
Superior efficient rechargeable lithium–air batteries using a bifunctional biological enzyme catalyst†
Linlin
Wang‡
 abc,
Yarong
Wang‡
c,
Yu
Qiao
c,
Shichao
Wu
c,
Xuanzhao
Lu
ab,
Jun-Jie
Zhu
*ab,
Jian-Rong
Zhang
*ab and
Haoshen
Zhou
*abc
abc,
Yarong
Wang‡
c,
Yu
Qiao
c,
Shichao
Wu
c,
Xuanzhao
Lu
ab,
Jun-Jie
Zhu
*ab,
Jian-Rong
Zhang
*ab and
Haoshen
Zhou
*abc
aCollege of Engineering and Applied Sciences, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: jjzhu@nju.edu.cn; jrzhang@nju.edu.cn
bState Key Laboratory of Analytical Chemistry for Life Sciences, Center of Energy Storage Materials and Technology, Nanjing University, Nanjing 210023, P. R. China
cResearch Institute for Energy Conservation, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1, Umezono, Tsukuba 305-8568, Japan. E-mail: hs.zhou@aist.go.jp
First published on 17th December 2019
Abstract
A biological enzyme – Laccase from Trametes versicolor (LacTv) – has been recognized as a highly efficient bifunctional catalyst in a rechargeable lithium–air battery system whose intrinsic pH change during discharge/charge can regulate the in situ recovery of the enzyme, making them an in situ synergistic match, enabling the battery to offer an extraordinarily flat discharge voltage of ∼3.75 V for 120 h, and remarkable durability (cycling steadily over 1100 h) with a narrow voltage gap of ∼0.24 V.
Broader contextLithium–air (Li–air) batteries are regarded as one of the most promising candidates for next-generation energy storage owing to their extremely high theoretical specific energy density. A key issue in further approaching the high theoretical energy density of lithium–air batteries lies in the search for an efficient bifunctional electrocatalyst of the air electrode that is stable in an acidic electrolyte and can withstand pH alterations. However, abiotic electrocatalysts inevitably suffer from decline in activity and stability caused by the intrinsic pH alteration of the Li–air battery system during cycling, even for precious Pt-based catalysts. Biological enzymes that dominate the oxygen-involving bioreactions exhibit vigorous activity over a broad pH range. Hereby, we originally presented an enzyme catalyzed lithium–air battery employing LacTv as a bifunctional biocatalyst, which shows catalytic activity and stability far exceeding that of the benchmark Pt/C. LacTv enables the battery to offer an extraordinarily flat discharge voltage of ∼3.75 V for 120 h, and remarkable durability (cycling steadily over 1100 h) with a narrow voltage gap of ∼0.24 V. Additionally, our electrochemical and spectroscopic results revealed that the intrinsic pH change of the battery during discharge/charge can in situ regulate the reversible transformation of the corresponding ORR/OER catalysis of LacTv for better serving the battery. |
As the energy density of the current benchmark lithium (Li)-ion batteries is fast-approaching its theoretical maximum (380 Wh kg−1),1–6 the development of the so-called post-Li-ion batteries for higher energy density and lower cost becomes ever more urgent,7–12 and Li–air batteries are currently regarded as one of the most promising candidates for next-generation energy storage owing to their extremely high theoretical specific energy density of nearly 3500 Wh kg−1 for nonaqueous and 3800 Wh kg−1 for aqueous ones.10,13–18 In contrast to the non-aqueous Li–air batteries,15,16 which are practically still a Li–O2 battery operating in a closed system,16–19 a hybrid Li–air battery can essentially be operated in the open air and offer a higher cell voltage as well as better ion transfer kinetics, thus leading to a higher system-level energy density due to the elimination of extra components for pure O2.15,18 If the battery starts with an acidic electrolyte, it will not only deliver energy at a higher initial discharge voltage but also avoid the formation of the solid compounds, such as Li2CO3, that occurs in an alkaline medium.15,18–20 The major problem here is that both the oxygen reduction reaction (ORR) in discharge and the oxygen evolution reaction (OER) in charge at the air electrode are sluggish and need to be catalyzed for a reasonable power output and energy efficiency, which necessitates a robust bifunctional catalyst that is catalytically active and chemically stable under the aqueous working conditions of the air electrode, i.e. in an electrolyte with its pH changing over a broad range. However, almost all known oxides and transition-metal alloy catalysts are unstable in acidic electrolytes and can only be used on air electrodes with alkaline aqueous electrolytes, which deliver much lower output cell voltages. So far, the only electrocatalysts stable in acidic electrolyte available are the Pt-based catalysts, which are not only economically untenable but also provide an unsatisfactory cycling performance.15,20–22 It would be ideal to start from a strong acidic electrolyte as it can deliver a higher energy density in general. Unfortunately, the currently available Li+-ion conducting solid electrolyte, namely Li1−xAlxTi2−x(PO4)3 (LATP) – a key component for aqueous Li–air batteries – is unstable under strong acidic conditions, seriously shortening the battery life.15,18,23 Thus, employing weak-acidic electrolyte with buffer capacity that is resistant to dramatic pH change will not only prolong the battery life but also boost the discharge capacity, but, the activity of the practical abiotic catalysts, even for Pt-based catalysts, is inferior in a weak-acidic electrolyte. Consequently, it is likely that a breakthrough in the development of rechargeable aqueous Li–air batteries will lie in seeking an efficient bifunctional electrocatalyst whose bifunctional activity is compatible with the intrinsic pH change of aqueous electrolytes in Li–air battery systems.
Enzymes are known to catalyze various kinds of chemical conversions under near ambient conditions, and are essentially environmentally benign, efficient in energy consumption, and selective towards the specific target compounds as compared to the abiotic catalysts.24–27 Specifically, the biological enzymes that dominate the oxygen-involving bioreactions can exhibit vigorous activity over a broad pH range. Fungal laccases are found to be particularly attractive as they have relatively high redox potentials (0.73–0.79 V vs. NHE)28–30 that are very close to that of the O2/H2O redox couple, exhibit electrocatalytic activity maxima under weak acidic conditions (pH 3–6)31–33 and are capable of direct electron transfer (DET).26,29,34,35 These features rendered them promising electrocatalysts for the ORR under weak acidic conditions.26,36,37 Under near neutral to alkaline conditions, most fungal laccases are found to become inactive toward ORR catalysis due to the inhibition of OH− ions.26,38 This situation might be favorable for the reverse reaction of the ORR, i.e. the OER,39,40 thus raising the intriguing possibility of making laccase-based enzymes as bifunctional–biocatalysts for rechargeable aqueous Li–air batteries.
In this contribution, as the first example, we demonstrate that the fungal laccase, originated from Trametes versicolor (LacTv), exhibits good catalytic activity for both the ORR and OER in a pH range of 3.5–7.4. Further evaluation reveals that this fungal laccase could indeed be an efficient bifunctional biocatalyst for establishing superior efficient rechargeable Li–air batteries with weak-acidic buffer as an electrolyte, which deliver a much higher voltage efficiency and exhibit superior durability even compared with that of the benchmark catalyst of Pt/C. Additionally, our study reveals that the electrocatalytic activity cycle toward ORR/OER of the LacTv can be regulated in situ by the intrinsically changing pH of the electrolyte during the discharge/charge processes in the Li–air battery we devised.
Results
Fig. 1 schematically depicts the configuration of the LacTv enhanced rechargeable Li–air battery. LacTv that is immobilized on the electrode works synergistically with the Li–air battery. Specifically, LacTv catalyzes the ORR during power output when the acidity of the electrolyte changes from weak-acidic to near-neutral while catalyzing the OER on charge when the electrolyte changes from near-neutral to weak-acidic. As the catalytic activity of LacTv toward the ORR is only stable in weak acidic media, it will decline when the acidity of the electrolyte approaches neutral. Unlike in fuel cells, where the catalytic activity of used enzyme is recovered via a separate step of immersion in its favorite media, the LacTv in our rechargeable battery recovers its full catalytic activity toward the ORR in situ during the charging process because the pH of the electrolyte intrinsically returns to the initial weak acidic conditions.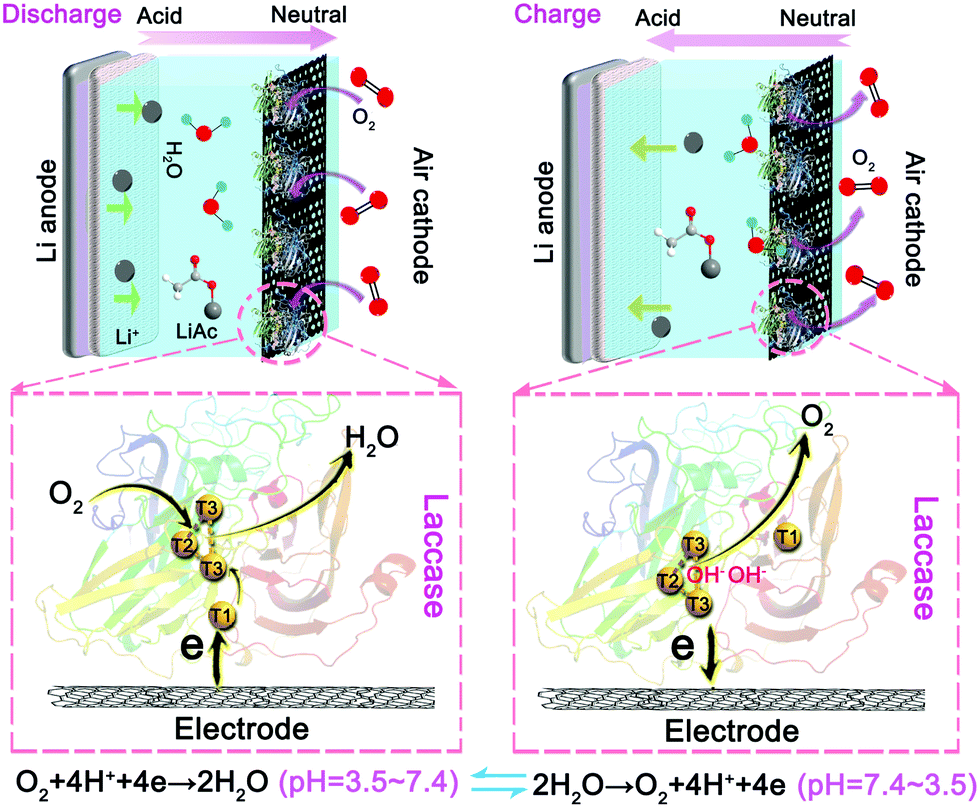 | ||
| Fig. 1 Schematic representation of the configuration and working principle of the LacTv catalytic rechargeable Li–air battery. | ||
A major challenge encountered in utilizing enzymatic electrodes is that the redox active sites of enzymes are usually buried deep within their insulating protein matrix, resulting in relatively long electron transfer (ET) distances between the active sites and the surfaces of the electrodes, hence the exclusively involved mediated electron transfer (MET) in the early research on enzymatic systems.25,26,41 It has been estimated that the maximum distance of electron tunneling between a donor and an accepter is 20 Å while a tunneling distance between redox active sites within a metalloenzyme should be less than 14 Å to support a sufficient electron transfer rate to avoid limiting the redox catalysis.24,42–44 Clearly, for an efficient DET, the prerequisites are: to choose a suitable enzyme and to place its active sites in close proximity to the surface of the electrode.
The fungal laccase from Trametes versicolor (LacTv) is considered to be one such suitable enzyme for DET in that its two active sites, as revealed by its crystal structure, are not too deeply buried within the enzyme to impede the possibility of establishing DET between the enzyme and an electrode (Fig. 2a).25,45 The two redox active sites, the mononuclear T1-Cu site and trinuclear T2/T3-Cu site, are embedded about 6.5 Å and 12 Å, respectively, beneath the enzyme surface, and are 12 Å away from each other.33,36,46 The advantage of LacTv in structure is beneficial for realizing DET between the enzyme and an electrode. Apart from that, the LacTv has the highest redox potential (0.79 V vs. NHE) so far reported for fungal laccases, and shows good catalytic activity for the ORR in a relatively wide pH range.47,48 As for reducing the ET distances between the active sites and the electrode surface, a frequently used strategy is to bind conductive nanomaterials on the electrode to form a porous 3D conducting network, onto which enzymes are immobilized.49 Carbon nanotubes are found to be particularly suitable for enzyme wiring owing to their excellent conductivity, high surface area and robustness.27,34,50,51 For these reasons, we chose to immobilize LacTv via covalent binding onto the surface of single walled carbon nanotubes (SWCNTs) (Fig. 2a) which are fixed on either glassy carbon or carbon paper electrodes. Considering that our LacTv-SWCNT electrode is designed for Li–air battery applications, it must maintain its catalytic activity during long time operation conditions, so the LacTv-SWCNT electrode was further crosslinked by glutaraldehyde, which is capable of forming multi-point covalent bonding with different functional groups (Fig. S1–S3, ESI†). Such crosslinking would add rigidity of the tertiary structure of the immobilized enzyme, consequently enhancing its mechanical as well as chemical stabilities. As the Fourier transform infrared spectroscopy analysis (FTIR) illustrated (Fig. 2b), after the carboxylated SWCNT electrode was incubated with LacTv, peaks at 1670 and 1500 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H vibrational modes of amide groups appeared significantly, confirming the covalent linkage between Lac and SWCNT. Furthermore, the X-ray spectroscopy element mapping of SWCNT and LacTv-SWCNT obtained from the Transmission Electron Microscopy (TEM) images also shows clearly that the N and O distribution in LacTv-SWCNT is obviously higher than that in the SWCNT. In particular, the N and O are found to distribute around the SWCNTs (Fig. 2c). Considering that LacTv is a protein, which is high in N and O, we can conclude that the LacTv is successfully immobilized on SWCNTs.
O and N–H vibrational modes of amide groups appeared significantly, confirming the covalent linkage between Lac and SWCNT. Furthermore, the X-ray spectroscopy element mapping of SWCNT and LacTv-SWCNT obtained from the Transmission Electron Microscopy (TEM) images also shows clearly that the N and O distribution in LacTv-SWCNT is obviously higher than that in the SWCNT. In particular, the N and O are found to distribute around the SWCNTs (Fig. 2c). Considering that LacTv is a protein, which is high in N and O, we can conclude that the LacTv is successfully immobilized on SWCNTs.
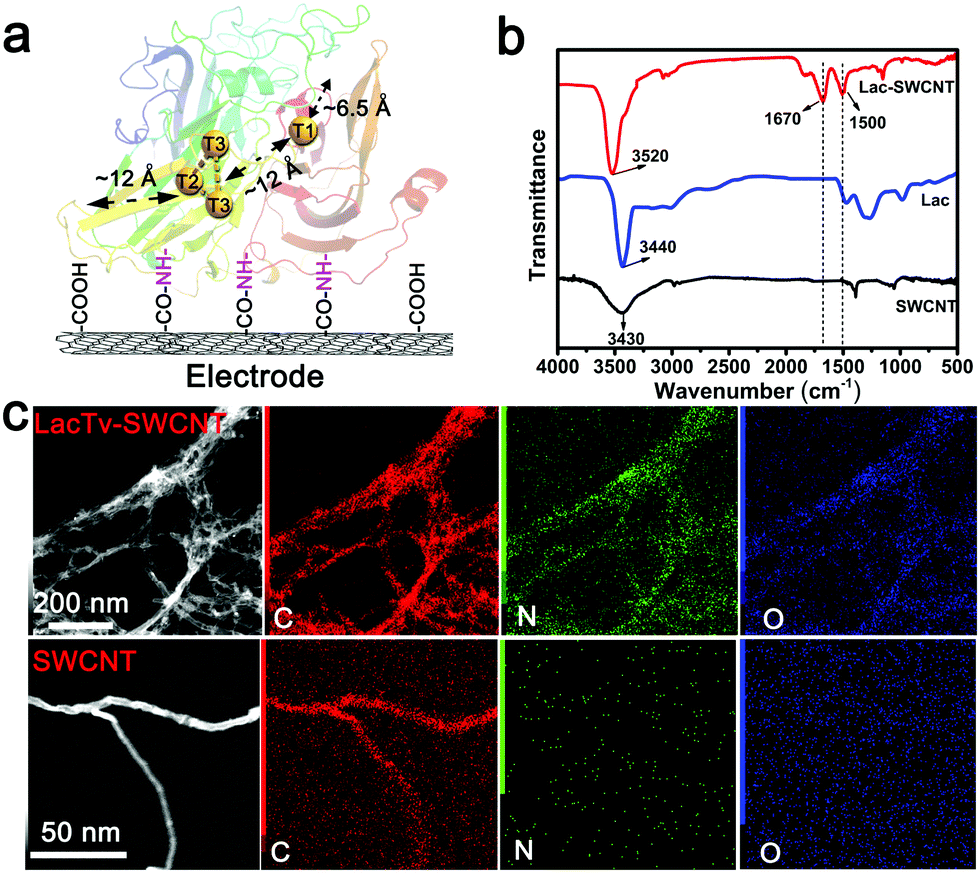 | ||
| Fig. 2 (a) Configuration representation of the Lac-SWCNT electrode. (b) FTIR analyses of SWCNTs (black curve), LacTv (blue curve) and Lac-SWCNTs (red curve). (c) TEM images and elemental mapping of SWCNTs and LacTv-SWCNTs. Dark field TEM images of LacTv-SWCNT (top, gray) and SWCNTs (bottom, gray); the corresponding energy-dispersive X-ray spectroscopy element mapping of C (red), N (green) and O (blue) in LacTv-SWCNT (top) and SWCNT (bottom) materials. | ||
A comparative study of the electrocatalytic activities of the Lac-SWCNTs, 5 wt% Pt/C (denoted as Pt/C) and carboxylated SWCNTs (denoted as SWCNTs) were examined via stationary cyclic voltammetry (CV) as well as linear sweeping voltammetry (LSV) on a rotating disk electrode (RDE) and rotating ring disk electrode (RRDE). The results will be discussed with potentials versus NHE.
For the CV measurement, the electrolyte was adjusted to pH 6.0, where reasonable catalytic activity of LacTv toward the ORR is expected to be observed.28,34 As Fig. 3a shows, the Pt/C electrode exhibits a cathodic peak at a potential of ∼0.48 V, while the Lac-SWCNT electrode shows a cathodic peak at potentials of ∼0.77 V, indicating that the Lac-SWCNT has superior catalytic activity toward the ORR as compared with that of Pt/C. In the case of SWCNTs, only a small cathodic peak at ∼0.26 V is observed, suggesting that the contribution from the SWCNTs in the Lac-SWCNT electrodes should be very limited. The CVs with different scan rates for Lac-SWCNTs (Fig. 3b) showed little potential shift in the cathodic peaks, signifying a fast electron transfer rate and a relatively slow mass diffusion at the electrode. In addition, the estimation of the electrochemical active surface area (ESCA) for LacTv-SWCNTs and Pt/C (Fig. S5, ESI†) revealed that the LacTv-SWCNTs possess a larger ESCA and thus can expose more active sites and promote the adsorption and activation of ORR-relevant species.
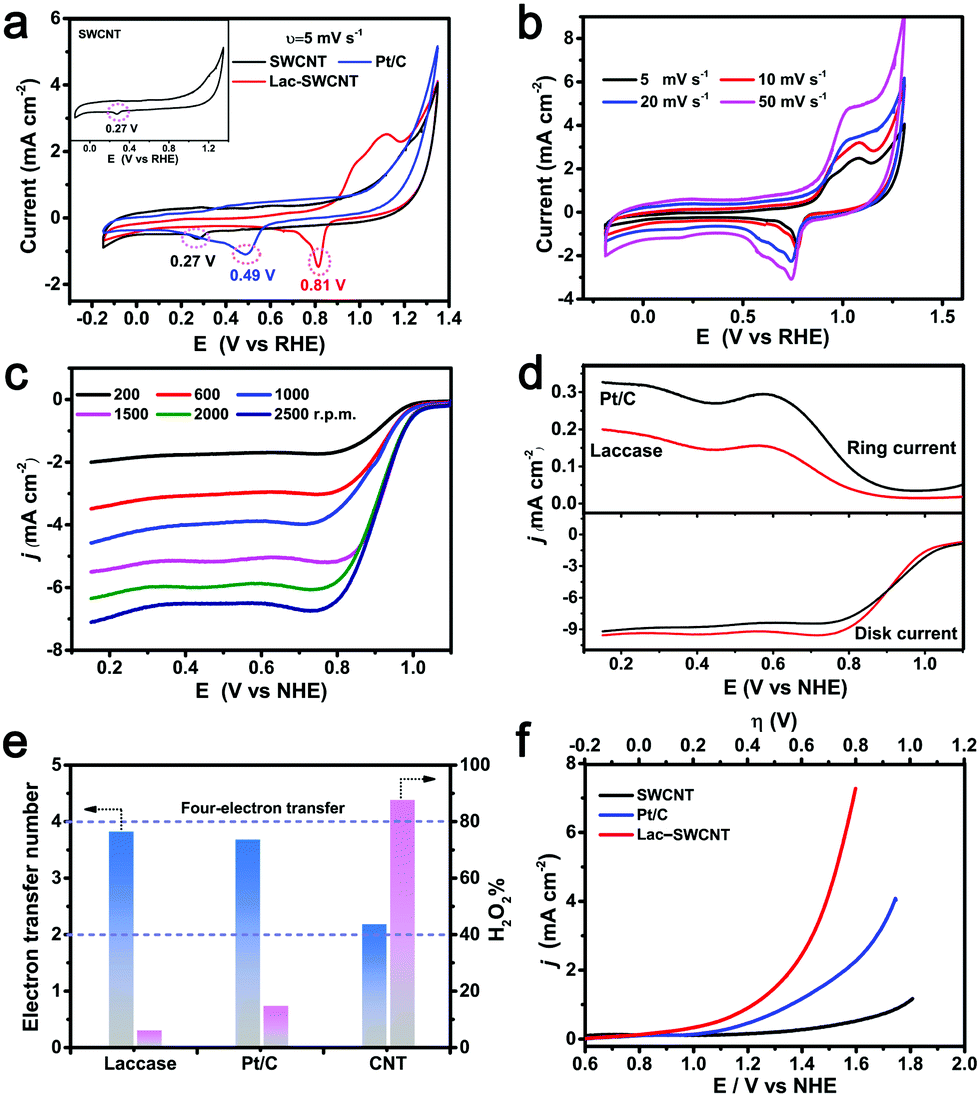 | ||
| Fig. 3 (a) CV curves of SWCNTs, Pt/C and Lac-SWCNTs in a 1 M LiAc-HAc electrolyte (pH = 6.0) saturated with O2 at a scanning rate of 5 mV s−1. (b) CV curves of Lac-SWCNTs at different scanning rates. (c) LSV curves of Lac-SWCNTs in 1 M LiAc-HAc buffer (pH = 3.5) saturated with O2 on a rotating-disk electrode at different rotating speeds. Scan rate (ν): 5 mV s−1. (d) RRDE evaluation of the ORR of Lac-SWCNTs (red) and Pt/C (black). ν = 5 mV s−1, rotating speed: 1600 rpm (e) The electron transfer number (N) and the peroxide yield (H2O2%) calculated according to the K–L equation. (f) The RDE probing of the OER activity of Lac-SWCNTs (red) and Pt/C (blue) and SWCNTs (black) at the rotating speed of 1600 rpm in 1 M LiAc-HAc buffer (pH = 7.4). | ||
For a deeper understanding of the catalytic mechanism of the Lac-SWCNTs, LSV was performed on RDE (Fig. 3b and c), where the oxygen diffusion limitation is excluded. The onset potential of the ORR catalyzed by Lac-SWCNTs was determined to be ∼0.98 V while those for Pt/C and SWCNTs were ∼0.96 and 0.76 V vs. NHE, respectively (Fig. S6, ESI†). Considering that the thermodynamic potential is 1.02 V with the electrolyte of pH 3.5, one would see that the ORR catalyzed by the Lac-SWCNTs and Pt/C can start at potentials very close to the thermodynamic one. In the case of SWCNTs, however, a considerable overpotential loss was observed. Based on the LSV curves swept at different rotating rates (200–2500 rpm), the electron transfer number of the ORR can be derived according to the Koutecky–Levich equations (Fig. S7, ESI†).52,53 The resultant electron transfer numbers are 3.9 for the Lac-SWCNT, 3.7 for the Pt/C and 2.3 for the SWCNT catalyzed ORRs (Fig. 3d), respectively, indicating that the ORR catalyzed by the Lac-SWCNTs and the Pt/C proceeded essentially via the fast direct 4-electron reduction pathway while the ORR catalyzed by the SWCNTs proceeds via a serial 2-electron pathway. This was further confirmed by the yield of the H2O2 intermediate, which is only ∼7.6% or ∼15% in the process of ORR catalyzed by the Lac-SWCNTs (Fig. 3e) or the Pt/C (Fig. S8, ESI†) but over 85% in the case of the SWCNT catalyzed ORR (Fig. S9, ESI†).
The catalytic activity toward the OER was investigated with the electrolyte adjusted to pH 7.4, where most laccases were found to become inactive toward ORR catalysis due to the inhibition of the OH− ions. Such a situation, however, seems to be favorable for the OER. Indeed, when 1.2 V potential was applied, we observed an OER current of ∼247 μA at the Lac-SWCNT electrode, indicating the catalytic activity toward the OER (Fig. 3f). Similarly, a smaller OER current (∼80 μA) has also been observed on the Laccase from Trametes hirsute.54 Moreover, compared with Pt/C, the Tafel slope for LacTv-SWCNT is apparently lower, further confirming the better OER kinetics of LacTv-SWCNT (Fig. S10, ESI†). Despite possessing bifunctional catalysis, Lac remains as a single functional biocatalyst due to the different acidity requirements toward catalyzing the ORR (weak acid) and OER (near-neutral). In addition, no further study has ever been reported on the catalytic behavior and chemical stability of laccase in a real rechargeable battery setting where the pH of the electrolyte is continuously changing, which is crucial for evaluating the viability of laccase as a bifunctional biocatalyst in rechargeable batteries.
We incorporated Lac-SWCNTs in an air electrode and investigated further the catalytic behavior and viability as a bifunctional biocatalyst for rechargeable Li–air batteries. It is well known that the catalytic activities of multicopper enzymes and Pt are strongly affected by the pH of the electrolytes, for example, Pt/C shows its best catalytic activity in strong acidic (pH ≤ 3) electrolytes while LacTv shows both good electrocatalytic activity for the ORR and good chemical stability in a pH range of ∼4–7.35,41,47,48 Besides, in an aqueous Li–air system with abiotic catalyst, the cell voltage is dependent on the intrinsically changing pH of the electrolyte during discharging/charging because water molecules are involved in the reactions at the air electrode. Therefore, we first investigated the effects of the pH on the electrocatalytic performances of the biocatalysts by running a 30 min/30 min discharge/charge process with the electrolytes of pH 3.5–7.4 (Fig. 4a). Surprisingly, for the Pt/C air electrode, the discharge (ORR) started from a cell voltage of ∼3.50 V vs. Li+/Li (pH 3.5), and then dropped dramatically with the increasing pH to 2.76 V vs. Li+/Li (pH 7.4) while the charge (OER) cell voltage only displayed a small gradual change, implying that the OER may be more favored on Pt/C within this pH range. In contrast, for the Lac-SWCNT air electrode, except for the relatively large voltage drop at pH 7.4, the discharge voltage kept so stable at ∼3.75 V vs. Li+/Li from pH 3.5 to pH 6.0 that no remarkable change was observed, while the charge voltage appeared to have a slight increase as the pH changed from pH 7.4 to pH 3.5. The overall very small discharge/charge voltage gap (∼0.25–0.40 V) makes the Lac-SWCNT an apparently ideal bifunctional biocatalyst for the ORR as well as OER in the pH range of pH 3.5 to pH 7.4.
 | ||
| Fig. 4 (a) The pH effect on the voltage of Li–air batteries based on Lac-SWCNTs (red) and Pt/C (black). Current density: 500 mA gcatalyst−1. (b) The full discharge–charge cycles and (c) the corresponding in situ pH change of the electrolyte of the rechargeable Li–air battery based on Lac-SWCNTs at a current density of 100 mA gcatalyst−1. (d) The first short term 10 h/10 h cycling profiles and (e) a comparison of the 10 h/10 h discharge/charge performances of the battery based on different air electrodes under a high current density of 500 mA gcatalyst−1 in a 1 M LiAc-HAc electrolyte with an initial pH value of 3.5. (f) The change of dissolved oxygen level in the electrolyte during a short term 10 h/10 h charge/discharge cycle (insertion). | ||
In order to examine the stability of its electrocatalytic activity, long term discharging/charging with the Lac-SWCNT air electrode was carried out. As shown in Fig. 4b and c, the cycle started with an electrolyte of pH 3.5 (Fig. 4c). At this point, LacTv exhibits high activity towards catalyzing the discharge reaction (ORR), thus allowing the battery to offer a high initial discharge voltage of ∼3.75 V vs. Li+/Li (Fig. 4b). Clearly, the discharge voltage nearly kept stable at ∼3.75 V for ∼120 h, although, the pH gradually increased to ∼5.1 upon continuous discharging for ∼120 h (Fig. 4c). Such favorable discharge stability is attributed to the stable and vigorous activity of LacTv over the pH range of ∼3.5 to ∼5.5 (Fig. 4a) and the retardation of the pH change by using a buffer LiAc-HAc electrolyte (Fig. S11, ESI†). The discharge voltage gradually dropped to ∼3.5 V from ∼3.75 V vs. Li+/Li after discharging for 160 h (Fig. 4c), where the pH of the electrolyte increased to ∼6.6 (Fig. 4c) and the catalytic activity of LacTv toward the ORR is inhibited (Fig. 4a). The battery was then switched to the charge process, during which the pH of the electrolyte would return to its initial value of ∼3.5 (Fig. 4c) after charging for ∼160 h, and evidently, the catalytic activity of LacTv toward the ORR was fully recovered in situ as a stable discharge voltage of ∼3.75 V vs. Li+/Li was delivered in the 2nd cycle of discharge (Fig. 4b). From the stable discharge/charge profiles that were separated only by a very narrow voltage gap (∼0.25 V), one can conclude that the LacTv is indeed a highly efficient bifunctional biocatalyst for both the ORR and OER in the Li–air batteries. Interestingly, the declined catalytic activity of the LacTv seems to have fully restored its vigor in situ when its environment changed gradually back to its initial state upon charging without any other treatment, implying that the catalytic cycle of the enzyme is synergistically matched with the discharge/charge cycle by the intrinsically changing pH of the electrolyte. Another intriguing phenomenon is that the discharge and charge profiles appear to have different responses toward the changing pH: On the one hand, the discharge profile appeared to have kept almost level at ∼3.75 V vs. Li+/Li, independent of the pH change of the electrolyte, except for a small region where the pH of the electrolyte is in the proximity of neutral (around pH 6.0 to 6.6); on the other hand, the charge profile appeared to be an uphill slope that was almost parallel to the thermodynamic change with pH, especially in the first cycle, with only a tiny charge overpotential of ∼0.12 V. This is actually in accordance with the results in Fig. 4d, where only snapshots of some key stages of the whole processes were captured. Such a phenomenon may have a significant implication, i.e. the discharge and charge could have proceeded via different ET pathways.
The durability of the Lac-SWCNT electrode was tested by 10 h/10 h discharge/charge cycling at a current density of 500 mA g−1 (Fig. 4d and e). The Lac-SWCNT electrode exhibited remarkably stable discharge/charge cycling performance with a small discharge/charge voltage gap of ∼0.24 V upon the 56 cycle operations (over 1100 hours) (Fig. 4e), indicating that the Lac-SWCNT not only retained its catalytic activity but was also well immobilized without leaching even after experiencing long term cycling. In contrast, Pt/C showed stable discharge/charge cycling performance up to 35 cycles, yet with a relatively large discharge/charge voltage gap of ∼0.7–0.9 V. Beyond that, however, the voltage gap became even larger due to the peeling of the Pt nanoparticles. The SWCNTs showed negligible catalytic activity toward either the ORR or OER in this instance. Fig. 4d displays the unfolded first cycle. Again, the charge process catalyzed by Lac-SWCNTs exhibits a unique profile that started with a very low charge cell voltage of 3.75 V (∼pH 5.6) and begins to increase after 3 h of charging. Fig. 4f gives the in situ change of dissolved oxygen level in the electrolyte during the charge/discharge cycle. A substantial increase in dissolved oxygen level was observed when applying a current density of 500 mA g−1 to the Lac-SWCNT air electrode and the dissolved oxygen level increased to 11.3 mg L−1 after charging for 10 hours. When switching the cycle to a discharge process for 10 hours, the dissolved oxygen level decreased to about 0.07 mg L−1. The results demonstrate evidently that the LacTv catalyze the OER and the ORR during charge and discharge processes, respectively.
Fig. 5 schematically summarizes the reversible in situ conversion of the ORR/OER activity of LacTv during the discharge/charge cycle in the Li–air battery which can explain the cycling voltage profiles. It has been proven that LacTv is a four-copper enzyme: one T1 Cu (redox potential ∼0.75–0.80 V vs. NHE) and a T2/T3 Cu cluster (redox potential ∼0.35–0.45 V vs. NHE) composed of a T2 Cu and a couple of binuclear T3 Cu.54,55 The T1 Cu, characterized by an intense ∼600 nm absorption that is resulted from a highly covalent S(Cys)–Cu bond, is the site of entry of electrons from various substrates or the electrode.
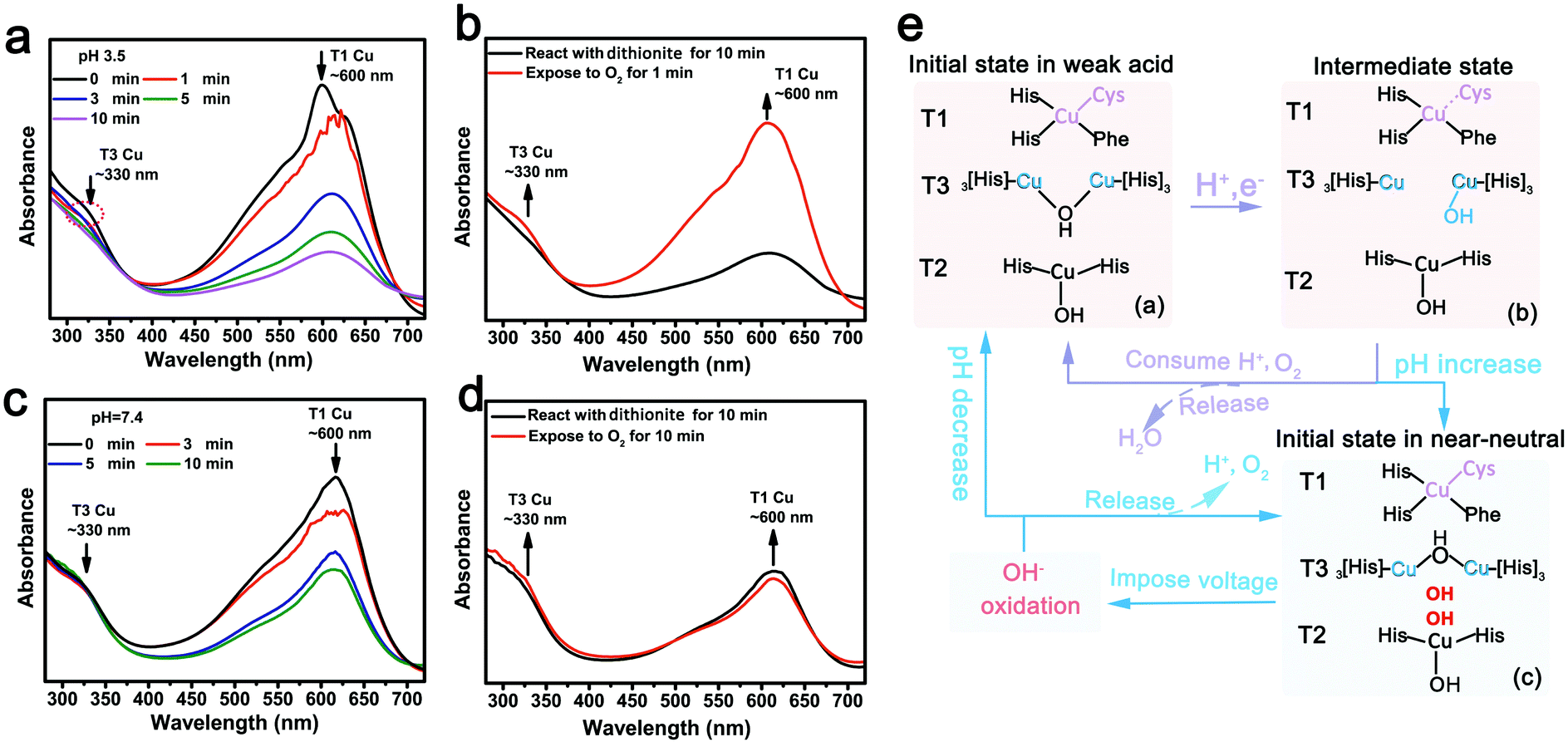 | ||
| Fig. 5 (a–d) Reductive titration of LacTv under anaerobic conditions. UV-vis spectra of LacTv obtained after adding dithionite for different times at pH 3.5 (a) and pH 7.4 (c). UV-vis spectra of LacTv in pH 3.5 (b) and pH 7.4 (d) obtained after adding dithionite for 10 min followed by feeding oxygen. (e) Schematic representation of the reversible in situ conversion of ORR/OER activity of LacTv during discharge/charge cycling in the Li–air battery. | ||
The T3 Cu in the T2/T3 cluster where oxygen can be reduced to water has an intense charge transfer (CT) absorption band at ∼330 nm, originating from a bridging hydroxide ligand.55 At pH 3.5, intense ∼600 nm and ∼330 nm absorption bands are observed (Fig. S13, ESI†), the initial state of the copper active center in LacTv can, therefore, be schematically described as Fig. 5e-(a). To further probe the catalytic intermediates of the copper active center in the LacTv, an anaerobic reductive titration has been performed, from which the electron distributions in the T1 Cu and T2/T3 Cu sites can be obtained by monitoring the added amount of low-potential dithionite with UV-vis absorption. In the absence of oxygen, the copper active centers reduced by dithionite remain stable without being oxidized, enabling such intermediate states of LacTv during ORR to be captured by monitoring their respective absorption changes in the UV-vis spectra. Fig. 5a shows the anaerobic reduction of the T3 Cu (∼330 nm) and the T1 Cu (∼600 nm). Considering that the electron donor, dithionite, is capable of reducing the T1 Cu but not T3 Cu on the minute scale, we can conclude that T1 Cu should be the first electron acceptor. The T1 Cu can quickly transfer an electron to the T2/T3 cluster, where oxygen is reduced to H2O through an intramolecular electron transfer (IET) path.25,56–58 More importantly, as Fig. 5a revealed, the intensity of T1 Cu UV-vis absorption at ∼600 nm decreased but did not disappear, indicating that T1 Cu was partially reduced and the partial fracture of the highly covalent S(Cys)–T1 Cu bond. The absorption of T3 Cu at ∼330 nm in the UV-vis spectra, originating from a bridging hydroxide ligand, disappeared after 10 min of anaerobic reduction, suggesting the complete fracture of the bridging hydroxide ligand at the T3 site in LacTv. Accordingly, we can describe the intramolecular electron transfer process in LacTv during the ORR as such: T1 Cu accepts an electron from the electrode and transfers it to T3, which undergoes an intermediate form as shown in Fig. 5e-(b). Furthermore, we found that the absorption bands at ∼330 nm and ∼600 nm restored to their initial intensity after feeding with O2, suggesting that the re-oxidation of T3 Cu occurred simultaneously with the reduction of O2 at this site. The oxidized T3 Cu can then reaccept an electron from T1 Cu to reduce another O2 molecule. Evidently, the LacTv undergoes an (a)–(b)–(a) cycle of states, as shown in Fig. 5e, during catalyzing the ORR in an acidic electrolyte, which is in accordance with previous studies.54,55 The stability of the vigorous catalytic activity of LacTv over a broad range of pH can be further confirmed with the results of absorbance measurement as shown in Fig. 4a and Fig. S14 (ESI†).
It is understood from the results shown in Fig. 5 that the mechanism of enzyme catalysis is totally different from that of a simple inorganic catalyst. An enzyme, which usually has several redox-active sites and involves inter- and intramolecular electron transfer, behaves more like a mediator immobilized on the electrode, and the ORR is actually mediated by the enzyme. Thus, the electrode potential of the air electrode catalyzed by an enzyme is dependent on the potential of the electron entry site of the enzyme rather than the potential of the ORR. A simple inorganic catalyst, which usually has no redox-active sites and does not involve any intramolecular electron transfer, provides only active sites for the ORR to directly proceed. So, the electrode potential of the air electrode catalyzed by a simple inorganic catalyst reveals the potential of the ORR. This explains why the ORR catalyzed by LacTv shows a flat discharge voltage in the pH range of 3.5 to 6.5 at 3.75 V vs. Li+/Li in Fig. 4a, b and d, which is well consistent with the redox potential of the T1–Cu (0.79 vs. NHE) since the potential of T1-Cu is independent of pH.29,32,38,59 In contrast, the ORR catalyzed by Pt/C shows a clear pH-dependent decline (Fig. 4a). The seemingly flat discharge/charge curves for Pt/C and SWCNT catalyzed ORR and OER resulted from the almost unchanged pH within 10 hours due to the retardation of the pH change by using a buffer LiAc-HAc electrolyte, which can be seen from Fig. 4c and also Fig. S11 (ESI†).
Conversely, the activity of LacTv toward the ORR has been inhibited at pH 7.4 (Fig. S14, ESI†). Hence, the discharge voltage dropped when the pH of the electrolyte increased to neutral upon continuous discharge for about 120 h (Fig. 4b and c). At pH 7.4, the intense absorption bands at ∼300 nm and ∼600 nm still exist without detectable decrease, demonstrating that the highly covalent S(Cys)–T1 Cu bond and the bridging hydroxide ligand at the T3 Cu site remain unchanged. But, in reductive titration, the T3 Cu absorption (at ∼330 nm) fails to show any change with the reduction of the T1 Cu (at ∼600 nm) (Fig. 5c), which means that the T3 Cu is unable to accept electrons from the T1 Cu, i.e. the affinity for an electron of T3 Cu declines. It is well known that the redox potential of T1 Cu (∼0.75–0.80 V vs. NHE) is apparently higher than that of the T2/T3 Cu cluster (∼0.35–0.45 V vs. NHE).60–62 However, our study demonstrated that the IET from T1 Cu to T2/T3 Cu is fast enough not to limit the LacTv catalysis under acidic conditions; only when the pH approaches 7.4 is the IET severely inhibited. This suggests that the electron affinity of the T2/T3 Cu site decreases when OH− accumulation occurs at the T2/T3 Cu cluster, turning the intramolecular electron transfer (IET) from T1 Cu to the T2/T3 Cu cluster into a thermodynamically uphill process, and hence, a rate limiting step.29,38,60 Thus, the discharge voltage drops when the pH of the electrolyte increases to near-neutral upon continuous discharge (Fig. 4b). The initial copper activity center of LacTv in neutral conditions can be depicted as state (c) in Fig. 5e. Owing to the reduced electron affinity of the T2/T3 Cu cluster at neutral conditions, a relatively low potential is needed to oxidize the OH− that has already attached to the T2/T3-Cu cluster, which manifested as a low charge cell voltage started at a neutral electrolyte (Fig. 4b and d). Differing from the flat discharge profiles, the charge cell voltage exhibits a pH-dependent changing trend, which matches the redox potential of the T2/T3 cluster that could be significantly affected by pH due to the existence of OH-bonding.
Noticeably, when recovering the pH back to 5.5 from 7.5, the LacTv exhibits the same vigorous activity as that of the LacTv at pH 5.5 (Fig. S14b, ESI†), proving that the declined activity of LacTv can be fully restored to its initial vigor in situ by simply recovering the initial environment. When the charge process reaches its end point, i.e. the initial state of discharge, the pH of the electrolyte also returns to the initial acidic conditions (Fig. 4c), which brings the catalytic activity of LacTv toward the ORR to its full capacity again. The battery thus reoffers a flat discharge voltage that is almost overlapped with that of the first cycle in Fig. 4b. Compared with the other recently reported hybrid Li–air batteries based on abiotic catalysts (Table S1, ESI†), our Li–air batteries incorporated with the highly-efficient bifunctional biocatalyst LacTv and buffer catholyte can offer an extraordinarily high discharge voltage of ∼3.75 V vs. Li+/Li due to the high redox potential of T1-Cu (∼0.79 V vs. NHE), and extraordinarily long cycle life (56 cycles, over 1120 h) due to the perfect compatibility between the efficient bifunctional activity of LacTv and the intrinsic pH change in the hybrid aqueous Li–air battery system during the discharge/charge cycle. The efficient bifunctional catalytic activity of LacTv demonstrated in our work not only proved that LacTv can be an efficient alternative catalyst but also provided a guidance model for searching for more applicable biocatalysts for advancing rechargeable Li–air batteries.
Conclusions
In conclusion, we recognized the first rechargeable Li–air battery incorporated with an enzyme bifunctional catalyst and a weak-acidic buffer as an electrolyte. The enzyme LacTv exhibits superior efficient bifunctional biocatalysis, far exceeding the catalytic activity of the benchmark Pt/C, for both the ORR and OER. In the rechargeable aqueous Li–air batteries, the LacTv catalyzes the discharge reaction (ORR) in a pH range of 3.5–6.0 during battery output while catalyzing the charge reaction (OER) in neutral conditions upon charging the battery. Benefiting from the highly active bifunctional catalysis of LacTv over a broad pH range, the rechargeable aqueous Li–air battery shows a highly stable and high discharge voltage (3.75 V) over a wide range of pH and a low charge voltage, resulting in superior voltage efficiency with narrow round-trip gap (0.24 V). In the meantime, the use of a buffer electrolyte can retard the pH change of the electrolyte, which maintains a suitable environment for the functioning of the enzyme for a prolonged time, thus leading to a higher energy density. We found that the intrinsically changing pH of the battery can regulate in situ the reversible conversion of the bifunctional catalysis of LacTv for better serving the batteries. So, if the initial and the terminal values of pH can be well selected and controlled, the catalytic activity cycle of the enzyme would work with the pH changing cycle during charge/discharge in perfect compatibility, thus delivering a remarkable durability (steadily cycled over 1100 h). This study not only renovates the practical applications of biological enzymes, but also exhibits an interdisciplinary development of biology and secondary energy storage.Conflicts of interest
The authors declare no conflicts.Acknowledgements
We gratefully appreciate the support from the National Natural Science Foundation of China (21775067, 21834004 and 21633003), and the International Cooperation Foundation from the Ministry of Science and Technology (2016YFE0130100). L. L. W. appreciates Program B for outstanding PhD Candidates of Nanjing University.Notes and references
- C.-Z. Zhao, et al. , Sci. Adv., 2018, 4, 3446–3454 CrossRef PubMed.
- S. Choi, T.-w. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
- P. Tan, B. Chen, H. R. Xu, H. C. Zhang, W. Z. Cai, M. Ni, M. L. Liu and Z. P. Shao, Energy Environ. Sci., 2017, 10, 2056–2080 RSC.
- F. S. Gittleson, R. E. Jones, D. K. Ward and M. E. Foster, Energy Environ. Sci., 2017, 5, 1167–1179 RSC.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- X. F. Yang, X. Li, K. Adair, H. M. Zhang and X. L. Sun, Electrochem. Energy Rev., 2018, 1, 239–293 CrossRef CAS.
- Y. J. Wang, B. Z. Fang, D. Zhang, A. J. Li, P. Wilkinson David, A. Ignaszak, L. Zhang and J. J. Zhang, Electrochem. Energy Rev., 2018, 1, 1–34 CrossRef CAS.
- X. B. Zhu, T. S. Zhao, Z. H. Wei, P. Tana and L. Ana, Energy Environ. Sci., 2015, 8, 3745–3754 RSC.
- Y. Liu, N. Li, S. C. Wu, K. M. Liao, K. Zhu, J. Yi and H. S. Zhou, Energy Environ. Sci., 2015, 8, 2664–2667 RSC.
- C. Xia, C. Y. Kwok and L. F. Nazar, Science, 2018, 361, 777–781 CrossRef CAS PubMed.
- N. Mahne, et al. , Nat. Energy, 2017, 2, 17036 CrossRef CAS.
- Z. Lyu, et al. , Chem. Soc. Rev., 2017, 46, 6046–6072 RSC.
- S. Feng, J. R. Lunger, J. A. Johnson and Y. Shao-Horn, Science, 2018, 361, 758 CrossRef CAS PubMed.
- D. Kundu, R. Black, E. J. Berga and L. F. Nazar, Energy Environ. Sci., 2015, 8, 1292–1298 RSC.
- P. He, T. Zhang, J. Jiang and H. Zhou, J. Phys. Chem. Lett., 2016, 7, 1267–1280 CrossRef CAS PubMed.
- C. T. Zhao, C. Yu, M. N. Banis, Q. Sun, M. D. Zhang, Y. L. Liu, Y. Zhao, H. W. Huang, S. F. Li, X. T. Han, B. W. Xiao, Z. X. Song, R. Y. Li, J. S. Qiu and X. L. Sun, Nano Energy, 2017, 34, 399–407 CrossRef CAS.
- C. T. Zhao, J. N. Liang, Q. Sun, J. Luo, Y. L. Liu, X. T. Lin, Y. Zhao, H. Yadegari, M. N. Banis, R. Y. Li, H. Huang, L. Zhang, Y. Yang, S. G. Lu and X. L. Sun, Small Methods, 2019, 3, 1800437 Search PubMed.
- A. Manthiram and L. Li, Adv. Energy Mater., 2015, 5, 1401302 CrossRef.
- Z. Chang, J. Xu and X. Zhang, Adv. Energy Mater., 2017, 7, 1700875 CrossRef.
- N. Imanishi and O. Yamamoto, Mater. Today, 2014, 17, 24–30 CrossRef CAS.
- L. Li and A. Manthiram, Adv. Energy Mater., 2014, 4, 1301795 CrossRef.
- A. A. Gewirth and M. S. Thorum, Inorg. Chem., 2010, 49, 3557–3566 CrossRef CAS PubMed.
- S. J. Visco, et al. , J. Solid State Electrochem., 2014, 18, 1443–1456 CAS.
- V. Hitaishi, et al. , Catalysts, 2018, 8, 192–198 CrossRef.
- K. Elouarzaki, D. Cheng, A. C. Fisher and J.-M. Lee, Nat. Energy, 2018, 3, 574–581 CrossRef.
- N. Mano and A. De Poulpiquet, Chem. Rev., 2017, 118, 2392–2468 CrossRef PubMed.
- J. H. Park, H. Xue, J. S. Jung and K. Ryu, Korean J. Chem. Eng., 2012, 29, 1409–1412 CrossRef CAS.
- D. M. Mate and M. Alcalde, Microb. Biotechnol., 2017, 10, 1457–1467 CrossRef CAS PubMed.
- M. Dagys, et al. , Energy Environ. Sci., 2017, 10, 498–502 RSC.
- C. J. Rodgers, C. F. Blanford, S. R. Giddens, P. Skamnioti, F. A. Armstrong and S. J. Gurr, Trends Biotechnol., 2010, 28, 63–72 CrossRef CAS PubMed.
- S. Liu, et al. , Adv. Mater., 2017, 29, 1700874 CrossRef PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- M. Tarasevich, A. Yaropolov, V. Bogdanovskaya and S. Varfolomeev, J. Electroanal. Chem. Interfacial Electrochem., 1979, 104, 393–403 CrossRef.
- F. Wu, L. Su, P. Yu and L. Mao, J. Am. Chem. Soc., 2017, 139, 1565–1574 CrossRef CAS PubMed.
- S. Shleev, V. Andoralov, D. Pankratov, M. Falk, O. Aleksejeva and Z. Blum, Electroanalysis, 2016, 28, 2270–2287 CrossRef CAS.
- C. Ji, J. Hou, K. Wang, Y. H. Ng and V. Chen, Angew. Chem., 2017, 129, 9894–9898 CrossRef.
- Y. Yan, W. Zheng, L. Su and L. Mao, Adv. Mater., 2006, 18, 2639–2643 CrossRef CAS.
- S. Shleev, et al. , Electroanalysis, 2012, 24, 1524–1540 CrossRef CAS.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed.
- M. Rasmussen, S. Abdellaoui and S. D. Minteer, Biosens. Bioelectron., 2016, 76, 91–102 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, Chem. Rev., 2013, 114, 3369–3380 CrossRef PubMed.
- C. C. Page, C. C. Moser and P. L. Dutton, Curr. Opin. Chem. Biol., 2003, 7, 551–556 CrossRef CAS PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47 CrossRef CAS PubMed.
- D. Ivnitski and P. Atanassov, Electroanalysis, 2007, 19, 2307–2313 CrossRef CAS.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS PubMed.
- F. Tonin, R. Melis, A. Cordes, A. Sanchez-Amat, L. Pollegioni and E. Rosini, New Biotechnol., 2016, 33, 387–398 CrossRef CAS PubMed.
- S. Kurniawati and J. A. Nicell, Bioresour. Technol., 2008, 99, 7825–7834 CrossRef CAS PubMed.
- N. D. Yates, M. A. Fascione and A. Parkin, Chem. – Eur. J., 2018, 24, 12164–12182 CrossRef CAS PubMed.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
- X. Liu, R. H. Hurt and A. B. Kane, Carbon, 2010, 48, 1961–1969 CrossRef CAS PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. N. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- D. E. Heppner, C. H. Kjaergaard and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 17788–17801 CrossRef CAS PubMed.
- C. H. Kjaergaard, S. M. Jones, S. Gounel, N. Mano and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 8783–8794 CrossRef CAS PubMed.
- M. Pita, et al. , J. Am. Chem. Soc., 2014, 136, 5892–5895 CrossRef CAS PubMed.
- C. H. Kjaergaard, S. M. Jones, S. B. Gounel, N. Mano and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 8783–8794 CrossRef CAS PubMed.
- P. Agbo, J. R. Heath and H. B. Gray, J. Am. Chem. Soc., 2014, 136, 13882–13887 CrossRef CAS PubMed.
- S. Clot, C. Gutierrez-Sanchez, S. Shleev, A. L. De Lacey and M. Pita, Electrochem. Commun., 2012, 18, 37–40 CrossRef CAS.
- F. Xu, J. Biol. Chem., 1997, 272, 924–928 CrossRef CAS PubMed.
- D. E. Heppner, C. H. Kjaergaard and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 17788–17801 CrossRef CAS PubMed.
- E. I. Solomon, et al. , Inorg. Chim. Acta, 1996, 243, 67–78 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02652e |
| ‡ L. L. Wang and Y. R. Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |