
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh carbonate ion conductance of a robust PiperION membrane allows industrial current density and conversion in a zero-gap carbon dioxide electrolyzer cell†
B.
Endrődi
 *a,
E.
Kecsenovity
a,
A.
Samu
a,
T.
Halmágyi
a,
S.
Rojas-Carbonell
b,
L.
Wang
b,
Y.
Yan
b and
C.
Janáky
*ac
*a,
E.
Kecsenovity
a,
A.
Samu
a,
T.
Halmágyi
a,
S.
Rojas-Carbonell
b,
L.
Wang
b,
Y.
Yan
b and
C.
Janáky
*ac
aDepartment of Physical Chemistry and Materials Science, Interdisciplinary Excellence Centre, University of Szeged, Aradi Square 1, Szeged, H-6720, Hungary. E-mail: janaky@chem.u-szeged.hu
bW7energy LLC, 200 Powder Mill Rd, E500-2440, Wilmington, DE 19803, USA
cThalesNanoEnergy Zrt, Alsó Kikötő sor 11, Szeged 6726, Hungary
First published on 21st September 2020
Abstract
A poly(aryl piperidinium)-based anion exchange membrane (PiperION) with high carbonate conductance is employed for CO2 electrolysis to CO in conjunction with a tailored electrolyzer cell structure. This combination results in unprecedentedly high partial current densities in zero-gap cells (jCO > 1.0 A cm−2), while maintaining high conversion (20–45%), selectivity (up to 90%) and low cell voltage (2.6–3.4 V).
Broader contextElectrochemical reduction of CO2 is a promising waste-to-wealth approach, as it converts a greenhouse gas into high value products, utilizing renewable energy. The CO2-to-CO conversion for example, generates a product (with approx. price of 700 USD per tonne) which can be readily used in the (petro)chemical value chain. To bring such technologies to the market, however, significant development is needed in the key performance indicators, leading to viable capital and operational expenditures. Novel catalysts, electrode assemblies, and cell configurations are all necessary to achieve economically appealing performance. While the majority of the scientific community is focusing on catalyst materials and reaction mechanisms, less emphasis has been devoted to cell structures and components. In this work, we presented the implementation of a novel anion exchange membrane in a zero-gap electrolyzer cell, which resulted in record high electrochemical performance. We uncovered the factors underpinning this exceptional performance and presented design concepts for future scale-up. |
Introduction
Electrochemical reduction of CO2 (CO2R) is an eminently attractive candidate technology for the green transformation of the chemical- and energy industries, by turning a harmful waste into valuable chemicals or energy vectors.1,2 To fulfill this promise however, efficient and low-cost electrolyzers need to be developed. In terms of capital expenditures (CapEx), the size and cost of the construction material of the electrolyzer cells/stacks must be minimized. This requires stable operation at high current densities (i.e., reaction rates) and electrolytes that do not require cell components with high corrosion resistance. As for the operating expenses (OpEx), a low cell voltage (i.e., high energy efficiency, EE) and high conversion are both crucial towards the overall cost structure of the product, considering the high fraction of the electricity input and product separation (from the reactant stream) costs. High selectivity is important from a product value perspective.3,4The increased research interest in the past decade in electrochemical valorization of CO2 has been centered mostly at understanding the reaction mechanism and studying new catalyst materials. Notably less attention was dedicated to the other components of the electrochemical cell, and the cell design itself. Gas diffusion electrode (GDE) based studies, feeding CO2 to the cathode in gas phase, have become more widespread only recently.5,6 This approach intensifies the CO2 reduction process by overcoming the mass transport limitation experienced in aqueous solutions.1
The first studies were performed in microfluidic cells, which employ a continuously flowing liquid electrolyte between the anode and the cathode GDE, while CO2 gas is fed from the back-side of the cathode. These studies provided information on the effect of the electrode mass loading, the size of the catalyst particles, electrolyte concentration, pH and composition, temperature, etc. Remarkable progress was observed in the partial current densities for product formation, most importantly for carbon monoxide (CO),7 ethylene8,9 and methane.10,11 While it seemed particularly challenging to reach 100–200 mA cm−2 current density a few years ago, operation at 1 A cm−2 partial current density for different products might be an achievable target in the near future.
At the same time, scale-up of microfluidic devices seems challenging largely due to the increased cross-talk of the two half reactions as the size of the electrolyzer cell increases. Furthermore, the precipitation of salts during operation seems inevitable due to the reaction between the alkaline electrolyte and the CO2 gas. Such precipitate formation hinders gas flow to the catalysts, which is detrimental in achieving long-term operation. Membrane separated electrolyzers circumvent the product mixing problem but add resistance to the system, thus lowering EE. To minimize the resistance increase (inspired by the PEM water electrolysis and fuel cell technologies), the concept of zero-gap electrolyzers evolved. In this configuration, the cathode GDE is pressed to the membrane, and (humidified) CO2 is directly fed to the catalyst through a gas diffusion layer (GDL). The schematic illustration of such electrochemical cells is shown in Fig. 1A.
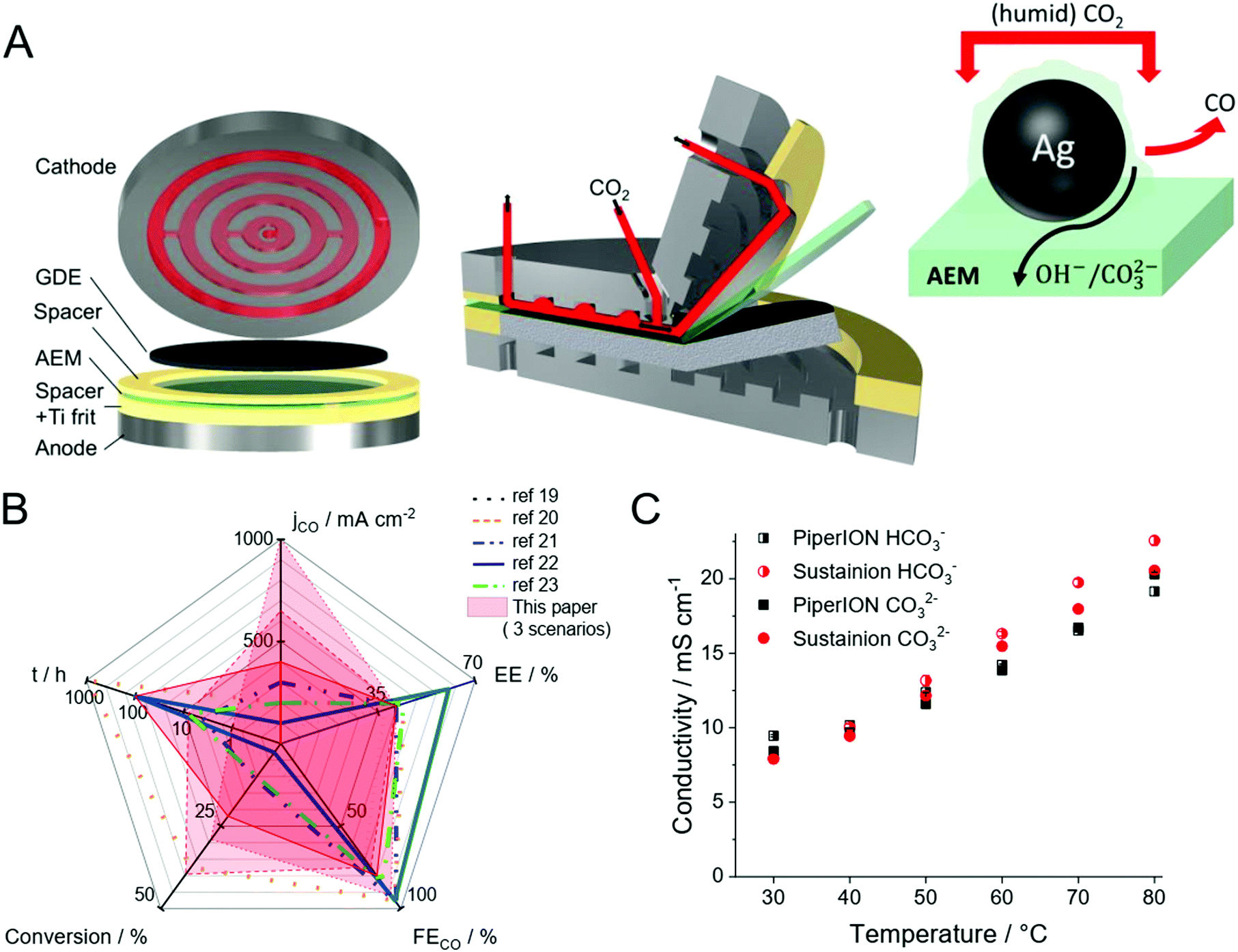 | ||
| Fig. 1 (A) 3D view of a zero-gap electrolyzer cell, together with the illustration of the triple-phase boundary. (B) Spider chart comparing the performance metrics constructed from the data published for zero-gap CO2 electrolyzers (lines), in each case showing the longest presented measurements.19–23 The results presented in this work are shown in red colored areas. Three different scenarios shown from this paper: solid: long-term operation at ΔU = 3.2 V with 0.01 mol dm−3 CsOH anolyte and u = 12.5 cm3 min−1 cm−2 CO2 feed rate; dashed: chronoamperometric measurement at ΔU = 3.4 V with 0.1 mol dm−3 CsOH anolyte and u = 12.5 cm3 min−1 cm−2 CO2 feed rate; dotted: chronoamperometric measurement at ΔU = 3.4 V, with 0.1 mol dm−3 CsOH anolyte and 25 cm3 min−1 cm−2 CO2 feed rate (C) comparison of membrane conductivities for HCO3− (half-filled) and CO32− (filled) ions, for 32 μm thick PiperION TP-85 and Sustainion® X37-50 membranes. | ||
If properly designed, zero-gap electrolyzers can offer operation at lower cell voltages, which results in higher EEs. Moreover, higher single pass conversion (number of formed CO molecules divided by the total number of CO2 molecules fed into the cell in this case) values are expected, as the CO2 gas can be supplied in the GDE more effectively. Interestingly, our literature survey on recent CO2-to-CO conversion studies employing zero-gap electrolyzer cells where at least 100 mA cm−2 partial current density was reported, didn’t reveal such pronounced advantages (Fig. 1B) (in each case the results of the longest presented experiment is shown on the spider chart, even if higher current density transient operation was also shown in the original publications). Instead, the overall trend is that zero-gap electrolyzers tend to operate with smaller current densities, but the total cell voltage and single pass conversion values are similar to studies using liquid catholyte.7,12–17 In general, jCO remains below 400 mA cm−2 in most studies with zero-gap electrolyzers (there is only one single report on jCO over 400 mA cm−2 employing the Sustainion® membrane18). Since very similar cathode GDEs (containing Ag nanoparticle catalysts), anode and anolytes are employed in most studies (irrespective of the cell architecture), we can assume that something else has been limiting the achievable current density in the zero-gap design. Among the multitude of possible factors, here we focus on the role of the membrane and the cell design.
In principle, cation-, anion-exchange and bipolar membranes can be used in zero-gap electrolyzers, however, the best performances were reported with those employing anion-exchange membranes (AEMs).1,24 At the same time, the knowledge, best practices and even the selection of AEMs are still limited in CO2R, compared to water electrolysis and fuel cell technologies. Furthermore, due to the very different operational conditions, new challenges are also faced, among which the presence of CO2 is the most important one.25–27 By analyzing the anode gas composition during CO2 reduction, HCO3− and CO32− were identified as majority charge carriers responsible for the ion conduction through the AEM (and not OH− such as in water electrolysis), due to the rapid formation of HCO3− and CO32− from OH− in a CO2 rich environment.28,29 This observation suggests that a membrane with high carbonate/bicarbonate conductivity is a prerequisite for high current density AEM CO2 electrolysis. This might seem counter-intuitive, as carbonate conduction leads to reactant loss to the anode, which on the system level invokes extra costs associated with the regeneration of the anolyte, and/or CO2 separation/loss from the anode gas. Cation exchange membranes or bipolar membranes might mitigate this issue, but studies with both high current density and energy efficiency are still lacking in such arrangements.19
In microfluidic cells, CO2 is continuously passed through a channel behind the GDE to supply reactant to the cathode (flow-by concept). In contrast, zero-gap electrolyzer cells encompass a GDE pressed between a current collector (having a gas-flow pattern) and the membrane. By carefully designing the gas-flow channels and controlling the compression of the GDE (i.e., maintain proper electronic contact without blocking the pore structure or breaking the structure) CO2 gas can be forced into the GDE (flow-through concept, Fig. 1A). This approach leads to a very high local CO2 concentration at the catalyst surface and increased reactant residence time, together allowing high single-pass conversion efficiency. In this configuration, a triple-phase boundary (the ionomer-coated catalyst/water/CO2 gas) is formed in direct contact with the membrane. Controlling the pressure of the reactant is an effective way to further increase the reaction rate, selectivity, and conversion efficiency.17,23,30 Importantly, zero-gap electrolyzers in principle allow operation at elevated pressure, because the membrane and the cathode GDE are supported from both sides. The best practices of the water electrolysis community can therefore be employed in this case, allowing simple system control. By contrast, in case of the microfluidic reactors, the liquid and gas pressures must be simultaneously and precisely controlled (not to break any components which are not mechanically supported from both sides), requiring very sophisticated equipment and protocols, which also makes scale-up challenging.
In this communication, we present how a new anion-exchange membrane (PiperION, see structure in Fig. S1, ESI†) with high carbonate-ion conductivity (Fig. 1C) was integrated into a custom zero-gap electrochemical cell (Fig. 1A). The synergistic combination of this novel AEM with a carefully tailored cell design led to unprecedented performance. Depending on the operational parameter, high current densities (over 1.0 A cm−2, with 39.1% EE and 29% single pass conversion) or over 39% single-pass conversion efficiency (0.65 A cm−2, 41.6% EE) can be achieved, as also visualized in the spider-chart (Fig. 1B).
Results and discussion
A custom-designed direct gas feed, zero-gap cell was employed in all experiments (Fig. 1A). The PiperION membrane was mounted between a cathode GDE (Ag nanoparticles immobilized on a carbon paper) and a 2 mm thick, IrOx-coated Ti frit anode, with the catalyst layers facing the membrane. Flow-channels were formed in the current collectors, directly pressing the electrodes together. Importantly, a CO2 gas inlet was formed in the center of the circular cathode current collector, while the outlet channel was on the perimeter. The cells were designed and assembled in a way to compress the cathode GDL by ca. 15%. This ensures proper electronic contact between the components without damaging the carbon paper structure and also forces the CO2 gas in the pore system (see the respective MicroCT image and electrochemical measurements in Fig. S2, ESI†). Further compression of the GDE causes unstable cell operation, while HER is favored over CO2R at lower compression ratios. All further experimental details are given in the ESI.†The electrochemical behavior of the cell was first probed by linear sweep voltammetry (LSV, Fig. S3, ESI†). Starting from ΔU ≈ 2.2 V, the onset voltage of the CO2R, there is a linear current increase. This value is very similar to the onset voltage found in our earlier study under similar experimental conditions, but with a different AEM.23 Performing the same measurements with both 0.1 M KOH and 0.1 M CsOH anolyte, about 20% larger currents were observed in the latter case, in agreement with earlier studies in microfluidic cells on the effect of the anolyte composition.31 Importantly, the current density reaches 200 mA cm−2 with the CsOH anolyte already at ΔU = 2.6 V, which was therefore chosen as the minimum cell voltage for our further experiments. All further data is presented for the CsOH anolyte throughout the manuscript.
The product distribution was analyzed during chronoamperometric measurements performed at different cell voltages (Fig. 2A and Fig. S4, ESI†). No detectable amount of liquid product formed, while CO and H2 were identified during these measurements, together accounting for the total 100% Faradaic current within the experimental error in all experiments. The CO formation rate (i.e., the partial current density, jCO) and the CO2 conversion increases with the cell voltage. The CO/H2 ratio remains around 4–7 in the investigated voltage range, resulting in 75–85% Faradaic efficiency (FE) for CO production. Under these conditions, the maximum CO formation partial current density was 630 ± 80 mA cm−2 (exceeding the results of all previous studies with zero-gap electrolyzers) and the full-cell energy efficiency was 40% (Fig. 1B). Importantly, this current density and CO2 feed rate (12.5 cm3 min−1 cm−2) translates to ∼40% single pass conversion efficiency, which was also validated by obtaining the complete CO2 balance of the process (see also Table S1, ESI†).
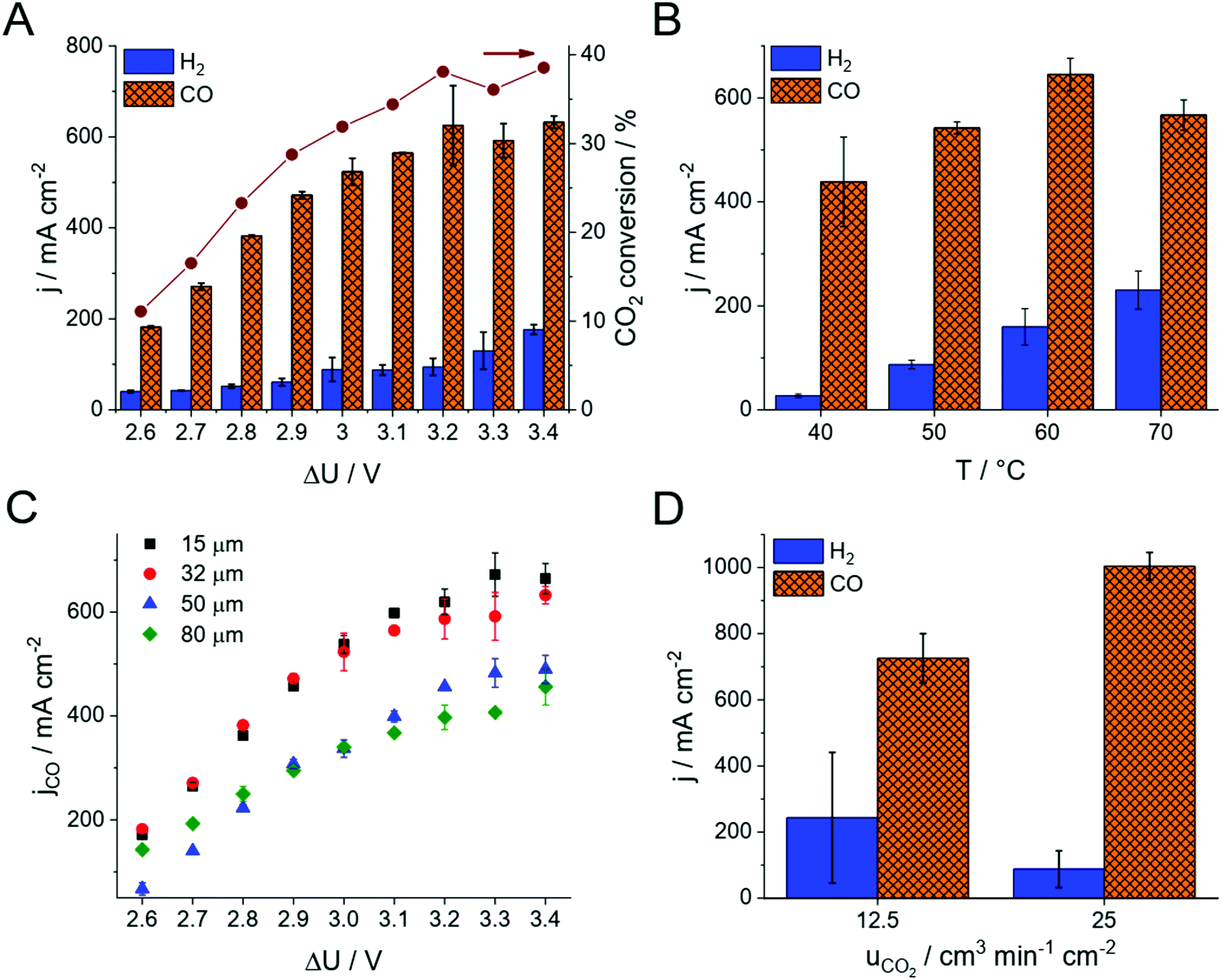 | ||
| Fig. 2 Partial current densities for CO and H2 formation during chronoamperometric measurements using 0.1 M CsOH anolyte, at (A) Tcell = 60 °C and different cell voltages (32 μm PiperION, 12.5 cm3 min−1 cm−2 CO2 feed rate) (B) at different cell temperatures at ΔU = 3.2 V (32 μm PiperION, 12.5 cm3 min−1 cm−2 CO2 feed rate) (C) at Tcell = 60 °C and different cell voltages, using PiperION membranes of different thickness (12.5 cm3 min−1 cm−2 CO2 feed rate), (D) at ΔU = 3.4 V, with different CO2 feed rates (15 μm PTFE-reinforced PiperION). The results shown in each panel were recorded on identical, but separate electrochemical cells. | ||
The same experiments were repeated at different cell temperatures (Fig. 2B). As concluded from these experiments, the total current density increases with the cell temperature. Over 60 °C however, the HER becomes favored, resulting in a decreased partial current density and FE for CO formation. Importantly, no performance fading was observed even at 70 °C cell temperature. This observation further extends the conclusion on the stability of the PiperION membrane, first demonstrated by a high temperature alkaline treatment for 2000 hours in 100 °C 1 M KOH, and long-term operation in a fuel-cell for 300 hours at j = 500 mA cm−2 with only ∼10% voltage loss.32 The stability in the presence of CO2 (and hence CO32− and HCO3−) is significant, because many AEMs lose their conductivity upon the interaction with these species.33 Importantly, the PiperION membrane shows a high conductivity even in the presence of these anions (Fig. 1C). The maximum achievable CO formation rate (jCO) scales inversly with the thickness of the applied membrane (Fig. 2C). Even though this is a trivial way of decreasing the cell resistance, hence increasing the electrolyzer performance, we have to highlight that even the thinnest studied membrane (15 μm, PTFE-reinforced) was found to be both mechanically and chemically very stable, and easy to handle during our experiments (see Discussion later).
We have continued our experiments with the thinnest PiperION membrane (15 μm, PTFE-reinforced), and increased the CO2 concentration in the GDE by doubling the CO2 feed rate to the cell (25 cm3 min−1 cm−2, and even further in case of the 32 μm thick membrane, see Fig. S5, ESI†). This extra feedstock enhanced the CO formation partial current density to 1004 ± 41 mA cm−2 (Fig. 2D, calculated from two parallel experiments, three GC data points recorded in each measurement). At the same time, the HER current density decreased (i.e., the parasitic HER was suppressed). The measured CO formation rate not only exceeds what has been reported for zero-gap electrolyzers so far, but it competes with the best results gathered in microfluidic cells, at a similar full cell energy efficiency. We have also found that achieving suitable reactant feed at such high current densities is not a trivial exercise, and careful cell engineering (e.g., gas flow management, GDL compression, see also Fig. S2, ESI†) becomes inevitable, especially when scaling-up the cell.
The stability of the electrolysis process (including that of the 15 μm thick, PTFE-reinforced PiperION membrane) was assessed using two different experimental conditions (Fig. 3A and B). In the first (milder) case, the cell was operated for 100 hours with 0.01 mol dm−3 CsOH anolyte, at 3.2 V with 12.5 cm3 min−1 cm−2 CO2 feed rate. In this case the FE fluctuated between 80 and 90% for CO production during the whole experiment. The water management of the cell stabilized in the first 10 hours of the experiment, accompanied by the continuous activation of the cell. Subsequently, the partial current density for CO production stabilized, and remained stable at jCO = 400 ± 15 mA cm−2. In this case, no performance fading was observed within the timeframe of the experiment, and no visible crystal formation (e.g., Cs2CO3, CsHCO3) occurred in the cathode GDE. The CO formation rate increased when more demanding conditions were employed (Fig. 3B). In this case after an initial current drop (from ∼1000 mA cm−2) relatively stable jCO = 700 ± 50 mA cm−2 was found over 24 hours, and the FE remained between 70–80% (similar data for the 32 μm thick membrane is shown in Fig. S6, ESI†). Importantly, to the best of our knowledge, this is the first demonstration of any CO2 electrolyzer operating for several hours at a CO formation current density above 0.7 A cm−2. Notably, the partial current density of the HER decreased continuously in the course of the experiment, indicating changes in the hydration conditions of the GDE. At such high current density operation we found that the electrolysis process is very sensitive to both temperature (Fig. 2B) and humidity (Fig. S7, ESI†). Both the total current density and the FE values change upon minor alterations of these factors. Two long-term electrolysis experiments on the very same cell, with identical components and experimental conditions, the only difference being the humidification of the CO2 stream, further confirmed this notion (Fig. S7, ESI†). When the cell was fed with humidified gas, a 50% higher jCO and a 10–15% larger FECO was observed compared to the non-humidified case. Furthermore, the cell operation was stable for an extended time in the humidified case (over 150 h, at jCO = 450 ± 50 mA cm−2). The effect of humidity is rationalized by our previous observations on the interdependency between carbonate-ion conductivity and hydration/dehydration processes of AEMs, and points towards future engineering challenges.33
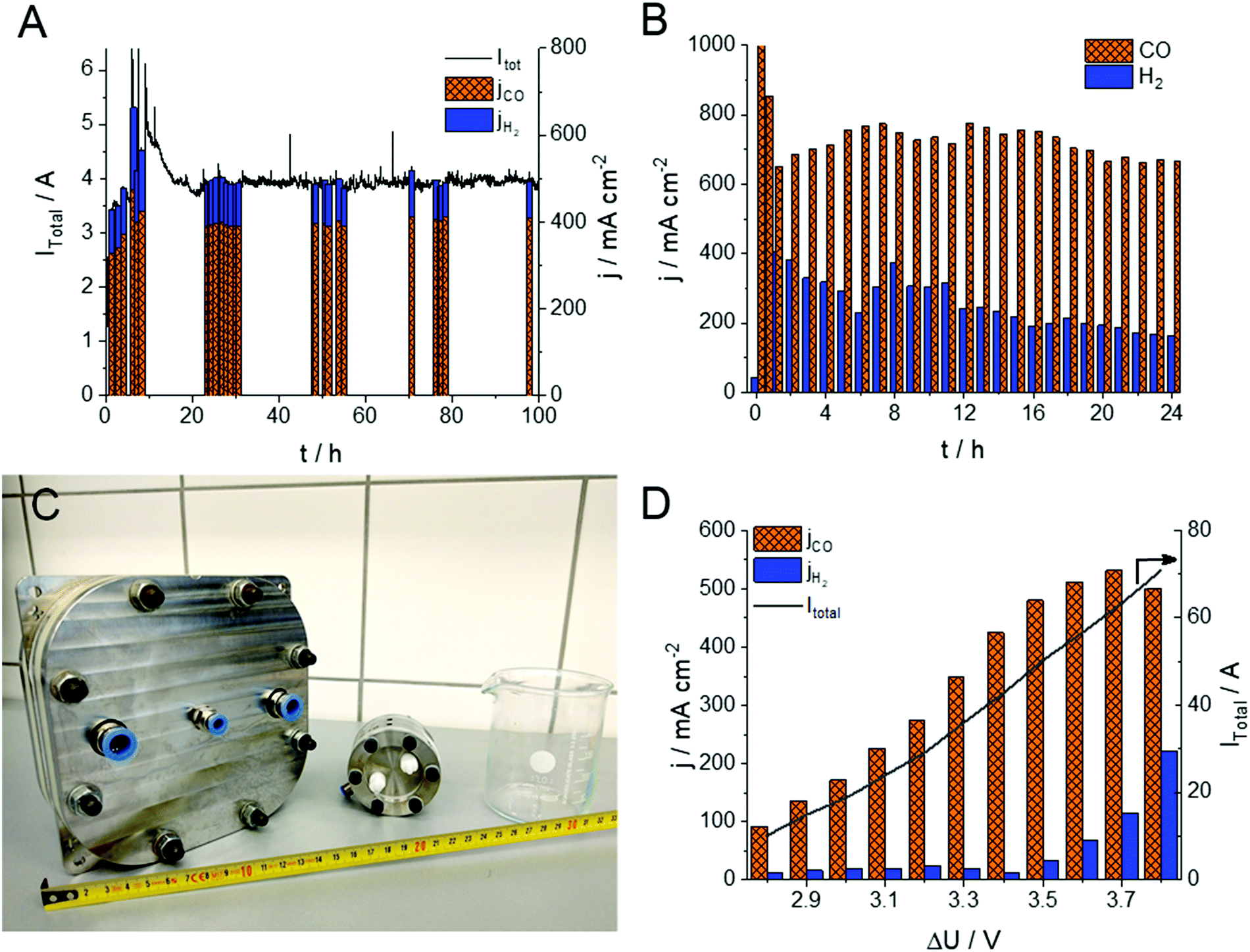 | ||
| Fig. 3 Chronoamperometric measurements at T = 60 °C with (A) 0.01 mol dm−3 CsOH anolyte and u = 12.5 cm3 min−1 cm−2 cathodic CO2 feed rate at ΔU = 3.2 V cell voltage and (B) 0.1 mol dm−3 CsOH anolyte and u = 25 cm3 min−1 cm−2 cathodic CO2 feed rate at ΔU = 3.4 V cell voltage. (C) Photograph of the A = 100 cm2 and A = 8 cm2 electrolyzer cells, used in this study, in comparison with a standard 250 cm3 beaker and a tape measure (cm scale). (D) Partial current densities for CO and H2 formation during chronoamperometric measurements operating the A = 100 cm2 cell with 0.1 M CsOH anolyte, at Tcell = 60 °C and different cell voltages (32 μm PiperION, 12.5 cm3 min−1 cm−2 CO2 feed rate). | ||
To demonstrate the scalability of the cell design, we have performed measurements in a larger electrolyzer cell (A = 100 cm2, Fig. 3C). Notably, we could achieve jCO values approaching those measured with the smaller active area electrolyzer cell (see Fig. 3D in comparison with Fig. 2A), although further optimization of the cell/process conditions is necessary to achieve higher energy efficiency (i.e., lower cell voltage). Overall, these results indicate that the presented zero-gap electrolyzer system might be an industrially viable strategy to simultaneously ensure high product formation rate, conversion and selectivity.
To identify the reasons behind the high CO formation rates, the same cell was assembled with different, widely used commercially available AEMs. Importantly, everything except the AEM was identical in these studies (i.e., GDEs, compression ratio, etc.). The electrochemical performance of the cells was assessed by chronoamperometry (Fig. 4A) and electrochemical impedance spectroscopy (EIS) measurements (Fig. 4B). Comparing the total current density in these cells under identical conditions (T = 60 °C, 0.1 mol dm−3 CsOH anolyte, ΔU = 3.0 V), a great difference was seen among the different membranes. The current density remains less than 10% of what is measured with the PiperION TP-85 (32 μm thick) membrane for the Fumasep-FAB-PK-130 and Selemion-AMV membranes and was less than 2/3 of that with the PTFE supported Sustainion® X37-50 (class T) membrane. As the same catalysts, cell hardware and test protocol were applied, it is safe to assume that the discrepancy is caused by the difference in membrane and ionomer conductance. To verify this notion, EIS traces were recorded (at 3.0 V), and major differences were shown (Fig. 4B). The high frequency intercept of these curves, which is directly related to the membrane resistance, shows the same difference as the current densities. It is about an order of magnitude higher for the Selemion and Fumasep membranes (4–5 Ω cm2), and more than twice as large for the Sustainion® membrane (0.85 Ω cm2) as compared to the PiperION membrane (0.36 Ω cm2). In addition, the semicircles related to both half reactions also shrunk. As the catalysts, gas flow, and the electrolyte are identical, this latter trend shall be related to the catalyst/ionomer interface. As the next step, we uncovered what is the charge transporting species in the process. For this purpose, the anode gas composition was analyzed at various voltages. As shown on the example of the trace recorded during an electrolysis at 3.0 V (same condition as in Fig. 4A), the anode gas composition stabilizes at 66% CO2 and 34% O2 (Fig. 4C, this result was also validated by GC-BID measurements). This is very similar to what was observed in a similar cell with Sustainion® membrane,28,29 confirming that the negative charges are exclusively transported through the PiperION membrane by CO32− ions. The overall mass balance is depicted in Fig. 4D, while detailed calculations are discussed in the ESI.†
 | ||
| Fig. 4 (A) Chronoamperometric curves and (B) EIS traces of electrolyzers assembled with different commercial membranes, operated with T = 60 °C, 0.1 mol dm−3 CsOH anolyte, ΔU = 3.0 V, with u = 12.5 cm3 min−1 cm−2 CO2 feed rate. (C) A representative anode gas analysis curve recorded during chronoamperometric measurements shown in Fig. 3. (D) Illustration of the mass balance of the electrolysis process, where the width of the blue arrows is proportional to the magnitude of the different fluxes at 40% conversion, while the black arrows only indicate the directions of the different fluxes. | ||
At this stage we hark back to Fig. 1C, where almost identical carbonate (and bicarbonate) ion conductivity were determined for PiperION (32 μm) and Sustainion® X37-50. Conductivity values are intrinsic properties of the membrane, and therefore independent from the thickness (i.e., they are normalized with the membrane thickness) (Fig. S8, ESI†). This also means that a similar electrochemical behavior is expected if membranes of similar thickness are employed. This notion was confirmed on the example of 50 μm thick membranes, where similar jCO values were recorded (Fig. S9, ESI†). As a further implication, if thinner membranes can be employed, the overall resistance of the cell will be diminished. This is indeed the case here: cells assembled with PiperION membrane (which is much thinner compared to Sustainion® X37-50; 32 μm vs. 50–70 μm (in hydrated form, according to our own measurements)) have notably smaller resistance as deduced by EIS (Fig. 4B). Furthermore, this resistance scales with the membrane thickness (Fig. S10, ESI†).
Even if a thin AEM is employed, it needs to be mechanically robust to withstand tension and creasing that occurs during manufacturing and operation of the electrolyzer. A qualitative test of the membrane resilience is shown in Fig. S11 (ESI†). By creasing a dry PiperION film into a compact ball, and then unfolding it, no cracks appear. In contrast, the Sustainion® membrane broke into multiple pieces when folded. Dynamic mechanical analysis was also performed to quantify the advanced mechanical properties of the PiperION membrane (Fig. S12, ESI†). Due to the plasticizer character of water on the AEMs, the samples were tested both dry and wet. For the dry PiperION membrane, the stress at break was 56 MPa and a strain at break of 113%. It was not possible to test the dry Sustainion® membrane, as it broke into pieces. For the wet PiperION the elongation at break increased to 365%, with the stress at break decreasing to 20.4 MPa. Finally, the wet Sustainion® showed an elongation at break of 38%, with the stress at break of 2.1 MPa.
Finally, if the contribution of the thinner AEM would only be a serial resistance, similarly high-performance operation could be achieved with other (less conductive) membranes, at higher voltages (although via sacrificing EE). This is clearly not the case here, because the operation of such CO2 electrolyzer cells is much more complex. Ion-transport through the AEM, heat-, gas-, and liquid management are all affected by the applied voltage, not mentioning the selectivity issues (CO2R vs. HER). These trends further highlight the benefits of using thinner AEMs.
Conclusions
Record high performance was achieved in terms of partial current density (>1.0 A cm−2), while maintaining high conversion (20–45%), selectivity (up to 90%) and moderate cell voltage (2.6–3.4 V). Overall, the mechanical robustness of PiperION membrane allows the use of thin membranes (down to 15 μm) with high carbonate ion conductance, which is the most important underlying factor for the unprecedented electrochemical CO2 reduction performance. In combination with a proper and easily scalable zero-gap electrolyzer cell design, which allows proper gas feed, this approach might pave the road to the industrial implementation of CO2 valorization (to CO and beyond, see Fig. S13, ESI†). Finally, we note that the generalization of these results shall be handled with caution. Membranes which underperform in a zero-gap configuration, might be very attractive in cells employing a liquid catholyte (e.g., above 200 mA cm−2 current densities for CO production were achieved with the Selemion membrane on Ni–N–C catalysts.34).Conflicts of interest
There are two pieces of intellectual property, filed by the authors of this paper and their institutions. Patent application number: PCT/HU2019/095001 (University of Szeged and ThalesNano Zrt) and USA granted patent number: US10290890B2, currently being nationalized in other countries under the PCT/US17/024615 (W7Energy LLC). Specifically, the patent applications cover the details of the cell architecture, description of the individual components of the cell, and the electrolysis process, while the granted patent protects the PiperION membrane.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 716539). The research was supported by the National Research, Development and Innovation Office (NKFIH) through the FK-132564 project, and by the “Széchenyi 2020” program in the framework of GINOP-2.2.1-15-2017-00041 project. The development of the PiperION membranes and ionomers is supported by the U.S Department of Energy, Advanced Research Projects Agency-Energy under Award No. DE-AR0001149. The authors thank Dr Jared Nash (W7energy) for the measurement of the mechanical properties of the membranes presented in this study, and Dr Dániel Sebők (University of Szeged) for recording micro-CT images.References
- B. Endrődi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar and C. Janáky, Prog. Energy Combust. Sci., 2017, 62, 133–154 Search PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 Search PubMed.
- S. Verma, B. Kim, H.-R. “Molly” Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 Search PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 Search PubMed.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 Search PubMed.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 Search PubMed.
- S. S. Bhargava, F. Proietto, D. Azmoodeh, E. R. Cofell, D. A. Henckel, S. Verma, C. J. Brooks, A. A. Gewirth and P. J. A. Kenis, ChemElectroChem, 2020, 7, 2001–2011 Search PubMed.
- M. G. Kibria, C.-T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. García de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 Search PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 Search PubMed.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 Search PubMed.
- X. Wang, A. Xu, F. Li, S. F. Hung, D. H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 Search PubMed.
- S. Ma, Y. Lan, G. M. J. Perez, S. Moniri and P. J. A. Kenis, ChemSusChem, 2014, 7, 866–874 Search PubMed.
- E. J. Dufek, T. E. Lister, S. G. Stone and M. E. McIlwain, J. Electrochem. Soc., 2012, 159, F514–F517 Search PubMed.
- S. Ma, R. Luo, J. I. Gold, A. Z. Yu, B. Kim and P. J. A. Kenis, J. Mater. Chem. A, 2016, 4, 8573–8578 Search PubMed.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2018, 3, 193–198 Search PubMed.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 Search PubMed.
- J. P. Edwards, Y. Xu, C. M. Gabardo, C.-T. Dinh, J. Li, Z. Qi, A. Ozden, E. H. Sargent and D. Sinton, Appl. Energy, 2020, 261, 114305 Search PubMed.
- Z. Liu, H. Yang, R. Kutz and R. I. Masel, J. Electrochem. Soc., 2018, 165, J3371–J3377 Search PubMed.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 Search PubMed.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 Search PubMed.
- R. Wang, H. Haspel, A. Pustovarenko, A. Dikhtiarenko, A. Russkikh, G. Shterk, D. Osadchii, S. Ould-Chikh, M. Ma, W. A. Smith, K. Takanabe, F. Kapteijn and J. Gascon, ACS Energy Lett., 2019, 4, 2024–2031 Search PubMed.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 Search PubMed.
- B. Endrödi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, ACS Energy Lett., 2019, 4, 1770–1777 Search PubMed.
- O. G. Sánchez, Y. Y. Birdja, M. Bulut, J. Vaes, T. Breugelmans and D. Pant, Curr. Opin. Green Sustainable Chem., 2019, 16, 47–56 Search PubMed.
- Y. Zheng, T. J. Omasta, X. Peng, L. Wang, J. R. Varcoe, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2019, 12, 2806–2819 Search PubMed.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 Search PubMed.
- Y. Zheng, G. Huang, L. Wang, J. R. Varcoe, P. A. Kohl and W. E. Mustain, J. Power Sources, 2020, 467, 228350 Search PubMed.
- G. O. Larrazábal, P. Strøm-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288 Search PubMed.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 Search PubMed.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 Search PubMed.
- M. R. Thorson, K. I. Siil and P. J. A. Kenis, J. Electrochem. Soc., 2013, 160, F69–F74 Search PubMed.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 Search PubMed.
- X. Luo, S. Rojas-Carbonell, Y. Yan and A. Kusoglu, J. Membr. Sci., 2020, 598, 117680 Search PubMed.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee02589e |
| This journal is © The Royal Society of Chemistry 2020 |