
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput and reliable determination of 13 haloacetic acids and dalapon in water and evaluation of control strategies†
Cristina
Postigo
 *a,
Pere
Emiliano
b and
Fernando
Valero
b
*a,
Pere
Emiliano
b and
Fernando
Valero
b
aWater, Environmental and Food Chemistry Unit (ENFOCHEM), Department of Environmental Chemistry, Institute of Environmental Assessment and Water Research (IDAEA-CSIC), C/Jordi Girona 18-26, 08034 Barcelona, Spain. E-mail: cprqam@cid.csic.es; Fax: +34 932 045 904; Tel: +34 934 006 100
bDepartment of R+D+i & Process Control, Ens d'Abastament d'Aigua Ter-Llobregat (ATL), Sant Martí de l'Erm 30, 08970, Sant Joan Despí, Barcelona, Spain
First published on 26th May 2020
Abstract
A simple, fast, highly-sensitive and selective method for the determination of 13 HAAs and dalapon in water has been optimized and validated. The method is based on large volume injection (200 μL) and analyte determination with liquid chromatography coupled to negative electrospray ionization-high resolution mass spectrometry (LVI-LC-ESI(−)-HRMS). High throughput is possible due to minimum sample manipulation and short analysis time (16 min in total). This is the first analytical LC-MS-based method that covers the whole suite of HAAs for which analytical standards are available and dalapon, and thus, represents a less costly option than ion-chromatography-based technologies developed for the same purpose. The method provided satisfactory trueness (91–120%) and precision (<17%) values for all analytes, except for CAA. Matrix effects, always in the form of ionization suppression effects, were not relevant (<25%), except in the case of CAA, and they were all well compensated with the use of internal standard calibration. This methodology allows quantifying HAAs in tap waters at concentrations below 1 μg L−1, except in the case of DBCAA and TCAA (3 μg L−1) and CAA and DCBAA (6 μg L−1). Thus, the presented analytical approach is satisfactory for the routine monitoring of HAA5 in drinking waters and obtaining additional knowledge on the formation and occurrence of other HAAs and dalapon that may be of relevance to ensure the provision of safe drinking water in the future. The concentrations of some of the brominated HAAs in chlorine-quenched disinfected water stored in the dark at −20 °C for seven days decreased between 26 and 46%, and thus, water samples should be analysed within 24 hours of their collection. As part of the validation method, the optimized approach was applied to evaluate two strategies to control HAA concentrations in water, i.e., lowering the water pH during the coagulation-flocculation step to improve process efficiency and using a household water pitcher filtration unit to remove HAAs in tap water.
Water impactThe methodology presented represents an analytical improvement over previous methods to determine haloacetic acids in water, in terms of reliability and high throughput. This class of contaminants has been included in the European Drinking Water proposal that will be approved in the next months, and thus, HAAs will have to be systematically monitored in drinking water in all European countries. |
1. Introduction
Chemical disinfection of water is a widespread practice to prevent waterborne diseases. Chlorination, particularly, is widely applied not only to produce drinking water and ensure sanitation of swimming pools and reclaimed water, but also to prevent biofilm growth on membranes and industrial cooling water systems, and to avoid the spread of invasive species through the release of ballast water by commercial shipping vessels. Since the 1970s, it is well known that the reaction between the organic matter naturally present in water with chlorine or any other chemical disinfectant results in the unintended formation of disinfection byproducts (DBPs).1,2 After trihalomethanes, haloacetic acids (HAAs) have been reported as the second most abundant class of DBPs that forms during water chlorination.3,4 This, together with their reported toxicity,5,6 has motivated their regulation in drinking water by the US EPA,7 and their inclusion in the new Drinking Water Directive drafted by the European Union,8 with a parametric value of 60 μg L−1 for HAA5 (i.e., the sum of monochloroacetic acid (CAA), monobromoacetic acid (BAA), dichloroacetic acid (DCAA), dibromoacetic acid (DBAA), and trichloroacetic acid (TCAA)). Maximum HAAs concentrations have also been established in various guidelines for drinking water quality. For instance, Canadian guidelines set a maximum acceptable concentration of HAA5 of 80 μg L−1,9 and the World Health Organization (WHO) proposes guideline values for CAA, DAA, and TCAA of 20 μg L−1, 50 μg L−1 and 200 μg L−1, respectively.10 Following WHO guidelines, maximum desirable values for chloroacetic acids in drinking water have been established in Australia (150 μg L−1 for CAA, and 100 μg L−1 for DCAA and TCAA),11 New Zealand (equal to WHO guidelines),12 China (50 μg L−1 for DCAA and 100 μg L−1 for TCAA),13 and Japan (20 μg L−1 for CAA, and 30 μg L−1 for DCAA and TCAA).14 Thus, to comply with current HAAs regulations, and upcoming restrictive regulations in drinking water, e.g., public health goals drafted in California for individual HAA5 in the range 0.03–53 μg L−1,15 reliable, highly sensitive, and high throughput analytical methodologies need to be in place for the routine monitoring of the whole spectrum of HAAs in water.Due to their physical–chemical properties, the determination of HAAs in water is a challenge from an analytical point of view. This class of DBPs has traditionally been measured in water using approaches based on US EPA methods16–18 that rely on the use of gas chromatography (GC) coupled to electron capture detection (ECD). To improve the selectivity of the analytical methods, variations that use mass spectrometry (MS) detection instead of ECD have been also employed.3,19–21 In any case, due to the polar nature and low volatility of HAAs, their analysis by GC–ECD or GC-MS requires increasing their volatility by converting them into their corresponding methyl esters. The agents commonly used for this derivatization step are the hazardous diazomethane gas or acidic methanol. In addition to being highly time-consuming, the derivatization process may negatively affect method performance in terms of reproducibility and requires highly-trained lab personnel to conduct the analyses.
Furthermore, the high polarity (Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow = 0.22–1.68 and water solubility = 1 × 103–1 × 106 mg L−1) and acidic properties (pKa values between 0.7 and 3.1) of HAAs dare their extraction from water (Table S1 in ESI†). Thus, for their extraction, water samples need to be acidified to pH values below 1 (ref. 3, 18 and 21–23) or specific sorbent materials (e.g., anionic exchange resins) are required.24 In either case, extraction protocols have to be exclusively and carefully developed to achieve acceptable performance levels. Several methodologies focused on the direct injection of the water sample into the analytical instrument have been also proposed to bypass the extraction step.25–33 The main advantage of direct injection methods is that sample treatment is reduced to internal standard addition and sample filtration, and consequently, the total analysis time is shortened.
Kow = 0.22–1.68 and water solubility = 1 × 103–1 × 106 mg L−1) and acidic properties (pKa values between 0.7 and 3.1) of HAAs dare their extraction from water (Table S1 in ESI†). Thus, for their extraction, water samples need to be acidified to pH values below 1 (ref. 3, 18 and 21–23) or specific sorbent materials (e.g., anionic exchange resins) are required.24 In either case, extraction protocols have to be exclusively and carefully developed to achieve acceptable performance levels. Several methodologies focused on the direct injection of the water sample into the analytical instrument have been also proposed to bypass the extraction step.25–33 The main advantage of direct injection methods is that sample treatment is reduced to internal standard addition and sample filtration, and consequently, the total analysis time is shortened.
The development of direct injection methods requires the use of highly selective analytical techniques, like those consisting of chromatographic separation of the sample components and MS determination of the target analytes. Chromatography has been achieved with ion exchange columns (ion chromatography, IC)29–32 or reversed-phase columns (reversed-phase liquid chromatography, RPLC, or LC) packed with a silica-based sorbent modified to enhance the retention of polar analytes.24,26,28,33,34
IC–MS/MS has been successfully used for the quantitative analysis of up to 13 HAAs and dalapon in water at levels in the low μg L−1 range (LOQs below 1 μg L−1).29,32,35 The main concerns associated with the use of this technique for HAA analysis are, in addition to its high cost, the potential interferences of other anions commonly present in water matrices in the MS signal, and the complexity of the technique. The use of LC-MS/MS to determine HAAs in water has gained attention in recent years.24,26,33,36,37 This may be linked to advances in LC technology, that allowed the development of silica-based stationary phases with increased affinity for polar compounds (e.g., Luna C18 Polar from Phenomenex,26 Acclaim HAAs from Thermo Fisher Scientific33 or HSS T3 from Waters).24,27 LC retention and separation of HAAs in the past, due to their high polar character, required the use of ion-pairing reagents in the mobile phase, specifically dibutylamine38 or trimethylamine.39,40 Of all the LC-MS-based methodologies to determine HAAs in water reported in the literature,24–28,33,34,36,37,41,42 none of them covers the whole suite of HAAs (13 chlorinated, brominated, and iodinated HAAs), and in some cases, details provided for method reproduction are scarce or method performance in real samples was poorly addressed.
In this context, the current work aimed at developing and fully validating a direct injection LC-high resolution MS (HRMS) method for the simple and high-throughput analysis of trace levels of 13 HAAs and dalapon in water. The method was applied to evaluate two strategies to control HAAs in water: i) improving the efficiency of the coagulation-flocculation process by lowering the water pH and hence reducing the formation potential of DBPs of the water, and ii) using a commercial household water pitcher filtration unit to remove HAAs from tap water. Furthermore, the stability of HAAs in chlorine-quenched samples stored at −20 °C for seven days was also investigated.
2. Material and methods
2.1. Chemicals and reagents
High purity analytical standards (>90%) were provided by CanSyn (Toronto, Canada) and Sigma Aldrich (Barcelona, Spain) (see Table S1† for further details and physical–chemical properties of target HAAs). A working standard mixture containing all target analytes at a concentration of 40 μg mL−1 was prepared after dilution of stock standard solutions (2000 μg mL−1 in the case of the Cl and Br-HAAs mixture, and 1000 μg mL−1 in the case of individual iodo-acids and dalapon) in methanol and stored at −20 °C in the dark for not longer than one month. This solution was further diluted with LC-grade water (J. T. Baker, Fisher Scientific, Madrid, Spain) to 200 μg L−1, 20 μg L−1, and 2 μg L−1 to prepare calibration solutions and samples used in the validation study. An aqueous internal standard (IS) solution containing 2,3-dibromopropanoic acid (DBPA) at a concentration of 200 μg L−1 was also prepared.Ascorbic acid and formic acid (FA) reagent-grade were purchased in Sigma-Aldrich. Other solvents used, e.g., acetonitrile and methanol were provided by Fisher Scientific and were Optima™ LC-MS grade.
2.2. Direct injection LC-ESI-HRMS analysis of HAAs
In the validated analytical approach, 200 μL of the water sample containing 0.1% FA and 10 μg L−1 of IS was injected on a Luna® Omega Polar C18 100 Å column (100 × 4.6 mm ID, 3 μm particle size) preceded by a security guard cartridge Polar C18 (4 × 3 mm ID) (Phenomenex, Barcelona). Elution of the HAAs from the column was achieved with an acidic mobile phase consisting of water and acetonitrile both with 0.1% FA, after applying a linear gradient of the organic solvent at a constant flow rate of 1 mL min−1. Optimum LC retention of the analytes was obtained with isocratic conditions during the first 3 minutes of the chromatographic run (5% of acetonitrile), a slow increase to 20% of the organic constituent in two minutes and then, a fast increase to 100% in two additional minutes. Pure organic conditions were maintained for 3 min, and then initial conditions were achieved in 1.50 min and maintained during 4.5 min for column re-equilibration. The total analysis time was 16 min. LC injection and separation was performed with a SCIEX Exion LC™ AD system that incorporates a Shimadzu FCV-11AL reservoir selection valve and a 0.5 mL injection loop (Sciex, Redwood City, CA, US). The auto-sampler temperature was maintained at 12 °C.MS analysis was conducted with a SCIEX X500R QTOF system. Target analytes and sample components were ionized using electrospray (ESI) in the negative polarity mode with a Turbo V™ source. Source conditions were ion spray voltage of −3500 V, source temperature of 650 °C, and nitrogen gas delivery pressures of 45 psi for the curtain gas, 60 psi for the atomizing gas, and 45 psi for the auxiliary gas. Mass accuracy was achieved with hourly calibrations, i.e., every four samples, during the acquisition batch by automated infusion of trifluoroacetic acid through the calibrant delivery system. Mass calibration ensured a mass resolution of about 30000 at the low m/z working range. MS acquisition was done using a high-resolution multiple reaction monitoring (MRMHR) workflow. It consists of a TOF-MS scan over the m/z range 50–350 Da (125 ms of accumulation time; −10 V declustering potential (DP) and −10 V collision energy (CE)), and a TOF-MS/MS experiment that measured one selected reaction monitoring (SRM) transition for each HAA. For this, the pseudomolecular ion is filtered in the quadrupole, and accurate mass measurement of the (selected) product ion is obtained with the TOF analyzer. In this second experiment, 40 ms of accumulation time was used to monitor each HR-SRM in scheduled time windows. Detailed LC-MS/MS conditions are shown in Table 1. Data acquisition, qualitative and quantitative data treatment was performed using SCIEX OS™ software version 1.4 (Sciex, Redwood City, CA, U.S.). The mass window was set to 20 mDa for data processing and compound quantitation.
| Analyte | t R (min) | TOF MS (m/z 50–350) | TOF MS/MS (HRMS scan after fragmentation of the parent ion) | Ion ratioa | ||
|---|---|---|---|---|---|---|
| Parent ion | Product ion | CE (V) | ||||
| a Quantification ion/confirmation ion. | ||||||
| CAA | 2.4 | 92.9743 | 92.97 | 34.9693 | −15 | 2.1 |
| BAA | 2.9 | 136.9238 | 136.92 | 78.9189 | −15 | 1.0 |
| IAA | 4.6 | 184.9099 | 184.91 | 126.9050 | −15 | 1.0 |
| DCAA | 2.0 | 126.9353 | 126.93 | 82.9461 | −10 | 1.0 |
| DBAA | 2.7 | 216.8330 | 216.83 | 172.8430 | −15 | 1.0 |
| DIAA | 6.5 | 310.8070 | 310.81 | 266.8172 | −10 | 2.4 |
| BCAA | 2.3 | 170.8848 | 170.88 | 78.9189 | −25 | 1.0 |
| BIAA | 3.9 | 262.8204 | 262.82 | 126.9050 | −35 | 1.1 |
| CIAA | 3.1 | 218.8709 | 218.87 | 126.9050 | −20 | 1.6 |
| TCAA | 4.2 | 116.9067 | 116.91 | 34.9693 | −10 | 0.4 |
| TBAA | 6.5 | 250.7536 | 250.75 | 78.9189 | −20 | 1.1 |
| DCBAA | 4.8 | 162.8540 | 162.85 | 78.9189 | −10 | 0.7 |
| DBCAA | 5.7 | 206.8034 | 206.80 | 78.9189 | −15 | 1.0 |
| DPN | 4.3 | 140.9511 | 140.95 | 34.9693 | −25 | 0.4 |
| IS | 7.7 | — | 78.92 | 78.9189 | −20 | — |
The accurate mass measurement of the pseudomolecular ion and one SRM transition provides 5.5 identification points in total. This value is well above the 3 identification points recommended by the EU 2002/657/EC Commission Decision to confirm chemical residues in live animals and animal products.43 The sole acquisition of one HR-SRM transition provides indeed the identification points required by the aforementioned legislation. Analyte identification was done according to its retention time, and its HRMS signals. The HRMS signal selected for quantification was the one that provided the highest response and/or was the least affected by background noise at low concentrations in real water samples. Following these selection criteria, the area of the product ion was used for quantification of most HAAs, while the area of the pseudomolecular ion was used for confirmation. There were only a few exceptions, namely CAA and TBAA, for which the pseudomolecular ion (TOF-MS signal) provided a better sensitivity and was affected by lower background noise than the HR-SRM transition (TOF-MS/MS signal).
2.3. Method performance
The analytical method was validated in terms of linearity, accuracy (trueness) and precision, sensitivity, and matrix effects. Method validation was conducted in two real aqueous matrices of different conductivity, i.e., clarified surface water (1200 μs cm−1) collected at a drinking water treatment plant located in the Barcelona area and tap water (600 μs cm−1).Method linearity was evaluated in LC-grade water and real aqueous matrices. For this, 12 calibration solutions containing the target HAAs at concentrations ranging from 0.03 to 100 μg L−1 and the IS at a fixed concentration of 10 μg L−1 were prepared in each matrix. After analysis of these calibration solutions, IS-based calibration curves were constructed following weighted least-squares linear regression models by plotting the HAA:IS peak area ratio against the HAA:IS concentration ratio and using 1/x as a weighting factor. The linearity of the method for each HAA was expressed as the goodness of fit, i.e., the coefficient of determination (R2), of the calibration data to the linear model over the analyte-specific linearity range.
Accuracy and precision of the method were appraised from the analysis of real aqueous matrices fortified in sextuplicate (n = 6) with the HAAs at three concentration levels: low or 0.5 μg L−1 (RL), medium or 5 μg L−1 (RM), and high 50 μg L−1 (RH). Accuracy was expressed as trueness, i.e., the closeness of the average measured value to the theoretical value. Precision was calculated as the relative standard deviation (RSD) of the trueness values at a given concentration level. Background concentrations of HAAs in the water matrices used in the method validation study were subtracted for trueness and precision calculations. In this regard, HAAs were found only in tap water: BAA (0.91 μg L−1), DBAA (6.42 μg L−1), TBAA (3.8 μg L−1), BCAA (1.2 μg L−1), DCAA (0.35 μg L−1), and DCBAA (3.1 μg L−1).
Method sensitivity was expressed through the analyte limit of detection (LOD), and limit of quantification (LOQ). These values were visually estimated from the analysis of low-level fortified matrices, i.e., the lowest points of the matrix-matched calibration curves prepared. LODs and LOQs were the analyte concentration that provided a signal-to-noise ratio of 3 and 10, respectively.
The extent of matrix effects in real water matrices was evaluated by comparing the slopes of the weighted linear regression models obtained using the external standard method in matrix-matched calibration curves with that of an LC-grade water-based calibration curve.
2.4. Sample collection and sample preparation
The method was applied to evaluate the formation potential of HAAs of the Llobregat River water after a coagulation-flocculation process based on aluminum polychloride implemented at two operating water pH values at plant scale. One of the clarifiers operated at pH 7.8, which is within the plant operational range (7.5–7.8), whereas the other one was operated at lower pH (7.1). Water pH in the clarifiers was adjusted by introducing CO2 (natural pH of the water >8). Characteristics of the source and clarified waters in terms of total organic carbon (TOC), turbidity, and UV absorbance at 254 nm are provided in Table S2 as ESI.† Lowering the water pH during the coagulation-flocculation process reduces aluminum solubility and hence, the residual concentrations of dissolved aluminum in water. However, this may have undesired effects on the HAA formation potential of the water that needs to be investigated. For this, 2 L water samples were collected at the outlet of the aforementioned clarifiers. Each water sample was chlorinated by adding 8 mg L−1 of free chlorine, according to the water chlorine demand, and transferred to four individual 500 mL amber glass bottles that were incubated at 25 °C. At different chlorine contact times (i.e., 0, 24, 48, 72 h) one bottle of each water sample was taken for analysis. For this, the residual chlorine (Table S3 in ESI†) was quenched with ascorbic acid (2.5 mg L−1 of ascorbic acid per 1 mg L−1 of free chlorine was added), and part of the volume was transferred to 125 mL glass bottles that were kept at −20 °C in the dark until analysis, that took place within one week of collection. Further details on the performance of HAAs formation potential tests have been provided elsewhere.3Additionally, the use of a commercial household water pitcher filtration unit to remove HAAs from tap water was investigated (Jata Hogar, model RE123x4 from Electrodomésticos JATA SA, Navarra, Spain). The filter tested had been in use for two weeks, and therefore it was at its half-life. It consisted of activated carbon mixed with an ion-exchange resin. No further details on its composition were obtained from the manufacturer. Water samples before and after filtration were analyzed. For sample collection, the cold tap water from a household kitchen faucet was opened for approximately five minutes and then, a 125 mL amber glass bottle that already contained ascorbic acid was filled without being overfilled. Next, tap water was poured into an empty pitcher filtration unit and then, filtered water was collected into a 125 mL amber glass bottle with ascorbic acid. These samples were kept under 4 °C during transport to the laboratory and analyzed within 24 h of collection time.
All sampling material and glassware used in the formation potential tests were pretreated overnight with a concentrated solution of chlorine (about 100 mg L−1) and rinsed with distilled water before use to avoid potential artifacts.
For analysis, 1.5 mL of homogenized sample was transferred into a 2 mL-vial and 75 μL of the IS solution (200 μg L−1) was added as well as 2 μL of concentrated formic acid. The water samples were not filtered because they were coarse particle-free. However, it should be a step to consider in other matrices (e.g., surface water or effluent wastewater) to avoid problems in the analytical system.
3. Results and discussion
3.1. Method optimization
Optimization of LC-MS/MS conditions was performed by on-column injection (10 μL) of the HAA standard mixture and individual solutions at a concentration of 200 μg mL−1. TOF-MS full scan analysis was conducted to identify molecular ions and information-dependent acquisition (IDA) experiments were done to obtain HAA fragmentation. Electrospray ionization of mono- and di-halogenated species resulted in the formation of the corresponding pseudomolecular ion via deprotonation of the carboxylic group [M − H]−. In the case of trihalogenated HAAs, the pseudomolecular ion formed resulted from their decarboxylation in the ionization source [M-COOH]− (Table 1 and Fig. S1 in ESI†). Adduct ion formation was not observed in any case. Electrospray ionization of HAAs with the Turbo V™ source also required to slightly lower the normal operating voltage to maximize MS signal and avoid excessive in-source fragmentation of these compounds. The least electronegative halogen of the pseudomolecular ion was the main product ion formed after fragmentation: iodine in the case of I-containing HAAs, bromine in the case of Br-containing HAAs, and chlorine in the case of Cl-containing HAAs. Exceptions were observed for DCAA, DBAA, and DIAA, for which the main product ion was formed after the loss of the carboxyl group [M-COOH]−. The IS was monitored with a pseudotransition (Br → Br) because no pseudomolecular ion could be observed for this compound.Optimum conditions for LC retention and separation of HAAs were achieved by testing different chromatographic columns, injection volumes, and mobile phase compositions, flows, initial conditions, and gradients. As for the chromatographic columns, two different columns were tested, Purospher® C8 column (125 × 4 mm, 5 μm, Merck, Darmstadt, Germany) and Luna® Omega Polar C18 (100 × 4.6 mm, 3 μm), in both cases preceded with guard columns of the same packing materials. Both columns showed a similar retention capacity for HAAs. However, the Luna® Omega Polar C18 provided better peak resolution, and overall sharper peaks at a lower mobile phase flow (1.0 mL min−1vs. 1.2 mL min−1) (Fig. S2†). This column is designed to enhance the retention of highly polar compounds and its sorbent is stable in 100% aqueous media. This has also been the choice for HAA separation in previous studies.26,39
A variety of mobile phase compositions using acetonitrile or methanol as the organic solvent and with and without acid were also tested. The acidification of the mobile phase was needed to enhance the retention of the analytes in the chromatographic column and provide neutral species that can be easily deprotonated during ionization. Acetonitrile was finally selected as organic solvent because it provided sharper peaks and better peak resolution than methanol, despite that methanol enhanced HAA ionization, and hence their MS signal (Fig. S3†). In previously published methods, both organic solvents were used for separation with FA concentrations ranging from 0.05 to 0.5%.26–28,37,41,42 Although with less frequency, acetic acid or ammonium acetate buffer have been also used as mobile phase modifiers.25,33,34,36 The retention of the analytes using different chromatographic mobile phase initial conditions (0–10% of organic solvent) was also evaluated. The best chromatographic performance for the most polar analytes was obtained with 5% of acetonitrile in the mobile phase.
The comparison of the chromatographic peaks obtained after the injection of different volumes (100 μL to 500 μL) of an aqueous standard solution containing the HAA mix allowed setting an optimal injection volume of 200 μL (data not shown). Injection of fortified real tap water samples also revealed the need for acidifying samples with 0.1% FA before injection.
3.2. Method performance
The performance of the optimized method was evaluated as described in section 2.3. The results obtained are summarized in Table 2, and the LVI-LC-ESI(−)-MS/MS peaks obtained in a tap water sample fortified with the targeted HAAs at a concentration of 5 μg L−1 of the target compounds are shown in Fig. 1.| Linearity | Trueness (repeatability) | Sensitivity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSW | TW | CSW | TW | |||||||||
| Range [μg L−1] | R 2 | R L 0.5 μg L−1 | R M 5 μg L−1 | R H 50 μg L−1 | R L 0.05 μg L−1 | R M 5 μg L−1 | R H 50 μg L−1 | LOD [μg L−1] | LOQ [μg L−1] | LOD [μg L−1] | LOQ [μg L−1] | |
| CSW: Clarified surface water, TW: tap water. | ||||||||||||
| CAA | 0.6–100 | 0.9959 | <LOD | 39 (25) | 76 (6.7) | <LOD | 47 (16) | 76 (6.4) | 3 | 6 | 3 | 6 |
| BAA | 1–100 | 0.9975 | <LOD | 97 (4.5) | 100 (4.7) | <LOD | 104 (6.2) | 94 (7.5) | 0.6 | 1 | 0.6 | 1 |
| IAA | 0.6–100 | 0.9985 | 114 (14) | 108 (1.9) | 107 (5.0) | 96 (5.6) | 104 (1.5) | 101 (7.3) | 0.3 | 0.6 | 0.3 | 0.6 |
| DCAA | 0.6–100 | 0.9987 | <LOD | 107 (3.2) | 103 (4.4) | 118 (6.8) | 103 (5.9) | 104 (8.2) | 0.3 | 0.6 | 0.3 | 0.6 |
| DBAA | 0.6–100 | 0.9958 | 113 (17) | 102 (8.4) | 104 (7.8) | 109 (10) | 105 (11) | 92 (9.2) | 0.3 | 0.6 | 0.3 | 0.6 |
| DIAA | 0.06–60 | 0.9939 | 115 (11) | 116 (2.8) | 120 (2.7) | 109 (3.2) | 116 (3.0) | 105 (6.8) | 0.03 | 0.06 | 0.1 | 0.3 |
| BCAA | 0.3–100 | 0.9990 | 106 (5.9) | 103 (7.2) | 100 (6.8) | 99 (14) | 96 (4.8) | 91 (7.1) | 0.3 | 0.6 | 0.1 | 0.3 |
| BIAA | 0.3–100 | 0.9966 | 98 (11) | 103 (9.8) | 112 (5.8) | 115 (11) | 102 (11) | 104 (11) | 0.3 | 0.6 | 0.3 | 0.6 |
| CIAA | 0.3–100 | 0.9980 | 124 (12) | 110 (5.4) | 112 (8.5) | 116 (4) | 113 (4.5) | 103 (9.6) | 0.1 | 0.3 | 0.1 | 0.3 |
| TCAA | 3–100 | 0.9920 | <LOD | 109 (3.7) | 109 (6.4) | <LOD | 107 (10) | 95 (4.9) | 1 | 3 | 1 | 3 |
| TBAA | 1–100 | 0.9919 | <LOD | 115 (5.7) | 133 (6.2) | <LOD | 113 (3.4) | 115 (3.8) | 1 | 3 | 0.6 | 1 |
| DCBAA | 6–100 | 0.9923 | <LOD | 98 (5.2) | 111 (8.6) | <LOD | 112 (8.8) | 100 (3.4) | 3 | 6 | 3 | 6 |
| DBCAA | 3–100 | 0.9942 | <LOD | 108 (4.3) | 116 (4.4) | <LOD | 112 (9.8) | 104 (3.2) | 1 | 3 | 1 | 3 |
| DPN | 1–100 | 0.9983 | <LOD | 107 (3.3) | 110 (3.8) | <LOD | 114 (8.0) | 108 (9.1) | 1 | 3 | 0.6 | 1 |
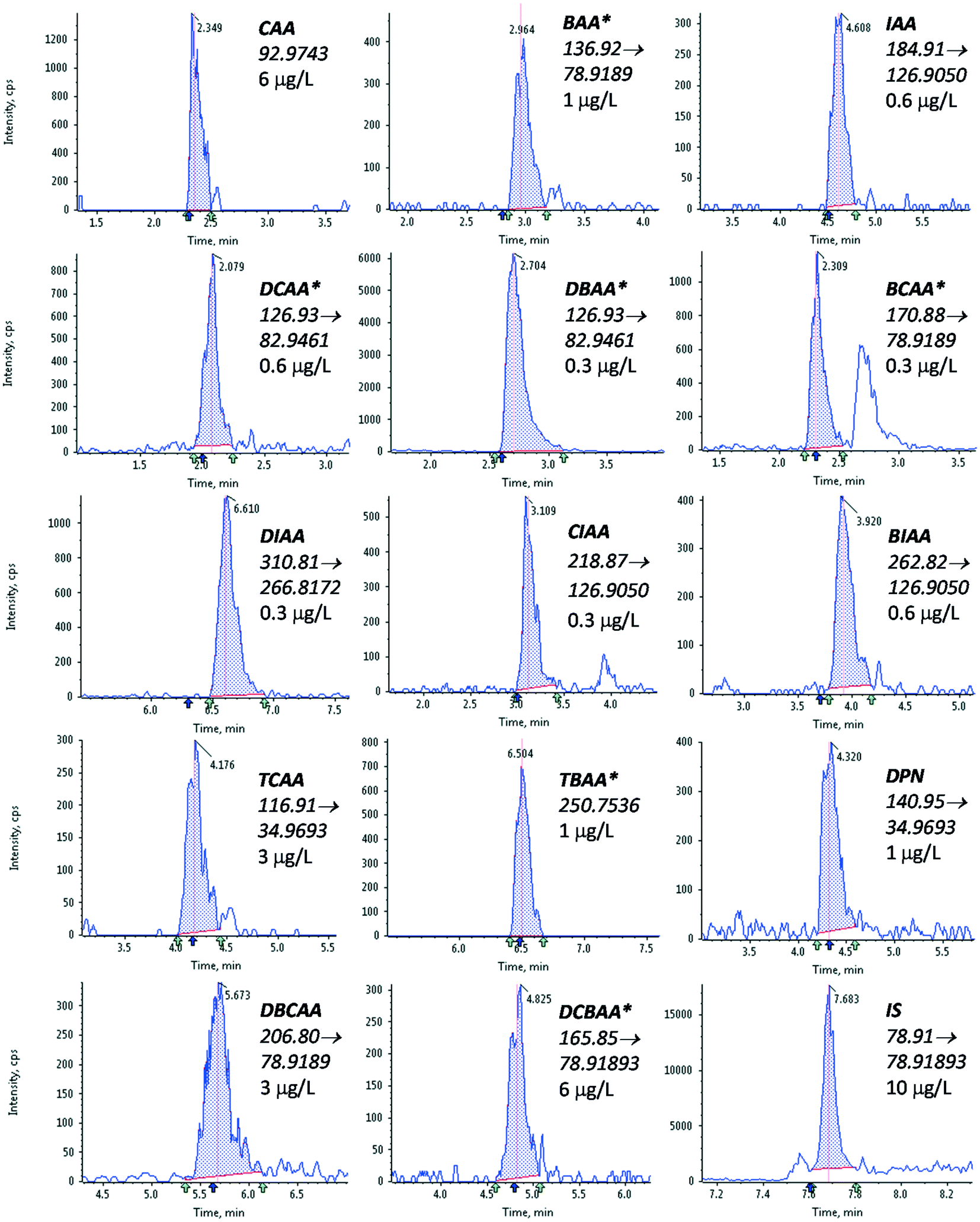 | ||
| Fig. 1 Chromatograms of HAAs after LVI-LC-ESI(−)-MS/MS analysis of tap water fortified at the analyte LOQ. *HAAs present in tap water before fortification (see section 2.3), and thus, showing peaks that correspond to higher concentrations than the LOQs indicated in the figure. | ||
The method was found to be linear for all analytes in all investigated matrices, with calibration curves expanding from the analyte LOQ in a specific matrix to the upper limit of the linearity range (100 μg L−1), except in the case of DIAA that was linear up to 60 μg L−1, because of its high instrumental sensitivity. Weighted linear regression models and corresponding coefficients of determination (R2) in all water matrices tested were constructed after interday injection in duplicate of matrix-matched calibration curves (Tables 2 and S4 in ESI†). Only those calibration points where the measured concentration did not deviate more than 20% of the theoretical concentration were included in the models.
The accuracy of the concentrations measured in clarified surface water and tap water was overall satisfactory, with trueness values between 91 and 120% for all analytes and concentration levels investigated, except in the case of TBAA at the highest concentration level tested in tap water (133%) and CAA that presented in general low trueness values at all concentrations in both investigated matrices (39–76%). Trueness values were obtained after quantification of fortified samples with LC-grade water-based calibration curves, free of HAA background concentrations. The precision of trueness values (n = 6) was below 17% for all analytes, except for CAA in clarified surface water at the low concentration level tested (25%). The poor performance of the method for CAA is attributed to both its poor sensitivity and the effect of matrix interferences on its ionization. Matrix effects in the investigated aqueous matrices are summarized in Fig. 2. They were calculated from the slopes obtained in matrix-matched calibration curves by the external standard method. Suppression ionization effects were observed for all analytes except for TBAA in clarified surface water. Excluding CAA, the analysis of the remaining HAAs was not strongly affected by other matrix components (<25%). Trueness figures indicate that the IS used corrected well these effects and allow the quantification with LC-grade water-based calibration curves.
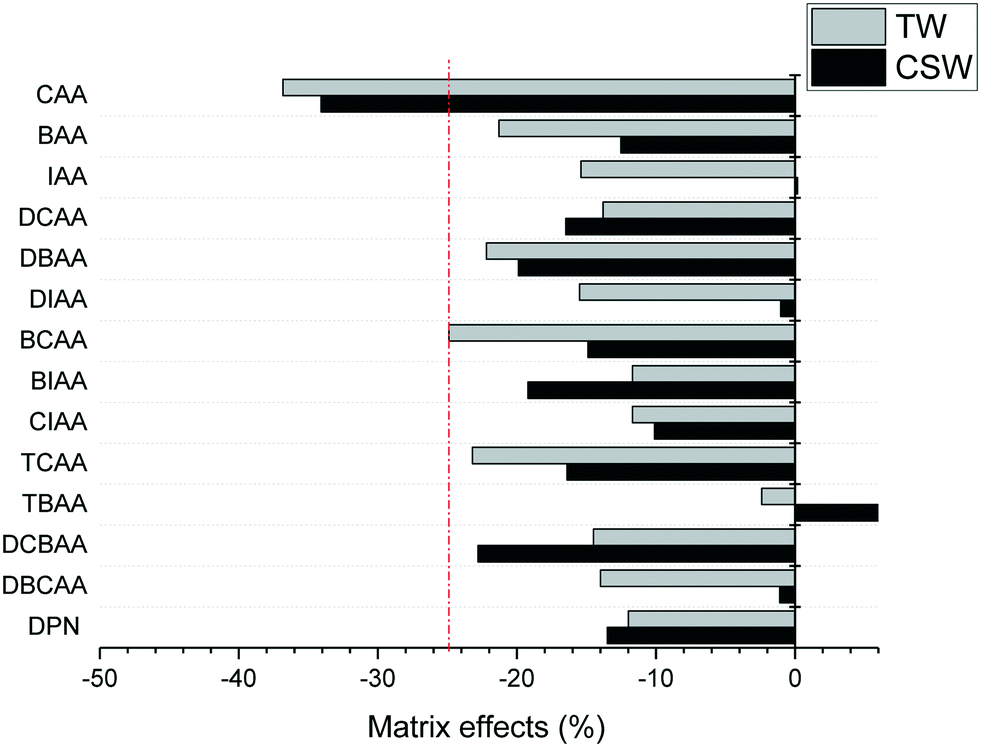 | ||
| Fig. 2 Matrix effects observed in LVI-LC-ESI(−)-MS/MS analysis of HAAs in clarified surface water (CSW) and tap water (TW). | ||
Overall the worst sensitivity was observed for the trihalogenated HAAs and the monohalogenated BAA and CAA. Particularly, CAA and DCBAA were the HAAs with the highest LOQs (6 μg L−1) in real water matrices. The remaining HAAs presented LODs in tap water and clarified surface water between 0.03 and 1 μg L−1 and LOQs between 0.06 and 3 μg L−1. Despite the relative high LOQs obtained for few of the HAAs in tap water (6 μg L−1 for CAA and DCBAA, and 3 μg L−1 for TCAA and DBCAA) and given that maximum concentrations for individual HAAs are not enforced in current drinking water regulations and only the sum of selected HAAs species is limited (∑HAA5 <60 μg L−1 in the US7 and the new European Drinking Water Directive proposal8), the method here described is suitable for the regular monitoring of these substances at drinking water facilities with a chlorine-based disinfection treatment. It also allows the monitoring of CAA, DCAA, and TCAA below WHO guidelines (20, 50, and 200 μg L−1, respectively).10
Compared to other methods previously published for the analysis of this class of DBPs, the approach here presented allows the simultaneous determination of dalapon and the 13 HAAs for which analytical standards are available in an extremely short analysis time (16 min per sample vs. 20–25 min (ref. 22, 26 and 34) in other LC-MS approaches or 27 min to 65 min in IC-MS-based methods29–32). Shorter analysis times (<10 min) were only reported for UPLC-MS approaches.25,27
3.3. Analysis of HAAs in water samples
:1, ascorbic acid:chlorine) at −20 °C was investigated. For this, LC-grade water and tap water were fortified with the HAA mixture at a concentration of 5 μg L−1, so that all analytes could be targeted. Vials in triplicate for each scenario were prepared, and those corresponding to time 0 were analyzed and the remaining were kept in the dark at −20 °C and analyzed after 1 day (or 2 days in the case of LC-grade water samples) and 7 days of storage time. The stability of HAAs was assessed by trueness values observed in these samples and results are summarized in Fig. 3 and S4 in ESI.† As shown, after one day and seven days of storage at −20 °C, HAA concentrations in finished water decreased significantly with a confidence level of 0.05 (*) and even 0.01 (**) (after t-student test) (Fig. 3). Although for most compounds the concentrations after 7 days decreased only by less than 22%, higher decrease rates were observed for the brominated HAAs DBAA (46%), DBCAA (38%), TBAA (35%), and DCBAA (26%). The area of the IS used did not differ significantly during the storage time and thus it cannot correct for analyte losses in time (data not shown). Similar behaviors were observed in LC-grade water; however, the decrease rate after 7 days was in all cases below 22% (Fig. S4†). Overall, the analysis of water samples within 24 hours of their collection is recommended to avoid potential changes in the HAA mixture. Residual chlorine quenching is also required to avoid the increase of HAA concentrations, as reported elsewhere.26
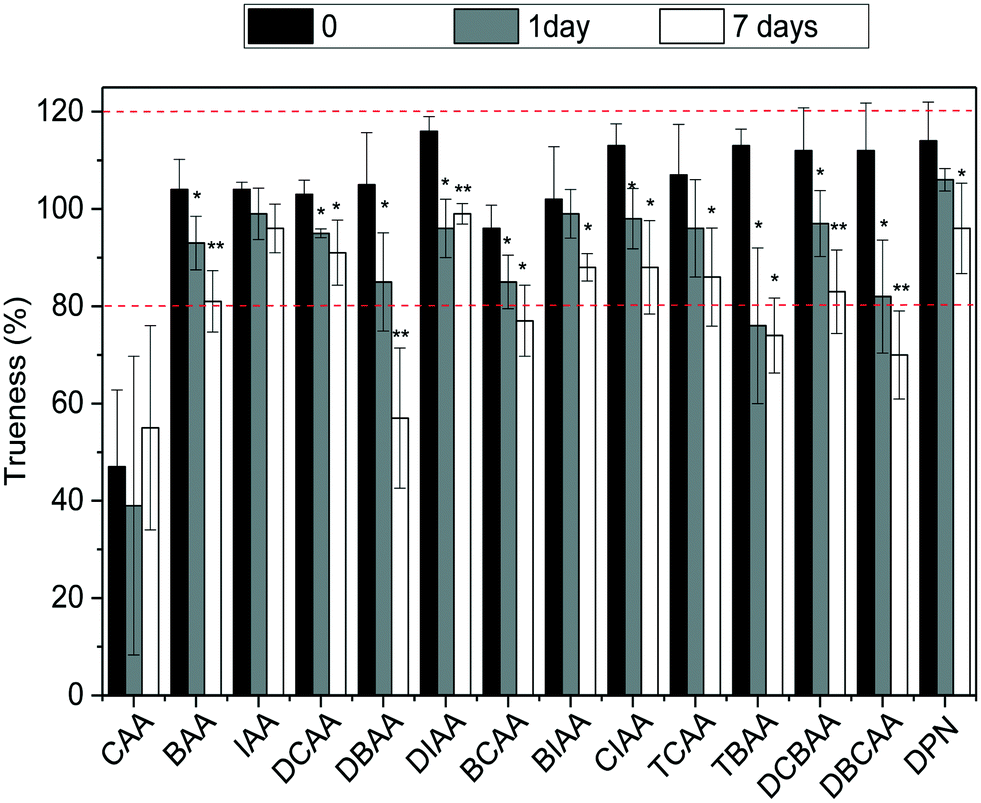 | ||
| Fig. 3 Trueness values of HAAs and dalapon in tap water fortified at a concentration of 5 μg L−1 and stored at −20 °C for 7 days. Error bars indicate RSD values of n = 3 measurements. | ||
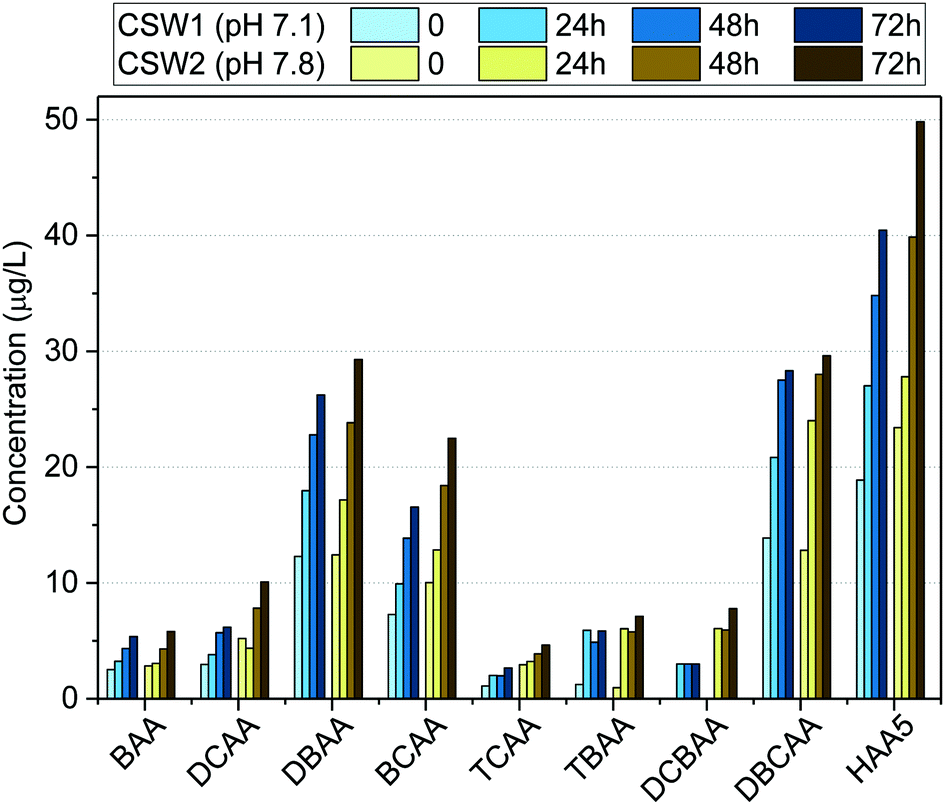 | ||
| Fig. 4 Formation potential of HAAs in clarified surface water (CSW) after clarification at different water pH values. | ||
The effect of pH on HAA formation during chlorination has been reported to be compound-specific.44–48 Previous works indicated the increase of trihalogenated acids at decreasing water pH values.48 This may explain the low difference observed in the formation potential of TBAA and DBCAA at the investigated clarified waters. Moreover, due to the high bromide levels in the source water, the formation potential of brominated DBPs such as BAA, DBAA, TBAA, DBCAA, and BCAA was reduced only 5%, whereas that of other species like DCBAA, TCAA, and DCAA was reduced between 40 and 60% when clarified at acidic pH. Although decreasing the water pH may have a positive effect in reducing the formation potential of other DBP classes (e.g., trihalomethanes),45,46,48 it may enhance the formation of nitrogen-containing DBPs, such as nitrosamines, or chloropicrin, which are known to have toxic effects on organisms,49,50 when nitrosating agents are present also in the water.51
Finally, the method was applied to investigate the removal of HAAs in tap water after being filtered with a household water pitcher filtration unit (Jata Hogar, model RE123x4). Results, shown in Fig. 5, revealed the presence of low concentrations of HAA5 in tap water (5.7 μg L−1). Brominated species prevailed in the HAA mixture because the household is served by a water treatment facility that treats highly brominated surface water. The most abundant HAAs species detected were DBAA (4.5 μg L−1), TBAA (3.4 μg L−1) and DBCAA (1.9 μg L−1). Trace amounts of BAA, DCAA, and BCAA were also found (<1 μg L−1). After filtration, total HAAs levels were found to decrease by 56% (HAA5 decreased by 54%). The filter was overall more effective to remove bromine-DBPs than chlorine-DBPs detected.
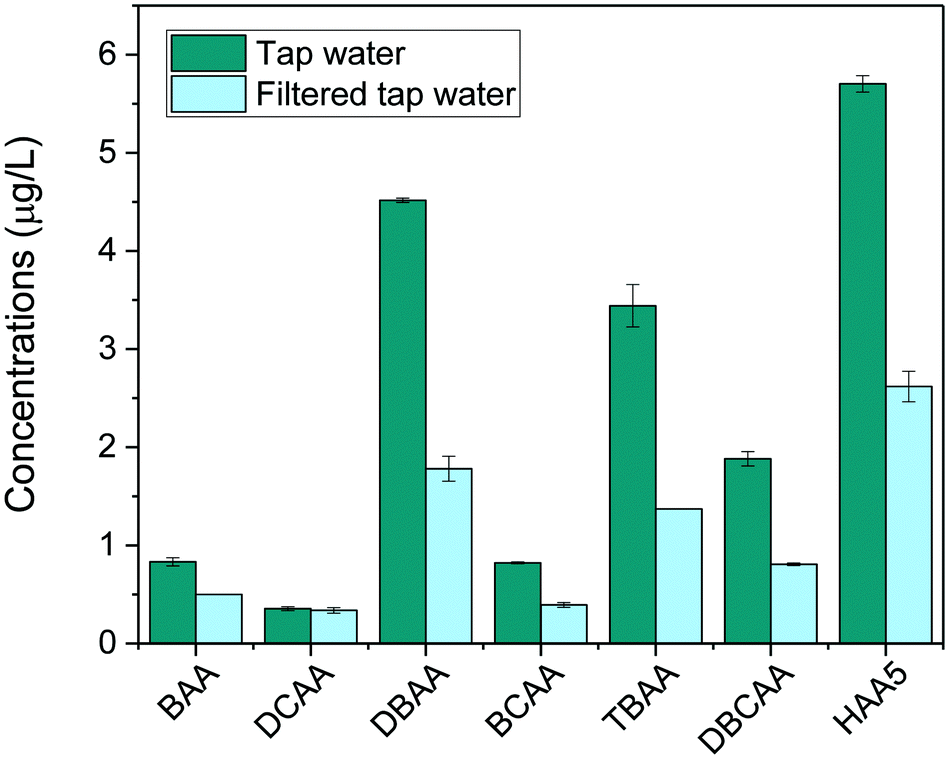 | ||
| Fig. 5 Average concentrations (n = 2) of HAAs in tap water and after filtration with a household water pitcher filtration unit (Jata Hogar, model RE123x4). | ||
4. Conclusions
A simple, fast, highly-sensitive and selective method for the determination of 13 HAAs and dalapon in water has been optimized and validated. The method is based on large volume injection (200 μL) and analyte determination with LC-ESI(−)-HRMS. High throughput is possible due to minimum sample manipulation (IS and FA addition) and short analysis time (16 min in total). Satisfactory results in terms of trueness (91–120%) and precision (< 17%) were obtained for all analytes except CAA (trueness = 39–76% and precision < 25%), also the HAA affected the most by matrix effects. This method allows quantifying the target HAA in tap waters at concentrations below 1 μg L−1, except in the case of DBCAA and TCAA (3 μg L−1) and CAA and DCBAA (6 μg L−1). Considering current drinking water regulations and the guidelines proposed for chlorinated HAAs, the method sensitivity is sufficient for the routine monitoring of HAA5 in drinking waters, and to obtain information on the occurrence of dalapon and the other HAAs for which analytical standards are available. Furthermore, the acquisition of a full HRMS scan allows the retrospective analysis of the data to search for other haloacids or DBPs amenable to negative electrospray ionization in the investigated samples.As part of the validation process, the method was applied to evaluate two strategies to control HAA concentrations in water. Lowering the water pH (from 7.8 to 7.1) during the coagulation-flocculation process improves the efficiency of this practice and results in a reduced formation potential of HAAs of the clarified waters. However, it could also enhance the potential of the water to form toxic DBPs during the disinfection process. Therefore, all aspects associated with changes in the water treatment train need to be carefully investigated before taking action. On the other hand, the use of a household water pitcher filtration unit also decreased total HAA concentrations in tap water. However, it is important to highlight that the performance of these household filtration devices depends on the type of filter and extent and conditions of use (amount of liters filtered, the temperature of the water, etc.).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CP acknowledges support from Fundación General del Consejo Superior de Investigaciones Científicas (FGCSIC) through the 2nd edition of the ComFuturo Programme. This work was supported by the Government of Catalonia (Consolidated Research Groups 2017 SGR 01404) and the Spanish Ministry of Science and Innovation (Project CEX2018-000794-S). Phenomenex is acknowledged for the gift of the HPLC column and AB Sciex for LC-MS instrumentation loan. Nicola Montemurro and Roser Chaler, from IDAEA-CSIC, are acknowledged for assisstance in the use of the analytical instrumentation.References
- J. J. Rook, Formation of haloforms during chlorination of natural waters, Water Treat. Exam., 1974, 23(2), 234–243 Search PubMed.
- T. A. Bellar, J. J. Lichtenberg and R. C. Kroner, Occurrence of organohalides in chlorinated drinking waters, J. - Am. Water Works Assoc., 1974, 66(12), 703–706 CrossRef CAS.
- C. Postigo, P. Emiliano, D. Barceló and F. Valero, Chemical characterization and relative toxicity assessment of disinfection byproduct mixtures in a large drinking water supply network, J. Hazard. Mater., 2018, 359, 166–173 CrossRef CAS PubMed.
- H. S. Weinberg, S. W. Krasner, S. D. Richardson and J. A. D. Thruston, The Occurrence of Disinfection By-Products (DBPs) of Health Concern in Drinking Water: Results of a Nationawide DBP Occurrence Study, National Exposure Research Laboratory, Office of Research and Development, U.S. Environmental Protection Agency, Athens, GA, 2002, Contract No.: EPA/600/R-02/068 Search PubMed.
- S. D. Richardson, M. J. Plewa, E. D. Wagner, R. Schoeny and D. M. Demarini, Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: a review and roadmap for research, Mutat. Res., 2007, 636(1–3), 178–242 CAS.
- S. D. Richardson, F. Fasano, J. J. Ellington, F. G. Crumley, K. M. Buettner and J. J. Evans, et al., Occurrence and Mammalian Cell Toxicity of Iodinated Disinfection Byproducts in Drinking Water, Environ. Sci. Technol., 2008, 42(22), 8330–8338 CrossRef CAS PubMed.
- USEPA, United States Environmental Protection Agency ( 2010), Comprehensive Disinfectants and Disinfectoin Byproducts Rules (Stage 1 and Stage 2): Quick Reference Guide. EPA816-F-10-080, Office of Water, Retrieved from https://bit.ly/2xXmFHP, Accessed March 2020.
- 5813/20 - Outcome of proceedings - Proposal for a Directive of the European Parliamentn and of the Council on the quality of water intended for human consumption (recast), Retrieved from: https://bit.ly/2UFotwK, Accessed in March 2020.
- Health Canada (2008) Guidelines for Canadian Drinking Water Quality: Guideline TechnicalDocument — Haloacetic Acids, Water, Air and Climate Change Bureau, Healthy Environmentsand Consumer Safety Branch, Health Canada, Ottawa, Ontario, Retrieved from https://bit.ly/3bookEH, Accessed in 2020 Search PubMed.
- WHO, World Health Organization ( 2017), Guidelines for drinking-water quality, 4th edition, incorporating the 1st addendum. Retrieved from https://bit.ly/2J7IHu3, Accessed in March 2020 Search PubMed.
- NHMRC, NRMMC (2011) Australian Drinking Water Guidelines Paper 6 National Water Quality Management Strategy, Version 3.4. updated October 2017, National Health and Medical Research Council, National Resource Management Ministerial Council, Commonwealth of Australia, Canberra, Retrieved from https://bit.ly/2WySDVf, Accessed in March 2020 Search PubMed.
- Ministry of Health (2008) Drinking-watger standards for New Zealand 2005 (revised 2008), ellington: Ministry of Health, Retrieved from https://bit.ly/2Ud7umx, Accessed in March 2020 Search PubMed.
- National Standard of the People's Republic of China, GB 5749–2006 Standards fro Drinking Water Quality, Retrieved from https://bit.ly/3aoXlcp, Accessed in March 2020 Search PubMed.
- Ministry of Health, Labour and Welfare of Japan, 2015, Diagram 13- Drinking Water Quality Standards in Japan, Retrieved from https://bit.ly/3biXT3l, Accessed in March, 2020 Search PubMed.
- California Environmental Protection Agency (2020) First Public Reciw Draft: Haloacetic Acids in Drinking Water, OEHHA, Office of Environmental Health Hazard Assessement, Retrieved from https://bit.ly/2WDzl14, Accessed in March 2020 Search PubMed.
- USEPA, United States Environmental Protection Agency (1992), “Method 552.1: Determination of haloacetic acids and dalapon in drinking water by ion-exchange liquid-solid extraction and gas chromatography with an electron capture detector”; Revision 1.0. in Methods for the Determination of Organic Compoudns in Drinking Water, Supplement II, EPA/600/R-92/129, pp. 143–172, Retrieved from https://bit.ly/3afd79r. Accessed March 2020 Search PubMed.
- USEPA, United States Environmental Protection Agency (1995) Method 552.2 Determination of haloacetic acids and dalapon in drinking water by liquid-liquid extraction, derivatization and gas chromatography with electron capture detector, Revision 1.0. in Methods for the Determination of Organic Compoudns in Drinking Water, Supplement III, EPA/600/R-95/131, Retrieved from https://bit.ly/2Udvlmk, Accessed March 2020 Search PubMed.
- USEPA, United States Environmental Protection Agency (2003) Method 522.3 Determination of haloacetic acids and dalapon in drinking water by liquid-liquid microextraction derivatization, and gas chromatography with electron capture detection, Revision 1.0, EPA 815-B-03-002. Retrieved from https://bit.ly/399Cb0d, Accessed March 2020 Search PubMed.
- I. Kristiana, A. Lethorn, C. Joll and A. Heitz, To add or not to add: The use of quenching agents for the analysis of disinfection by-products in water samples, Water Res., 2014, 59, 90–98 CrossRef CAS PubMed.
- E. S. Franco, V. L. Pádua, M. D. V. R. Rodriguez, D. F. Silva, M. Libânio and M. C. Pereira, et al., A simple liquid-liquid extraction-gas chromatography-mass spectrometry method for the determination of haloacetic acids in environmental samples: Application in water with Microcystis aeruginosa cells, Microchem. J., 2019, 104088 CrossRef CAS.
- A. Kinani, J. Olivier, A. Roumiguières, S. Bouchonnet and S. Kinani, A sensitive and specific solid-phase extraction–gas chromatography–tandem mass spectrometry method for the determination of 11 haloacetic acids in aqueous samples, Eur. J. Mass Spectrom., 2018, 24(5), 375–383 CrossRef CAS PubMed.
- X. Liu, X. Wei, W. Zheng, S. Jiang, M. R. Templeton and G. He, et al., An Optimized Analytical Method for the Simultaneous Detection of Iodoform, Iodoacetic Acid, and Other Trihalomethanes and Haloacetic Acids in Drinking Water, PLoS One, 2013, 8(4), e60858 CrossRef CAS PubMed.
- C. Zhao, Y. Fujii, J. Yan, K. H. Harada and A. Koizumi, pentafluorobenzyl esterification of haloacetic acids in tap water for simple and sensitive analysis by gas chromatography/mass spectrometry with negative chemical ionization, Chemosphere, 2015, 119, 711–718 CrossRef CAS PubMed.
- S. Hu, T. Gong, J. Ma, Y. Tao and Q. Xian, Simultaneous determination of iodinated haloacetic acids and aromatic iodinated disinfection byproducts in waters with a new SPE-HPLC-MS/MS method, Chemosphere, 2018, 198, 147–153 CrossRef CAS PubMed.
- L. Meng, S. Wu, F. Ma, A. Jia and J. Hu, Trace determination of nine haloacetic acids in drinking water by liquid chromatography-electrospray tandem mass spectrometry, J. Chromatogr. A, 2010, 1217(29), 4873–4876 CrossRef CAS PubMed.
- C. Planas, Ó. Palacios, F. Ventura, M. R. Boleda, J. Martín and J. Caixach, Simultaneous analysis of 11 haloacetic acids by direct injection-liquid chromatography-electrospray ionization-triple quadrupole tandem mass spectrometry and high resolution mass spectrometry: occurrence and evolution in chlorine-treated water, Anal. Bioanal. Chem., 2019, 411(17), 3905–3917 CrossRef CAS PubMed.
- J. Duan, W. Li, J. Si and D. Mulcahy, Rapid determination of nine haloacetic acids in water using ultra-performance liquid chromatography-tandem mass spectrometry in multiple reactions monitoring mode, Anal. Methods, 2011, 3, 1667–1673 RSC.
- Y. Li, J. S. Whitaker and C. L. McCarty, Analysis of iodinated haloacetic acids in drinking water by reversed-phase liquid chromatography/electrospray ionization/tandem mass spectrometry with large volume direct aqueous injection, J. Chromatogr. A, 2012, 1245, 75–82 CrossRef CAS PubMed.
- M. C. Bruzzoniti, L. Rivoira, L. Meucci, M. Fungi, M. Bocina and R. Binetti, et al., Towards the revision of the drinking water directive 98/83/EC, Development of a direct injection ion chromatographic-tandem mass spectrometric method for the monitoring of fifteen common and emerging disinfection by-products along the drinking water supply chain, J. Chromatogr. A, 2019, 1605, 360350 CrossRef CAS PubMed.
- H. B. Teh and S. F. Y. Li, Simultaneous determination of bromate, chlorite and haloacetic acids by two-dimensional matrix elimination ion chromatography with coupled conventional and capillary columns, J. Chromatogr. A, 2015, 1383, 112–120 CrossRef CAS PubMed.
- H. Shi and C. Adams, Rapid IC-ICP/MS method for simultaneous analysis of iodoacetic acids, bromoacetic acids, bromate, and other related halogenated compounds in water, Talanta, 2009, 79(2), 523–527 CrossRef CAS PubMed.
- S. Wu, T. Anumol, J. Gandhi and S. A. Snyder, Analysis of haloacetic acids, bromate, and dalapon in natural waters by ion chromatography–tandem mass spectrometry, J. Chromatogr. A, 2017, 1487, 100–107 CrossRef CAS PubMed.
- Application Note 590, (2016) Quantification of haloacetic acids int ap wate rusing dedicated HAA LC column with LC-MS/MS detection - Thermo Fisher Scientific, Inc. Available at: https://bit.ly/2QGWMmd, Accesed in March 2020 Search PubMed.
- C.-Y. Chen, S.-N. Chang and G.-S. Wang, Determination of Ten Haloacetic Acids in Drinking Water Using High-Performance and Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry, J. Chromatogr. Sci., 2009, 47(1), 67–74 CrossRef CAS.
- USEPA, United States Environmental Protection Agency (2009), Method 557: Determination of Haloacetic Acids Bromate and Dalapon in Drinking Water by Ion Chromatography Electrospray Ionization Tandem Mass Spectrometry (IC-ESI-MS/MS) Office of water, 815-B-09-012. Retrieved from https://bit.ly/3dhcSN0, Accessed in March 2020 Search PubMed.
- L. Alexandrou, C. Bowen and O. A. H. Jones, Fast analysis of multiple haloacetic acids and nitrosamines in recycled and environmental waters using liquid chromatography-mass spectrometry with positive-negative switching and multiple reaction monitoring, Anal. Methods, 2019, 11(30), 3793–3799 RSC.
- X. Geng, X. Li, J. Ye, E. Cudjoe and F. Qin, Rapid Analysis of Haloacetic Acids in Drinking Water Using UHPLC/MS/MS, Perkin Elmer, Inc. Available at https://bit.ly/2JaVTyc. Accessed in March 2020, 2018 Search PubMed.
- M. C. Prieto-Blanco, M. F. Alpendurada, P. López-Mahía, S. Muniategui-Lorenzo, D. Prada-Rodríguez and S. MacHado, et al., Improving methodological aspects of the analysis of five regulated haloacetic acids in water samples by solid-phase extraction, ion-pair liquid chromatography and electrospray tandem mass spectrometry, Talanta, 2012, 94, 90–98 CrossRef CAS PubMed.
- Z. Wu and R. B. Cody, Determination of haloacetic acids in Water by LC/MS, Analytical Instrument Division, JEOL, USA, Available at: https://bit.ly/33E3gaV, Accessed in March 2020, 2004 Search PubMed.
- R. Loos and D. Barceló, Determination of haloacetic acids in aqueous environments by solid-phase extraction followed by ion-pair liquid chromatography-electrospray ionization mass spectrometric detection, J. Chromatogr. A, 2001, 938(1–2), 45–55 CrossRef CAS PubMed.
- N. Cullum, Monitoring for haloacetic acids in treated waters using direct aqueous injection on the agilent 6460 LC/QQQ, Agilent Technologies, Inc., Available at https://bit.ly/2J9nRdF, Accessed in March 2020, 2013 Search PubMed.
- Application Data Sheet no. 43. (2014), High speed analysis of haloacetic acids in tap water using triple quadrupole LC-MS/MS, Shimadzu Corporation, Retrieved from: https://bit.ly/33H1xSf, Accessed in March 2020 Search PubMed.
- 2002/654/EC, Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Official Journal of the European Communities L 221/8. Retrieved from https://bit.ly/33GsMMA, Accessed in March 2020 Search PubMed.
- A. D. Nikolaou, S. K. Golfinopoulos, T. D. Lekkas and G. B. Arhonditsis, Factors affecting the formation of organic by-products during water chlorination: A bench-scale study, Water, Air, Soil Pollut., 2004, 159(1), 357–371 CrossRef CAS.
- +12 G. Hua and D. A. Reckhow, DBP formation during chlorination and chloramination: Effect of reaction time. pH, dosage, and temperature, J. - Am. Water Works Assoc., 2008, 100(8), 82–95 CrossRef CAS.
- B. Ye, W. Wang, L. Yang and J. Wei, E X. Factors influencing disinfection by-products formation in drinking water of six cities in China, J. Hazard. Mater., 2009, 171(1), 147–152 CrossRef CAS PubMed.
- S. Kucukcongar, M. F. Sevimli and E. Yel, DBP formation and speciation in a central Anatolian dam water depending on pH, TOC level, fraction and chlorine dose, Global NEST J., 2013, 15(4), 447–456 CAS.
- P. Roccaro, G. V. Korshin, D. Cook, C. W. K. Chow and M. Drikas, Effects of pH on the speciation coefficients in models of bromide influence on the formation of trihalomethanes and haloacetic acids, Water Res., 2014, 62, 117–126 CrossRef CAS PubMed.
- WHO, World Health Organization (2003), Chloropicrin in drinking water - background document for develoment of WHO-Guidelines for drinking-water quality. WHO/SDE/WSH/03.04/52 Retrieved from https://bit.ly/2JmsPDL, Accessed in March 2020 Search PubMed.
- WHO, World Health Organization (2008), N-Nitrosodimethylamine in drinking water - background document for develoment of WHO-Guidelines for drinking-water quality, WHO/HSE/AMR/08.03/8 Retrieved from https://bit.ly/39jxCRg, Accessed in March 2020 Search PubMed.
- T. Bond, J. Huang, M. R. Templeton and N. Graham, Occurrence and control of nitrogenous disinfection by-products in drinking water – A review, Water Res., 2011, 45(15), 4341–4354 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ew00296h |
| This journal is © The Royal Society of Chemistry 2020 |