
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
FSI-inspired solvent and “full fluorosulfonyl” electrolyte for 4 V class lithium-metal batteries†
Weijiang
Xue
 a,
Zhe
Shi
b,
Mingjun
Huang
c,
Shuting
Feng
d,
Chao
Wang
a,
Fei
Wang
b,
Jeffrey
Lopez
e,
Bo
Qiao
ce,
Guiyin
Xu
a,
Wenxu
Zhang
c,
Yanhao
Dong
a,
Rui
Gao
a,
Yang
Shao-Horn
*ef,
Jeremiah A.
Johnson
*c and
Ju
Li
*ab
a,
Zhe
Shi
b,
Mingjun
Huang
c,
Shuting
Feng
d,
Chao
Wang
a,
Fei
Wang
b,
Jeffrey
Lopez
e,
Bo
Qiao
ce,
Guiyin
Xu
a,
Wenxu
Zhang
c,
Yanhao
Dong
a,
Rui
Gao
a,
Yang
Shao-Horn
*ef,
Jeremiah A.
Johnson
*c and
Ju
Li
*ab
aDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: liju@mit.edu
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
cDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: jaj2109@mit.edu
dDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
eResearch Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: shaohorn@mit.edu
fDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
First published on 20th November 2019
Abstract
High-voltage rechargeable lithium-metal batteries (LMBs) require electrolytes that are compatible with both the Li metal anode (LMA) and the metal-oxide cathode. Herein, by imitating the fluorosulfonyl imide group from a well-known LMA-compatible salt, lithium bis(fluorosulfonyl) imide (LiFSI), we come up with an organic solvent dimethylsulfamoyl fluoride (FSO2NC2H6), a fluorosulfonamide (FSA) with two methyl substituents, to develop a new “full fluorosulfonyl” (FFS) electrolyte. Remarkably, it enables a highly reversible LMA with an excellent initial coulombic efficiency (CE) ∼91%, and rapidly approaching 99% within only 10 cycles, with average CE outperforming the well-known LMA-compatible fluoroethylene carbonate (FEC)-based electrolyte. Furthermore, benefitting from its high anodic stability against the oxidative LiNi0.6Mn0.2Co0.2O2 (NMC622) and LiMn2O4 (LMO) surfaces, the Li‖NMC622 cell retains 89% of its original capacity after 200 cycles using a limited Li excess anode. This electrolyte design strategy opens a new avenue for exploring new medium-concentration organic electrolytes for 4 V class lithium-metal batteries (LMBs).
Broader contextThe energy density of rechargeable batteries can be enhanced by pairing a lithium metal anode (LMA) with a commercially available Ni-rich NMC cathode (e.g. NMC622). However, it is a great challenge for the electrolyte to have good compatibility with both the highly-reductive LMA and the oxidative cathode. In this work, we develop a new FFS electrolyte by imitating the fluorosulfonyl imide group from a well-known LMA-compatible salt—LiFSI, to utilize an “FSI-inspired solvent” dimethylsulfamoyl fluoride (FSO2NC2H6) and also dissolving LiFSI in it. FFS electrolyte enables a highly reversible LMA, outperforming the well-known FEC-based electrolyte. The Li‖NMC622 and Li‖LiMn2O4 cells exhibit high capacity retention using a limited Li resource, demonstrating 4 V class LMBs. |
Introduction
Current lithium-ion batteries (LIBs) based on a lithium transition metal oxide cathode (LiCoO2, LiMn2O4, etc.) and a graphite anode (theoretical specific capacity of 372 mA h g−1) are approaching their energy density ceilings1,2 (∼300 W h kg−1), unable to satisfy the increasing demands from electric vehicles, electronic devices, and other markets. A promising approach to boost energy density >400 W h kg−1 is to turn to rechargeable LMBs3–7 based on a LMA and a commercially available Ni-rich NMC (LiNixM1−xO2, M = Mn, Co and x ≥ 0.6) with higher capacity and lower cost than LiCoO2.LMA8 has attracted much attention due to its largest theoretical capacity (3860 mA h g−1) and lowest electrochemical potential (−3.04 V vs. the standard hydrogen electrode) of all possible anodes. It is still a great challenge, however, to achieve stable and efficient cycling of LMBs, which requires both dendrite-free deposition and high Li plating/stripping coulombic efficiency (CE), defined as the ratio between the amount of Li ions plated onto a Li-inventory free substrate (usually Cu) and the amount that can be stripped later. The CE, or rather the coulombic inefficiency9 (CI![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 1–CE), is often used to assess the reversible Li consumption per cycle. To survive for hundreds of cycles using practical industrial battery parameters including lean electrolyte and small negative to positive electrode capacity ratio (N/P ratio), a CE above 99% or even 99.9% would be needed.10–12 Even though the highest CE value achieved is often reported, the average CE (〈CE〉) during entire cycling is more reliable for characterizing the exhaustion of cyclable Li inventory, which could be improved by increasing the initial CE (ICE) and stabilized CE (SCE) as well as by minimizing the essential cycle number (ECN) from ICE to SCE. Imperfect CE (<100%) is often attributed to reactions between the organic electrolyte and the highly reductive LMA surface, forming a passivation layer called the solid–electrolyte interphase (SEI).
1–CE), is often used to assess the reversible Li consumption per cycle. To survive for hundreds of cycles using practical industrial battery parameters including lean electrolyte and small negative to positive electrode capacity ratio (N/P ratio), a CE above 99% or even 99.9% would be needed.10–12 Even though the highest CE value achieved is often reported, the average CE (〈CE〉) during entire cycling is more reliable for characterizing the exhaustion of cyclable Li inventory, which could be improved by increasing the initial CE (ICE) and stabilized CE (SCE) as well as by minimizing the essential cycle number (ECN) from ICE to SCE. Imperfect CE (<100%) is often attributed to reactions between the organic electrolyte and the highly reductive LMA surface, forming a passivation layer called the solid–electrolyte interphase (SEI).
In order to achieve high-efficiency cycling of high-voltage LMBs, the electrolyte must satisfy the following requirements: (1) it must be able to rapidly form a stable, mechanically robust, ionically conductive SEI to isolate the remaining liquid electrolyte from the highly reactive LMA and accommodate large volume changes, thus improving the ICE, ECN and SCE thus yielding a high 〈CE〉; (2) it should favor compact and flat Li deposits, that reduce dendritic growth, improving safety. (3) It must have sufficient anodic stability to support the high voltage cathode (>4.3 V), and do not corrode the Al current collector. Traditional ether/carbonate-based electrolytes do not fulfill all of these requirements because they are either not sufficiently oxidatively stable (>4.0 V) or they are not compatible with LMA (CE < 90%).11 Electrolyte components were reported to greatly affect CE and the Li deposition morphology include LiNO3,13,14 lithium polysulfides,13 Cs+,15 SnTFSI,16 dimethoxyethane (DOL).17 High concentration electrolytes18–23 (HCEs) have also been reported to be an effective way to expand the electrochemical stability window, which benefits both the LMA and the high-voltage cathodes. In this regard, LiFSI, which features a “magic” anion FSI group,24,25 is a popular salt due to its high solubility in many organic solvents and its unique ability to form SEI layers with LiF as a significant component. LiF is mechanically robust,26 has an extremely large bandgap, and so a very thin LiF layer is able to stop electron tunneling and further electrolyte decomposition.11,27,28 The SEI derived from liquid electrolyte decomposition is dominated by the competing reactions between LMA and solvent molecules and salt anions.29 Since most solvent molecules are coordinated with Li+ in HCEs, SEI formation may shift from solvent-dominated decomposition in dilute electrolytes to anion-dominated decomposition18,19,30 in HCE. This means the good CE in LiFSI-based HCEs may be governed by reactions between the FSI− with Li metal. For example, a breakthrough came by incorporating 4 M (M denotes molarity, mol-salt in L-solution, mol L−1) LiFSI in ether solvents,31 which enables high-rate cycling of the LMA with high CE. More recently, our group proposed a novel concept of using highly fluorine-donated electrolyte11 based on 7 m LiFSI in FEC solvent to achieve a reversible 5 V class LMB, where we found the LiF-rich SEI greatly reduces the occurrence of high-aspect ratio Li metal whiskers and promote the formation of equiaxed Li metal grains. Finally, using a high concentration (10 M) of LiFSI in dimethyl carbonate (DMC) led to a great success by significantly improving the Li-compatibility of traditionally dilute (1 M) DMC-based electrolyte.32
In spite of their promising features, the practical use of HCEs is hindered8,33 by their high viscosity, poor ability to wet nonpolar polyolefin separators, and the high cost of Li salts. Although a new class of localized high concentration electrolytes (LHCEs)34–36 obtained by diluting HCEs with “inert” co-solvents may ultimately overcome these challenges, new highly compatible electrolytes for both LMAs and high-voltage cathodes that do not employ high salt concentrations are still urgently needed. This clearly calls for new organic solvents with excellent LMA-compatibility. It has been empirically known that fluorinated solvents14,32–35,37,38 generally exhibit high Li CEs. However, the 〈CE〉s achieved with these systems are still not satisfactory. An explicit molecular design strategy is needed to develop new electrolytes that can fulfill the above requirements and thereby enable high-voltage LMBs.
Given the beneficial properties of the FSI− which has two fluorosulfonyl groups (Fig. 1a), we sought to identify neutral solvent molecules that bear one or more fluorosulfonyl groups similar to FSI−, are liquids at room temperature, are able to dissolve sufficient amounts of Li salts, and have wide electrochemical stability windows. These “FSI-inspired solvents,” when mixed with LiFSI, could produce a new “full fluorosulfonyl” (FFS) electrolytes. After some searching, we identified commercially available N,N-dimethylsulfamoyl fluoride (FSO2NC2H6, FSA) colorless liquid as a promising solvent, that consists of one fluorosulfonamide group with two methyl substituents (Fig. 1a). Sulfonyl fluorides can readily release F atoms, having attracted significant interests in organic synthesis: for example, they are key reagents for recently developed sulfur(VI) fluoride exchange (SuFEx) click reaction,39,40 providing unique reactivity for drug discovery and chemical biology.41,42 A medium concentration FFS electrolyte was obtained by dissolving 2.5 m LiFSI and 0.2 m LiPF6 in FSA (m denotes molality, number of moles of salt per kg of pure solvent). The viscosity and Li+ diffusion coefficient of the FFS electrolyte at 25 °C are 11.875 mPa s and 2.4 (±0.1) × 10−9 m2 s−1 (Fig. S1 and Table S1, ESI†), respectively, which are comparable to the LHCEs.34,35 This novel electrolyte successfully yields compact and flat Li deposits that suppress Li dendrite growth. From both density functional theory (DFT) calculations and experimental analyses, FSA is revealed to possess higher Li-compatibility than another well-known LMA-compatible solvent FEC, which is effective in forming desirable SEI layers.9 FFS electrolyte enables a high Li reversibility as verified by an excellent ICE ∼91%, 〈CE〉 ∼99.03% (averaged from 1st to 400th cycles), and ECN of only 10 cycles to reach CE > 99%. Furthermore, thanks to its high anodic stability >4.5 V, the Li‖NMC622 cell with a limited Li resource exhibits a high capacity retention of 89% after 200 cycles. This “FFS” electrolyte design strategy opens an avenue for exploring new organic electrolytes for 4 V class LMBs.
 | ||
| Fig. 1 Design strategy for the FFS electrolyte. (a) The FFS electrolyte is composed of FSA (solvent) and LiFSI (salt) with fluorosulfonyl groups in both components; (b) schematic diagram of Li growth and SEI formation mechanism in the FFS electrolyte. Relaxed structure and their associated differential charge density plot of FSA (c and d) and FEC (e and f) upon their adsorption on the Li(100) surface, as determined from DFT calculations. The binding energies of Li–FSA and Li–FEC are 0.6 eV and 0.4 eV, respectively. | ||
Results and discussion
As a well-known Li-compatible carbonate, FEC is widely used as an electrolyte additive14,43–45 or sole solvent11 for improving the Li CE, due to its ability to form compact LiF-rich SEI that covers the LMA surface and stops further electron tunneling and electrolyte decomposition. Theoretically, FEC and FSA molecules have similar lowest unoccupied molecular orbital (LUMO) levels (Fig. S2, ESI†) calculated by first-principles density functional theory (DFT). In order to evaluate their electrochemical reduction window in an actual electrolyte environment, linear sweep voltammetry (LSV) was performed on electrolytes with 2.5 m LiFSI in FEC and FSA. Although FEC and FSA exhibit similar stability against reduction, the baseline current of the cell using the FSA-based electrolyte is much larger than that using the FEC-based electrolyte (Fig. S3, ESI†). This result indicates that FSA is kinetically more reactive with LMA than FEC which agrees with the fast reaction of FSI group with Li metal by cleavage of the S–F bond.46 DFT calculations were also performed to gain further understanding of their respective reaction pathways. As depicted in Fig. 1c and e, the primary chemical bond formed during the adsorption is F–Li in both cases. The F–C bond in FEC remains strong, however, after the anchoring of FEC onto the Li surface (Fig. 1d). This result stands in contrast to the case of FSA adsorption where the F–S interaction is obviously weakened upon adsorption (Fig. 1f). The higher binding energy (Eb) for FSA (Eb = 0.60 eV) compared to FEC (Eb = 0.40 eV) suggests that the F in FSA is easier to be donated towards the formation of LiF than that in FEC. Therefore, it would be expected that both FSA and LiFSI can contribute to the formation of LiF-rich SEI layers (Fig. 1b).To better evaluate the influence of these solvents on the reversibility of the LMA, dilute electrolytes are preferable since the Li CE is dominated by reactions between Li and solvents rather than Li and salts. Thus, we first performed Li plating-stripping on Cu foil in 1 m LiFSI in FSA or FEC (“LFF” in short for LiFSI in 100% FEC). It is apparent that 〈CE〉 in the FFS electrolyte (97.87%) is much higher than that in the LFF electrolyte (95.36%, Fig. S4, ESI†), thus FSA exhibits an intrinsically higher Li metal CE than FEC in accordance with the simulation results (Fig. 1). As the LiFSI concentration increases from 1 m to 2.5 m (the room-temperature solubility limit) in FSA, the CE is further improved (Fig. 2a). At a current density of 0.5 mA cm−2, the LMA exhibits a very high reversibility in the FFS electrolyte, which is reflected by the very high ICE ∼ 91% and 〈CE〉 ∼ 99.03% (averaged over 400 cycles) while the CE quickly reaches 99% within only 10 cycles (ECN of 10). In sharp comparison, the LFF electrolyte achieves much lower ICE ∼ 89%, 〈CE〉 ∼ 97.72%, and ECN of 164 cycles (to reach >99%); while the standard electrolyte (“SE”, commercial carbonate-based electrolyte, 1 M LiPF6 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 EC/DMC/DEC by weight ratio) for commercial LIBs shows even poorer compatibility with LMA (〈CE〉 ∼ 51.74%). To our knowledge, the 〈CE〉 of our FFS electrolyte is the highest ever reported for non-ether-based electrolytes (Table S2, ESI†) and its high anodic stability beyond 4.5 V (to be discussed) makes it a very good candidate for LMBs with Ni-rich cathodes. While conventional ether-based electrolytes (except HCEs21,38 and LHCEs36) are well known for their high LMA-compatibility, their electrochemical stability window is limited on the high-voltage end.11,32
:1:1 EC/DMC/DEC by weight ratio) for commercial LIBs shows even poorer compatibility with LMA (〈CE〉 ∼ 51.74%). To our knowledge, the 〈CE〉 of our FFS electrolyte is the highest ever reported for non-ether-based electrolytes (Table S2, ESI†) and its high anodic stability beyond 4.5 V (to be discussed) makes it a very good candidate for LMBs with Ni-rich cathodes. While conventional ether-based electrolytes (except HCEs21,38 and LHCEs36) are well known for their high LMA-compatibility, their electrochemical stability window is limited on the high-voltage end.11,32
 | ||
| Fig. 2 Electrochemical performances of the Li plating/stripping in different electrolytes, FFS electrolyte: 2.5 m LiFSI + 0.2 m LiPF6 in FSA (0.2 m LiPF6 was added to increase anodic stability. Details are discussed in the paragraph related to anode stability.); LFF electrolyte: 2.5 m LiFSI + 0.2 m LiPF6 in FEC; SE electrolyte: 1 M LiPF6 in 1:1:1 EC/DMC/DEC by weight ratio. (a) Li plating/stripping CEs evaluated by Li‖Cu coin cells; (b) The comparison of the cumulative irreversible capacity over 200 cycles between FFS and LFF electrolytes calculated from (a); (c) the voltage profiles and overpotential evolution of the Li‖Cu cell using FFS electrolyte. (d) Li plating/stripping from Li‖Li symmetric cells and corresponding voltage profiles (e and f) in FFS and SE electrolytes. All cells were cycled at a current density of 0.5 mA cm−2 with a capacity of 0.5 mA h cm−2. | ||
An alternative comparison between FFS and LFF electrolytes is presented in Fig. 2b, where the amount of irreversible Li consumption was integrated over number of cycles.9,22 As of 200 cycles, the repeated Li plating/stripping in LFF electrolyte loses nearly twice the amount of reversible Li inventory compared to FFS electrolyte. In addition, Fig. 2c shows the voltage profiles of the Li‖Cu cells at a current density of 0.5 mA cm−2. The overpotential at the 100th cycle is only 42 mV with a very slight increase to 54 mV at the 200th cycle and keeping almost constant afterwards (57 mV at the 350th cycle), suggesting that the SEI on the Li surface in FFS electrolyte is very thin and stable against cycling. Furthermore, the long-term cycling stability of Li‖Li symmetric cells was also evaluated (Fig. 2d). The overpotential of the Li‖Li cell in FFS electrolyte remains at a very low and stable value for 1100 h while in SE electrolyte the overpotential is much higher (Fig. 2e) initially and it fluctuates considerably after 800 h (Fig. 2d and f). The Li plating/stripping in FFS electrolyte remained stable with high efficiency even at higher areal capacities of 1 and 1.25 mA h cm−2 (Fig. S5, ESI†). These results demonstrate that the high reversibility of LMA in FFS electrolyte could be attributed to the robust and stable SEI layers that could suppress LMA porosity and dendrite growths and minimize reactions between Li and the electrolyte.
For SE, a considerable amount of tangled needle-like whiskers with large aspect ratios are observed on the surface of cycled LMAs (Fig. S6a, ESI†). Such high-surface area LMA with harmful parasitic reactions lead to a low CE and serious safety concerns. After switching to LFF electrolyte, the morphology of Li deposits changes to whiskers with smaller aspect ratio (Fig. 3a). Notably, unlike the above SE and LFF electrolytes, the Li metal deposits in FFS electrolyte are larger, more uniform and compact particles with 5–15 μm diameters, without sharp features (Fig. 3b). Such a morphology would significantly reduce not only short-circuit related safety problems but also the growth in porosity and reactive surface area, giving rise to a much higher CE. X-ray photoelectron spectroscopy (XPS) analysis was further conducted to study the SEI components formed on the LMA in different liquid electrolytes. The comparison of F 1s spectra in Fig. 3c clearly reveals that the SEI in FFS electrolyte features a much higher LiF content than that in LFF electrolyte, which is consistent with the discussion about the reaction pathways in Fig. 1. Considering the salt concentration is the same in both solvents, this again confirms that FSA has a much higher propensity to donate F to the Li surface, which agrees with our calculations in Fig. 1. The amounts of carbon and oxygen species (C–H, C–C, C–O and C–O3) are lower in the FFS-derived SEI (Fig. S7, ESI†). Moreover, another pronounced difference in surface chemistry is the obvious presence of lower-valence sulfur species47 (S−/S2−) in the FFS-derived SEI layer (Fig. 3d), which could improve the Li+ conductivity of SEI layers;48 no such species exist in the SEI with LFF and SE (Fig. S6b, ESI†) electrolytes. Compared to oxygen species, the larger ionic radius and greater atomic polarizability of the sulfide ions lead to weaker covalent bonds with Li+, facilitating cation mobility and thus higher Li+ conductivities in sulfide-based solid electrolytes49,50 than in oxide-based solid electrolytes. Previous works showed that the reduction of the FSI anion to S−/S2− components was only observed when a very high LiFSI concentration (10 M) in DMC32 was reached, but here only 2.5 m LiFSI (intermediate concentration of salt) was used. Therefore, we propose that such S−/S2− components could be derived from the reduction of the FSA solvent component of our FFS electrolyte rather than from the LiFSI.
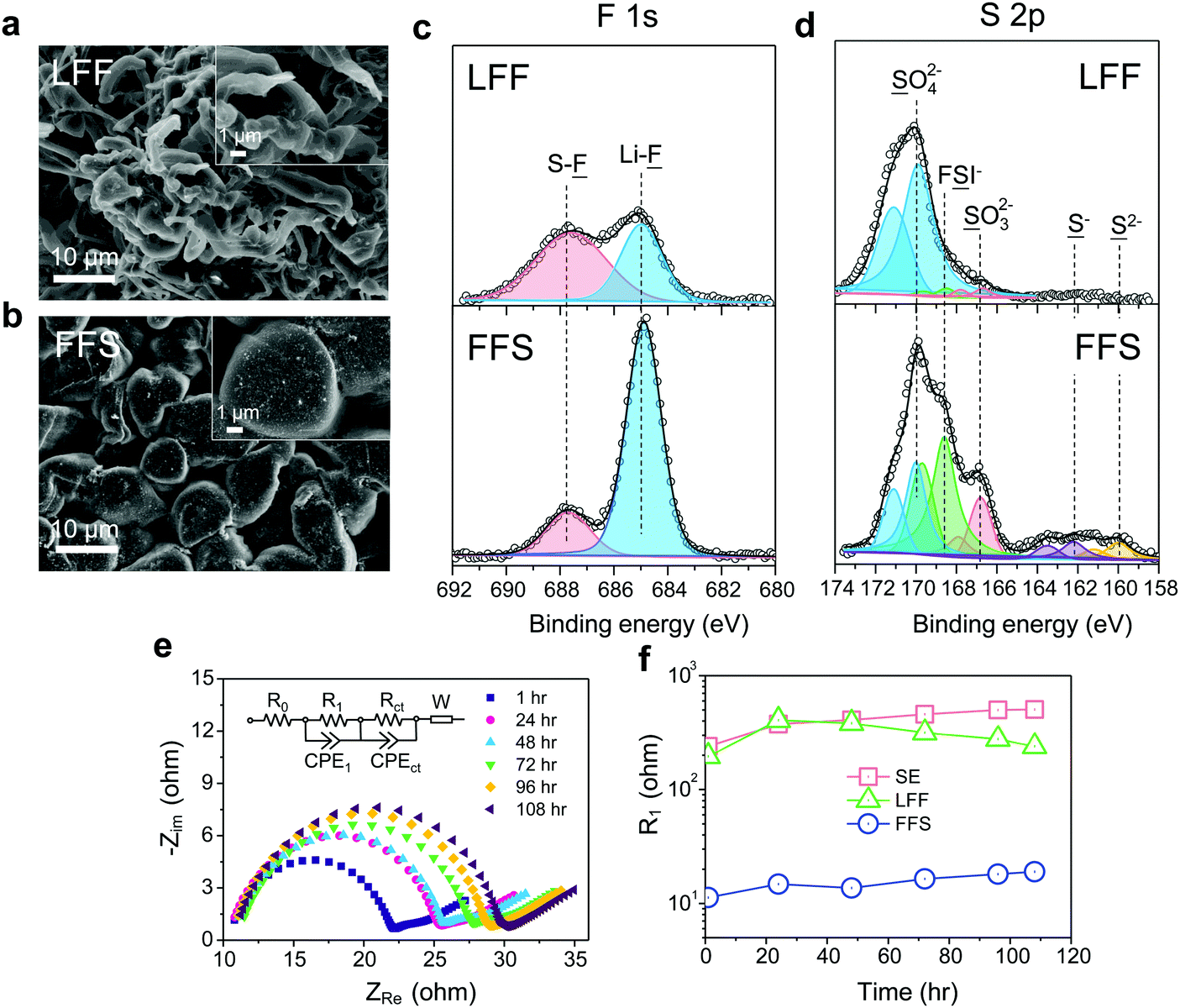 | ||
| Fig. 3 Morphology, surface chemistry and stability analysis of the cycled LMA and SEI layers in different electrolytes. SEM figures of the cycled LMA on Cu substrates in LFF (a) and FFS (b) electrolytes (10 cycles, 0.5 mA cm−2, 0.5 mA h cm−2) with high resolution SEM images inset. XPS analysis of the SEI components, F 1s (c) and S 2p (d) spectra are presented. The dotted lines in S 2p spectra only point out the S 2p3/2 doublets. (e) The evolution of impedance spectra of the Li‖Li symmetric cell with resting time in FFS electrolyte; (f) fitted R1 of the Li‖Li cells at different resting time in FFS, LFF and SE electrolytes. | ||
To evaluate the stability of SEI layers formed in these different electrolytes, we performed electrochemical impedance spectroscopy51 (EIS) on Li‖Li symmetric cells as a function of storage time (Fig. 3e, f and Fig. S8, ESI†). The Nyquist plots were fitted to an equivalent circuit composed of an electrolyte ohmic resistance (R0) with resistances//constant phase elements (R//CPE) and a Warburg element (W). The semi-ellipses from high to medium frequency could be mainly attributed to the resistance (R1) of SEI layer while the second semi-ellipses at medium frequency range are often resulting from the charge transfer resistance (Rct). It is noted that in FFS electrolyte the R1 on fresh Li metal is one order magnitude lower than those in the other two electrolytes (Fig. 3f), suggesting a highly ionic conductive nature of the FFS-derived SEI in accordance with the surface chemistry analysis. R1 slightly increases in FFS electrolyte after 108 h storage while that in SE electrolyte increases much faster, further demonstrating that FFS electrolyte enables the formation of stable SEI layer. After resting for 108 h, large voltage slopes at initial cycles were observed when cycling the Li‖Li cells in LFF and SE electrolytes due to large resistances that must be overcome. In contrast, there is almost no voltage slope in FFS electrolyte (Fig. S9, ESI†).
An electrolyte for LMBs with Ni-rich NMC must exhibit good anodic stability, requiring both high oxidative resistance to >4.3 V and no Al current collector corrosion. The anodic stability of FSA with different salt concentration was evaluated by LSV with Li metal and Al foils as the counter and working electrodes, respectively (Fig. S10, ESI†). The Al oxidation corrosion is effectively suppressed by increasing the LiFSI concentration from 1 m to 2.5 m, which agrees well with the classic interpretation.19 In order to gain a better anodic stability, 0.2 m LiPF6 was also added due to its excellent ability to prevent Al corrosion.52 Thereupon, the oxidation onset is further pushed beyond 4.5 V, exhibiting a good stability within the operating electrochemical window of NMC622 (3–4.3 V). The rate performances of Li‖NMC622 cells in FFS, LFF and SE electrolytes (Fig. 4a) show that the FFS electrolyte outperforms the commercial SE electrolyte, especially at higher rates. The cell using FFS electrolyte exhibits smaller polarization (Fig. 4b and Fig. S11, ESI†) with higher reversible capacities of 166.7 mA h g−1 (C/2) and 156.9 mA h g−1 (1C) compared to 160.6 mA h g−1 (C/2) and 147.7 mA h g−1 (1C) using SE electrolyte. Compared to FFS electrolyte, the cell using LFF electrolyte exhibits similar rate performance at C/4 and C/2, but lower capacity (154 mA h g−1) at 1C (Fig. 4a and Fig. S11, ESI†).
 | ||
| Fig. 4 Electrochemical performances of the Li‖NMC622 cells in FFS, LFF and SE electrolytes. (a) Rate performance of the Li‖NMC 622 cells using FFS, LFF and SE electrolytes. (b) Voltage profiles of the Li‖NMC 622 cells using FFS electrolyte at C/4, C/2 and 1C. Voltage profiles of the Li‖NMC 622 cells using FFS (c) and SE (d) electrolytes at the 1st, 2nd, 50th, 100th and 150th cycle and cycling performance (e). The N/P ratio is ∼7.6. | ||
To demonstrate the effect of Li reversibility on the cycling of LMBs, a thin Li foil (60 μm) was used. The cell using SE electrolyte shows higher polarization and faster capacity decay compared to the one using FFS electrolyte, indicated by the charge–discharge voltage profiles in Fig. 4c and d. Moreover, drastic differences in cycling stability were observed between these two electrolytes (Fig. 4e). The cell using FFS electrolyte exhibits a quite high capacity retention of 89% after 200 cycles while the capacity and CE of the cell using SE electrolyte drops sharply at ∼80 cycles, which matches well with an obvious increase in the resistance originating from LMA (Fig. S12, ESI†). Such sharp drop is due to the rapid exhaustion of cyclable Li inventory when using a poor Li-compatible SE electrolyte. Compared to FFS electrolyte, LFF electrolyte exhibits slightly lower cycling retention of 86% after 200 cycles. Furthermore, we also evaluated the performance of “anode-free” full cells (Cu‖NMC622) using different electrolytes (Fig. S13, ESI†). While the capacity of the cell with SE electrolyte quickly drops to nearly zero after 27 cycles, LFF electrolyte shows a better capacity retention of 22.7% after 45 cycles. In contrast, the cell with FFS electrolyte retains 50.8% of its original capacity after 45 cycles indicating its good compatibility with both the LMA and NMC622. The well-maintained morphology of NMC622 particles also demonstrates the good compatibility of FFS electrolyte with NMC622 cathode (Fig. S14, ESI†). Furthermore, a LiF-rich cathode electrolyte interface (CEI) is also observed after cycling in FFS and LFF electrolytes (Fig. S15, ESI†), derived from the oxidation of the FSA molecules or salt anion leading to formation of a good passivation layer on the cathode surface and eliminating parasitic reactions between the electrolyte and cathode.11,32,33,37 Furthermore, FFS electrolyte also exhibits good compatibility with some other high-voltage cathode materials like LiMn2O453 (Fig. S16, ESI†).
Conclusions
A new electrolyte design strategy inspired by transplanting the useful features of the FSI anion to neutral organic solvent molecule leads to the discovery of a “full fluorosulfonyl” electrolyte (FFS) based on the commercially available FSA molecule. The FSA-based FFS electrolyte shows excellent Li compatibility and does not need high salt concentration to achieve highly reversible Li plating/stripping, which avoids the high viscosity, poor wettability and high cost issues. The FFS electrolyte enables a highly reversible LMA with an excellent initial CE ∼ 91%, which rapidly approaches 99% within only 10 cycles. The 〈CE〉 over 400 cycles outperforms the well-known LMA-compatible FEC-based electrolyte and is the highest 〈CE〉 ever reported for non-ether-based electrolytes. Furthermore, the FFS electrolyte satisfies the high anodic stability requirement for LMBs with Ni-rich NMC and spinel LiMn2O4. Benefitting from its excellent compatibility with both the anode and cathode and the current collector, the Li‖NMC622 cell retains 89% of its original capacity after 200 cycles using a limited Li resource. This electrolyte design strategy opens a new avenue for exploring new intermediate-concentration organic electrolytes for 4 V class LMBs.Experimental procedures
Materials
LiNi0.6Mn0.2Co0.2O2 (NMC622) cathodes were provided by Argonne National Laboratory with areal loading of ∼1.6 mA h cm−2. LiMn2O4 powder and conductive carbon (Super C65) were purchased from MTI Co. LiFSI salt was supplied by KISCO Co. Ltd which was vacuum dried at 80 °C overnight prior to use. The FSA and FEC solvents were purchased from Sigma Aldrich and Alfa Aesar, respectively. All the solvents were treated by molecular sieves for 3 days before use. The commercial standard electrolyte 1 M LiPF6 in 1:1:1 EC/DMC/DEC by weight ratio was purchased from BASF Corporation. All solvents and salts were stored and handled in a glove box filled with ultra-high purity Argon with O2 and H2O level <1 ppm. The electrolytes were prepared by molality (“m”, mol-salt in kg-solvent, mol kg−1). “M” denotes molarity (mol-salt in L-solution, mol L−1). The 60 μm-thick Li on Cu foil (12.2 mA h cm−2, Fig. S17, ESI†) was supplied by China Energy Lithium Co. Ltd. Please find an overview of all abbreviations in Supplementary List, ESI.†
Characterization
The morphology and microstructure were investigated by Zeiss Merlin High-resolution SEM. Phase composition of the NMC622 cathodes before and after cycling was studied by Rigaku Smartlab XRD. Surface chemistry was analyzed by Physical Electronics Versaprobe II X-ray Photoelectron Spectrometer (XPS). For the characterization of samples after cycling, CR2032 coin cells were disassembled in the glove box. Cathodes and anodes were isolated, then washed by dimethyl ether (DME) three times and dried under vacuum for at least two hours before testing. Specially-designed transfer vessel was used to transfer the samples into XPS chamber to avoid the contact with air. The temperature dependencies of viscosity and density were measured using a SVM3001 viscometer (Anton Paar). Pulse-field gradient (PFG) sequence was applied to the FFS electrolyte using a Bruker Avance 500 MHz NMR spectrometer to evaluate the Li+ diffusion coefficient. The NMR peak intensity of 7Li (0 ppm, corresponding to Li+) as functions of the increasing gradient were analyzed using the following equations:| I = I0 exp[−(γGδ)2D(Δ – δ/3)] | (1) |
| G = Gmax·g | (2) |
Electrochemical measurements
LSV was conducted using Gamry electrochemical work station to measure the anodic stability window at a scan rate of 15 mV s−1 using two-electrode configuration (CR 2032 coin cells) with Al foil and Li strip as the working and counter electrodes, respectively. The electrochemical reduction stability of electrolytes was evaluated by LSV at a scan rate of 0.2 mV s−1 using Li‖Cu two-electrode configuration. EIS measurements were performed on Li‖Li symmetric and Li‖NMC622 cells. CR 2032 coin cells (MTI Co.) were assembled and cycled galvanostatically on a Landt CT 2001A battery cycler at room temperature. Li stripping/plating CE was measured based on Li‖Cu cells where a given amount of Li was plated on the Cu foil substrate (MTI Co.) and then stripped until the potential reached 1 V vs. Li/Li+. Li‖Li symmetric cells were also assembled to perform Li stripping/plating between two Li foils. A slurry of 84 wt% LiMn2O4 with 8 wt% Super C65 and 8 wt% PVDF was coated on Al foil (MTI Co.) followed by drying and rolling. Li‖NMC622 and Li‖LiMn2O4 cells were assembled with 60 μm-thick Li on Cu foil (China Energy Lithium Co., Ltd). Celgard 2300 separator was used except for the LFF electrolyte where glass fiber separator was used due to its poor wettability. To standardize the experiments, 40 μL electrolyte was added to each coin cell. For anode free (Cu‖NMC622) cells, 4 mA h cm−2 Li was pre-deposited on Cu foil and then fully stripped to remove possible oxidation layers. The anode free cells were cycled at C/10 charge and C/3 discharge rate.First-principles simulations
Density functional theory (DFT) calculations were used to study the reactivity of F in FEC and FSA. A slab of 4 Li layers (the bottom 1 layer frozen during optimization) was constructed to model the BCC Li (001) surface based on a 4 × 4 × 4 supercell. In all cases, a vacuum region of 15 Å in the direction perpendicular to the Li surface was kept. The Perdew–Burke–Ernzerhof54 exchange-correlational functional and the projector augmented wave method55 as implemented in the Vienna Ab Inito Simulation Package56 was used in our calculations. The DFT-D3 was employed to include the van der Waals interactions.57 A plane wave basis set with an energy cutoff of 520 eV was used to expand the electronic wavefunctions. Due to the large amount of atoms involved in our model, the Brillouin zone integration was performed on a 1 × 1 × 1 Monkhorst–Pack k-point mesh.58 Atomic coordinates were relaxed until the total energy change was smaller than 1 × 10−4 eV. When calculating energy levels of FSA and FEC, including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), B3LYP59 and HSE60 level of theories are utilized. We use binding energy (Eb) to measure the adsorbing ability. Eb is defined as the difference between the total energy of the molecule-adsorbed system (Etotal) and the energy sum of isolated solvent molecule (FEC or FSA) and a clean Li substrate: Eb Emolecule + Esubstrate – Etotal, where a larger value indicates greater adsorbing strength.3 Differential charge density (Δρ) is adopted to describe the charge transfer and bond formation during in the adsorption process. Δρ is defined by subtracting the charge density of the molecule-adsorbed system (ρtotal) by the charge density of isolated molecule and Li substrate: Δρ ρtotal – ρLi – ρsubstrate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to acknowledge the NMC622 cathodes and LiFSI salt provided by US DOE CAMP Facility, Argonne National Laboratory and KISCO Co. Ltd, respectively. This work made use of the MRSEC Shared Experimental Facilities supported by the National Science Foundation under award number DMR-1419807. We acknowledge support by Samsung Advanced Institute of Technology and NSF ECCS-1610806. Jeffrey Lopez acknowledges support by an appointment to the Intelligence Community Postdoctoral Research Fellowship Program at the Massachusetts Institute of Technology, administered by Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the Office of the Director of National Intelligence.References
- W. Xue, L. Miao, L. Qie, C. Wang, S. Li, J. Wang and J. Li, Curr. Opin. Electrochem., 2017, 6, 92–99 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS.
- W. Xue, Z. Shi, L. Suo, C. Wang, Z. Wang, H. Wang, K. P. So, A. Maurano, D. Yu, Y. Chen, L. Qie, Z. Zhu, G. Xu, J. Kong and J. Li, Nat. Energy, 2019, 4, 374–382 CrossRef CAS.
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2019, 4, 551–559 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- C. Niu, H. Pan, W. Xu, J. Xiao, J.-G. Zhang, L. Luo, C. Wang, D. Mei, J. Meng, X. Wang, Z. Liu, L. Mai and J. Liu, Nat. Nanotechnol., 2019, 14, 594–601 CrossRef CAS.
- W. Xue, D. Yu, L. Suo, C. Wang, Z. Wang, G. Xu, X. Xiao, M. Ge, M. Ko and Y. Chen, Matter, 2019, 1, 1047–1060 CrossRef.
- S. S. Zhang, ACS Appl. Energy Mater., 2018, 1, 910–920 CrossRef CAS.
- Y. Jin, S. Li, A. Kushima, X. Q. Zheng, Y. M. Sun, J. Xie, J. Sun, W. J. Xue, G. M. Zhou, J. Wu, F. F. Shi, R. F. Zhang, Z. Zhu, K. P. So, Y. Cui and J. Li, Energy Environ. Sci., 2017, 10, 580–592 RSC.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2017, 3, 16–21 CrossRef.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS.
- C. Fang, X. Wang and Y. S. Meng, Trends Chem., 2019, 1, 152–158 CrossRef.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS.
- X. Q. Zhang, X. Chen, X. B. Cheng, B. Q. Li, X. Shen, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 5301–5305 CrossRef CAS.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS.
- Z. Tu, S. Choudhury, M. J. Zachman, S. Wei, K. Zhang, L. F. Kourkoutis and L. A. Archer, Nat. Energy, 2018, 3, 310–316 CrossRef CAS.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- J. Zheng, J. A. Lochala, A. Kwok, Z. D. Deng and J. Xiao, Adv. Sci., 2017, 4, 1700032 CrossRef.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- L. Suo, Y. S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef.
- X. Ren, L. Zou, S. Jiao, D. Mei, M. H. Engelhard, Q. Li, H. Lee, C. Niu, B. D. Adams, C. Wang, J. Liu, J.-G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 896–902 CrossRef CAS.
- F. Qiu, X. Li, H. Deng, D. Wang, X. Mu, P. He and H. Zhou, Adv. Energy Mater., 2018, 9, 1803372 CrossRef.
- J. Alvarado, M. A. Schroeder, T. P. Pollard, X. Wang, J. Z. Lee, M. Zhang, T. Wynn, M. Ding, O. Borodin, Y. S. Meng and K. Xu, Energy Environ. Sci., 2019, 12, 780–794 RSC.
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef.
- I. A. Shkrob, T. W. Marin, Y. Zhu and D. P. Abraham, J. Phys. Chem. C, 2014, 118, 19661–19671 CrossRef CAS.
- J. Zhao, L. Liao, F. Shi, T. Lei, G. Chen, A. Pei, J. Sun, K. Yan, G. Zhou, J. Xie, C. Liu, Y. Li, Z. Liang, Z. Bao and Y. Cui, J. Am. Chem. Soc., 2017, 139, 11550–11558 CrossRef CAS.
- Z. L. Brown, S. Jurng, C. C. Nguyen and B. L. Lucht, ACS Appl. Energy Mater., 2018, 1, 3057–3062 CrossRef CAS.
- M. J. Zachman, Z. Tu, S. Choudhury, L. A. Archer and L. F. Kourkoutis, Nature, 2018, 560, 345–349 CrossRef CAS.
- X.-Q. Zhang, X. Chen, L.-P. Hou, B.-Q. Li, X.-B. Cheng, J.-Q. Huang and Q. Zhang, ACS Energy Lett., 2019, 4, 411–416 CrossRef CAS.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang, H. C. Kim, Y. Qi, W. Yang, F. Pan, J. Li, K. Xu and C. Wang, J. Am. Chem. Soc., 2017, 139, 18670–18680 CrossRef CAS PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS.
- X. Fan, L. Chen, X. Ji, T. Deng, S. Hou, J. Chen, J. Zheng, F. Wang, J. Jiang, K. Xu and C. Wang, Chem, 2018, 4, 174–185 CAS.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- C. Wang, Y. S. Meng and K. Xu, J. Electrochem. Soc., 2018, 166, A5184–A5186 CrossRef.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 9466–9470 CrossRef CAS.
- J. Dong, L. Krasnova, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- P. K. Chinthakindi and P. I. Arvidsson, Eur. J. Org. Chem., 2018, 3648–3666 CrossRef CAS.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- E. Markevich, G. Salitra, F. Chesneau, M. Schmidt and D. Aurbach, ACS Energy Lett., 2017, 2, 1321–1326 CrossRef CAS.
- Y. Okuno, K. Ushirogata, K. Sodeyama and Y. Tateyama, Phys. Chem. Chem. Phys., 2016, 18, 8643–8653 RSC.
- B. Liu, J.-G. Zhang and W. Xu, Joule, 2018, 2, 833–845 CrossRef CAS.
- A. Budi, A. Basile, G. Opletal, A. F. Hollenkamp, A. S. Best, R. J. Rees, A. I. Bhatt, A. P. O’Mullane and S. P. Russo, J. Phys. Chem. C, 2012, 116, 19789–19797 CrossRef CAS.
- S. Moon, H. Park, G. Yoon, M. H. Lee, K.-Y. Park and K. Kang, Chem. Mater., 2017, 29, 9182–9191 CrossRef CAS.
- H. Chen, A. Pei, D. Lin, J. Xie, A. Yang, J. Xu, K. Lin, J. Wang, H. Wang, F. Shi, D. Boyle and Y. Cui, Adv. Energy Mater., 2019, 9, 1900858 CrossRef.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- R. Tatara, P. Karayaylali, Y. Yu, Y. Zhang, L. Giordano, F. Maglia, R. Jung, J. P. Schmidt, I. Lund and Y. Shao-Horn, J. Electrochem. Soc., 2018, 166, A5090–A5098 CrossRef.
- J. Zheng, M. H. Engelhard, D. Mei, S. Jiao, B. J. Polzin, J.-G. Zhang and W. Xu, Nat. Energy, 2017, 2, 17012 CrossRef CAS.
- M.-J. Lee, E. Lho, P. Bai, S. Chae, J. Li and J. Cho, Nano Lett., 2017, 17, 3744–3751 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02538c |
| This journal is © The Royal Society of Chemistry 2020 |