx@Ag/AgCl, which reveals that AgCl has a spherical structure of about 1 μm in diameter. Compared with the fresh sample, the overall structure of the used TiOx@Ag/AgCl (Fig. 1b) did not change significantly after cycling, indicating the stability of TiOx@Ag/AgCl during degradation. Nevertheless, some particles appear on the surfaces of the used TiOx@Ag/AgCl, indicating that Ag/AgCl decomposed into Ag nanoparticles under electron beam irradiation. The high angle annular dark field (HAADF) image in Fig. 1d shows that TiOx@Ag/AgCl has a diameter of around 500 nm. The shape and size distribution of Ag NPs on the AgCl surface are quite different. Therefore, the SPR effect of Ag NPs would span a wide wavelength range rather than occurring at a specific wavelength, allowing TiOx@Ag/AgCl to absorb visible light over a wide range. In addition, the high resolution TEM image (Fig. 1c) shows that the lattice spacing of 0.24 nm corresponds to the (111) plane of Ag.26 The energy dispersive X-ray (EDX) elemental mapping images (Fig. 1e) confirm the existence of Ag, Cl and Ti in TiOx@Ag/AgCl. The EDX quantitative analysis results (Table S1†) show the mass fraction (wt%) of Ti, Ag and Cl in TiOx@Ag/AgCl.
 |
| | Fig. 1 (a) SEM images of fresh TiOx@Ag/AgCl and (b) the used sample TiOx@Ag/AgCl after 10 cycles; (c) HAADF image of TiOx@Ag/AgCl; (d) HRTEM image of TiOx@Ag/AgCl; (e) HAADF-STEM-EDX elemental mapping images of TiOx@Ag/AgCl. | |
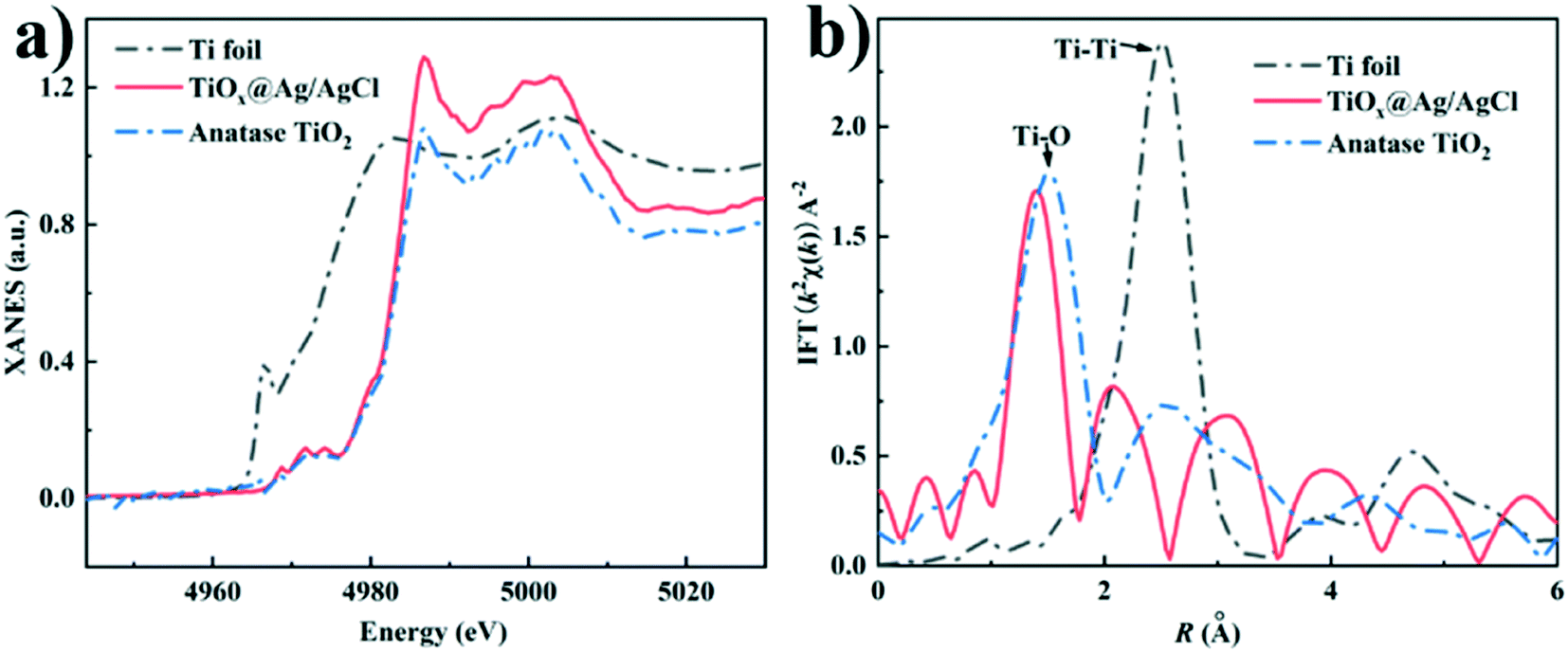
To further clarify the chemical state and coordination environment of the Ti atoms in TiOx@Ag/AgCl, Ti K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) characterizations were carried out (Fig. 2). The K edge white line (WL) intensities increase in the order of Ti foil, TiOx@Ag/AgCl and anatase TiO2, indicating that the valence state of Ti in TiOx@Ag/AgCl is between zero and positive tetravalence (Fig. 2a). The evolution of different peaks in the FT EXAFS spectra (Fig. 2b) indicates varying titanium coordination environments: Ti–O bonding located at 1.3 Å–1.5 Å and Ti–Ti bonding located at 2.56 Å. The Ti foil exhibits a strong peak at 2.56 Å, which is assigned to direct Ti–Ti bonding. In anatase, the main peak at around 1.3 Å indicates Ti–O bonding. In contrast, there is dominant Ti–O bonding in TiOx@Ag/AgCl with no obvious Ti–Ti bonding, indicating the absence of Ti metal clusters. Combined with the EDX mapping results, there was no obvious Ti signal gathering region exceeding one nanometre. We have ample reasons to infer that the Ti element in TiOx@Ag/AgCl exists in the form of ultra-small subnano clusters TiOx.
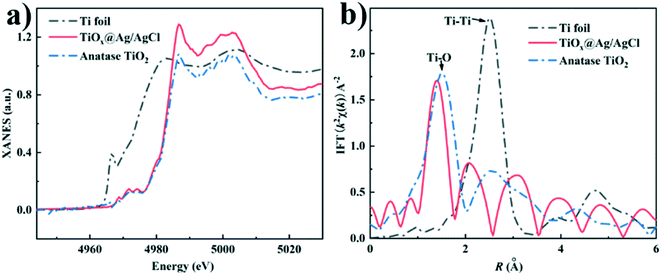 |
| | Fig. 2 (a) Ti K-edge XANES spectra of TiOx@Ag/AgCl, anatase TiO2 and Ti foil; (b) Fourier transform EXAFS spectra of TiOx@Ag/AgCl, anatase TiO2 and Ti foil. | |
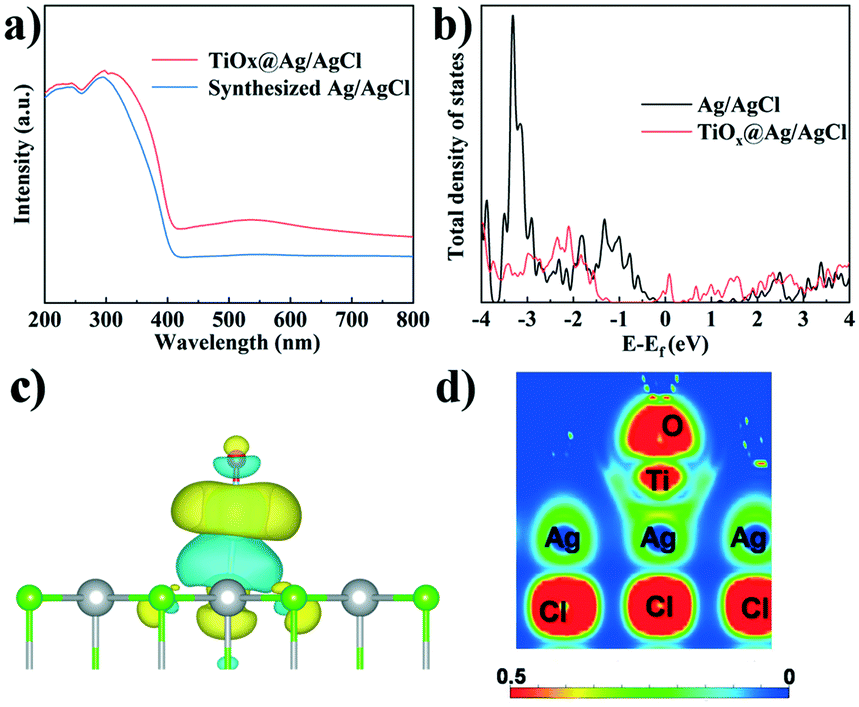
The UV-vis spectra of TiOx@Ag/AgCl and synthesized Ag/AgCl are shown in Fig. 3a, which show a change in the SPR signal of Ag NPs and different absorption onsets. The UV-vis absorption spectra red shifted and the full spectrum light absorption ability showed prominent enhancement when TiOx was loaded on Ag/AgCl. To further investigate the effect of ultra-small subnano TiOx cluster modification, the PL spectra of TiOx@Ag/AgCl were collected. The PL spectra reveal charge carrier behaviours, such as the migration, transfer, and recombination rates of the photogenerated electron–hole pairs in semiconductors. Fig. S2† shows the PL spectra of TiOx@Ag/AgCl and synthesized Ag/AgCl. The emission spectra are in the range of 360–520 nm, and the location of the emission peaks is similar but the intensities are different. The fluorescence intensity of TiOx@Ag/AgCl is significantly lower than that of synthesized Ag/AgCl, indicating that the introduction of TiOx inhibits the recombination of electrons and holes.
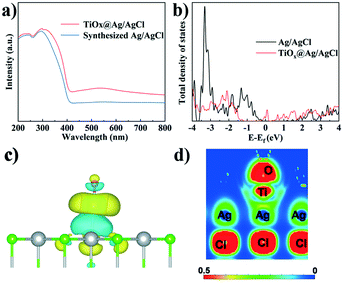 |
| | Fig. 3 (a) UV-vis absorption spectra; (b) DOS of TiOx@Ag/AgCl and Ag/AgCl; 3D (c) and 2D (d) charge density distributions of TiOx@Ag/AgCl. | |
Fig. S3† shows the FTIR spectra of Ag/AgCl and TiOx@Ag/AgCl. The bands at 3462 cm−1, 1624 cm−1, 1384 cm−1 and 1075 cm−1 were observed in synthetic Ag/AgCl, which are attributed to O–H stretching, O–H bending, C–H characteristic vibration of alkyl halide groups and C–O stretching, respectively.27,28 As TiOx is loaded on the surface of AgCl, the O–H stretching band shifts to 3445 cm−1 and the O–H bending blue-shifts to 1635 cm−1, indicating that the twin hydroxyl groups in the sample act as a stabilizer for Ag NPs and AgCl.
In this work, the calculated total density of states (Fig. 3b) shows that additional states are created in the gap region (∼0 eV) of AgCl upon the decoration with TiOx, rationalizing the experimentally observed red shift of the UV-vis absorption spectra. Moreover, the charge density difference (Fig. 3c) shows remarkable charge redistribution at the interface of TiOx/AgCl, indicating strong bonding between TiOx and AgCl. Accordingly, the photoinduced electrons can be efficiently transferred via the Ti–Ag bonds, thus facilitating the separation of photogenerated electron–hole pairs. The EIS result of TiOx@Ag/AgCl shows that the impedance becomes smaller when TiOx is supported on Ag/AgCl (Fig. S4†), suggesting the efficient interfacial charge transfer. These results are in accordance with the DFT simulation results.
Photocatalytic characterization
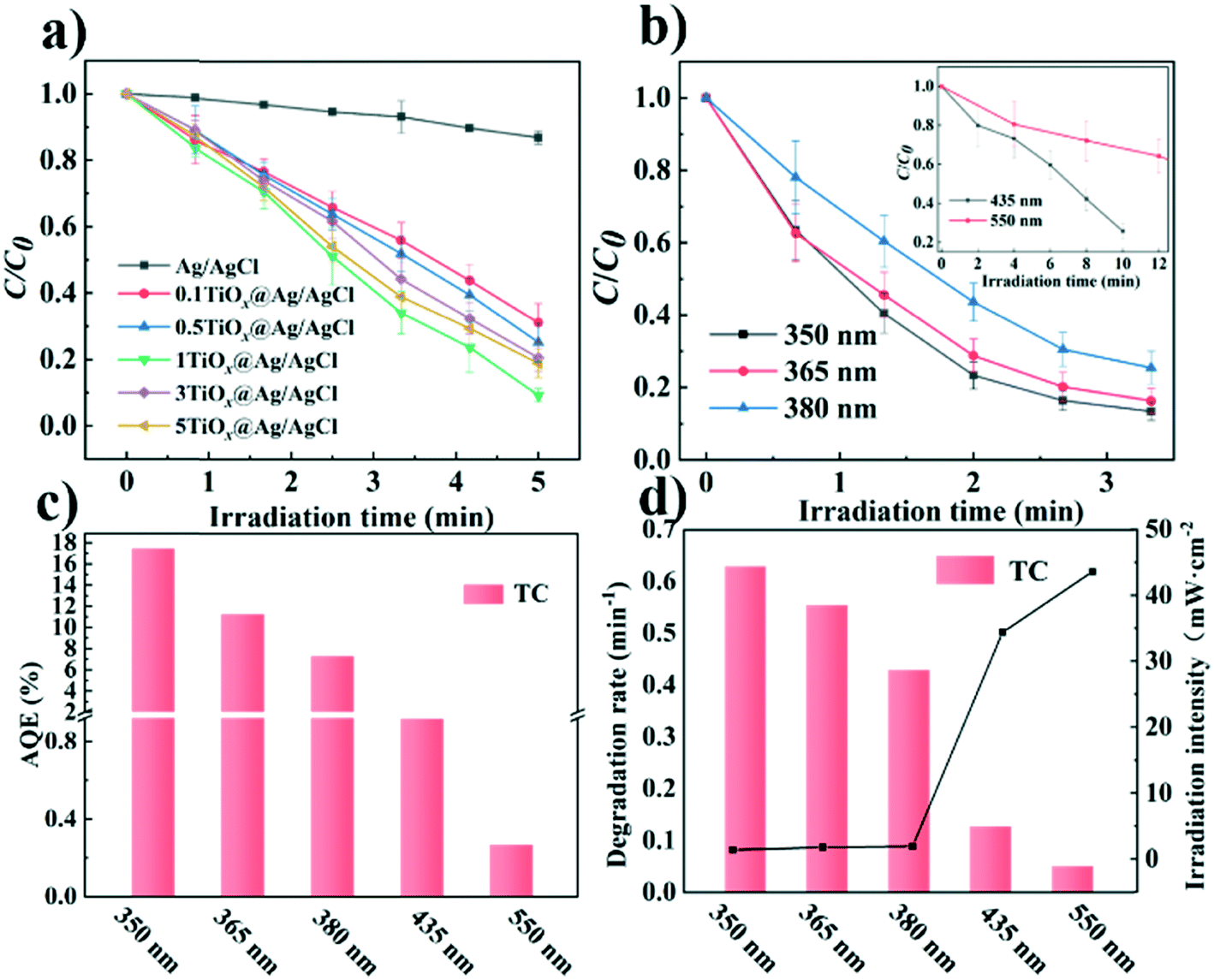
The photocatalytic performance of the catalysts was evaluated by the photocatalytic degradation of TC under simulated sunlight irradiation (Fig. 4). The photocatalyst was mixed with a TC solution and kept in the dark before being exposed to light irradiation to reach adsorption–dissociation equilibrium. The temporal evolution of the absorption spectra of the aqueous solutions of TC in the presence of TiOx@Ag/AgCl under simulated sunlight irradiation is shown in Fig. S5.†Fig. 4a shows the photocatalytic activity of X-TiOx@Ag/AgCl (X = 0.1, 0.5, 1, 3, 5) and Ag/AgCl under simulated sunlight irradiation for the degradation of TC. The degradation by Ag/AgCl, as the blank test, shows slight degradation in the absence of TiOx loading. When the Ti concentration increases to 0.1, the photoactivity is enhanced notably. Furthermore, we investigated the optimal Ti loading, with the Ti concentration of up to 1. The photocatalyst showed the fastest degradation rate within 5 min with TC removal rate exceeding 80%. All the TiOx@Ag/AgCl samples show better photocatalytic performance than Ag/AgCl. Compared with various TiOx@Ag/AgCl, TC was degraded almost completely within 5 min while Ag/AgCl degraded only 13%. To investigate the role of the wavelength of incident light in a wide range across ultraviolet and visible light, we studied the photocatalytic performance for the degradation of TC under 350, 365, 380, 435, and 550 nm wavelength light irradiation. TiOx@Ag/AgCl demonstrates a remarkable degradation rate for the photo-removal of TC under ultraviolet irradiation, and the pollutant was almost degraded within 5 min. Even when the incident light wavelength increased to 435 nm, the degradation time is less than 20 min (Fig. 4b). The dependence of the TC adsorption efficiency on TiOx@Ag/AgCl in the dark is shown in Fig. S6.†Fig. 4c shows the apparent quantum efficiency (AQE) of TC degradation via TiOx@Ag/AgCl as the photocatalyst under various wavelengths of incident light irradiation. The state-of-the-art photocatalyst shows an unparalleled advantage in terms of TC photodegradation AQE, which is almost one order of magnitude higher than others. As shown in Fig. 4d, TiOx@Ag/AgCl exhibits high reaction rate constants of above 0.4 min−1 for TC degradation in the UV region and above 0.05 min−1 under 435 nm irradiation, which has surpassed most of the plasmonic photocatalysts reported previously (Table S2†).
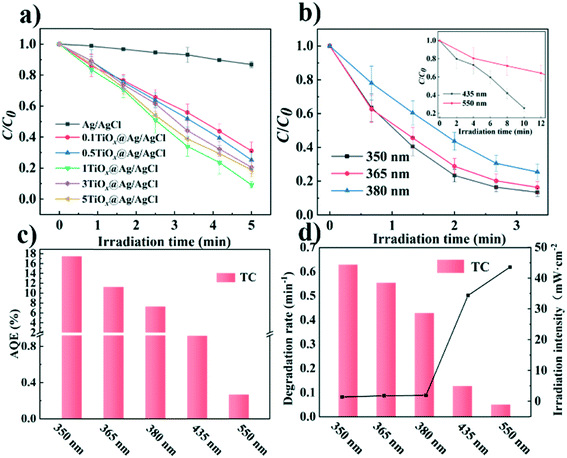 |
| | Fig. 4 (a) Photocatalytic degradation of TC by various materials; (b) photocatalytic degradation of TC under light irradiation of different wavelengths; (c) apparent quantum efficiency (AQE) of TiOx@Ag/AgCl under light irradiation of different wavelengths; (d) the corresponding reaction rate constant k of TiOx@Ag/AgCl under light irradiation of different wavelengths. | |
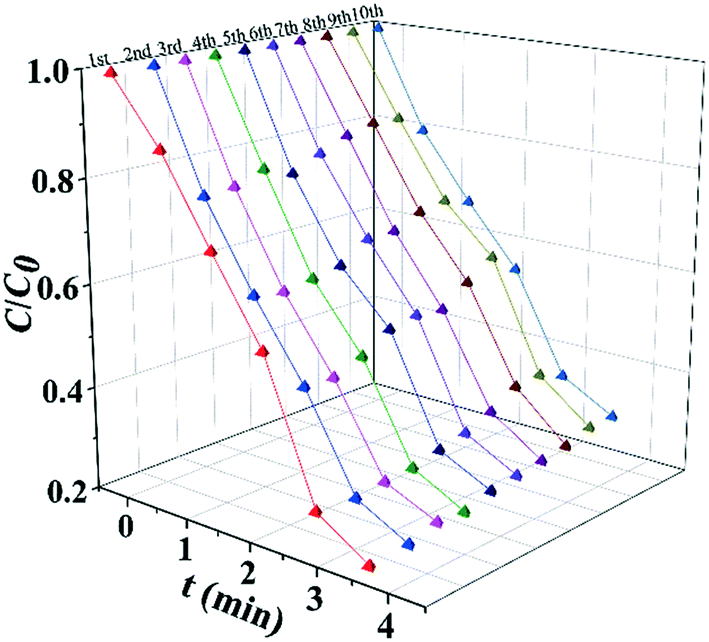
The results of long term stability photodegradation tests over TC are shown in Fig. 5. TiOx@Ag/AgCl exhibits outstanding long term stability even after 10 cycles of TC degradation without obvious performance loss, which has surpassed a number of AgX (X = Cl, Br, or I) based plasmonic photocatalysts. The XRD and SEM results also reveal that TiOx@Ag/AgCl remained stable during the degradation of TC. Furthermore, we collected the HAADF images of TiOx@Ag/AgCl under different times of electron beam irradiation to investigate the in situ dynamic process of Ag NP growth (Fig. S7†). It can be seen that Ag NPs gradually precipitated from the TiOx@Ag/AgCl surface under electron beam irradiation, and Ag NPs grew gradually with the extension of irradiation time.
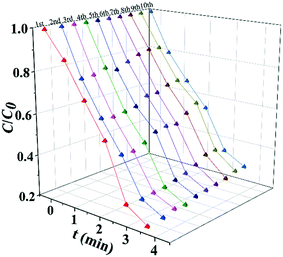 |
| | Fig. 5 Photocatalytic degradation cycling runs for photocatalytic TC degradation in the presence of TiOx@Ag/AgCl. | |
In order to better understand the mechanism of the photodegradation of TC over TiOx@Ag/AgCl under visible light irradiation, we studied the photodegradation process in the presence of different scavengers. We used TEA as an h+ scavenger, TBA as an ˙OH scavenger, p-benzoquinone as an ˙O2− scavenger, and KBrO3 as an e− scavenger.29–34 The scavengers were added before the solution was irradiated. As shown in Fig. S8,† significant inhibition of the photocatalytic effect after the addition of scavenger TEA (0.065 mL) is observed, which suggests that the photogenerated holes (h+) are the major reactive species in our study. The effect of ˙O2− was evaluated using p-benzoquinone (10 mg). The moderate degradation rate suggests that ˙O2− plays an auxiliary role in the photodegradation process. We used TBA (1.25 mL) to remove ˙OH. A small decrease in the photodegration of MO indicates that ˙OH plays a minor role in the overall degradation in our study. The effect of e− was confirmed using KBrO3 (1 mmol). A small increase in the photodegration of TC indicates that removing e− promotes the degradation process. In addition, the control experiments show practically no degradation of TC, suggesting that TiOx@Ag/AgCl is responsible for the photodegradation. In order to further confirm the type of free radicals, the ESR signals of the radicals were spin-trapped by 5,5-dimethyl-1-pyrroline N-oxide (DMPO). The results of ESR are shown in Fig. S9,† which reveals that the samples have no ESR signals of the radicals in the dark. The ESR signal peaks of ˙O2− and ˙OH appeared after being irradiated by simulated sunlight for 6 min. Therefore, we believe that the sample produces ˙O2− and ˙OH under light irradiation.
Total organic carbon (TOC) was considered as a criterion for determining whether the photodegradation was carried out thoroughly. TOC tests were performed every 1 min and the results are shown as Fig. 6. Approximately 58.8% of TOC was removed after 4 min of irradiation with a Xe lamp loaded with an AM1.5 filter. The result shows that most of the TOC has been degraded into small molecules (Fig. S10†) and TC has been finally mineralized. The possible pathways of the degradation of TC were proposed in Fig. S11.†
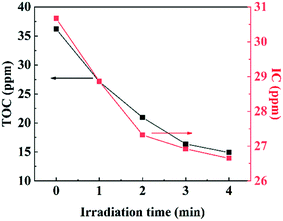 |
| | Fig. 6 The IC and TOC concentrations during tetracycline photodegradation. | |
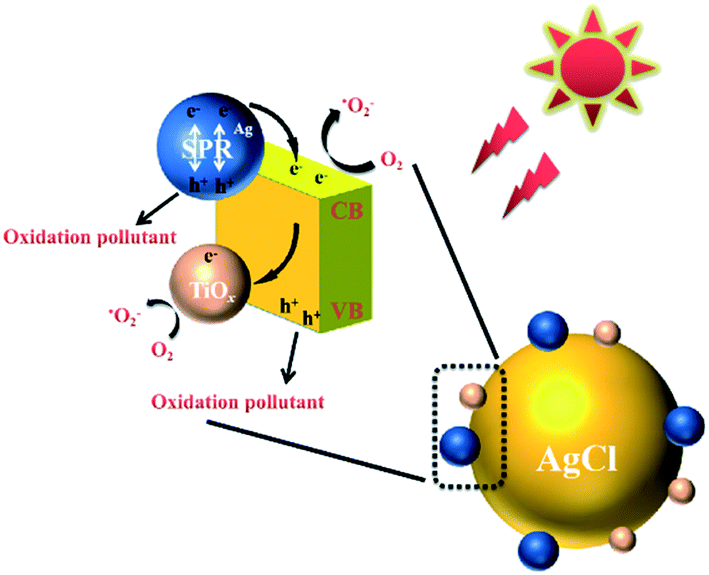
The proposed reaction pathway for the degradation of TC with the TiOx@Ag/AgCl composite under simulated sunlight irradiation is shown in Fig. 7. The Ag NPs that grow on the surface of TiOx@Ag/AgCl exhibit the SPR effect so that the photocatalyst will have a broad extinction band range to the visible spectrum. Even under visible light irradiation, Ag NPs can concentrate the photons into a tiny volume and transfer the irradiative energy to AgCl; in other words, lots of “hot electrons” will flow from Ag NPs to AgCl. At the same time, when the wavelength of incident light is short enough, the electrons of the valence band are transferred to the conduction band and the holes remain in the valence band. There is strong bonding between TiOx and AgCl; therefore, the photoinduced electrons can be efficiently transferred via the Ti–Ag bonds, thus facilitating the separation of photogenerated electron/hole pairs. This distinctive phenomenon is in favour of the charge transformation and suppression of the charge recombination rate. According to the above discussion, the following reactions could be proposed for the removal of TC:
| | | Ag + hν → hot (e− + h+) | (1) |
| | | Hot e− (Ag) → hot e− (AgCl) | (2) |
| | | TiOx@Ag/AgCl + hν → TiOx@Ag/AgCl (e− + h+) | (3) |
| | | TC + h+ → Deg. Product (Dominant) | (6) |
| | | TC + ˙O2− → Deg. Product (Major) | (7) |
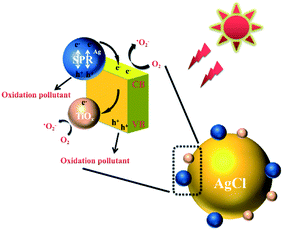 |
| | Fig. 7 Schematic diagram of the photocatalytic degradation of TC in the presence of TiOx@Ag/AgCl. | |
Conclusions
In summary, for the first time, we have prepared a high-stability and high-activity ultra-small subnano TiOx cluster loaded Ag/AgCl composite. The SPR effect excited by Ag NPs broadens the absorption range to the visible region, leading to the excellent properties for the degradation of TC. In addition, it is found that the introduction of TiOx effectively accelerates the separation of electron–hole pairs. This study provides a new strategy for designing advanced photocatalysts.
Author contributions
X. Zhou, X. Han, C. Wang, K. Wang and W. He conceived and designed this work. W. He synthesized the samples and wrote the first draft of the manuscript. K. Wang and X. Han obtained the TEM images and EXAFS results. Z. Zhu helped with the synthesis of samples. Y. Feng, H. Zou and K. Zhou analyzed the XRD data. Z. Hu and K. Lv carried out the density functional theory calculations. Y. Duan helped with electrochemical testing. X. Zhou, X. Han, C. Wang, L. Gan, K. Wang and W. He analysed the results and revised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is financially supported in part by the National Natural Science Foundation of China (Grant No. 11344010, 11404044, 51472036, and 51772035) and the Fundamental Research Funds for the Central Universities (CQDXWL-2013-Z010, 106112017CDJQJ308821 and 2018CDJDWL0011).
Notes and references
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef.
- B. Yu, Y. Zhou, P. Li, W. Tu, P. Li, L. Tang, J. Ye and Z. Zou, Nanoscale, 2016, 8, 11870–11874 RSC.
- L. Guo, Z. Yang, K. Marcus, Z. Li, B. Luo, L. Zhou, X. Wang, Y. Du and Y. Yang, Energy Environ. Sci., 2018, 11, 106–114 RSC.
- H. Yu, X. Huang, P. Wang and J. Yu, J. Phys. Chem. C, 2016, 120, 3722–3730 CrossRef CAS.
- Z. Hu, K. Li, X. Wu, N. Wang, X. Li, Q. Li, L. Li and K. Lv, Appl. Catal., B, 2019, 256, 117860 CrossRef CAS.
- M. Liu, R. Inde, M. Nishikawa, X. Qiu, D. Atarashi, E. Sakai, Y. Nosaka, K. Hashimoto and M. Miyauchi, ACS Nano, 2014, 877229–877238 Search PubMed.
- Y. Gao, J. Lin, Q. Zhang, H. Yu, F. Ding, B. Xu, Y. Sun and Z. Xu, Appl. Catal., B, 2018, 224, 586–593 CrossRef CAS.
- A. Meng, L. Zhang, B. Cheng and J. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 5581–5589 CrossRef CAS PubMed.
- D. Wang, Y. Li, G. Puma, C. Wang, P. Wang, W. Zhang and Q. Wang, Chem. Commun., 2013, 49, 10367–10369 RSC.
- X. Wang, J. Yu, C. Fu, T. Li and H. Yu, Appl. Surf. Sci., 2019, 494, 740–748 CrossRef CAS.
- C. Zhang, H. Hua, J. Liu, X. Han, Q. Liu, Z. Wei, C. Shao and C. Hu, Nano-Micro Lett., 2017, 9, 49 CrossRef PubMed.
- X. Liang, P. Wang, M. Li, Q. Zhang, Z. Wang, Y. Dai, X. Zhang, Y. Liu, M. Whangbo and B. Huang, Appl. Catal., B, 2018, 220, 356–361 CrossRef CAS.
- J. Cai, X. Wu, S. Li and F. Zheng, Appl. Catal., B, 2017, 201, 12–21 CrossRef CAS.
- Q. Wang, K. Wang, L. Zhang, H. Wang and W. Wang, Appl. Surf. Sci., 2019, 470, 832–839 CrossRef CAS.
- S. Cao, Y. Li, B. Zhu, M. Jaroniec and J. Yu, J. Catal., 2017, 349, 208–217 CrossRef CAS.
- L. Li, Z. Deng, L. Yu, Z. Lin, W. Wang and G. Yang, Nano Energy, 2016, 27, 103–113 CrossRef CAS.
- B. Cao, G. Li and H. Li, Appl. Catal., B, 2016, 194, 42–49 CrossRef CAS.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543–2546 RSC.
- J. Wang, Z. Wang and Z. Zhu, Appl. Catal., B, 2017, 204, 577–583 CrossRef CAS.
- G. Bi, J. Wen, X. Li, W. Liu, J. Xie, Y. Fang and W. Zhang, RSC Adv., 2016, 6, 31497–31506 RSC.
- M. Zhang, R. Sun, Y. Li, Q. Shi, L. Xie, J. Chen, X. Xu, H. Shi and W. Zhao, J. Phys. Chem. C, 2016, 120, 10746–10756 CrossRef CAS.
- Z. H. Shah, J. Wang, Y. Ge, C. Wang, W. Mao, S. Zhang and R. Lu, J. Mater. Chem. A, 2015, 3, 3568–3575 RSC.
- J. Yu and J. Ran, Energy Environ. Sci., 2011, 4, 1364 RSC.
- H. Zhao, H. Zhang, G. Cui, Y. Dong, G. Wang, P. Jiang, X. Wu and N. Zhao, Appl. Catal., B, 2018, 225, 284–290 CrossRef CAS.
- C. Wang, A. Li, C. Li, S. Zhang, H. Li, L. Hu, Y. Feng, K. Wang, Z. Zhu, R. Shao, Y. Chen, P. Gao, S. Mao, J. Huang, Z. Zhang and X. Han, Adv. Mater., 2019, 1903491 CrossRef.
- Q. Liu, C. Zeng, L. Ai, Z. Hao and J. Jiang, Appl. Catal., B, 2018, 224, 38–45 CrossRef CAS.
- X. Wang, Z. Jia, F. Liu, H. Liang, X. You, K. Wang, X. Lou, W. Shuang, L. Xiao, B. Cai and L. Yang, RSC Adv., 2016, 6, 48985–48994 RSC.
- D. Ashok Kumar, V. Palanichamy and S. M. Roopan, J. Photochem. Photobiol., B, 2014, 138, 302–306 CrossRef CAS.
- W. Jones, D. J. Martin, A. Caravaca, A. M. Beale, M. Bowker, T. Maschmeyer, G. Hartley and A. Masters, Appl. Catal., B, 2019, 240, 373–379 CrossRef CAS.
- W. Peng, Y. Lin, Z. Wan, H. Ji, W. Ma and J. Zhao, Catal. Today, 2018, 11, 038 Search PubMed.
- J. Fu, G. Z. Kyzas, Z. Cai, E. A. Deliyanni, W. Liu and D. Zhao, Chem. Eng. J., 2018, 335, 290–300 CrossRef CAS.
- A. Al-Asfar, Z. Zaheer and E. S. Aazam, J. Photochem. Photobiol., B, 2018, 185, 143–152 CrossRef CAS.
- S. J. Armaković, S. Armaković, N. L. Finčur, F. Šibul, D. Vione, J. P. Šetrajčić and B. F. Abramović, RSC Adv., 2015, 5, 54589–54604 RSC.
- R. Scotti, M. D'Arienzo, F. Morazzoni and I. R. Bellobono, Appl. Catal., B, 2009, 88, 323–330 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S11. See DOI: 10.1039/c9cy01876j |
| ‡ These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Kangle
Lv
a,
Kangle
Lv