DOI:
10.1039/D0DT03047C
(Paper)
Dalton Trans., 2020,
49, 16683-16692
Functionalised phosphonate ester supported lanthanide (Ln = La, Nd, Dy, Er) complexes†
Received
31st August 2020
, Accepted 13th October 2020
First published on 19th October 2020
Abstract
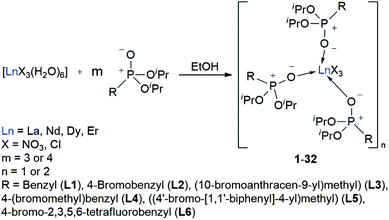
A series of phosphonate ester supported lanthanide complexes bearing functionalities for subsequent immobilisation on semiconductor surfaces are prepared. Six phosphonate ester ligands (L1–L6) with varying aromatic residues are synthesised. Subsequent complexation with lanthanide chloride or -nitrate precursors (Ln = La, Nd, Dy, Er) affords the corresponding mono- or dimeric lanthanide model complexes [LnX3(L1–L3 or L5–L6)3]n (X = NO3, Cl; n = 1 (Nd, Dy, Er), 2 (La, Nd)) or [LnCl2Br(L4-Br)2(L4-Cl)]n (n = 1 (Nd, Dy, Er), 2 (La, Nd)) (1–32). All compounds are thoroughly characterised, and their luminescence properties are investigated in the visible and NIR spectral regions, where applicable.
Introduction
Lanthanide ions already find widespread application in lighting, sensing and display technologies, due to their outstanding photoluminescence properties.1–3
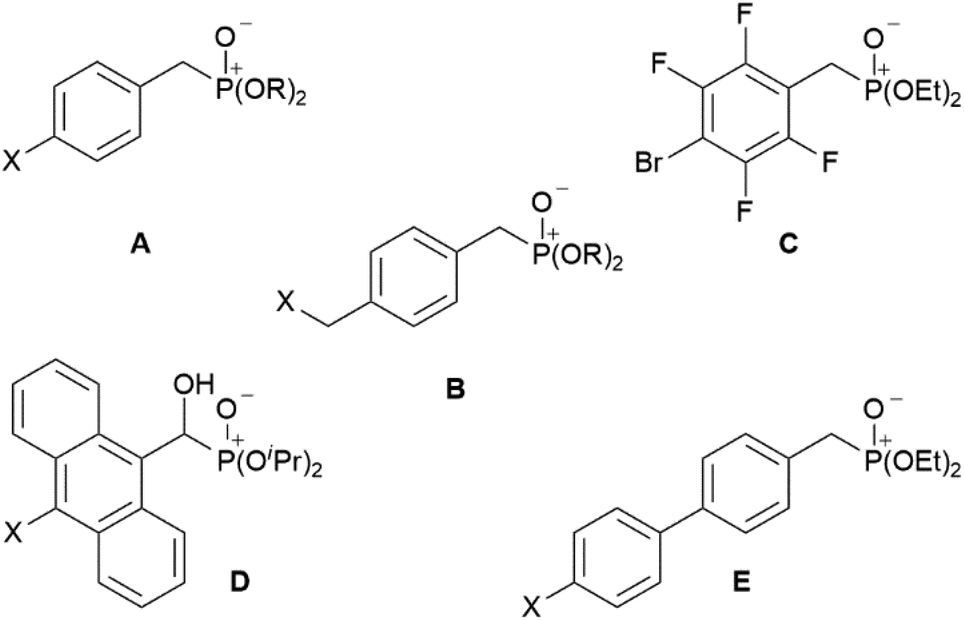
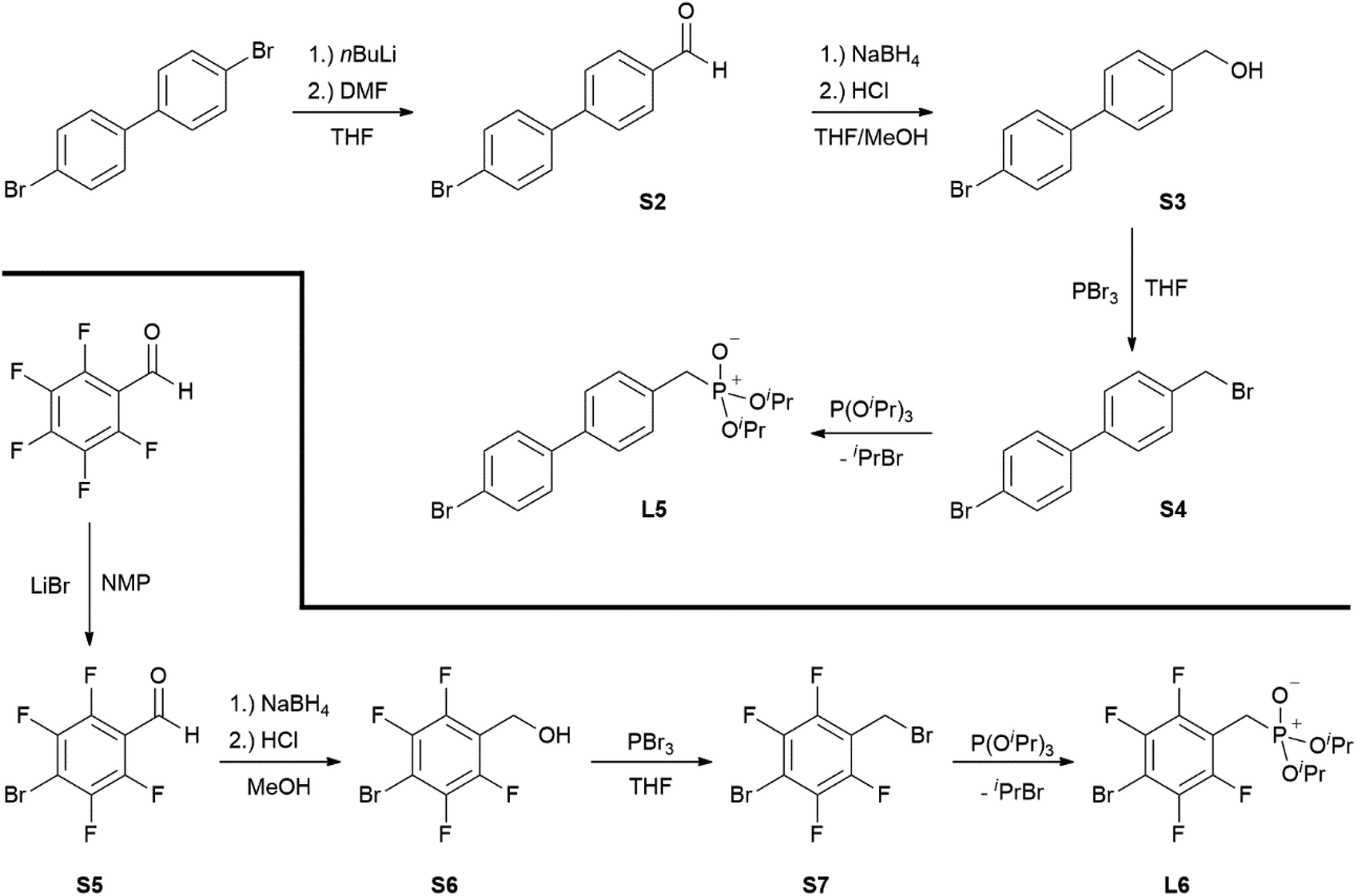
Bis-phosphonic acid ester ligands have already been successfully shown to be versatile building blocks in transition metal organic frameworks (MOFs).4 In contrast to their carboxylic acid congeners, the key advantage of phosphonate esters originates from their lower vibrational frequencies resulting in reduced non-emissive excited state quenching and enhanced quantum yields.5 In case of lanthanide-based coordination polymers also complexes with improved luminescence properties have been obtained.6 Our group has focussed on this topic during the last years preparing a variety of efficiently luminescent lanthanide based MOFs.7–9 Moreover, different poly-fluoroaryl substituted mono-10,11 and bis-phosphanes,12 and phosphinic acids13 with enhanced rigidity have been prepared and investigated in terms of reactivity and coordination behaviour. Now, to obtain distinct molecular lanthanide complexes with enhanced excited state lifetimes in the context of a future attachment to semiconductor surfaces, our research focusses on preparation of aryl-substituted mono phosphonate ester ligands. A reactive functionality at the para-position with respect to the phosphonate substituent shall give additional access to future substitution reactions. Selected related aryl-bound mono-phosphonate esters are summarised in Chart 1. These range from differently para-substituted ligands A and B (X = H, Cl, Br; R = Me, Et, iPr, tBu),14–16 over polyfluorinated species C,17 to anthracene D (X = H, Cl) and 1,1′-biphenyl derivates E (X = H, Me).14,18
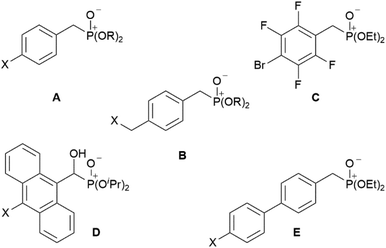 |
| | Chart 1 Selected known aryl-substituted phosphonate ester ligands A–E. | |
Results and discussion
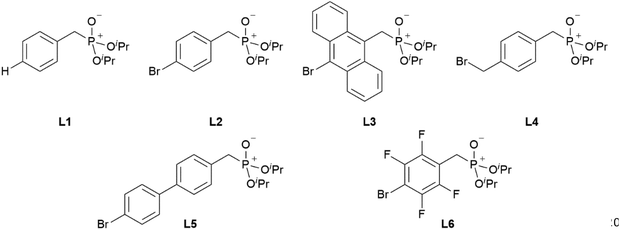
Synthesis of ligands L1–L6
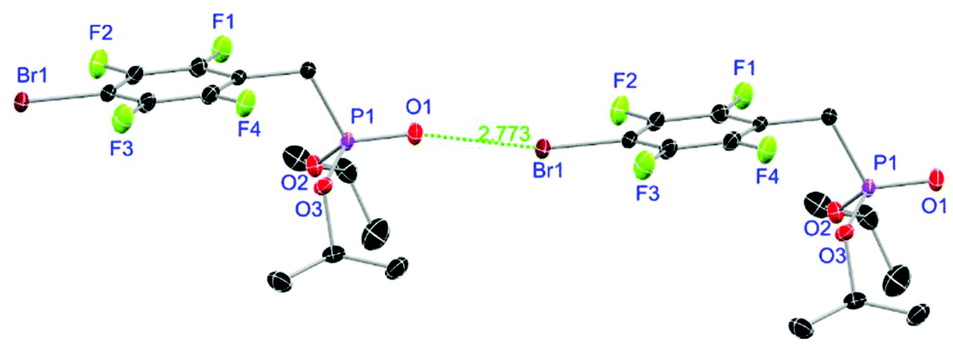
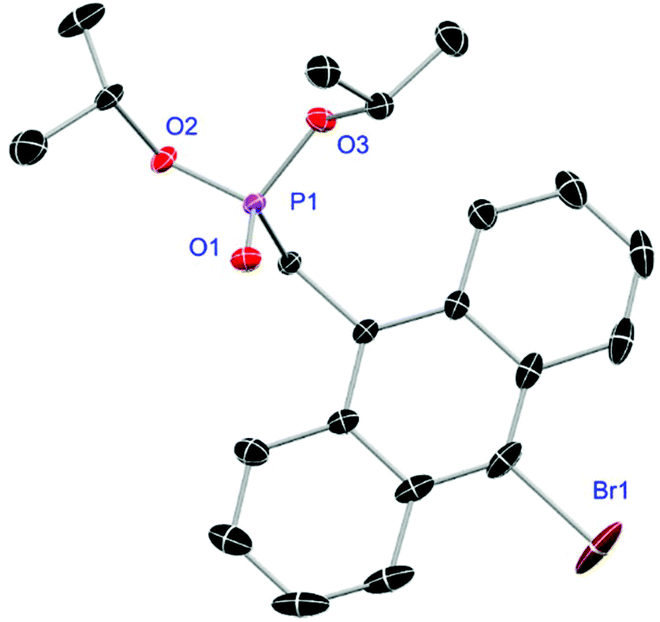
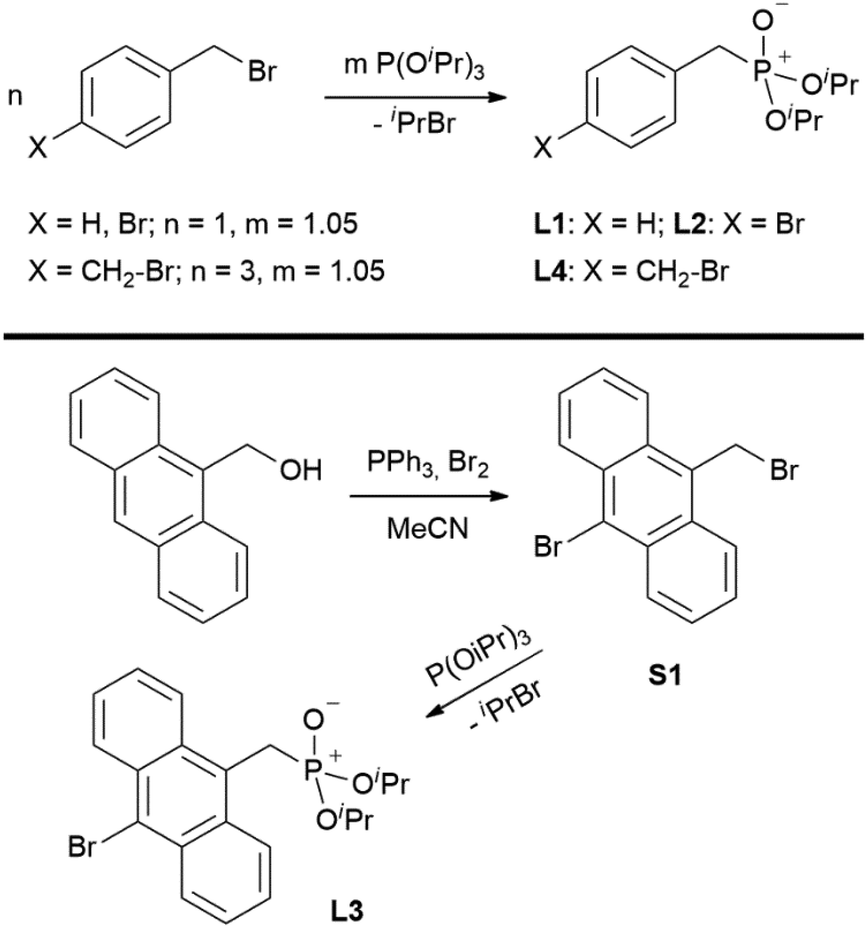
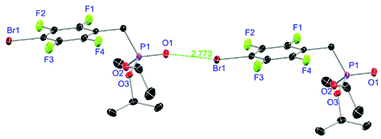
Ligands L1, L2 and L4 are prepared by straight forward Michaelis–Arbuzov19 reactions from the corresponding benzyl-bromide starting materials in excellent yields of 90% and 99% for L1 and L2, respectively (Scheme 1, top). L4 is prepared according to a modified literature procedure starting from a threefold excess of 1,4-bis(bromomethyl)benzene treated with 20 h of reflux in dry toluene to promote formation of the mono-phosphorylated product.20L4 is obtained in a good yield of 58% after workup. All three ligand species are obtained as colourless or pale-yellow oils. To obtain a ligand with increased bulkiness and antenna effect, anthracene-based L3 is prepared in a two-step synthesis starting with a modified double bromination reaction of 9-(hydroxymethyl)anthracene.21 Subsequent Michaelis–Arbuzov reaction of the obtained 9-bromo-10-(bromomethyl) anthracene (S1) gives L3 as a yellow powder in an excellent overall yield of 80% (Scheme 1, bottom). Crystals suitable for SCXRD experiments were grown from vapor diffusion of pentanes into a saturated solution of L3 in THF. L3 crystallises in the monoclinic space group P21/c containing one molecule in the asymmetric unit (Fig. 2). Phosphonate ester L5 is synthesised in a good overall yield of 36% via a four-step procedure starting from 4,4′-dibromo-1,1′-biphenyl (Scheme 2, top; vide infra). The starting materials S2, S3 and S4 are prepared according to modified literature protocols.22,23S4 is then phosphorylated with P(OiPr)3 to give L5 under release of iPrBr as a colourless oil which solidifies upon standing. Unfortunately, only crystals of poor quality were obtained from this solid material. Electron withdrawing substituents like fluorides are known to improve the photoluminescence properties and quantum yields of polyconjugated organic ligands and derived metal complexes due to lowered HOMO as well as LUMO energy levels and thus electron injection.24,25 Hence, also polyfluorinated phosphonate ester ligand L6 was prepared for subsequent lanthanide complexation (Scheme 2, bottom, vide infra). Compounds S5 and S6 are synthesised according to modified protocols.26 Starting from pentafluoro benzaldehyde, a bromide substituent is introduced at the para position via a nucleophilic aromatic substitution to yield S5 followed by reduction to alcohol S6. Subsequent bromination with PBr3 and Michaelis–Arbuzov reaction gives S7 and L6, respectively. After workup, L6 is obtained in a good overall yield of 45% as a colourless oil which solidifies upon standing. L6 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) hosting one molecule in the asymmetric unit. Moreover, a short intermolecular Br⋯O distance of only 2.77 Å is observed between the sp2 hybridised O-atom of the phosphonate moieties and the para Br substituents of neighbouring molecules which remains 0.58 Å under the sum of the van der Waals radii of 3.35 Å (ref. 27) of both atoms (Fig. 1). This can only be rationalised by the formation of a so-called halogen bond for which first crystallographic evidence was provided by Hassel as early as 1954.28,29 Here, the solid-state structure of bromine-1,4-dioxanate revealed a comparable strong intermolecular Br⋯O interaction with a distance of 2.71 Å. Halogen bonds are strong, specific and directional interactions including a significant charge transfer.30 Hence, due to the short Br⋯O distance in L6 the present halogen bonds can be assigned to the inner-sphere complexes of the type [BX]+⋯Y− according to Mulliken.31–33 Despite that, also many analogies have been drawn between halogen- and the much weaker hydrogen bonds,30 and many applications have been found in polymer science.34 An intrinsic feature of the halogen bond is the [BX]+⋯Y− bond vector being nearly linear.29 The corresponding C–Br⋯O and P
hosting one molecule in the asymmetric unit. Moreover, a short intermolecular Br⋯O distance of only 2.77 Å is observed between the sp2 hybridised O-atom of the phosphonate moieties and the para Br substituents of neighbouring molecules which remains 0.58 Å under the sum of the van der Waals radii of 3.35 Å (ref. 27) of both atoms (Fig. 1). This can only be rationalised by the formation of a so-called halogen bond for which first crystallographic evidence was provided by Hassel as early as 1954.28,29 Here, the solid-state structure of bromine-1,4-dioxanate revealed a comparable strong intermolecular Br⋯O interaction with a distance of 2.71 Å. Halogen bonds are strong, specific and directional interactions including a significant charge transfer.30 Hence, due to the short Br⋯O distance in L6 the present halogen bonds can be assigned to the inner-sphere complexes of the type [BX]+⋯Y− according to Mulliken.31–33 Despite that, also many analogies have been drawn between halogen- and the much weaker hydrogen bonds,30 and many applications have been found in polymer science.34 An intrinsic feature of the halogen bond is the [BX]+⋯Y− bond vector being nearly linear.29 The corresponding C–Br⋯O and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Br angles in L6 of 167.4° and 166.5° deviate significantly from 180° but are in a similar range like in comparable literature structures.35 This deviation is most likely due to a crystallographic packing effect causing the formation of an advantageous ladder-structure between adjacent molecules. Nonetheless, both angles are nearly identical classifying this interaction as a type I halogen bond.35 In comparison to related bis-phosphonate esters (anthracene-based I,9 and tetra-fluorophenyl-based II
O⋯Br angles in L6 of 167.4° and 166.5° deviate significantly from 180° but are in a similar range like in comparable literature structures.35 This deviation is most likely due to a crystallographic packing effect causing the formation of an advantageous ladder-structure between adjacent molecules. Nonetheless, both angles are nearly identical classifying this interaction as a type I halogen bond.35 In comparison to related bis-phosphonate esters (anthracene-based I,9 and tetra-fluorophenyl-based II![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36), ligands L3 and L6 show comparable structural features (Table 1, vide infra). The P1–C1 and P1–O1 distances differ only marginally between 1.79–1.81 Å and 1.46–147 Å, respectively. A similar tendency can be found for the respective P1–C1–C2 and O1–P1–C1 angles. Interestingly, L3 shows in both cases the widest ((114.22(2)° and 116.07(1)°) and L6 the narrowest angles ((113.60(2)° and 112.97(1)°) within the row. The 31P-NMR shifts of L1–L6 are in-between 19.0 ppm (L6) and 24.4 ppm (L5) (Table 2) and are located within the range of related compounds found in the literature.7–9
36), ligands L3 and L6 show comparable structural features (Table 1, vide infra). The P1–C1 and P1–O1 distances differ only marginally between 1.79–1.81 Å and 1.46–147 Å, respectively. A similar tendency can be found for the respective P1–C1–C2 and O1–P1–C1 angles. Interestingly, L3 shows in both cases the widest ((114.22(2)° and 116.07(1)°) and L6 the narrowest angles ((113.60(2)° and 112.97(1)°) within the row. The 31P-NMR shifts of L1–L6 are in-between 19.0 ppm (L6) and 24.4 ppm (L5) (Table 2) and are located within the range of related compounds found in the literature.7–9
 |
| | Fig. 1 Molecular structure of L6 with schematic halogen-bond. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 1. | |
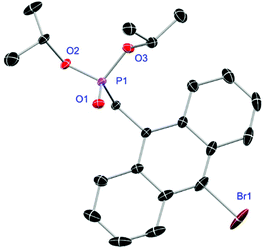 |
| | Fig. 2 Molecular structure of L3. Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 1 (vide infra). | |
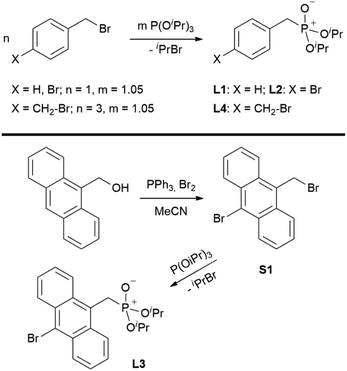 |
| | Scheme 1 Syntheses of ligands L1, L2, L4 (top) and L3 (bottom). | |
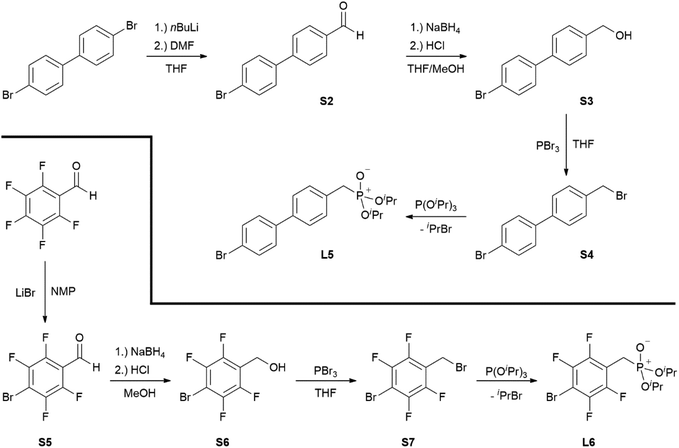 |
| | Scheme 2 Four-step preparations of phosphonate ester ligands L5 (top) and L6 (bottom). | |
Table 1 Selected bond lengths [Å] and angles [°] for L3 and L6 and related literature compounds I and II. I and II contain two molecules in the asymmetric unit thus averaged values are given
| Compound |
P1–C1 |
P1–O1 |
P1–C1–C2 |
O1–P1–C1 |
|
L3
|
1.805(2) |
1.4679(2) |
114.22(2) |
116.07(1) |
|
L6
|
1.803(3) |
1.4653(2) |
113.60(2) |
112.97(1) |
|
I
|
1.7978(2) |
1.4633(1) |
113.64(1) |
115.37(1) |
|
II
|
1.7900(2) |
1.4690(2) |
113.67(1) |
113.29(8) |
Table 2
31P-NMR chemical shifts [ppm] of ligands L1–L6 and the derived lanthanum(III) complexes 1, 5, 9, 13, 17, 25, 29 [LaX3(L1–L3 or L5–L6)3]2 (X = NO3, Cl) and 21 [LaCl2Br(L4-Br)2(L4-Cl)]2
| Ligand |
[LaCl3(L1–L3 or L5–L6)3]2 or [LaCl2Br(L4-Br)2(L4-Cl)]2 |
[La(NO3)3(L1–L6)3] |
|
L1: 24.3 |
24.2 |
23.2 |
|
L2: 23.6 |
23.2 |
22.3 |
|
L3: 23.0 |
23.0 |
— |
|
L4: 24.0 |
23.73, 23.67 |
— |
|
L5: 24.4 |
24.0 |
— |
|
L6: 19.0 |
19.0 |
— |
Lanthanide complexes 1–32
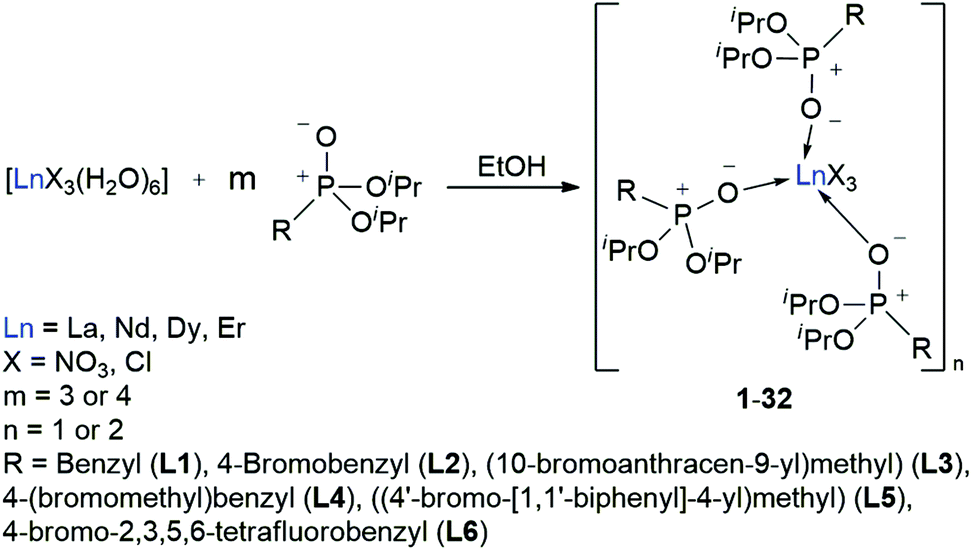
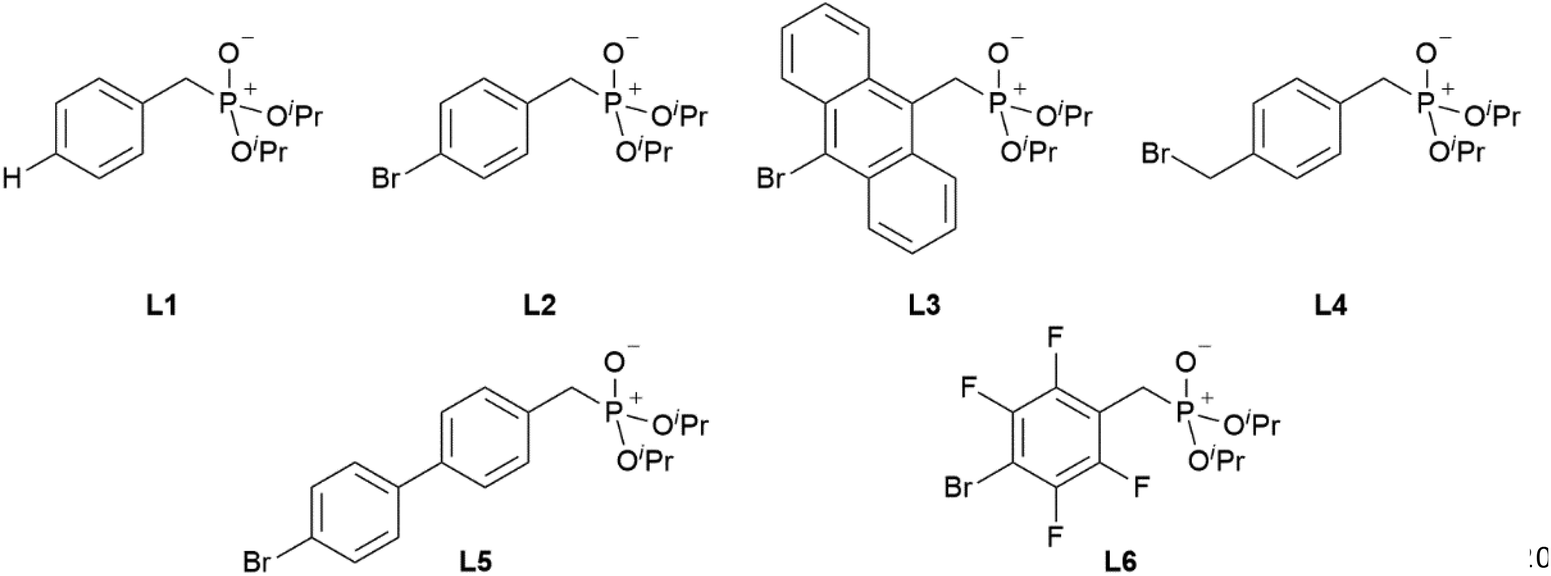
With the desired phosphonate ester ligands L1–L6 in hand, synthesis of derived lanthanide complexes (Ln = La, Nd, Dy, Er) is performed in EtOH starting from [LnX3(H2O)6] precursors (Scheme 3, vide infra). To have recourse to NMR spectroscopy for a proper characterisation of the products, all preparations have first been carried out with the diamagnetic La3+ derivative. For X = NO3 or the use of the steric demanding anthracene-based L3, only three equivalents of the phosphonate esters are needed to replace all aqua ligands in the starting material and to obtain product IR spectra without residual OH-bands. In contrast, in the case of X = Cl or the less bulky L1, L2, L4–L6, four equivalents of the ligands are needed to oust the H2O substituents and to obtain products featuring no OH-bands in the IR spectra. For a ligand overview, see Chart 2.
 |
| | Scheme 3 Synthesis of the L1–L6 supported lanthanide complexes 1–32. | |
 |
| | Chart 2 Overview of the phosphonate ester ligands L1–L6 used within this work. | |
After reaction, the excess of the phosphonate esters is removed by thorough extraction with pentanes so that in almost all cases mono- or dimeric complexes of the form [LnX3(L)3]n (X = NO3, Cl, n = 1 (Nd, Dy, Er), 2 (La, Nd)) are obtained in a quantitative yield. For L4, complexes of the form [LnCl2Br(L4-Br)2(L4-Cl)]n (n = 1 (Nd, Dy, Er), 2 (La, Nd)) are found due to an Br–Cl exchange between one ligand and the lanthanide chloride precursor.
In comparison to the chlorides, the NO3− anions are bulkier and act as two-dentate chelate ligands. This might cause the [LnCl3(H2O)6] precursors to be prone to take up more than six H2O molecules into their coordination sphere over time resulting in an aggravated permanent replacement of the water molecules. Despite of the phosphonate esters L1–L6 being potential three-dentate chelate ligands, all obtained lanthanide complexes exhibit only a mono-dentate coordination by the oxygen atom of the P+–O− moieties like it is observed in related structures.7–9 Except for the L4- and L5-supported complexes 21 and 25, the 31P-NMR signal of the free ligands experiences a 0.1–0.4 ppm shift to higher field upon complex formation with the lanthanum chloride precursors (Table 2). This effect is even more pronounced upon complexation with the lanthanum nitrates causing a high-field shift of 1.1 ppm (1) and 1.3 ppm (9), respectively. Due to a Br–Cl exchange between one ligand and the lanthanum chloride, two 31P signals at 23.73 and 23.67 ppm in a ratio of 1:2 are observed for the L4-based La3+ complex 21. Two sets of signals in the same ratio are also found in the corresponding 1H- and 13C-NMR spectra and are assigned to the two ligand modifications. Even in the MALDI-MS spectrum, aggregates with bromide as well as chloride substituted ligands are detected (see ESI†). In general, the nitrate supported complexes 1–4 and 9–12 easily solidify while most of the chloride substituted complexes tend to form viscous waxes that do not solidify or only solidify after some time. Because of that, no crystal structure determination could be carried out for the L4- (21–24) and L5-based complexes 25–28.
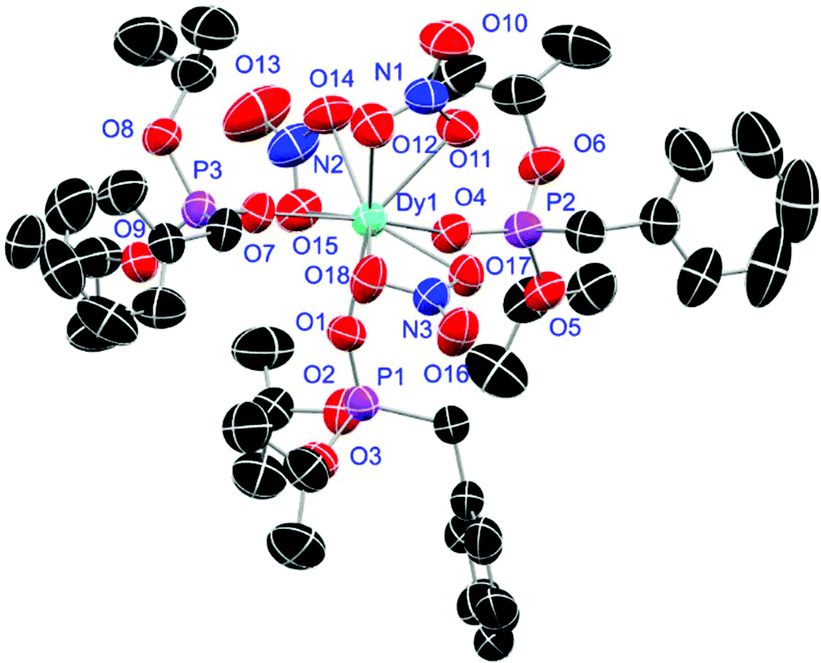
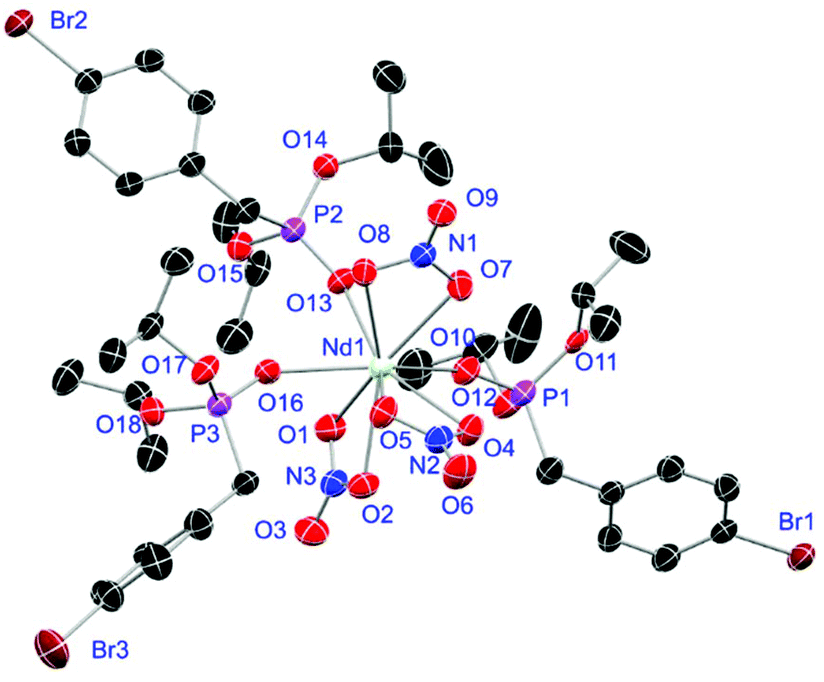
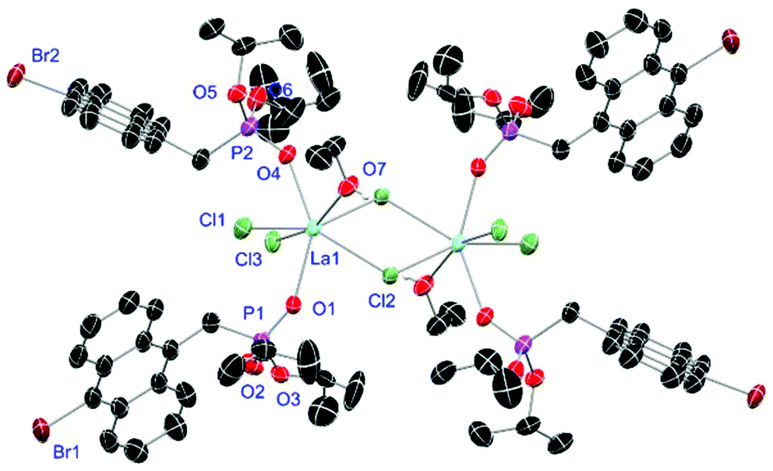
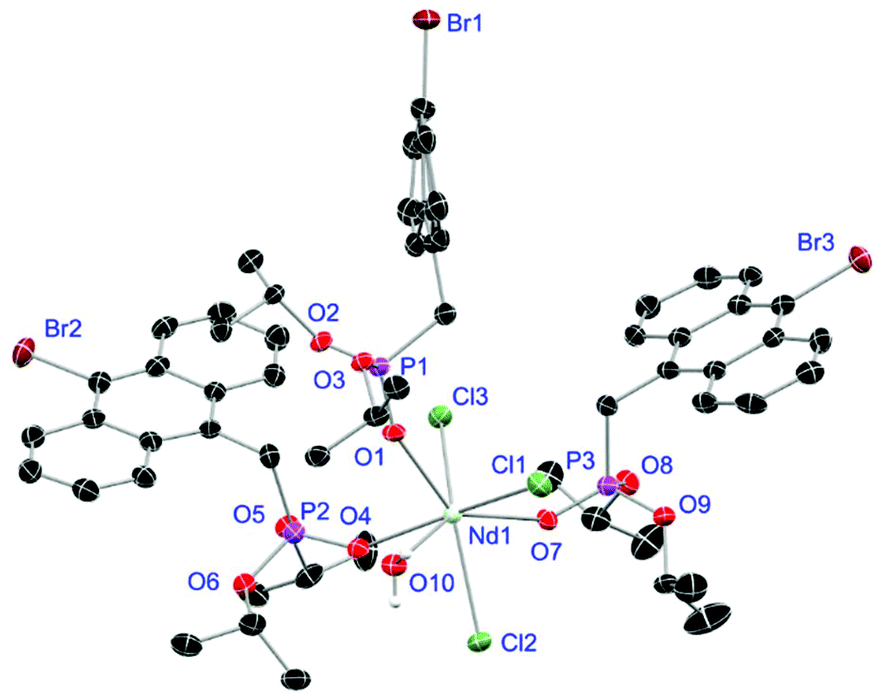
Crystals of L1- and L2-based compounds 2, 3, 5, 10, 13 and 14 are grown from slow evaporation of saturated DCM solutions or from obtained wax-like material that crystallised after some time. The lanthanide nitrate complexes 2 (see ESI†), 3 (Fig. 3) and 10 (Fig. 4) form monomers and show a nine-fold coordination in the solid-state. Just like in L6, halogen bonds are formed in complex 10. In this case between the bromides (Br2) and the outwards titled NO (O9) oxygen atoms of a NO3− anion of adjacent molecules. In contrast to L6, there is less lack of electron density at the bromides thus a weaker halogen bond is formed. The O⋯Br distance of 3.06 Å is elongated but still shorter than the sum of the van der Waals radii of both atoms (3.35 Å (ref. 27)). The present C–Br⋯O angle of 169.1° is in a similar range like in L6. However, the NO⋯Br angle of 108.3° indicates a certain perpendicular arrangement of the nitrate moiety with respect to the bromide substituent assigning this interaction rather as a type II halogen bond.35 The L1- and L2-based lanthanide chloride compounds 5 (Fig. 5), 13 (Fig. 6) and 14 (see ESI†) show seven-fold, pentagonal-bipyramidal coordination forming chloride bridged dimers in the solid-state with an averaged M–M distance of 4.78 Å. A comparison of the L1- and L2-based Nd3+ nitrate complexes 2 and 10 and the La3+ chloride compounds 5 and 13 shows a shortening of the M–O distances in both cases when switching from L1 to L2 (2.381(6) (2) to 2.372(2) Å (10) and 2.433(2) (5) to 2.409(4) Å (13)). The same trend can be seen for the M–Cl distances of the non-bridging chlorides in 5 and 13 (2.821(6) to 2.750(2) Å) while the bond length of the bridging chlorides (2.924(6) to 2.914(2) Å) seems to be less effected by an increasing steric demand of the ligand periphery in L2. Apparently contrary trends are obtained for the P+–O− distances and the M–O–P angles. The P+–O− bond length in 2 and 10 is unaffected while the M–O–P angle is slightly narrowed from 154.6(4)° in 2 to 152.4(2)° in complex 10. In contrast, the P+–O− distance decreases from 1.486(2) Å in 5 to 1.463(5) Å in 10 while the M–O–P angle is significantly increased from 151.3(1)° to 159.4(3)°.
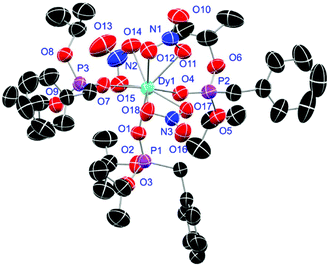 |
| | Fig. 3 Molecular structure of [Dy(NO3)3(L1)3] (3). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 3 (vide infra). | |
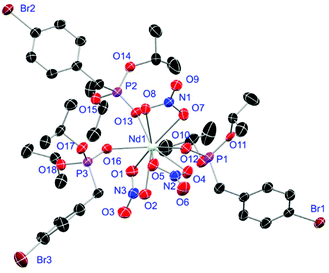 |
| | Fig. 4 Molecular structure of [Nd(NO3)3(L2)3] (10). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 3 (vide infra). | |
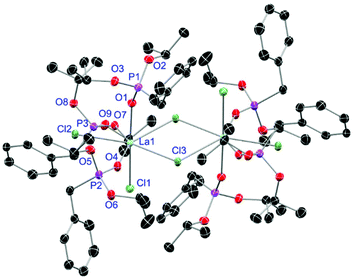 |
| | Fig. 5 Molecular structure of [LaCl3(L1)3]2 (5). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 3 (vide infra). | |
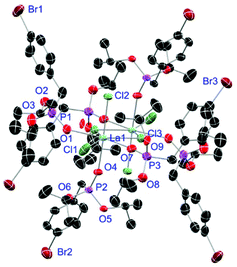 |
| | Fig. 6 Molecular structure of [LaCl3(L2)3]2 (13). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. Structural data are given in Table 3. | |
Table 3 Selected bond lengths [Å] and angles [°] of L1-based complexes 2, 3, 5, L2-based 10, 13, 14, L3-based 17–20 and L6-based 31 and 32. Distances to solvent oxygen atoms are not considered
| Complex |
M–O/M–O(NO3) |
M–Cl/M–(μ-Cl) |
P+–O− |
M–O–P |
| [Nd(NO3)3(L1)3] (2) |
2.381(6)/2.531(6) |
— |
1.483(7) |
154.6(4) |
| [Dy(NO3)3(L1)3] (3) |
2.297(2)/2.455(2) |
— |
1.476(2) |
155.6(1) |
| [LaCl3(L1)3]2 (5) |
2.433(2) |
2.821(6)/2.924(6) |
1.486(2) |
151.3(1) |
| [Nd(NO3)3(L2)3] (10) |
2.372(2)/2.538(2) |
— |
1.483(3) |
152.4(2) |
| [LaCl3(L2)3]2 (13) |
2.409(4) |
2.750(2)/2.914(2) |
1.463(5) |
159.4(3) |
| [NdCl3(L2)3]2 (14) |
2.378(9) |
2.725(3)/2.910(3) |
1.480(9) |
160.2(6) |
|
17 as [LaCl3(L3)2 (EtOH)]2·EtOH |
2.421(3) |
2.802(1)/2.873(1) |
1.483(3) |
149.2(2) |
|
18 as [NdCl3(L3)3 (H2O)] |
2.378(4) |
2.764(1) |
1.483(4) |
147.9(2) |
|
19 as [DyCl3(L3)2 (EtOH)]·2EtOH |
2.253(3) |
2.612(1) |
1.489(3) |
153.1(2) |
| [ErCl3(L3)3] (20) |
2.230(4) |
2.616(1) |
1.493(4) |
150.1(3) |
|
31 as [DyCl2(L6)3 (H2O)]Cl |
2.256(6) |
2.574(3) |
1.477(6) |
167.0(4) |
|
32 as [ErCl2(L6)3 (H2O)]Cl |
2.209(6) |
2.521(2) |
1.457(6) |
166.9(4) |
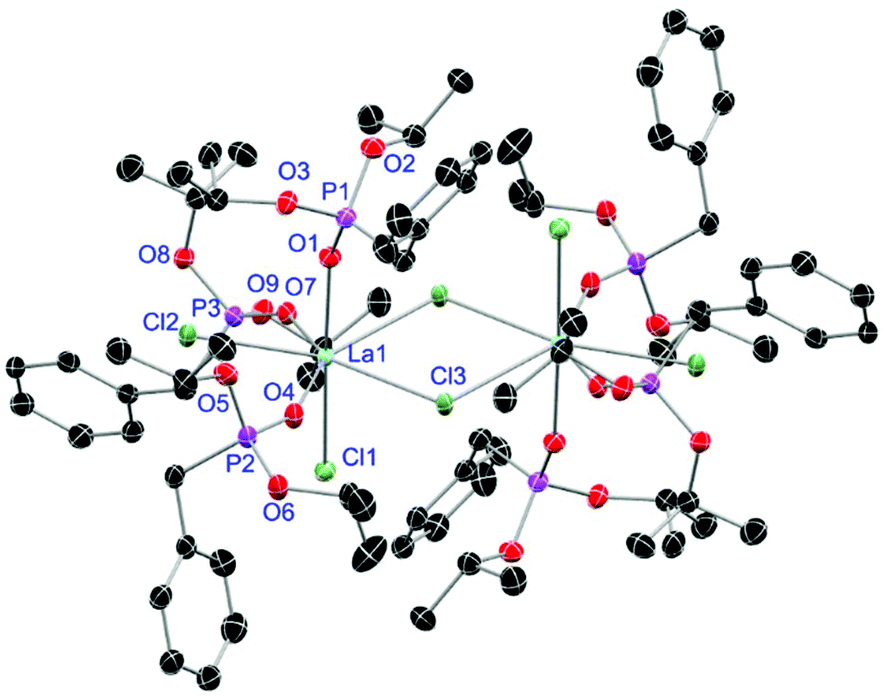
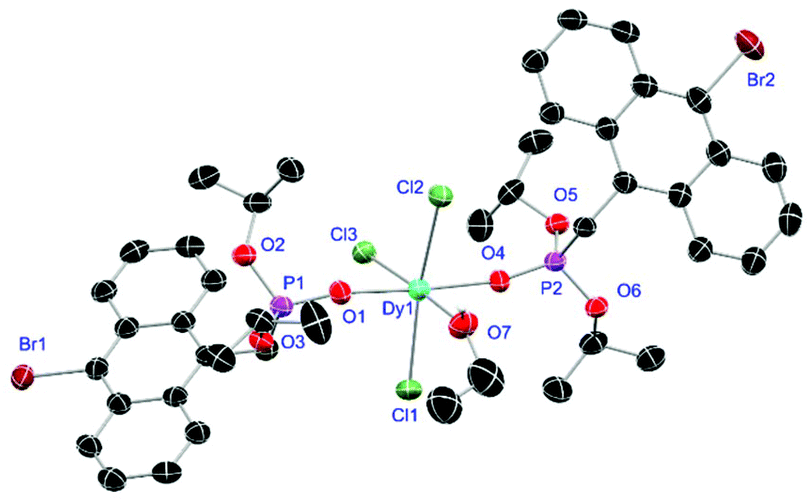
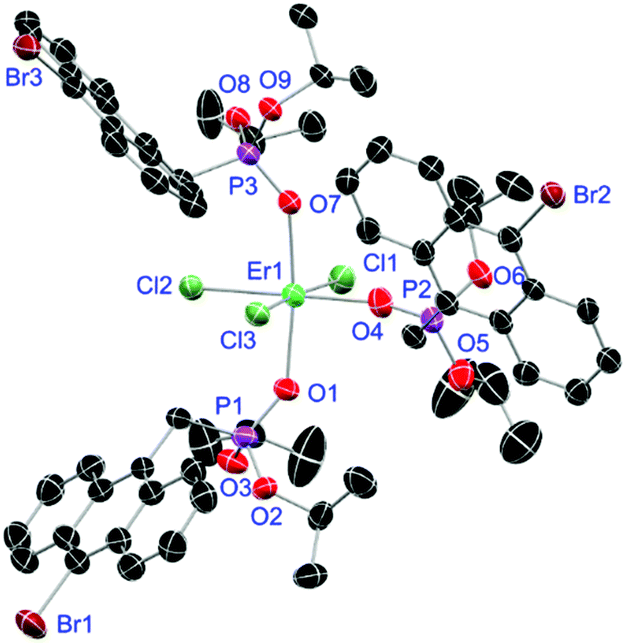
For the L3-based complexes, crystals suitable for X-ray diffraction experiments could only be obtained from saturated EtOH solutions at −20 °C (17–19) or from vapor diffusion of pentanes into a saturated THF solution (20). Just like complexes 5, 13 and 14, compound 17 shows a seven-fold coordination forming a chloride bridged dimer in the solid-state (Fig. 7, vide infra). Owing to the increased steric demand of L3, it is most likely that the preferred dimer formation only can occur upon exchange of an equivalent L3 with an EtOH molecule per La3+ ion. Complex 18 also shows a seven-fold pentagonal-bipyramidal coordination but forms a monomer because of three attached bulky molecules of L3 (Fig. 8, vide infra). Since all obtained products show no residual OH-bands in the IR spectrum, the additionally attached H2O molecule to suit the coordination sphere of the Nd3+ cation has to origin from airborne water during the crystallisation process. Just like in 17, a phosphonate ester L3 is substituted by an EtOH molecule in complex 19 (Fig. 9). A monomeric octahedral coordination is most likely preferred due to the smaller ionic radius of the Dy3+ ion. The same coordination mode can be observed for complex 20 (Fig. 10). The coordination sphere is made up by three chloride anions and three L3 oxygen atoms. Consistent with a step-wise decrease of the effective ionic radii of the cations: La3+ (103 pm) > Nd3+ (98 pm) > Dy3+ (91 pm) > Er3+ (89 pm),37 the M–O and M–Cl distances are successively decreasing from 2.421(3) Å and 2.802(1) Å in 17, over 2.378(4) Å and 2.764(1) Å (18) and 2.253(3) Å and 2.612(1) Å (19) to 2.230(4) Å and 2.616(1) Å in complex 20. The P+–O− bond lengths show a contrasting trend by slightly increasing from 1.483(3) Å in 17 to 1.493(4) Å in 20. Moreover, the acutest M–O–P angles amongst all synthesised complexes are found in 17 (149.2(2)°) and 18 (147.9(2)°).
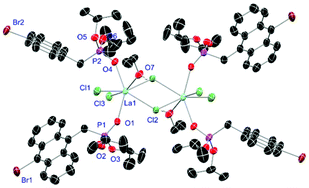 |
| | Fig. 7 Molecular structure of 17, crystallised as [LaCl3(L3)2(EtOH)]2·EtOH. Anisotropic displacement parameters are depicted at the 50% probability level. Except for freely refined hydrogens, these and a co-crystallised EtOH molecule are omitted for clarity. Structural data are given in Table 3. | |
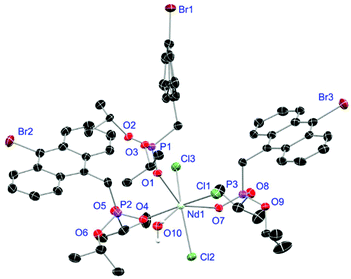 |
| | Fig. 8 Molecular structure of 18, crystallised as [NdCl3(L3)3(H2O)]. Anisotropic displacement parameters are depicted at the 50% probability level. Except for freely refined hydrogens, these are omitted for clarity. Structural data are given in Table 3. | |
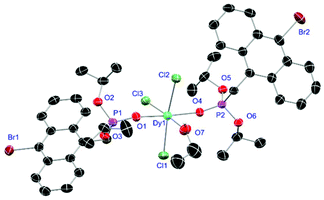 |
| | Fig. 9 Molecular structure of 19, crystallised as [DyCl3(L3)2(EtOH)]·2EtOH. Anisotropic displacement parameters are depicted at the 50% probability level. Except for freely refined hydrogens, these and two co-crystallised EtOH molecules are omitted for clarity. Structural data are given in Table 3. | |
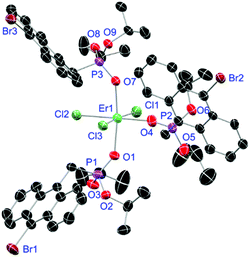 |
| | Fig. 10 Molecular structure of [ErCl3(L3)3] (20). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms and a co-crystallised THF molecule are omitted for clarity. Structural data are given in Table 3. | |
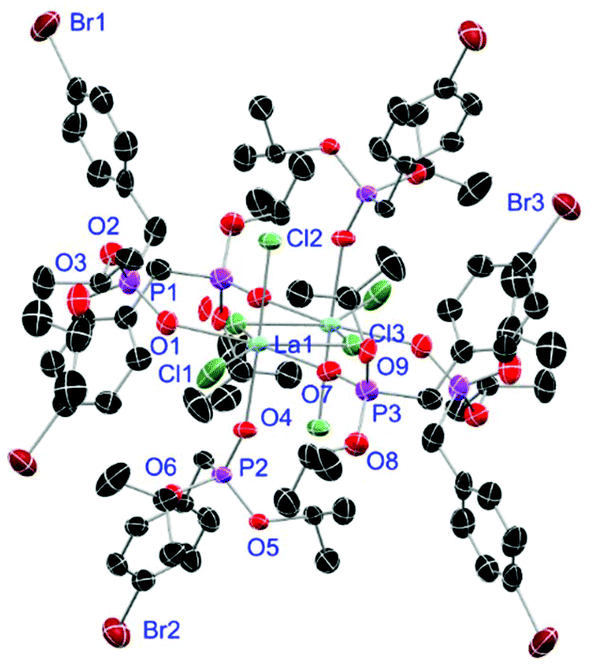
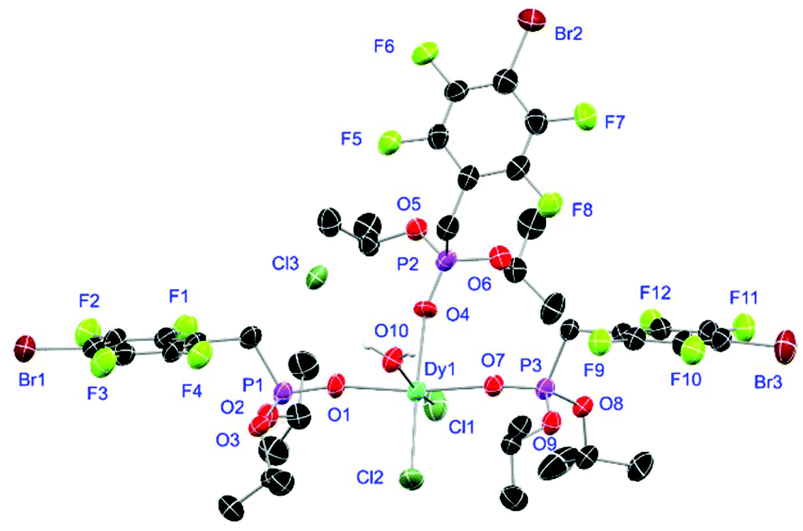
Complexes 31 and 32 are obtained as slowly crystallising waxes. They are isostructural and crystallise as pseudo-octahedrally coordinated monomers (Fig. 11). A chloride anion is replaced by a water substituent in both molecules resulting in the Cl− to function now as counterion forming a partially solvent separated ion pair. Just like in complex 18, the IR spectra of both complexes show no residual OH-bands after workup. In contrast to the free ligand L6, no halogen bonds are formed between the complex molecules in 31 and 32 due to the phosphonate esters now donating electron density towards the lanthanide cations. On the one hand, both compounds show with 2.256(6), 2.574(3) and 1.477(6) Å (31) and 2.209(6), 2.521(2) and 1.457(6) Å (32) the shortest M–O, M–Cl and P+–O− distances among the crystallised complexes. On the other hand, this automatically causes the widest M–O–P angles of 167.0(4)° (31) and 166.9(4)° (32), respectively.
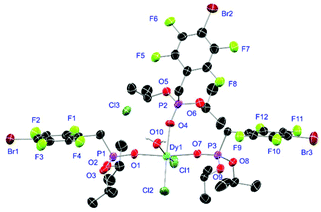 |
| | Fig. 11 Molecular structure of 31, crystallised as [DyCl2(L6)3(H2O)]Cl. Anisotropic displacement parameters are depicted at the 50% probability level. Except for freely refined hydrogens, these are omitted for clarity. Structural data are given in Table 3 (vide supra). | |
Solid-state absorption properties
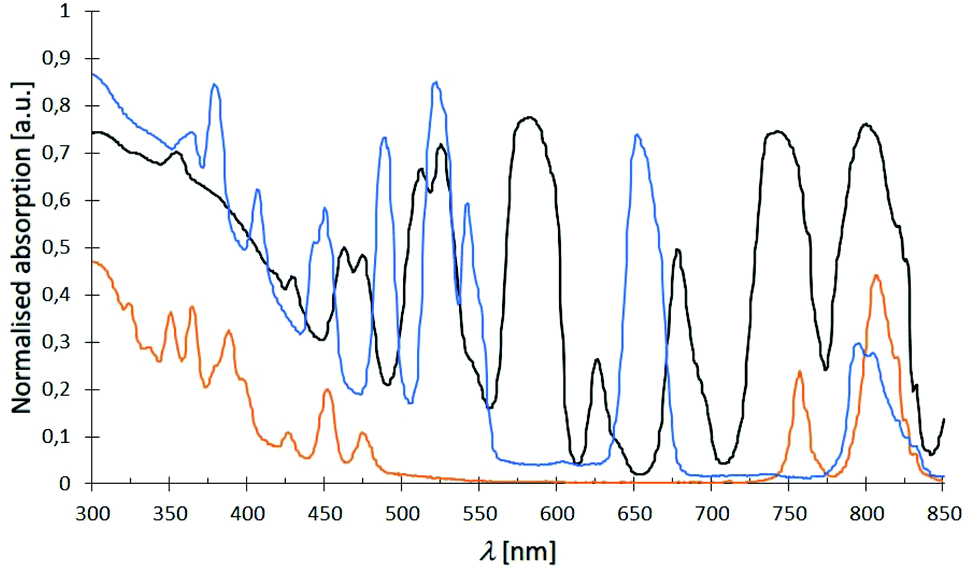
The absorption spectra of complexes 1–32 in the range of 300–850 nm essentially look the same showing expected sharp absorption bands of the Nd3+ (λabs, max = 584 nm), Dy3+ (λabs, max = 811 nm) and Er3+ (λabs, max = 526 nm) cations, respectively. Some exemplary spectra of complexes 2 (black), 3 (orange) and 4 (blue) are summarised in Fig. 12 indicating all three cations to share an absorption around 800 nm (for detailed data, see ESI†). The obtained spectra only differ by an either more or less strong ligand absorption in the blue region that is most pronounced for the L3-based complexes 17–20.
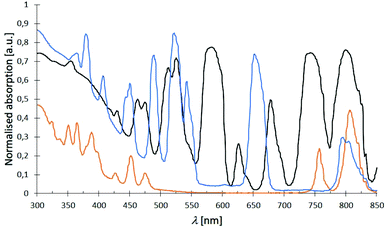 |
| | Fig. 12 Examplary normalised absorption spectra of complexes [Nd(NO3)3(L1)3] (2) (black), [Dy(NO3)3(L1)3] (3) (orange) and [Er(NO3)3(L1)3] (4) (blue) at room temp. with typically sharp Nd3+, Dy3+ and Er3+ absorption bands. | |
Emission properties of the Dy3+ complexes
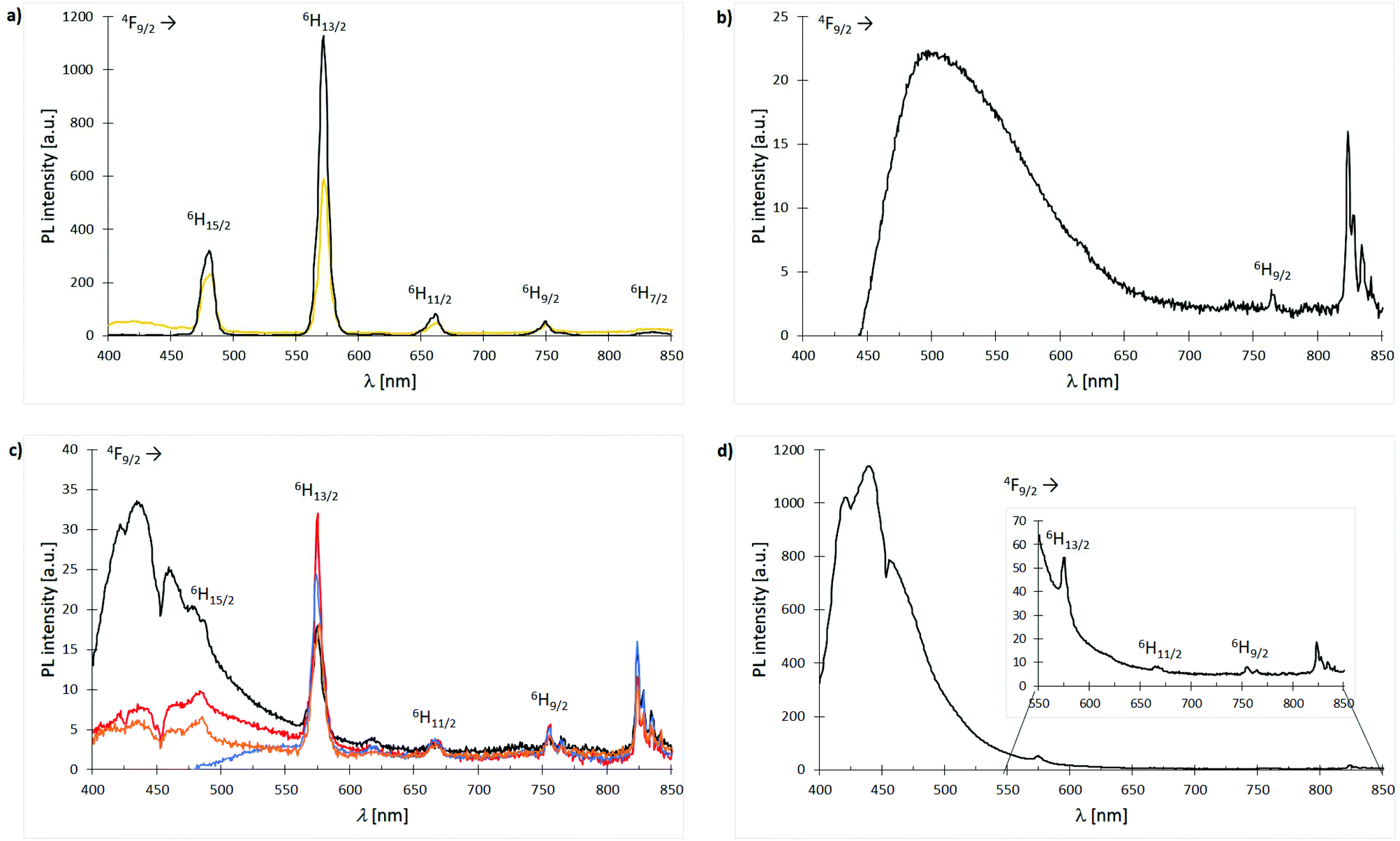
Owing to Laporte-forbidden 4f–4f-transitions, all complexes are expected to show no or only weak metal emissions. An applied excitation at 366 nm affords only in case of the Dy3+ compounds sharp emission bands in the range of 300–850 nm. Nonetheless, the Dy3+ nitrate based complexes 3 and 11 exhibit relatively strong white light emission of the cation with quantum yields of 9% and 13%, respectively which is most likely due to a ligand-to-metal energy transfer (Fig. 13(a)). In turn, the obtained Dy3+ chloride-based complexes 7, 15, 19, 23, 27 and 31 show the expected very weak emissions with no evidence for bands affiliated to ligand-to-metal energy transfer or modified ligand emission. This indicates an enhanced stabilisation of long living excited states within the latter systems. The only difference between the nitrate- and chloride-based Dy3+ complexes is the anions themselves. Hence, this deviating luminescence behaviour can only be rationalised by the NO3− anions promoting a ligand-to-metal energy transfer and fast fluorescence processes. In general, the emission spectrum of anthracene ligand L3-based complex 19 is dominated by the weak but broad ligand emission between 450–650 nm (blue to green) covering most of the metal emissions (Fig. 13(b)). The emission spectra of the Dy3+ chloride complexes 7, 15, 23 and 31 supported by ligands L1, L2, L4 and L6, respectively are very similar exhibiting very weak to weak ligand emissions between 400 to 550 nm and some of the weak Dy3+ emission bands can be assigned (Fig. 13(c), vide infra). The spectrum of complex 27 is dominated by a very strong emission of the 1,1′-biphenyl-based phosphonate L5 between 400–550 nm (blue) covering the 4F9/2 → 6H15/2 emission band of the cation (Fig. 13(d)). After enlargement of the area between 550–850 nm, still some of the remaining, weak Dy3+ emissions could be visualised and assigned.
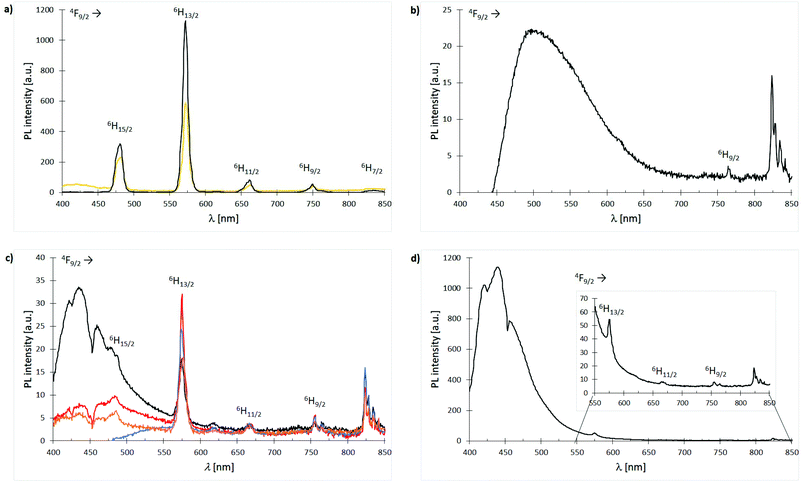 |
| | Fig. 13 Emission spectra (λexc = 366 nm) of Dy3+ complexes 3, 7, 11, 15, 19, 23, 27 and 31 at room temp.: (a) spectra overlay of complexes [Dy(NO3)3(L1)3] (3) (bright orange) and [Dy(NO3)3(L2)3] (11) (black); (b) spectrum of complex [DyCl3(L3)3] (19); (c) spectra overlay of complexes [DyCl3(L1)3] (7) (black), [DyCl3(L2)3] (15) (red), [DyCl2Br(L4-Br)2(L4-Cl)] (23) (blue) and [DyCl3(L6)3] (31) (dark orange); (d) spectrum of complex [DyCl3(L5)3] (27). | |
Emission properties of the Nd3+ complexes
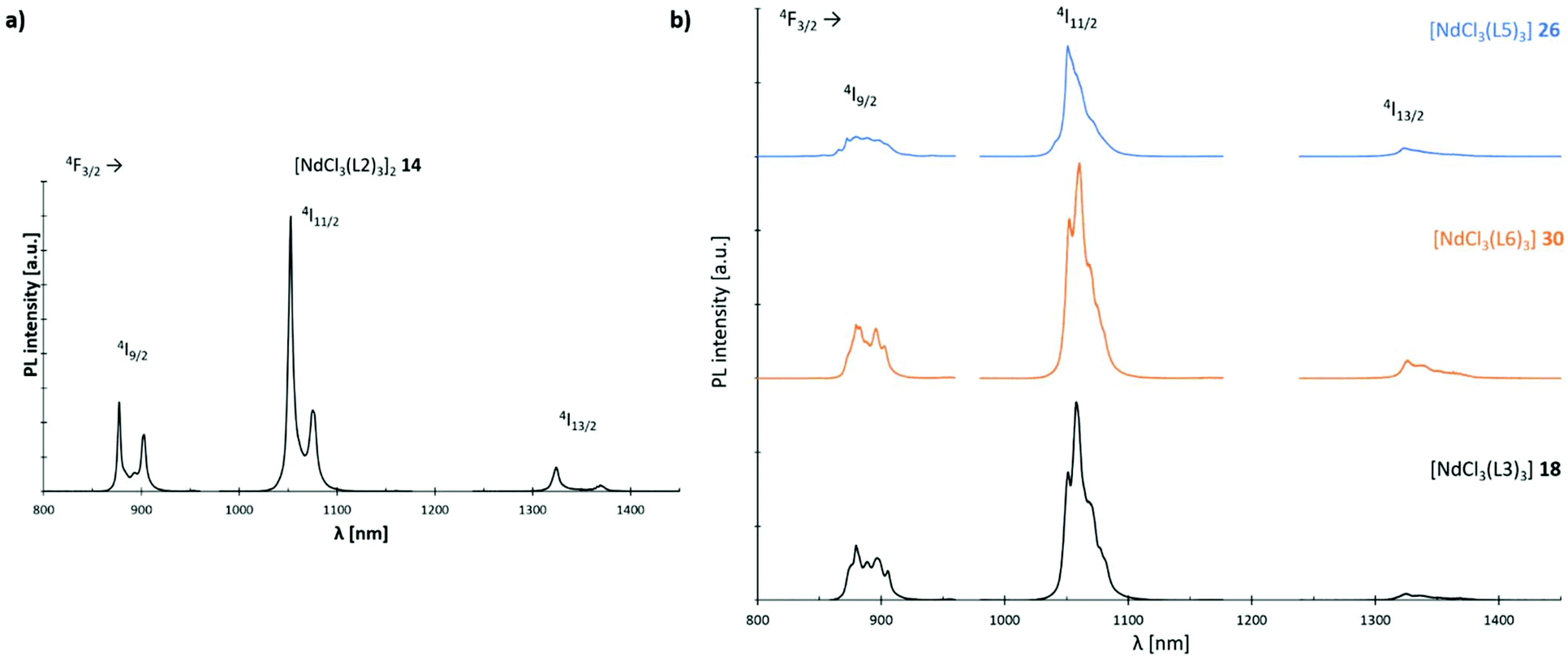
The luminescence properties of L2, L3; L5 and L6 supported Nd3+ chloride-based complexes 14, 18, 26 and 30 are investigated using micro-photoluminescence (μ-PL) spectroscopy. The emission spectra feature the characteristic transitions of the Nd3+ ion in the NIR region (Fig. 14). Three narrow emission bands are illustrated which are centred at ca. 890, 1060 and 1320 nm, that correspond to the 4F3/2 → 4I9/2, 4F3/2 → 4I11/2 and 4F3/2 → 4I13/2 transitions, respectively. The band at 1060 nm is predominant in all four cases. Interestingly, only complex 14 (Fig. 14(a)) exhibits a distinct signal splitting of 25 nm (4F3/2 → 4I9/2, 4F3/2 → 4I11/2) and 47 nm (4F3/2 → 4I13/2), respectively in its PL-spectrum. This feature is most likely due to a preferential formation of dimeric structures in L2-based 14 resulting in a Nd–Nd distance of 4.80 Å. Electrostatic ligand–metal interactions are known to influence lanthanide ion f-shell electrons,38 and as a result to affect their fluorescence response.39–41 In a recent study by Guillou et al., a similar splitting was observed for Eu3+ clusters and tentatively assigned to electronic orbital interactions between f-orbitals of the rare-earth metal centres and, in the case of this cluster, the oxygen-centred orbitals of an adjacent oxo-ligand.42 Such an electrostatic ligand effect could also be observed for the electronic structure of a variety of mono-lanthanide species by Furet et al.38 Hence, the less pronounced splitting in the monomeric complexes 18, 26 and 30 can most likely be assigned as well to such a type of interaction.
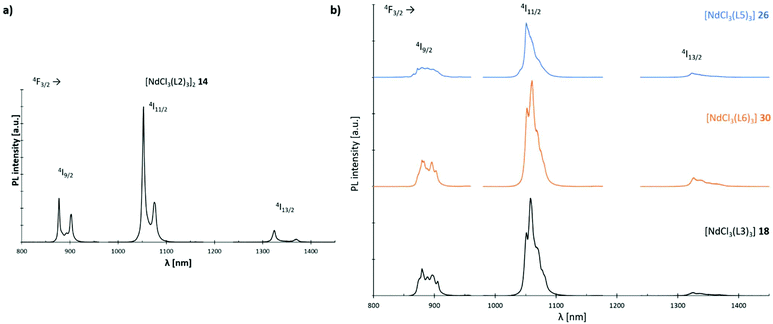 |
| | Fig. 14 Emission spectra (λexc = 750 nm) of Nd3+ complexes 14, 18, 26 and 30 at room temp.: (a) spectrum of complex [NdCl3(L2)3]2 (14); (b) stacked spectra of complexes [NdCl3(L3)3] (18) (black), [NdCl3(L5)3] (26) (blue) and [NdCl3(L6)3] (30) (orange). | |
Experimental section
X-ray diffraction experiments were performed with either a STOE IPDS 2 with an image plate (Ø 34 cm) using a Mo-GENIX source (λ = 0.71073 nm) or a STOE StadiVari instrument with DECTRIS PILATUS 200 K using a Mo-GENIX source (λ = 0.71073 nm). All structures were solved using the dual space method (SHELXT)43 and were refined against F2 with SHELXL44 or OLEX2.45 Additional programs used for structural analysis include Mercury46 and Platon.47 CCDC 2021050–2021063† contain the supplementary crystallographic data for this paper.
For μ-PL experiments, the laser (λexc = 750 nm) is focused on the complexes by a microscope objective (NA = 0.7) to a spot size of ∼1 μm. The same objective was used to collect the emission from the complexes. The luminescence is then spectrally filtered by a 0.75 m focal length spectrometer equipped with liquid nitrogen-cooled CCD and InGaAs detectors. The μ-PL spectra were taken at room temperature with a laser excitation power of 6 mW.
Conclusions
The preparation of six phosphonate ligand systems (L1–L6) carrying differently substituted aryl residues as well as of 32 derived lanthanide complexes (1–32) has been presented. For L1, L2 and L4 modified literature known procedures are introduced while phosphonates L3, L5 and L6 represent completely new ligand systems with an additional bromo functionality for further functionalisation. As a highlight, the tetrafluoro aryl substituted phosphonate L6 shows a unique two-dimensional type I halogen bond network in the solid-state. Substitution reactions with [LnX3(H2O)6] (Ln = La, Nd, Dy, Er; X = NO3−, Cl−) precursor compounds in EtOH yielded the derived model complexes of the form [LnX3(L1–L3 or L5–L6)3]n (X = NO3, Cl; n = 1 (Nd, Dy, Er), 2 (La, Nd)) or [LnCl2Br(L4-Br)2(L4-Cl)]n (n = 1 (Nd, Dy, Er), 2 (La, Nd)) which have been thoroughly characterised. Unfortunately, not all the obtained crystal structures obey to this formula due to additional solvent coordination accompanied by partial ligand replacement during the crystallisation process. The obtained crystal structures show the lanthanide nitrate-based ones to form monomers in the solid-state. The lanthanide chloride-based compounds preferably form chloride bridged dimers for the bigger La3+ cation and the less sterically demanding phosphonates L1, L2 and L6, and tend to form monomers in case of the smaller Dy3+ and Er3+ ions and the bulky anthracene-based ligand L3. For the medium sized Nd3+, monomeric as well as dimeric species are observed. Moreover, compound [Nd(NO3)3(L2)3] (10) is the only complex within this work to show the formation of a two-dimensional halogen bond network in the solid-state which was assigned to a type II halogen bond interaction due to a more perpendicular arrangement of the respective moiety. Absorption spectra of complexes 1–32 have been recorded showing the expected sharp lanthanide absorption bands in all cases. The recording of emission spectra (λexc = 366 nm) of the obtained Dy3+ model compounds shows the respective nitrate-based derivatives 3 and 11 to exhibit a relatively strong white metal emission with quantum yields of 9% and 13%, respectively. In contrast, the chloride-based complexes show only very weak emissions at this excitation wavelength. Without speculation potential reasons for this behaviour must remain elusive until more detailed studies such as excited state lifetime measurements along with quantum chemical calculations shed more light on this issue in the future. The recorded μ-PL-spectra (λexc = 750 nm) of the Nd3+ chloride-based complexes 14, 18, 26 and 30 exhibit a similar behaviour showing weak metal emissions in the NIR region. Nonetheless, interesting splitting features tentatively assigned to electronic orbital interactions between f-orbitals of the Nd3+ centres and the oxygen-centred orbitals of adjacent oxo-ligands are observed.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The federal state of Hesse, Germany is kindly acknowledged for financial support of the SMolBits project within the LOEWE program.
Notes and references
- O. Guillou, C. Daiguebonne, G. Calvez and K. Bernot, Acc. Chem. Res., 2016, 49, 844–856 CrossRef CAS.
- J.-C. Bünzli, Nat. Chem., 2010, 2, 696–696 CrossRef.
- B. Samson, A. Carter and K. Tankala, Nat. Photonics, 2011, 5, 466–467 CrossRef CAS.
- J.-W. Zhang, C.-C. Zhao, Y.-P. Zhao, H.-Q. Xu, Z.-Y. Du and H.-L. Jiang, CrystEngComm, 2014, 16, 6635–6644 RSC.
- Y. Hasegawa, Y. Kimura, K. Murakoshi, Y. Wada, J.-H. Kim, N. Nakashima, T. Yamanaka and S. Yanagida, J. Phys. Chem., 1996, 100, 10201–10205 CrossRef CAS.
- L. Zhang, W. Dou, W. Liu, C. Xu, H. Jiang, C. Chen, L. Guo, X. Tang and W. Liu, Inorg. Chem. Commun., 2015, 59, 53–56 CrossRef CAS.
- K. Krekić, E. Käkel, D. Klintuch, D. Bloß and R. Pietschnig, Z. Anorg. Allg. Chem., 2018, 644, 149–154 CrossRef.
- K. Krekić, D. Klintuch and R. Pietschnig, Chem. Commun., 2017, 53, 11076–11079 RSC.
- K. Krekić, D. Klintuch, C. Lescop, G. Calvez and R. Pietschnig, Inorg. Chem., 2019, 58, 382–390 CrossRef.
- A. Orthaber, M. Fuchs, F. Belaj, G. N. Rechberger, C. O. Kappe and R. Pietschnig, Eur. J. Inorg. Chem., 2011, 2011, 2588–2596 CrossRef.
- A. Orthaber, F. Belaj and R. Pietschnig, J. Organomet. Chem., 2010, 695, 974–980 CrossRef CAS.
- A. Orthaber, F. Belaj, J. H. Albering and R. Pietschnig, Eur. J. Inorg. Chem., 2010, 2010, 34–37 CrossRef.
- A. Orthaber, J. H. Albering, F. Belaj and R. Pietschnig, J. Fluor. Chem., 2010, 131, 1025–1031 CrossRef CAS.
- T. Huang, T. Chen and L.-B. Han, J. Org. Chem., 2018, 83, 2959–2965 CrossRef CAS.
- K. Baczko, W.-Q. Liu, B. P. Roques and C. Garbay-Jaureguiberry, Tetrahedron, 1996, 52, 2021–2030 CrossRef CAS.
- P. Li, M. Zhang, M. L. Peach, H. Liu, D. Yang and P. P. Roller, Org. Lett., 2003, 5, 3095–3098 CrossRef CAS.
- H. C. Li, Y. P. Lin, P. T. Chou, Y. M. Cheng and R. S. Liu, Adv. Funct. Mater., 2007, 17, 520–530 CrossRef CAS.
-
B. A. Arbuzov, V. M. Zoroastrova and G. A. Tudril, Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1970, pp. 90–95 Search PubMed.
- A. E. Arbuzov, J. Russ. Phys.-Chem. Soc., 1906, 38, 687–671 Search PubMed.
- B. Moreau, P.-A. Jaffrès and D. Villemin, Org. Prep. Proced. Int., 2002, 34, 549–552 CrossRef CAS.
- J. H. Clements and S. E. Webber, J. Phys. Chem. B, 1999, 103, 9366–9377 CrossRef CAS.
- F. Leroux, T. U. Hutschenreuter, C. Charrière, R. Scopelliti and R. W. Hartmann, Helv. Chim. Acta, 2003, 86, 2671–2686 CrossRef CAS.
- H. Sato, J. A. Bender, S. T. Roberts and M. J. Krische, J. Am. Chem. Soc., 2018, 140, 2455–2459 CrossRef CAS.
- F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC.
- Y. Hattori, T. Kusamoto, T. Sato and H. Nishihara, Chem. Commun., 2016, 52, 13393–13396 RSC.
- A. Franzke and A. Pfaltz, Synthesis, 2008, 245–252 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- O. Hassel and J. Hvoslef, Acta Chem. Scand., 1954, 8, 873 CrossRef CAS.
- O. Hassel, Science, 1970, 170, 497–502 CrossRef CAS.
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef.
- R. S. Mulliken, J. Phys. Chem., 1952, 56, 801–822 CrossRef CAS.
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- R. S. Mulliken, J. Am. Chem. Soc., 1950, 72, 600–608 CrossRef CAS.
- G. Berger, J. Soubhye and F. Meyer, Polym. Chem., 2015, 6, 3559–3580 RSC.
- L. Brammer, G. Mínguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- F. C. Krebs and T. Jensen, J. Fluor. Chem., 2003, 120, 77–84 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- E. Furet, K. Costuas, P. Rabiller and O. Maury, J. Am. Chem. Soc., 2008, 130, 2180–2183 CrossRef CAS.
- D. Van der Voort, G. J. Dirkens and G. Blasse, J. Phys. Chem. Solids, 1992, 53, 219–225 CrossRef CAS.
- D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS.
- S. K. Mahesh, P. P. Rao, M. Thomas, T. L. Francis and P. Koshy, Inorg. Chem., 2013, 52, 13304–13313 CrossRef CAS.
- F. Le Natur, G. Calvez, J.-P. Guégan, L. Le Pollès, X. Trivelli, K. Bernot, C. Daiguebonne, Ch. Neaime, K. Costuas, F. Grasset and O. Guillou, Inorg. Chem., 2015, 54, 6043–6054 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures & detailed spectroscopic data, NMR spectra, X-ray data, PL spectra. CCDC 2021050–2021063. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03047c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Artur
Lik
a,
Miriam
Gerstel
b,
Clemens
Bruhn
a,
Johann Peter
Reithmaier
a,
Artur
Lik
a,
Miriam
Gerstel
b,
Clemens
Bruhn
a,
Johann Peter
Reithmaier