
Engineering an alcohol dehydrogenase with enhanced activity and stereoselectivity toward diaryl ketones: reduction of steric hindrance and change of the stereocontrol element†
Kai
Wu
ab,
Zhijun
Yang
ab,
Xiangguo
Meng
ab,
Rong
Chen
a,
Jiankun
Huang
a and
Lei
Shao
 *bc
*bc
aSchool of Pharmacy, Shanghai University of Medicine & Health Sciences, 279 Zhouzhu Highway, Pudong New Area, Shanghai 201318, China
bMicrobial Pharmacology Laboratory, Shanghai University of Medicine & Health Sciences, 279 Zhouzhu Highway, Pudong New Area, Shanghai 201318, China. E-mail: shaolei00@gmail.com
cState Key Laboratory of New Drug and Pharmaceutical Process, Shanghai Institute of Pharmaceutical Industry, 285 Gebaini Rd., Shanghai 200040, China
First published on 21st January 2020
Abstract
Steric hindrance in the binding pocket of an alcohol dehydrogenase (ADH) has a great impact on its activity and stereoselectivity simultaneously. Due to the subtle structural difference between two bulky phenyl substituents, the asymmetric synthesis of diaryl alcohols by bioreduction of diaryl ketones is often hindered by the low activity and stereoselectivity of ADHs. To engineer an ADH with practical properties and to investigate the molecular mechanism behind the asymmetric biocatalysis of diaryl ketones, we engineered an ADH from Lactobacillus kefiri (LkADH) to asymmetrically catalyse the reduction of 4-chlorodiphenylketones (CPPK), which are not catalysed by the wild type (WT) enzyme. Mutants seq1–seq5 with gradually increased activity and stereoselectivity were obtained through iterative “shrinking mutagenesis.” The final mutant seq5 (Y190P/I144V/L199V/E145C/M206F) demonstrated the highest activity and excellent stereoselectivity of >99% ee. Molecular simulation analyses revealed that mutations may enhance the activity by eliminating steric hindrance, inducing a more open binding loop and constructing more noncovalent interactions. The pro-R pose of CPPK with a halogen bond formed a pre-reaction conformation more easily than the pro-S pose, resulting in the high ee of (R)-CPPO in seq5. Moreover, different halogen bonds formed due to the different positions of chlorine substituents, resulting in opposite substrate binding orientation and stereoselectivity. Therefore, the stereoselectivity of seq5 was inverted toward ortho- rather than para-chlorine substituted ketones. These results indicate that the stereocontrol element of LkADH was changed to recognise diaryl ketones after steric hindrance was eliminated. This study provides novel insights into the role of steric hindrance and noncovalent bonds in the determination of the activity and stereoselectivity of enzymes, and presents an approach producing key intermediates of chiral drugs with practical potential.
Introduction
Alcohol dehydrogenases (ADHs, EC 1.1.1.1) constitute a large family of NAD(P)(H)-dependent oxidoreductases. Multiple ADHs efficiently catalyse asymmetric bioreduction for chiral pharmaceutical intermediate production with great industrial potential.1 Biocatalyst activity and stereoselectivity are two key properties. Based on previous studies involving stereocontrol mechanisms,2,3 the stereoselectivity of ADHs originating from the preference of the specific binding orientation of prochiral ketones and steric hindrance in binding pockets plays an important role in determining the binding orientation of substrates. The prochiral ketones bearing substituents differ in size and bind to small and large binding pockets of ADHs. Therefore, ADHs generally demonstrate high activity and stereoselectivity toward bulky–small ketones. However, when an ADH catalyses diaryl ketones bearing two bulky substituents with similar phenyl groups, its low activity and stereoselectivity are often observed.4,5Diaryl ketones can be asymmetrically reduced to their corresponding chiral alcohol. Optically pure diaryl alcohols are important pharmaceutical intermediates.6 Among these alcohols, (R)-(4-chlorophenyl)(phenyl)methanol (CPPO) is the critical intermediate in the production of laevorotatory cloperastine, which can be prepared through the symmetric reduction of diaryl ketone CPPK. Levocloperastine is faster acting and causes greater reduction in the intensity and frequency of coughing of recipients compared with racemic cloperastine.7 The chemical synthesis of chiral diaryl alcohols often involves expensive Ru catalysts or resolving agents.8 Therefore, the alternative strategy for production is bio-reduction by ADHs, which is considered the more ecologically and economically viable route for synthesis.
Due to the important role of steric hindrance in chiral recognition and activity, from the view of rational design, it can be inferred that the elimination of steric hindrance in the binding pocket has a double effect of providing improved ligand–protein interactions with the enhancement of activity toward bulky CPPK, and potentially low stereoselectivity, unless a different stereocontrol element takes over. In this study, we aimed to create an ADH variant with enhanced activity and stereoselectivity toward CPPK without property trade-offs. The molecular basis of an ADH from Lactobacillus kefiri DSM 20587 (LkADH), which has a resolved crystal structure,2 was selected for engineering. Naturally occurring LkADH shows a preference for the reduction of bulky–small ketones as opposed to bulky–bulky ketones. It is difficult for the phenyl group of CPPK to bind to a small pocket, which results in an inactive binding conformation. Reduction using LkADH wild-type (WT) only produced a conversion of less than 0.1%. This result further suggests that steric hindrance prevents optimal substrate interactions. Moreover, the subtle differences between the two phenyl groups of CPPK result in difficulty in chiral recognition for LkADH. In this study, we constructed focused libraries consisting of limited amino acids with sites selected in or around the catalytic pocket to screen variants with enhanced activity and stereoselectivity. A 5-point mutant (seq5) with reduced steric hindrance and dramatically increased properties was obtained. The molecular docking and dynamic simulations of the WT and mutants further revealed the origin of enhanced activity and stereoselectivity. Moreover, huge differences in chiral recognition between the WT and seq5 were discovered, which indicated that the stereocontrol element can be converted in the engineered ADH.
Experimental section
Materials
Ketones, racemic sec-alcohol standards, and NADPH used in this study are commercially available. The conversion ratio and enantiomeric excess after bioreduction were analysed using chiral high-performance liquid chromatography (HPLC; Chiralcel OB-H/AD-H/OD-H column [250 × 4.6 mm]). The KOD-Plus-neo DNA polymerase was obtained from TOYOBO CO., Ltd. Restriction enzyme Dpn I was procured from Thermo Scientific. Oligonucleotide synthesis and DNA sequencing were conducted by GENEWIZ. The TIANprep Mini plasmid kit used for plasmid extraction was procured from Tiangen Biotech.Mutant construction, expression, and screening
Active pocket iterative mutagenesis is based on the information regarding the protein active pocket structure, with iterative cycles of mutagenesis being performed at rationally chosen sites using smaller glycine, alanine, cysteine, serine, proline, and valine residues to replace the original residues. The full-length LkADH gene was expressed using pET-21b, and the recombinant plasmid was used as the PCR template for “shrinking mutagenesis.” The obtained PCR products were digested using DpnI to remove parent plasmids. The digested PCR products were transformed into Escherichia coli DH5α cells. Plasmids containing the mutant gene were then extracted and retransformed into E. coli BL21 (DE3) cells for mutant enzyme expression.The gene encoding LkADH (accession number: AY267012) from Lactobacillus kefiri DSM 20587 was expressed using pET-21b. A single transformant was cultured at 37 °C for 12 h and was then transferred to 100 mL fresh Luria–Bertani (LB) medium and cultured at 37 °C. The culture was induced by adding 0.1 mM isopropyl β-D-1-thiogalactopyranoside when its optical density at 600 nm reached 0.6. After induction at 20 °C for 20 h, resting cells were harvested by centrifuging at 8500g for 10 min and were then resuspended in 100 mM Tris-HCl (pH 7.6) with a cell concentration of 80 mg·mL−1.
Resting cells expressing mutants were used as biocatalysts in the reaction of CPPK to determine the conversion and ee. The screening reaction mixture (500 μL) included 200 μL of suspended resting cells expressing mutants, 25 μL of 5 mM NADP stock, and 50 μL of 200 mM CPPK dissolved in isopropanol. The reaction mixture was incubated at 30 °C with rotation at 200 rpm for 30 min. The reaction was terminated by adding 1 mL isopropanol and incubating the mixture at 65 °C for 10 min. The samples were analysed after filtering by HPLC to determine the conversion ratio and ee.
Purification of LkADH and mutants
For purifying WT and seq1–5, the respective genes flanked by NdeI and HindIII sites were expressed using pET-28a with an N-terminal His-tag. Resting cells expressing mutant LkADH were disrupted by sonication, and cell debris/inclusion bodies were removed by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C for 15 min. A soluble cell-free extract was loaded onto a nickel column that was pre-equilibrated with 50 mM Tris-HCl (pH 7.6) binding buffer. The bound recombinant enzyme was eluted using a binding buffer containing increasing concentrations of imidazole (20–200 mM), according to a standard protocol. The purified enzyme was obtained using 200 mM imidazole. The purified enzyme was further used to determine its specific activity and stereoselectivity.
000g at 4 °C for 15 min. A soluble cell-free extract was loaded onto a nickel column that was pre-equilibrated with 50 mM Tris-HCl (pH 7.6) binding buffer. The bound recombinant enzyme was eluted using a binding buffer containing increasing concentrations of imidazole (20–200 mM), according to a standard protocol. The purified enzyme was obtained using 200 mM imidazole. The purified enzyme was further used to determine its specific activity and stereoselectivity.
Measurement of the activity and ee of WT and seq5
Specific activities of WT and seq5 were spectrophotometrically assayed by measuring the change in NADPH absorbance at 340 nm for 1 min. The reaction system contained the appropriate weights of the purified enzymes, 0.625 mM NADPH, 5 mM substrates, and 100 mM Tris-HCl (pH 7.6) buffer, at a final reaction volume of 200 μl. The kinetic parameters of WT and seq5 to CPPK were determined by increasing the substrate concentration from 0.3 to 10 mM at 30 °C. Initial velocities at different substrate concentrations were used to generate a Lineweaver–Burk plot (1/v vs. 1/[S]). The product ee of the corresponding 1a–9a was determined by chiral HPLC. The chiral analysis methods and retention time of R and S enantiomers are listed in Table S3.† The absolute configuration was determined by comparing the retention time with previous reports.Molecular docking and MD simulations
The crystal structure of LkADH was obtained from the Protein Data Bank (PDB ID 4RF2).2 Virtual mutation was conducted using Discovery Studio 4.0. Substrate docking was performed using the Autodock 4.2 program. CPPK, 2-chloro-1-phenylethanone, and 4-chloro-1-phenylethanone were docked into the substrate-binding pocket of the LkADH–NADPH complex by flexible docking. 3D-coordinates (mol2 files) of substrates were downloaded from the ZINC database. AutoDock tools (ADT, version 1.5.6) were used for enzyme and substrate preparation. To encompass the entire substrate-binding pocket, the docking box was set to a size of 50 × 50 × 50 grid points with a grid spacing of 0.375 Å. The box centre was set as X = 21.557, Y = −29.525, and Z = 26.307. To obtain a reasonable initial conformation for MD, residues S143, Y156, V144, C145, L195, V196, V199, F206, and Y249 of seq5 were set as flexible. The obtained conformations, in which the carbonyl oxygen had hydrogen bond interactions with Tyr156 and the distance between carbonyl carbon and NADPH-C4 atoms was less than 4.5 Å, were considered reasonable. The reasonable conformation with the lowest binding energy was selected for further analysis. The ligand interaction was analysed using the receptor–ligand interactions tool of Discovery Studio V4.0 (Biovia, USA).MD simulation was performed using the particle mesh Ewald molecular dynamics module implemented in the Amber 18 suite,9 with the ff14SB force field used for the protein system, and the GAFF force field used for the ligands.10 The NADPH parameters were obtained from the AMBER parameter database.11 The ANTECHAMBER module and Gaussian 09 were used to calculate the CPPK RESP atom charges. Hydrogen atoms and sodium ions (to neutralise the negative charges) were added to the protein using the tleap utility. Each simulation system was immersed in a cube of TIP3P explicit water, extending to 12 Å outside the protein on all sides. Water molecules were treated using the SHAKE algorithm, and the long-range electrostatic effects were considered using the particle mesh Ewald method. The binary complex of protein–NADPH without the substrate being bound, the ternary complex obtained from the molecular docking of seq1–NADPH–CPPK, and complexes with the pro-R/S docking poses of the seq5–NADPH–CPPK were treated as follows. The water molecules and ions were relaxed to minimise the energy during the 10000 minimisation steps with the protein and ligands restrained. The backbone of the protein was restrained with the other section relaxed to minimise the energy. The whole system was then minimised without the restraints during the 10000 minimisation steps. After energy minimisation, the system was gradually heated in the NVT ensemble from 0 to 303 K over 200 ps. This procedure was followed by 200 ps of NPT simulation at 303 K and 1 atm pressure using the Langevin dynamics algorithm with the complex constrained. Equilibration for 200 ps was performed with the complex constrained. All the positional constraints in energy minimisation and equilibration used a force constant of 2.0 kcal mol−1 Å−2. Three independent productive simulations of WT, seq1, and seq5 were performed without any restraints for 200 ns. A time-step of 2.0 fs was used, and coordinates of the system were saved every 2 ps during production. The pocket volumes were calculated using the POVME2 program.12 The WT, seq1, and seq5 used the same parameters to describe the binding pocket using POVME 2.0. Based on the algorithm of this program, the grid spacing was set as 0.5 for better accuracy. The centre of the point inclusion sphere was set as X = 45, Y = 42, and Z = 27 and the radius was 10 Å. The centre of the point exclusion sphere was set as X = 50, Y = 44, and Z = 38 and the radius was 6 Å. The default distance cutoff value was used. The convex-hull-clipping option was set as “True” to increase the accuracy. The centre of the contiguous pocket seed sphere was set as X = 45, Y = 42, and Z = 26 and the radius was 7 Å. The contiguous points criteria was set as 3.
To calculate the proportion of the pre-reaction conformations of the pro-S and pro-R poses, each pose obtained from the docking result underwent 4 ns of MD simulation with ten replicas. Distances were calculated with the cpptraj module. The plot of the probability density distribution of the distances of C4NADPH–C1CPPK and OHTyr156–O1CPPK was calculated using 2D kernel density estimation using OriginPro 2018. The binding free energies of CPPK in seq5 and seq1 were calculated from 1000 frames extracted from ten independent 4 ns simulations with stable root-mean-square deviation, using the generalised Born surface area method implemented using the MMPBSA.py script of Amber 18.13
Gram-scale preparation of (R)-CPPO and (S)-CPMO
We scaled-up the production (200 mL) of (R)-CPPO and (S)-CPMO to determine the practical application of the engineered enzyme seq5. The resting cells expressing seq5 were used for the scale-up preparation. The recombinant glucose dehydrogenase (GDH) from Bacillus subtilis CGMCC 1.1398 and glucose were used for NADPH recycling. The pH was automatically adjusted to 7.6 by titration with 2.0 M Na2CO3 solution. The bioreduction of CPPK was initiated at 30 °C by adding 17.3 g of CPPK dissolved in 60 mL methyl tert-butyl ether to 140 mL of 100 mM Tris-HCl (pH 7.6), which contained 0.25 mM NADP, 17.5 g glucose, 16 g resting cells expressing seq5, and 3.2 g resting cells expressing GDH. The preparation of (S)-CPMO was initiated at 30 °C by adding 17.4 g of 5a dissolved in 20 mL methanol to 180 mL of 100 mM Tris-HCl (pH 7.6), which contained 0.25 mM NADP, 16.4 g glucose, 10 g resting cells expressing seq5, and 3.2 g resting cells expressing GDH. The conversion ratio was monitored using HPLC and the reaction mixture was extracted with ethyl acetate after reaction completion.Results and discussion
Construction and screening of the mutagenesis library
CPPK is a typical bulky–bulky ketone, which is flanked by two phenyl rings. The two bulky phenyls may prevent binding near catalytic residues. The catalysis of LkADH involves a highly conserved triad consisting of Ser143–Tyr156–Lys160.2 Ser143 stabilises the substrate. Tyr156 acts as a general acid/base initiating catalysis by transferring its proton to the carbonyl group of the substrate. NADPH transfers hydride to carbonyl carbon atoms. Therefore, the distance between the substrate carbonyl oxygen and Tyr156-OH and the distance between the carbonyl carbon and NADPH-C4 are critical for catalysis. A rigid docking experiment was designed to explore the binding conformation of this inactive substrate. Large distances (distance OHTyr156–O1CPPK = 12.1 Å and C4NADPH–C1CPPK = 13.4 Å, respectively) were observed in the docking conformation, indicating steric hindrance by the side chains within the binding pocket preventing CPPK access and catalysis. Based on this analysis, the primary objective of our engineering project was to create a binding pocket permissive for CPPK to facilitate catalysis.Based on the structural information of the binding pocket and catalytic mechanism, we selected five residues around the catalytic triad as the target for “shrinking mutagenesis” with an iterative strategy aimed at the expansion of the original small binding pocket (Fig. S1†). Based on the CPPK hydrophobicity (logP = 3.57), van der Waals volumes, and charges of residues, the bulkier residues with large volumes (Tyr190/Ile144/Leu199/Glu145/Leu195) were selected for mutagenesis of aliphatic residues (Ala/Pro/Val/Gly) and polar residues (Ser/Cys) with smaller volumes. Basic and acidic amino acids were excluded because of their hydrophilicity. The van der Waals volumes of 20 residues are listed in Table S1.† Based on the crystal structure of LkADH, Leu199, Glu145, and Leu195 were in different α-helixes, and the Tyr190 was located in a loop region. Based on previous reports, Pro is the worst helix former due to its lack of an amide hydrogen for main chain hydrogen bonding and because of its unique geometry.14 Mutations to Pro at sites Leu199, Glu145, and Leu195 could destabilize the α-helix structure and result in inactive variants. However, proline is frequently found in turn and loop structures of proteins.15 Therefore, only the site in the loop region (Tyr190) was mutated into Pro, and the sites in the α-helixes (Leu199, Glu145, and Leu195) were excluded.
Iterative mutagenesis is a powerful approach for semi-rational design.16 With the structural information on the binding pocket, the sequence space could be reduced by selecting limited residues in or near the binding pocket. Beneficial mutations with additive and synergistic effects could be achieved using several rounds of mutagenesis.
Tyr190 was the residue with the largest volume (141 Å3) in the binding pocket. Therefore, it may be the key residue preventing the binding of the large groups in the small binding pocket. This site was targeted first as changes to this site may result in higher activity. Using resting cells expressing mutants, all six mutants at sites Y190 produced measurable CPPO. Among them, Y190G, Y190A, and Y190C demonstrated a conversion ratio of more than 30%, a dramatic increase when compared with WT. However, these mutants demonstrated low ee values of 3%, 30.5% and 31.3%, respectively. The Y190P (seq1) mutation demonstrated the highest ee of 72.1% with a medium conversion ratio of 20.1%. Based on this, we used Y190P as the template to perform the second cycle of mutagenesis at Ile144. The location of Ile144 was quite close to catalytic residue Ser143, which may be sensitive to residue change. Therefore, only similar small aliphatic mutations of Val, Ala, and Gly were constructed. Y190P/I144V (seq2) demonstrated an increased conversion ratio of 26.1%, and was the only active mutant at site I144. The I144G and I144A lost their activity toward CPPK. Gly is an amino acid that has a single hydrogen atom as its side chain, which results in the smallest VDW volume. However, it has also lost the necessary hydrophobic interactions for CPPK binding. Compared with Ala, Val demonstrated enhanced activity and stereoselectivity. This may be because of its relatively large hydrophobic isopropyl group providing better hydrophobic interactions. In a previous study, Val was also used for manipulating stereoselectivity in an epoxide hydrolase to achieve much better results than using Ala.17 The stereoselectivity of seq2 further increased from 72.1% to 89% ee.
Seq2 was then used as a template to mutate Leu199 obtaining seq3 (Y190P/Y144V/L199V). The conversion of seq3 increased to 31.3%, and ee increased to 94.7%. Using seq3 as a template, Glu145 was selected for mutagenesis, creating seq4 (Y190P/I144V/L199V/E145C). The stereoselectivity of seq4 was more than 99% ee, and the conversion ratio further increased to 33.9%. Using seq4 as a template, all six mutations demonstrated decreased activity at Leu195, indicating that leucine exhibits favourable interaction with CPPK.
To increase the activity of seq4 further, flexible docking analysis was performed. Based on the docking results of seq4 (template used for M206F), the CPPK in the pre-reaction conformation was found to interact with Met206 and Tyr249. Besides, as the CPPK is a hydrophobic compound with two aromatic rings, this indicates that increasing hydrophobic effect and constructing π–π interaction will probably help to stabilize the pre-reaction conformation. Among the amino acids, only Phe, Tyr, and Trp have an aromatic ring that allows them to form π–π interaction with CPPK. Moreover, Phe has the smallest van der Waals volume among the aromatic amino acids. Therefore, it was chosen for the site-directed mutation of M206F to construct π–π ‘T’-shaped interaction and enhance the hydrophobic effect. After virtual mutation, docking results demonstrated that the phenyl ring of CPPK interacted with Tyr249 and Phe 206 by π–π ‘T’-shaped interaction and formed a densely packed hydrophobic region. In addition, based on a previous report, the π–π interactions had been successfully designed to affect transition-state binding for catalysis.18 Therefore, from the view of rational design, site direct mutation of M206F was performed. As expected, seq5 (Y190P/I144V/L199V/E145C/M206F) further increased the conversion to 46% and maintained an ee value of more than 99%.
The conversion ratio and enantiomer excess of all the mutants created using iterative mutagenesis are listed in Table 1. Through five rounds of mutagenesis with minimal screening, we obtained five-point mutant seq5 (Y190P/I144V/L199V/E145C/M206F) with a dramatic enhancement of activity and stereoselectivity.
| Template | Mutation | Conversion ratio (%) | ee (%) | Configuration |
|---|---|---|---|---|
| WT | None | <0.1 | — | — |
| WT | Y190G | 31 | 3 | R |
| Y190A | 35 | 30.5 | R | |
| Y190S | 0.9 | — | R | |
| Y190C | 24.3 | 31.3 | R | |
| Y190P | 20.1 | 72.1 | R | |
| Y190V | 4.32 | 43 | R | |
| Seq1: Y190P | I144G | <0.1 | — | — |
| I144A | <0.1 | — | — | |
| I144V | 26.1 | 89 | R | |
| Seq2: Y190P/I144V | L199A | 4.2 | 90 | R |
| L199S | 4.5 | >99 | R | |
| L199C | 12.3 | 98.5 | R | |
| L199V | 31.3 | 94.7 | R | |
| L199G | <0.1 | — | — | |
| Seq3: Y190P/I144V/L199V | E145G | 2.2 | >99 | R |
| E145A | 27 | 95 | R | |
| E145S | 5.3 | 85.9 | R | |
| E145C | 33.9 | >99 | R | |
| E145V | <0.1 | — | — | |
| Seq4: Y190P/I144V/L199V/E145C | L195G | 8.9 | >99 | R |
| L195A | <0.1 | — | — | |
| L195S | 7.7 | >99 | R | |
| L195C | <0.1 | — | — | |
| L195V | <0.1 | — | — | |
| Seq4: Y190P/I144V/L199V/E145C | M206F | 46 | >99 | R |
Investigation of the origin of activity toward CPPK
To further confirm the gradual enhancement of catalytic efficiency, mutants seq1–5 were purified to allow the determination of the kinetic parameters (Fig. S2†). The parameters are listed in Table 2. The mutation of seq1 produced low but detectable activity compared to WT, indicating that the reduction of hindrance at Tyr190 was critical for increased activity. Seq1 paved the way for further enhancement in activity and stereoselectivity. The final mutant seq5 demonstrated a kcat 246 times higher than seq1. The kinetic characterisation of the variants indicated that the enhancement in catalytic efficiency was predominantly the result of an improvement in kcat. Based on the Michaelis–Menten kinetics, it was reasonable that a lower Km was obtained in seq1 than seq5 because the rate of the reaction step of seq1 is extremely slow relative to the rate of substrate binding. Saturation will thus be reached at fairly low substrate concentrations. In contrast, as the reaction step of seq5 is relatively fast, it will take a large quantity of substrate to reach saturation. An engineered epoxide hydrolase with an active tunnel expanded towards the bulky α-naphthyl glycidyl ether also showed an increased Km value and enhanced kcat compared with WT,19 which was similar to our findings.| Mutant | K m (mM) | V max (μmol min−1 mg−1) | k cat (min−1) |
|---|---|---|---|
| Seq1 (Y190P) | 0.54 | 0.0055 | 0.15 |
| Seq2 (Y190P/I144V) | 1.08 | 0.0426 | 1.2 |
| Seq3 (Y190P/I144V/L199V) | 1.13 | 0.0710 | 2.0 |
| Seq4 (Y190P/I144V/L199V/E145C) | 3.30 | 0.1316 | 3.8 |
| Seq5 (Y190P/I144V/L199V/E145C/M206F) | 3.14 | 1.2811 | 36.9 |
The enhanced activity implied that hindrance in the binding pocket of the WT enzyme was reduced, and that the pocket expanded to accept the bulkier CPPK substrate. To understand the differences in the enzymatic activity, the pocket volumes of WT and seq5 were calculated to be 163.25 Å3 and 203.63 Å3, respectively. The engineered seq5 reduced the steric hindrance and achieved an expanded binding pocket, as expected.
Considering the dynamic motion of the enzyme–NADPH complex, we further calculated the pocket volumes of WT, seq1, and seq5 during three independent MD simulations for 200 ns (in total 600 ns). As illustrated in Fig. 1A, significant differences in volume distribution were found. Pocket volume distribution analysis revealed that seq1 and seq5 exhibit larger average pocket volumes as well as broader distributions. The WT exhibits a smaller average pocket volume and a narrower distribution. However, this difference cannot be solely explained by the replacement of bulky residues with smaller ones. Furthermore, the representative structures of WT, seq1, and seq5 during simulation were overlaid and compared. The comparison of the secondary structure revealed that the substrate-binding loop (residues 190–210) was much more open in seq1 and seq5 than in the WT (Fig. 1B). The distance between Tyr156 and Leu195 also indicated that the proportion of the open state in seq1/seq5 was much more occupied than that in the WT during the simulations (Fig. S3†). The prolonged open state of the pocket induced by mutations may be the main reason for the dramatically increased pocket volume in seq1 and seq5.
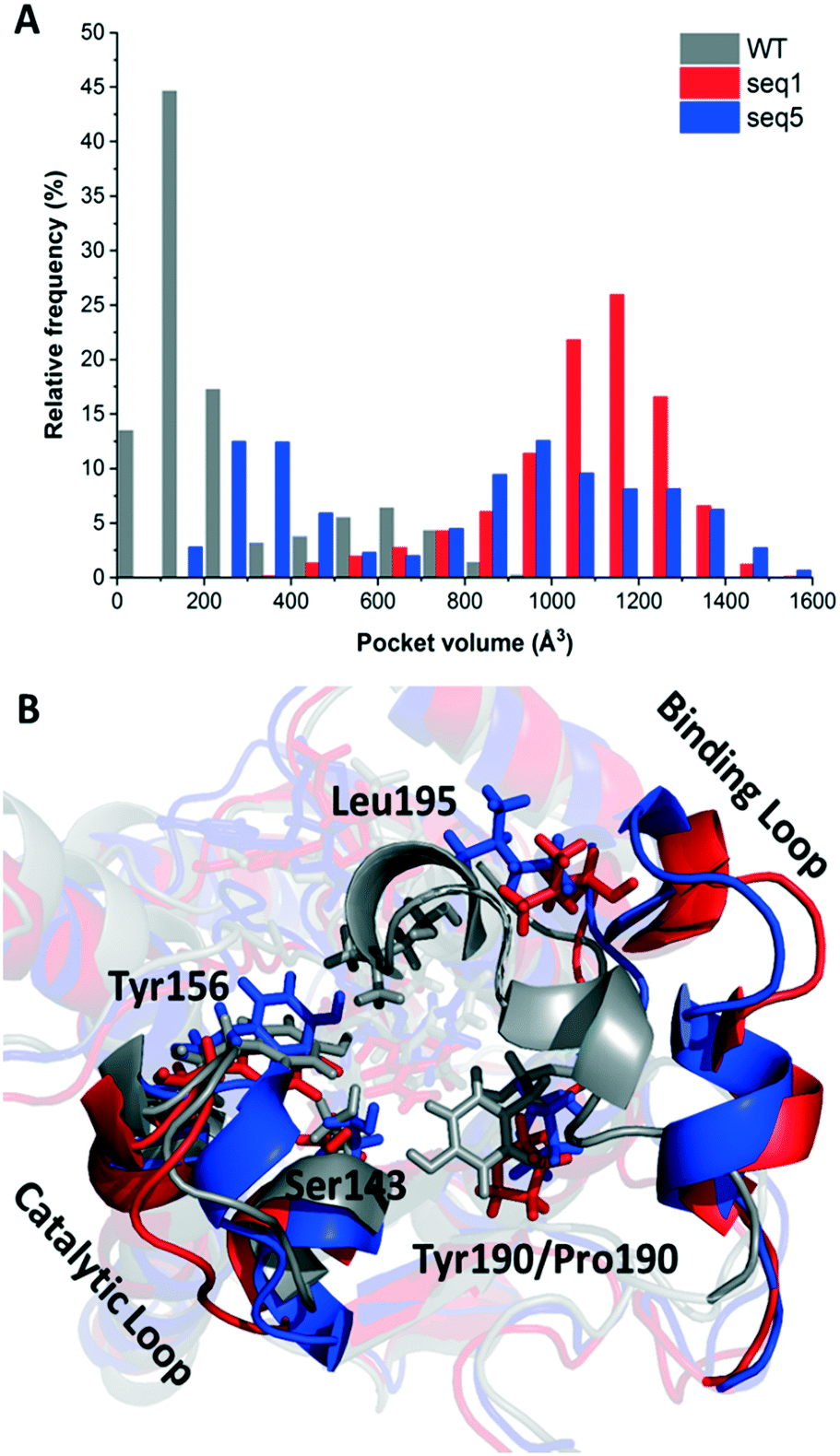 | ||
| Fig. 1 Curves of binding pocket volumes and overlays of representative snapshots for the substrate binding loop. A. Differences in pocket volumes of WT, seq1, and seq5 during simulations. B. Overlay of the representative structures of the binding loop of WT, seq1, and seq5. WT, seq1, and seq5 are coloured grey, red, and blue, respectively. | ||
The one amino acid substitution Y190P of seq1 with a dramatically increased volume suggests that proline was responsible for the prolonged open state of the binding pocket. Proline is often found in the secondary structure of the β-turn and is often used to induce a turn in the loop.20,21 This might be because the cyclic structure of proline gives rotation. The rotation could bend the binding loop with a greater probability of opening rather than closing, which further resulted in a large pocket volume. The different proportions of the closed or opened state of the binding pocket could be manipulated by different mutations. Contrary to mutation Y190P, the previously reported mutation A94F promotes a slightly more closed binding loop because Phe94 exhibits van der Waals interaction with Leu195.2 The mobile loop regions located at the entrance of the active site pocket are often considered as the gates that control the entry and exit of substrates and products, in addition to shielding the active site from the bulk solvent during catalysis.22,23 Therefore, the results above indicate that reduced steric hindrance and a wider entrance for substrate access was obtained in seq1 and seq5.
Although a more open entrance conformation was achieved by seq1 than seq5, the activity of the single point mutant seq1 remained low. The expanded pocket caused by Y190P (seq1) provided basic and essential space for CPPK access, which resulted in detectable activity compared with WT. However, seq1 bound CPPK with relatively poor affinity compared to seq5 (binding free energy calculated to be −25.3 ± 2.9 vs. −18.1 ± 2.3 kcal mol−1). This suggests that the other four mutations contribute significantly to the enhanced affinity and activity in seq5. As illustrated in Fig. 2, the mutations in seq5 contributed to binding through different interactions. I144V/E145C located in the catalytic loop and Y190P/L199V/M206F located in the binding loop form the expanded pocket. They interact with substrates through energy-favouring interactions. In this conformation, two hydrogen bonds between O1CPPK and catalytic Try156/Ser143 are formed. Y190P interacts with CPPK via a π–alkyl interaction. A π–sulphur interaction was observed between E145C and the two phenyl groups. Moreover, the phenyl groups of CPPK interacted with M206F and Tyr249 in π–π stacking with a T shape, as was expected. The π–alkyl interaction observed between the Leu195 and L199V side chains and CPPK may synergistically influence CPPK binding. In addition, the amide oxygen of Leu195 forms a halogen bond with the para-chlorine of CPPK. These interactions favour substrate binding and anchor the phenyl in the expanded pocket, which dramatically increases the activity of LkADH. Therefore, in addition to the more open entrance, the favourable interactions between the substrate and residues that help stabilise substrate binding are also crucial for activity enhancement.
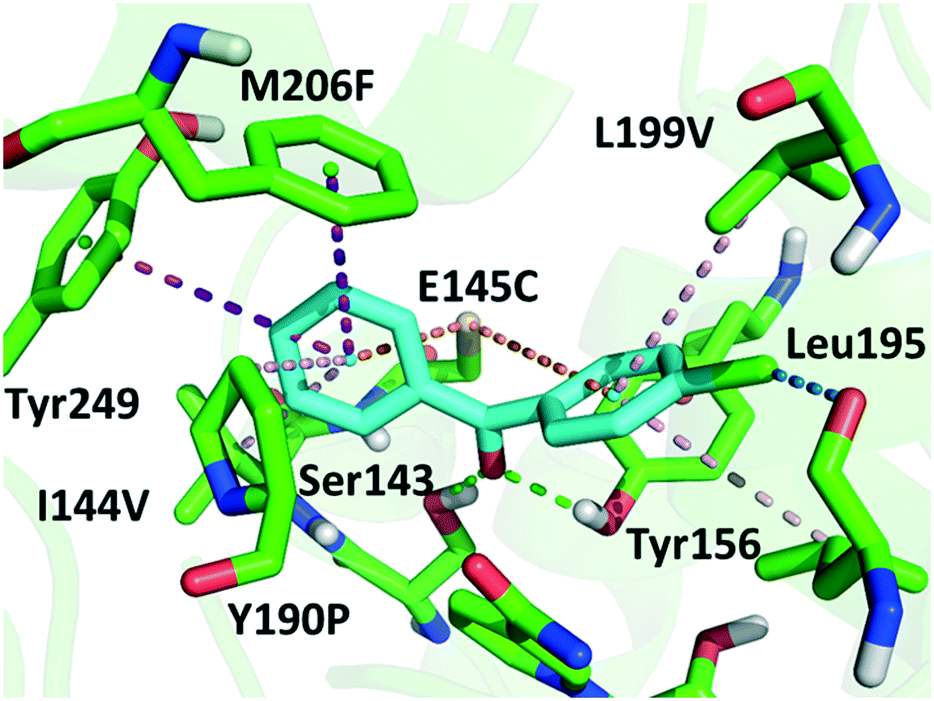 | ||
| Fig. 2 Docking conformation and interactions of CPPK in seq5. Binding mode of CPPK in seq5. The hydrogen bonds are shown in green; π–sulphur interactions in orange; π–π interactions in purple; the π–alkyl interaction in light pink; and the halogen bond in blue. | ||
We further compared the specific activities of the WT and seq5 to nine ketones. The specific activities for bulky–bulky ketones (1a–5a) and bulky–small ketones (6a–9a) are compared in Fig. 3. Compared to the WT, engineered seq5 demonstrated higher activity in all the tested substrates. Substrate 5a demonstrated the highest activity of seq5 among all the tested ketones. Its corresponding chiral alcohol, (S)-(4-chlorophenyl)(pyridin-2-yl)methanol (CPMO), is an important intermediate in the synthesis of the antihistamine drugs bepotastine and carbinoxamine.24 Meanwhile, the WT has no measurable activity toward 1a–5a (specific activity less than 0.001 μmol min−1 mg−1) and was only active toward small–bulky ketones. Furthermore, compared to ketones without substituents, electron-withdrawing chlorine substituent seemed to enhance the activity. Moreover, seq5 demonstrated gradually increasing activities from ortho-chlorine- to para-chlorine-substituted ketones. This indicates the importance of the chlorine position, which may affect substrate binding by noncovalent interaction. The expanded substrate scope further confirms that the engineered binding pocket reduced the hindrance near catalytic residues, which demonstrated a general increase in enzyme activity in different substrates.
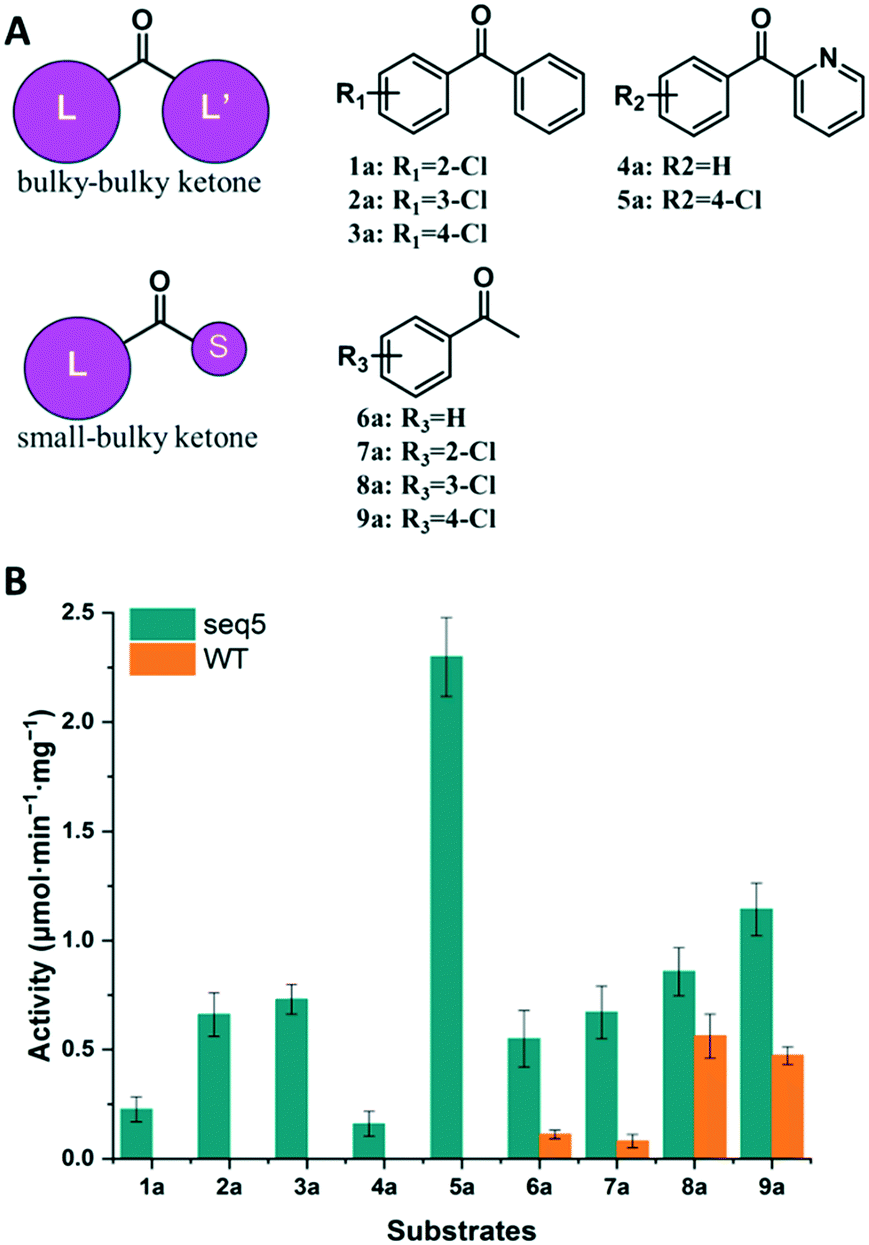 | ||
| Fig. 3 Activity determination of bulky–bulky and bulky–small ketones. (A) Structures of small–bulky and bulky–bulky ketones used in this study. (B) Comparison of the specific activities of seq5 and WT. | ||
Investigation of stereocontrol elements in mutant seq5
Steric hindrance, the determining factor of stereoselectivity in WT, was reduced in seq5. Due to the excellent stereoselectivity toward CPPK, it could infer that a different stereocontrol element played a role in seq5. Based on the Prelog rule of the chiral recognition of ADHs,3,25–28 the stereoselectivity of LkADH was determined by the binding orientation of the substrate (Fig. 4). The subtle structural differences between the two aromatic rings of CPPK render them extremely challenging targets for asymmetric catalysis. Therefore, for the substrate CPPK, in order to transfer hydride from the si-face, the binding pocket should be able to discriminate para-chlorine-substituted phenyl and non-substituted phenyl to prefer pro-R binding, which was key to the high stereoselectivity to CPPK. It is reasonable to presume that para-chlorine affects substrate binding orientation, and thus, stereoselectivity. Therefore, to explore the impact of chlorine substituent, the stereoselectivities of seq5 toward chlorine-substituted ketones with different positions (1a–3a; 7a–9a) and non-substituted ketones (4a and 6a) were determined (Fig. 5).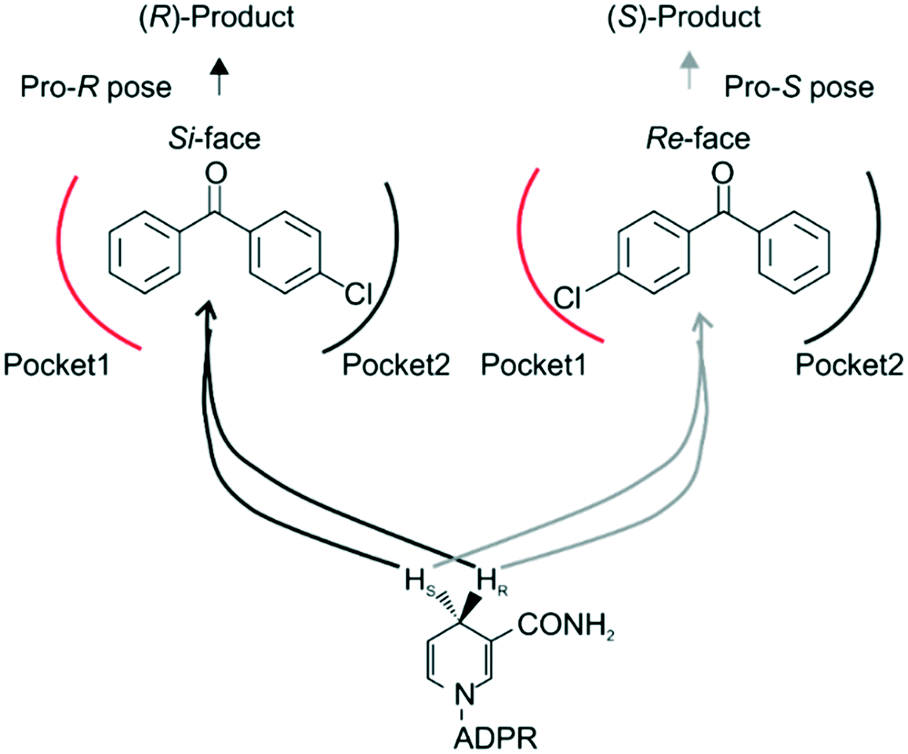 | ||
| Fig. 4 Binding orientation determined the stereoselectivity. ADPR, adenosine diphosphoribose. Based on the Prelog rule for predicting the stereo-preference of alcohol dehydrogenases (ADHs), the hydride from the coenzyme (NADH/NADPH) can attack the carbonyl of an asymmetrical ketone from either the re- or si-face to produce (R)- or (S)-products, respectively. In our study, the CPPK was expected to bind as the pro-R pose. | ||
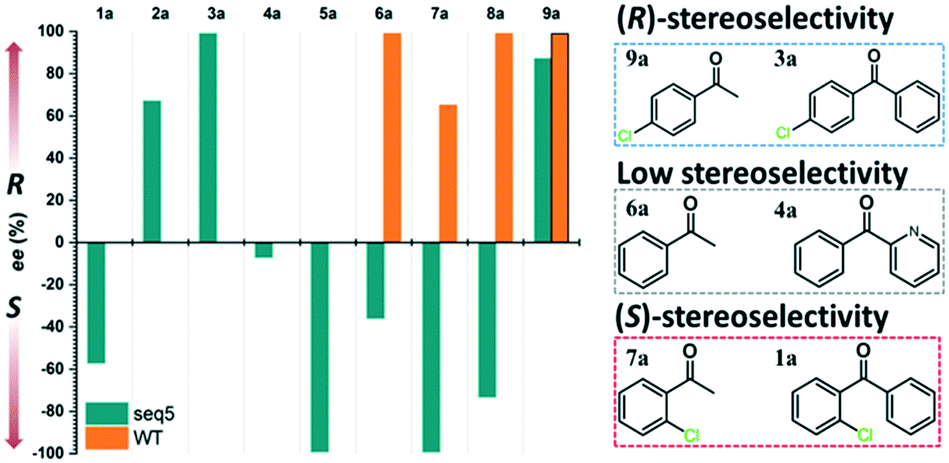 | ||
| Fig. 5 Stereoselectivities of seq5 for different substituted ketones. The chlorine position plays an important role in product stereo configuration. | ||
All the bulky–small ketones were reduced in the R enantiomer by WT, and three of them exhibited a relatively high ee of >99%. 2-Chloro-1-phenylethanone (10a) exhibited a relatively low ee of 65%. These results indicate that WT was an anti-Prelog-selective ADH that produced (R)-enantiomers, which is consistent with a previous report.29 The small and large binding pockets differ in size in the WT, which fixes the substrate-binding orientation. As a result, the substituents on the bulky group have almost no impact on stereoselectivity, as steric hindrance mainly determines the stereoselectivity.
Surprisingly, contrary to the strict anti-Prelog-selectivity of the WT, the stereoselectivity of seq5 was inverted toward some substrates, demonstrating an opposite stereo-preference. Seq5 was highly anti-Prelog selective for para-chlorine-substituted ketones, such as 3a (CPPK), 5a, and 9a, all producing (R)-enantiomers, except 5a. 5a was reduced to an (S)-enantiomer due to the presence of a pyridine group instead of phenyl, which is higher than a large phenyl group according to the Cahn–Ingold–Prelog (CIP) rule. However, the stereoselectivities inverted to Prelog selective when 2-chlorodiphenylketone (1a), 2-chloro-1-phenylethanone (7a), and 3-chloro-1-phenylethanone (8a) were reduced, mainly yielding (S)-enantiomers. Therefore, docking experiments were performed using 4-chloro-1-phenylethanone (9a) producing an (R)-enantiomer of 82% ee and 2-chloro-1-phenylethanone (7a) producing an (S)-enantiomer of >99% ee, which may reveal the reason for stereoselectivity inversion (Fig. 6). 4-Chloro-1-phenylethanone bound in the pro-R pose, the para-chlorine of which formed a halogen bond with Leu195, stabilising the pro-R pose (Fig. 6A). In contrast, 2-chloro-1-phenylethanone bound in the pro-S pose (Fig. 6B), the phenyl ring of which was located in the expanded pocket and interacted with Y249 and F206 as a π–π T-shape. Significantly, the ortho-chlorine formed halogen bonds with Gly189 and Pro188. The superimposition clearly illustrated the opposite binding orientation of two substrates, resulting in opposite enantiomers (Fig. 6C). Similar binding conformations of 2-chlorodiphenylketone (1a) producing an (S)-enantiomer of 57% ee and CPPK were also observed (Fig. S4†). These results further revealed that sufficient space was created for the inversion of binding orientation and the halogen bond controls the binding orientation in seq5.
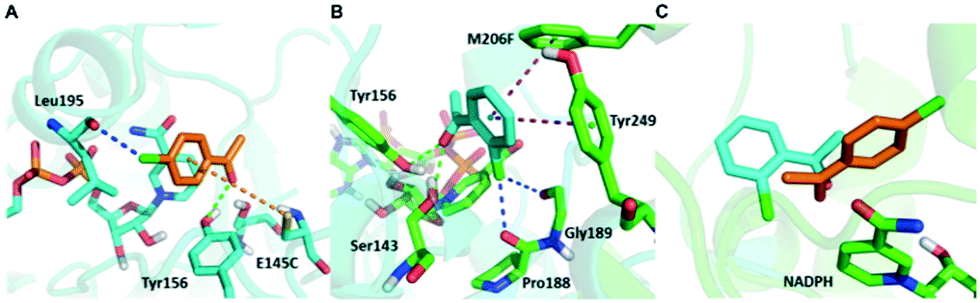 | ||
| Fig. 6 Opposite binding orientation of 2-chloro-1-phenylethanone and 4-chloro-1-phenylethanone in seq5. It was controlled by the different halogen bonds which led to stereoselectivity inversion. 2-chloro-1-phenylethanone and 4-chloro-1-phenylethanone are represented in cyan and orange, respectively. Halogen bonds are represented in blue; π–sulfur interactions in orange; π–π interactions in purple; hydrogen bonds in green. A. 4-Chloro-1-phenylethanone bound in the pro-R pose. Halogen bonds formed between para-chlorine and Leu195. B. 2-Chloro-1-phenylethanone bound in the pro-S pose. Halogen bond formed between ortho-chlorine and Gly189 or Pro188. C. Overlay of the two substrate conformations, which bound in the opposite orientation. | ||
In addition, it should be noted that ketones without any substitution on phenyl, such as phenyl-2-pyridinylmethanone (4a) and acetophenone (6a), only decreased with a low ee of 7% and 35.9%, respectively. When para-chlorine was attached, the ee of 5a and 9a dramatically increased to >99% and 82%, respectively. These significant increases and the inversion in stereoselectivity clearly indicate that the halogen bonds formed by chlorine substituent play an important role in the determination of stereoselectivity. This important halogen bond was also observed in the pro-R docking pose of CPPK but was absent in the pro-S docking pose (Fig. 2; Fig. S5 and S6†).
Very recently, during the engineering of KpADH from Kluyveromyces polyspora, high stereoselectivity was obtained using substrate 5a due to the opposite charge characteristics between chlorophenyl and pyridine substituents.24 KpADH was revealed to prefer the formation of the pro-R pose of 5a because of the electrostatic attraction formed between the positively charged pyridine substituent and the negatively charged Glu214.24 In the present study, no charged residues interacted with any substrate, which suggests a different chiral recognition mechanism in seq5. Halogen bonds are commonly found in protein–ligand complexes, with >2000 structures having been reported in recent years.30 They are analogous to hydrogen bonds and are highly directional and specific.31 They are also applied to control substrate selectivity in biocatalysis.32 In addition, although halogen bonds have been successfully applied in enantioseparations and chemical chiral catalysts,33–35 to the best of our knowledge, the role of the halogen bonds in determining stereoselectivity has not been reported in the area of biocatalysis. All the results that we obtained indicate that the elimination of steric hindrance in seq5 created sufficient space and appropriate conformation for phenyl ring inversion and new noncovalent bond formation. In this situation, differences in the sizes of groups had a limited impact on stereoselectivity, and the noncovalent halogen bond mainly determined the stereoselectivity. The main stereocontrol element changed in LkADH after engineering.
Based on the results above, seq5 prefers the pro-R binding of CPPK mainly because of the halogen bond between the para-chlorine and oxygen of Leu195. Moreover, the absence of a halogen bond with a pro-S conformation may cause difficulty in catalysis initiation. The catalysis mechanism of the ADH revealed that hydride transfer can happen when the distance between the NADPH-C4 atom and ketone carbonyl carbon is ≤4.5 Å, and the carbonyl oxygen forms a hydrogen bond with the side chains of the tyrosine catalytic residue.24,36–41 Therefore, the CPPK conformations in which the carbonyl oxygen formed hydrogen bonds (≤3.7 Å) with Tyr156 and the distance between the carbonyl carbon and NADPH-C4 atoms was less than 4.5 Å were defined as pre-reaction conformations. Both the docking conformations of the pro-R and pro-S poses were pre-reaction conformations (Fig. 7A and C). However, seq5 only produced an (R)-enantiomer, and previous reports have shown that docking analysis alone may provide an incomplete picture of ligand binding.42
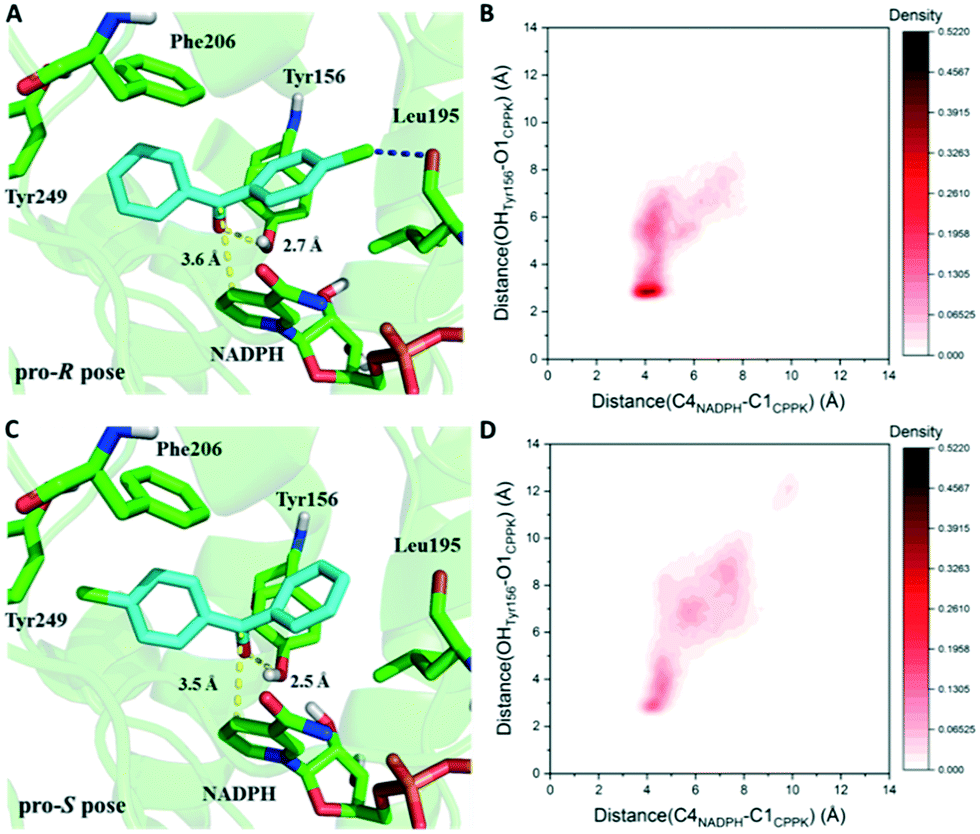 | ||
| Fig. 7 Comparison of two critical distances between the pro-R and pro-S poses. The yellow dashed line represents the initial distance obtained in the docking results. The deeper red color represents the higher probability of the corresponding conformations. A. Docking result obtained for the pro-R pose. B. The probability density distribution of pro-R conformations obtained from the simulations. The conformations of pro-R binding demonstrated a close contact with NADPH and Tyr156 simultaneously. C. Docking result obtained for the pro-S pose. D. The probability density distribution of pro-S conformations obtained from the simulations. The conformations of pro-S binding demonstrate relatively dispersed distances to Tyr156 and NADPH. | ||
Considering the dynamic motion of the substrate–enzyme complex, MD simulation was performed to further analyse the two critical distances and compare the proportions of the pre-reaction conformations of the pro-R and pro-S poses. As previous reports suggest that multiple short simulations achieve better correlation of stereoselectivity than a single long simulation,43,44 the two distances of the pro-R and pro-S conformations were monitored with ten independent 4 ns simulations (in total 40 ns). The binding free energy of the pro-S pose was calculated to be −22.1 ± 2.5 kcal mol−1, higher than that of the pro-R pose (−25.3 ± 2.9 kcal mol−1). Furthermore, as illustrated in Fig. 7B, the pro-R conformations of CPPK demonstrated a close and relatively stable distance to NADPH and Tyr156. However, it is difficult for the pro-S pose to sustain a stable close contact with NADPH and Tyr156, which implies that the probability of hydride transfer from the re-face was quite low (Fig. 7D). Remarkably, the proportion of the pre-reaction conformations with both distance C4NADPH–C1CPPK ≤ 4.5 Å and distance OHTyr156–O1CPPK ≤ 3.7 Å was 22.2% in the pro-R pose and 7.3% in the pro-S pose. The occurrence of more than three times of pre-reaction conformations of the pro-R pose than those of the pro-S pose indicated that seq5 prefers to transfer hydride from the si-face producing (R)-CPPO, which is consistent with the bio-reduction results. The above results suggest that the pro-R pose with a halogen bond can form pre-reaction conformations more easily than the pro-S pose, resulting in (R)-CPPO with high ee.
Scale-up preparation of (R)-CPPO and (S)-CPMO
With the highly stereoselective seq5, we scaled-up the production (200 mL) of (R)-CPPO and (S)-CPMO to determine the practical application of the engineered biocatalyst. Results of the scaled-up production demonstrate that both valuable diaryl alcohols can be easily prepared with high ee and complete conversion. The (R)-CPPO concentration was calculated to be 87.5 g L−1, and the (S)-CPMO was calculated to be 87.8 g L−1. The 200 mL reaction produced 15.3 g of (R)-CPPO with an ee of 99.3% and an isolated yield of 88.1%. The CPPO was confirmed by NMR analysis: 1H-NMR (600 MHz, CDCl3) δ: 7.33 (4H, d, J = 4.28 Hz), 7.27–7.29 (5H, m), 5.78 (1H, s); 13C-NMR (150 MHz, CDCl3) δ: 143.4, 142.2, 133.3, 128.7, 128.7, 128.6, 128.6, 127.9, 127.9, 127.8, 127.8, 126.5, 75.6. The chiral HPLC chromatograms of (R)-CPPO after a reaction period of 16 h are shown in Fig. S7.† The 200 mL reaction produced 14.8 g of (S)-CPMO with an ee of 98.1% and an isolated yield of 84.2%. The CPMO was confirmed by NMR analysis: 1H-NMR (600 MHz, CDCl3) δ: 8.55 (1H, d, J = 4.92 Hz), 7.62 (1H, ddd, J = 15.36, 7.62, 1.68 Hz), 7.29–7.32 (4H, m), 7.20 (1H, dd, J = 7.02, 5.16 Hz), 7.12 (1H, d, J = 7.86 Hz), 5.73 (1H, s); 13C-NMR (150 MHz, CDCl3) δ: 160.4, 147.9, 141.7, 137.0, 133.6, 128.7, 128.7, 128.4, 128.4, 122.6, 121.3, 74.3.Among the variants recently reported, seq5 demonstrated attractive substrate tolerance and satisfactory ee without additional optimisation (Table S2†). These results demonstrate that the bio-reduction approach using seq5 has potential for further large-scale applications.
Conclusions
In this study, we created an ADH variant with enhanced activity and stereoselectivity toward diaryl ketones without property trade-offs and investigated its molecular basis. A “shrinking mutagenesis” strategy was proposed to screen mutants with increased activity and stereoselectivity. The obtained seq5 could reduce the bulky–bulky model substrate CPPK and generate an (R)-enantiomer with a high ee of >99%. The activity and stereoselectivity were further investigated using five bulky–bulky ketones and four bulky–small ketones. Seq5 had higher activity than the WT toward all nine tested substrates. Molecular docking and MD simulations revealed that the original binding pocket was dramatically expanded, not only because of a smaller side chain but also the more open binding loop induced by Y190P. Therefore, the change in the pre-organisation state of seq5 facilitated the interaction of CPPK with catalytic residues. Moreover, all five constructed mutations provided energy favourable interactions and facilitated substrate binding in the pro-R pose. In the WT, stereoselectivity was mainly determined by steric effects. However, in seq5, after the elimination of steric hindrance, the main stereocontrol element probably changed to a halogen bond. The MD simulations further revealed that the formation of pre-reaction conformations would be easier with the pro-R pose with a halogen bond and π–π interaction than with the pro-S pose. These results indicate that the stereocontrol element of LkADH was changed to recognise diaryl ketones. To avoid property trade-offs, changing the stereocontrol element could be important for the simultaneous enhancement of the activity and stereoselectivity for ADHs. Furthermore, the obtained seq5 is a promising biocatalyst for the preparation of valuable chiral diaryl alcohols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by grants from the National Natural Science Foundation of China (No. 81773616), the Program of Shanghai Technology Research Leader (No. 17XD1423200) and the Seed Fund Program of the Shanghai University of Medicine & Health Sciences (SFP-18-22-07-002). We are also grateful to professors Jiahai Zhou and Hualei Wang for fruitful discussions.References
- Y. Ni and J. H. Xu, Biotechnol. Adv., 2012, 30, 1279 CrossRef CAS PubMed.
- E. L. Noey, N. Tibrewal, G. Jimenez-Oses, S. Osuna, J. Park, C. M. Bond, D. Cascio, J. Liang, X. Zhang, G. W. Huisman, Y. Tang and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E7065 CrossRef CAS PubMed.
- F. Qin, B. Qin, W. Zhang, Y. Liu, X. Su, T. Zhu, J. Ouyang, J. Guo, Y. Li, F. Zhang, J. Tang, X. Jia and S. You, ACS Catal., 2018, 8, 6012 CrossRef CAS.
- H. Li, D. Zhu, L. Hua and E. R. Biehl, Adv. Synth. Catal., 2009, 351, 583 CrossRef CAS.
- M. D. Truppo, D. Pollard and P. Devine, Org. Lett., 2007, 9, 335 CrossRef CAS PubMed.
- J. Lee, Y. Oh, Y. K. Choi, E. Choi, K. Kim, J. Park and M.-J. Kim, ACS Catal., 2014, 5, 683 CrossRef.
- P. Aliprandi, L. Cima and M. Carrara, Clin. Drug Invest., 2002, 22, 209 CrossRef CAS.
- Q. Liu, C. Wang, H. Zhou, B. Wang, J. Lv, L. Cao and Y. Fu, Org. Lett., 2018, 20, 971 CrossRef CAS PubMed.
- T.-S. Lee, D. S. Cerutti, D. Mermelstein, C. Lin, S. LeGrand, T. J. Giese, A. Roitberg, D. A. Case, R. C. Walker and D. M. York, J. Chem. Inf. Model., 2018, 58, 2043 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- N. Holmberg, U. Ryde and L. Bulow, Protein Eng., 1999, 12, 851 CrossRef CAS PubMed.
- J. D. Durrant, L. W. Votapka, J. Sorensen and R. E. Amaro, J. Chem. Theory Comput., 2014, 10, 5047 CrossRef CAS PubMed.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314M CrossRef PubMed.
- M. K. Kim and Y. K. Kang, Protein Sci., 1999, 8, 1492 CrossRef CAS PubMed.
- C. Nick Pace and J. Martin Scholtz, Biophys. J., 1998, 75, 422 CrossRef.
- T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891 CrossRef PubMed.
- Z. Sun, R. Lonsdale, X. D. Kong, J. H. Xu, J. Zhou and M. T. Reetz, Angew. Chem., Int. Ed., 2015, 54, 12410 CrossRef CAS PubMed.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637 CrossRef CAS PubMed.
- X. D. Kong, S. Yuan, L. Li, S. Chen, J. H. Xu and J. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15717 CrossRef CAS PubMed.
- I. Nilsson and G. von Heijne, J. Mol. Biol., 1998, 284, 1185 CrossRef CAS PubMed.
- B. K. Ho and R. Brasseur, BMC Struct. Biol., 2005, 5, 14 CrossRef PubMed.
- D. Ouedraogo, M. Souffrant, S. Vasquez, D. Hamelberg and G. Gadda, Biochemistry, 2017, 56, 2477 CrossRef CAS PubMed.
- A. Gora, J. Brezovsky and J. Damborsky, Chem. Rev., 2013, 113, 5871 CrossRef CAS PubMed.
- J. Zhou, Y. Wang, G. Xu, L. Wu, R. Han, U. Schwaneberg, Y. Rao, Y. L. Zhao, J. Zhou and Y. Ni, J. Am. Chem. Soc., 2018, 140, 12645 CrossRef CAS PubMed.
- C. M. Nealon, M. M. Musa, J. M. Patel and R. S. Phillips, ACS Catal., 2015, 5, 2100 CrossRef CAS.
- P. Liang, B. Qin, M. Mu, X. Zhang, X. Jia and S. You, Biotechnol. Lett., 2013, 35, 1469 CrossRef CAS PubMed.
- A. Li, L. Ye, X. Yang, C. Yang, J. Gu and H. Yu, Chem. Commun., 2016, 52, 6284 RSC.
- K. Wu, H. Wang, L. Chen, H. Fan, Z. Zhao and D. Wei, Appl. Microbiol. Biotechnol., 2016, 100, 8757 CrossRef CAS PubMed.
- A. Weckbecker and W. Hummel, Biocatal. Biotransform., 2006, 24, 380 CrossRef CAS.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267 RSC.
- A. R. Voth, P. Khuu, K. Oishi and P. S. Ho, Nat. Chem., 2009, 1, 74 CrossRef CAS PubMed.
- S. Jiang, L. Zhang, D. Cui, Z. Yao, B. Gao, J. Lin and D. Wei, Sci. Rep., 2016, 6, 34750 CrossRef CAS PubMed.
- P. Peluso, V. Mamane, E. Aubert and S. Cossu, J. Chromatogr. A, 2014, 1345, 182 CrossRef CAS PubMed.
- P. Peluso, V. Mamane, E. Aubert, A. Dessi, R. Dallocchio, A. Dore, P. Pale and S. Cossu, J. Chromatogr. A, 2016, 1467, 228 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, L. Ferreira, V. Félix and P. D. Beer, Chem. Commun., 2016, 52, 5527 RSC.
- V. Pace, Á. C. Cabrera, V. Ferrario, J. V. Sinisterra, C. Ebert, L. Gardossi, P. Braiuca and A. R. Alcántara, J. Mol. Catal. B: Enzym., 2011, 70, 23 CrossRef CAS.
- C. Filling, K. D. Berndt, J. Benach, S. Knapp, T. Prozorovski, E. Nordling, R. Ladenstein, H. Jornvall and U. Oppermann, J. Biol. Chem., 2002, 277, 25677 CrossRef CAS PubMed.
- V. Stojkovic, L. L. Perissinotti, D. Willmer, S. J. Benkovic and A. Kohen, J. Am. Chem. Soc., 2012, 134, 1738 CrossRef CAS PubMed.
- J. Deng, Z. Yao, K. Chen, Y. A. Yuan, J. Lin and D. Wei, J. Biotechnol., 2016, 217, 31 CrossRef CAS PubMed.
- J. An, Y. Nie and Y. Xu, Crit. Rev. Biotechnol., 2019, 39, 366 CrossRef CAS PubMed.
- M. A. Maria-Solano, A. Romero-Rivera and S. Osuna, Org. Biomol. Chem., 2017, 15, 4122 RSC.
- Y.-C. Chen, Trends Pharmacol. Sci., 2015, 36, 78 CrossRef PubMed.
- S. Genheden and U. Ryde, J. Comput. Chem., 2010, 31, 837 CAS.
- J. J. Perez, M. S. Tomas and J. Rubio-Martinez, J. Chem. Inf. Model., 2016, 56, 1950 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02444a |
| This journal is © The Royal Society of Chemistry 2020 |