
Diet control to achieve euglycaemia induces tau hyperphosphorylation via AMPK activation in the hippocampus of diabetic rats†
Dong Jun
Sung‡
 a,
Yun-Hee
Noh‡
b,
Jun-Ho
Lee
c,
Mingli
Jin
d,
Jin-Seoung
Kim
e and
Sang-Don
Han
*fg
a,
Yun-Hee
Noh‡
b,
Jun-Ho
Lee
c,
Mingli
Jin
d,
Jin-Seoung
Kim
e and
Sang-Don
Han
*fg
aDivision of Sport and Health Studies, College of Biomedical and Health Science, Konkuk University, Chungju, 27478, Republic of Korea
bDepartment of Biochemistry, Konkuk University School of Medicine, Seoul, 05029, Republic of Korea
cC & R Research, 06199, Seoul, Republic of Korea
dGachon Institute of Pharmaceutical Sciences, College of Pharmacy, Gachon University, 21936, Republic of Korea
eInternational Ginseng & Herb Research Institute, Chungnam, 32724, Republic of Korea
fDepartment of Medical Education, Konkuk University School of Medicine, Seoul, 05029, Republic of Korea. E-mail: hihanol@kku.ac.kr
gDepartment of Medicine, Chungbuk National University, Cheongju 28644, Republic of Korea
First published on 13th November 2019
Abstract
Alzheimer's disease (AD) is a chronic neurodegenerative disease, and typical pathologic findings include abnormally hyperphosphorylated tau aggregation and neurofibrillary tangles. Insulin resistance and hyperglycaemia have been proposed as risk factors for AD development. As the maintenance of optimal blood glucose level is an important indicator of diabetes mellitus (DM) treatment, diet control is essential. AMPK is a crucial sensor of cellular bioenergetics for controlling anabolic and catabolic metabolism. Since AMPK is a direct regulator of tau phosphorylation, we hypothesized that strict diet control to achieve euglycaemia affects tau protein phosphorylation through increased AMPK activity in the hippocampus of DM rats. To test this hypothesis, we generated insulin-deficient DM rats by subtotal pancreatectomy and the animals were categorized into the diet-restriction (R) group and ad libitum (AL) feeding group. We found that tau phosphorylation was significantly higher in the R group than that in the sham-control (C) or AL group. AMPK activity in the R group was significantly higher than that in the C or AL group, as expected. Furthermore, the R group showed more critical tau pathology in the hippocampus than the other groups. These results suggest that diet control to achieve euglycaemia in an insulin-deficient DM condition may be harmful because of the greater possibility of AD development through increased tau phosphorylation by AMPK activation in the hippocampus.
Introduction
Alzheimer's disease (AD) is characterized by progressive neuronal loss and synaptic injury. The primary pathologic hallmarks of AD are neurofibrillary tangles (NFTs) and amyloid beta (Aβ) plaques. NFTs are formed by intracellular aggregation of paired helical filaments, which are composed of a microtubule-associated protein known as tau that shows abnormal hyperphosphorylation.1 Aβ plaques are extraneuronal deposits of Aβ that develop through amyloid precursor protein cleavage by beta secretase and gamma secretase.2 Although much attention has recently been focused on Aβ as the causative agent in AD, NFT pathology correlates better with cognitive decline in AD than amyloid pathology.3Accumulating evidence suggests that diabetes mellitus (DM) is associated with the development of AD. An epidemiologic study demonstrated that DM patients have a high risk of developing AD, which is independent of the risk of vascular dementia.4 Moreover, patients with DM had a nearly 2-fold increased incident risk of AD5 and a 65% increase in the risk of developing AD compared to those without DM.6 Many experimental animal models of DM have also shown AD pathology, including tau hyperphosphorylation.7,8
DM is characterized by hyperglycaemia due to the combination of insulin resistance and impaired insulin secretion. The goal of DM treatment has been correction of hyperglycaemia to euglycaemia, or near-euglycaemia, because hyperglycaemia is associated with the development of DM complications and mortality.9,10 Thus, diet control is highly emphasized as a method of good glycaemic control to achieve and maintain euglycaemia in patients with DM.11,12 However, our previous study demonstrated that diet control to achieve euglycaemia reduces the weight of the heart, liver, skeletal muscle, and epididymal fat mass as well as body weight in insulin-deficient DM rats via excessive autophagy.13 Following this study, we also revealed that diet control to achieve euglycaemia is deleterious in the insulin-deficient state due to increased apoptosis and autophagy in the liver via AMPK activation.14
Meanwhile, recent studies have demonstrated that AMPK plays a role as a tau kinase in AD development, in which AMPK itself phosphorylates tau at Ser262 and Ser396, altering the microtubule binding of tau.15 AMPK is also activated abnormally in tangle- and pre-tangle-bearing neurons in AD and multi-tau phosphorylation sites.16 Therefore, we hypothesized that strict diet control to achieve euglycaemia during insulin deficiency affects tau protein hyperphosphorylation, one of the hallmarks of AD pathophysiology, through AMPK activation in the hippocampus of DM rats.
To test our hypothesis, first we generated insulin-deficient DM rats by subtotal pancreatectomy to mimic end-stage type 2 DM, where insulin deficiency is prominent because of the progressive loss of pancreatic beta cell mass and function.17,18 Also, we induced euglycaemia in the DM rats by diet control, where the DM rats were fed isocalories per kg body weight compared to the food amount in the sham-control rats, as we performed previously elsewhere.13 Concerning the protocol to achieve euglycaemia in DM rats using the isocaloric rate eaten by sham-control rats, the restriction rate was less strict than that used recently in a clinical study aimed at remission of type 2 diabetic patients, where the subjects were provided for 3–5 months with about one third of calories required daily by healthy adults.19 After achieving euglycaemia in the DM rats, we compared the levels of AMPK activation and tau phosphorylation in the hippocampus of the experimental groups to investigate whether diet control to achieve euglycaemia in insulin-deficient DM induces tau hyperphosphorylation through activated AMPK. In addition, we also examined the phosphorylation level of the Akt-GSK-3β axis, a typical pathway for the phosphorylation of tau protein that is regulated by insulin and protein phosphatase 2A (PP2A). PP2A is known to regulate AMPK activation and tau phosphorylation by dephosphorylation.20,21
Materials and methods
Animals
Eleven-week-old, specific pathogen-free male Sprague Dawley rats purchased from OrientBio (Sungnam, Korea) were used. All experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Animal, and the Institutional Animal Care and Use Committee at Konkuk University approved this study (approval ID: KU10075).Upon arrival, the rats were weighed and housed individually. For the measurement of daily food intake, the rats were housed individually until the end of the study. Our study consisted of 2 weeks of adaptation, 5 weeks of induction of diabetes after surgery, and 6 weeks of the diet control period. At 13 weeks of age, after 2 weeks of adaptation, the rats were divided into two groups as follows: 5 for the sham-control group (C group) and 11 for the pancreatectomized group (Px group). At 18 weeks of age, after 5 weeks of induction of diabetes after surgery, the Px group was divided into two groups as follows: 6 for the ad libitum group (AL group) and 5 for the calorie-restriction group (R groups). The rats were fed standard chow based on AIN-76A (Dyets Inc., Bethlehem, PA, USA) and had free access to tap water during the study. Throughout the whole study, the C and AL groups were fed ad libitum, while the R group was fed ad libitum only before the diet control period and subsequently fed the same rate of daily food intake (g per kg of body weight per day) as the C group during 6-weeks of the diet control period. All rats were kept under conditions maintained in a 12 h light–dark cycle (light on 08:00–20:00 h), at a temperature of 20–23 °C and at a relative humidity of 40–65%. On the last day of the study, the rats were anesthetized by CO2 gas after overnight fasting, weighed immediately, and humanely sacrificed.
Subtotal pancreatectomy
To generate an end-stage insulin-deficient diabetes model in adult rats, we performed a subtotal pancreatectomy at 13 weeks of age. Briefly, we opened the abdominal wall under a 0.7 mg per kg body weight of Zoletil 50 (Virbac, Carros, France) and 0.2 mg per kg body weight of Rompun (Bayer Korea, Ansan, Korea) anesthesia and removed the pancreatic tissue carefully with a cotton tipped applicator from the spleen to 1 mm away from the common bile duct without vascular injury. After the surgery, the rats were covered with sheets and were incubated under an infrared light to maintain body temperature in the normal range. The control rats underwent a sham operation without the removal of pancreatic tissue.Food intake, fasting blood glucose (FBG), and body weight
Food intake (g) was individually measured every day, and an average daily food intake (g day−1) was calculated weekly. FBG levels (mg dl−1) were measured at 9 am every other week after overnight fasting from tail vein blood using a portable glucometer (Caresense II, Gentrol Co., Incheon, Korea). Body weight was measured every week and on the last day of the study just before the rats were sacrificed. Every weekend, the average daily food intake rates (g kg−1 day−1) of individual rats were determined using the data for an average daily food intake (g day−1) and body weight (kg) of an individual rat, which were collected during the corresponding weekdays. Then the average daily food intake rate of the C group was calculated every weekend, which was used as the daily food intake rate for the R group during the next week.Plasma insulin and C-peptide analysis
Overnight fasting blood samples were taken from the inferior vena cava for the determination of insulin and C-peptide concentrations immediately before the excision of organs. Plasma insulin and C-peptide levels were analyzed using radioimmunoassay kits (Millipore, Billerica, MA, USA) according to the manufacturer's instructions, and the resulting radioactivities were measured using a γ-counter (Beckman Coulter, Brea, CA, USA).Preparation for the hippocampal tissue
At the end of the study, brain tissue was excised from the CO2-anesthetized rats and weighed immediately. Both sides of the hippocampus were rapidly separated and flash-frozen in liquid nitrogen for western blots. The frozen samples were ground to powder in liquid nitrogen and stored at −70 °C until use.Western blot analysis
The frozen-ground hippocampal tissue samples were homogenized in ice-cold buffer containing 25 mM HEPES, 25 mM benzamidine, 100 mM sodium fluoride, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 1% Triton X-100, 4 mM EDTA, 5 μl mL−1 of phosphatase inhibitor cocktail I, 5 μl mL−1 of phosphatase inhibitor cocktail II, and 5 μl mL−1 of protease inhibitor cocktail. After centrifugation at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400g for 30 min at 4 °C, the supernatants were collected, and the protein concentrations were measured by using a BCA kit (Pierce, Rockford, IL, USA) according to the manufacturer's instructions. The extracted proteins were separated on SDS polyacrylamide gels, i.e., 8% gels for AMPK, PP2A, tau, and Akt and 15% gels for GSK-3β. The separated proteins were transferred to nitrocellulose (Bio-Rad Laboratories, Inc., Hercules, CA, USA) membranes at 250 mA for 1 hour and 30 min.
400g for 30 min at 4 °C, the supernatants were collected, and the protein concentrations were measured by using a BCA kit (Pierce, Rockford, IL, USA) according to the manufacturer's instructions. The extracted proteins were separated on SDS polyacrylamide gels, i.e., 8% gels for AMPK, PP2A, tau, and Akt and 15% gels for GSK-3β. The separated proteins were transferred to nitrocellulose (Bio-Rad Laboratories, Inc., Hercules, CA, USA) membranes at 250 mA for 1 hour and 30 min.
The membranes were blocked by incubating them with 5% bovine serum albumin buffer for 1 hour at room temperature, and they were then incubated overnight with the primary antibodies (see Table S1†), including phospho-Thr172 AMPK (1:5000; Cell Signaling Technology, Inc., Danvers, MA, USA), phospho-PP2A (1:1000; Millipore, Bellerica, MA, USA), phospho-Ser199/202 tau (1:1000; Invitrogen, Camarillo, CA, USA), phospho-Ser396 tau (1:1000; Invitrogen), phospho-Ser473 Akt (1:5000; CST, Inc.), or phospho-Ser9 GSK-3β antibodies (1:1000; CST, Inc.) at 4 °C.
We also detected total AMPK (1:5000, CST, Inc.), total PP2A (1:1000; Millipore), total tau (1:5000; Invitrogen), total Akt antibody (1:5000; CST, Inc.), or total GSK-3β antibody (1:1000; CST, Inc.) activity. The membranes were then developed using a secondary antibody, horseradish peroxidase-conjugated anti-rabbit IgG (1:5000; CST, Inc.), followed by detection with enhanced chemiluminescence (GE Healthcare, Wauwatosa, WI, USA). The immunoreactive protein bands were quantified by using Multi Gauge version 3.1 (Fujifilm, Tokyo, Japan).
Histology
After the experimental rats (additional three animals for each group which were raised under the same experimental conditions, but were used only for histology) were perfused with cold PBS, followed by cold 4% paraformaldehyde, the brain was excised, fixed in 4% paraformaldehyde, embedded in paraffin, and sliced into 5 μm thick sections. The sections containing hippocampus were stained with crystal violet (CV).Immunohistochemistry
Briefly, the sections were deparaffinized, rehydrated and washed in PBS-TW, treated with 55% formic acid and 2% hydrogen peroxide, blocked with 10% FBS and incubated overnight at 4 °C with phospho-Tau (1:500, Invitrogen). Then, the sections were sequentially incubated with peroxidase-conjugated anti-rabbit IgG (1:500; CST, Inc.). Peroxidase activity was developed with 0.05% 3,3′-diaminobenzidine (Sigma) and 0.01% hydrogen peroxide until the reaction products were visualized (brown color).
Statistical analysis
Data are presented as the mean ± SD. Statistical analysis was performed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). We compared the differences among the experimental groups in food intake rate, fasting glucose level, and change of body weight at each measurement time by using one-way ANOVA with Tukey's post hoc test. In addition, the interaction (time × treatment) in Fig. 1 was analyzed by repeated measured ANOVA. P values of less than 0.05 were considered statistically significant.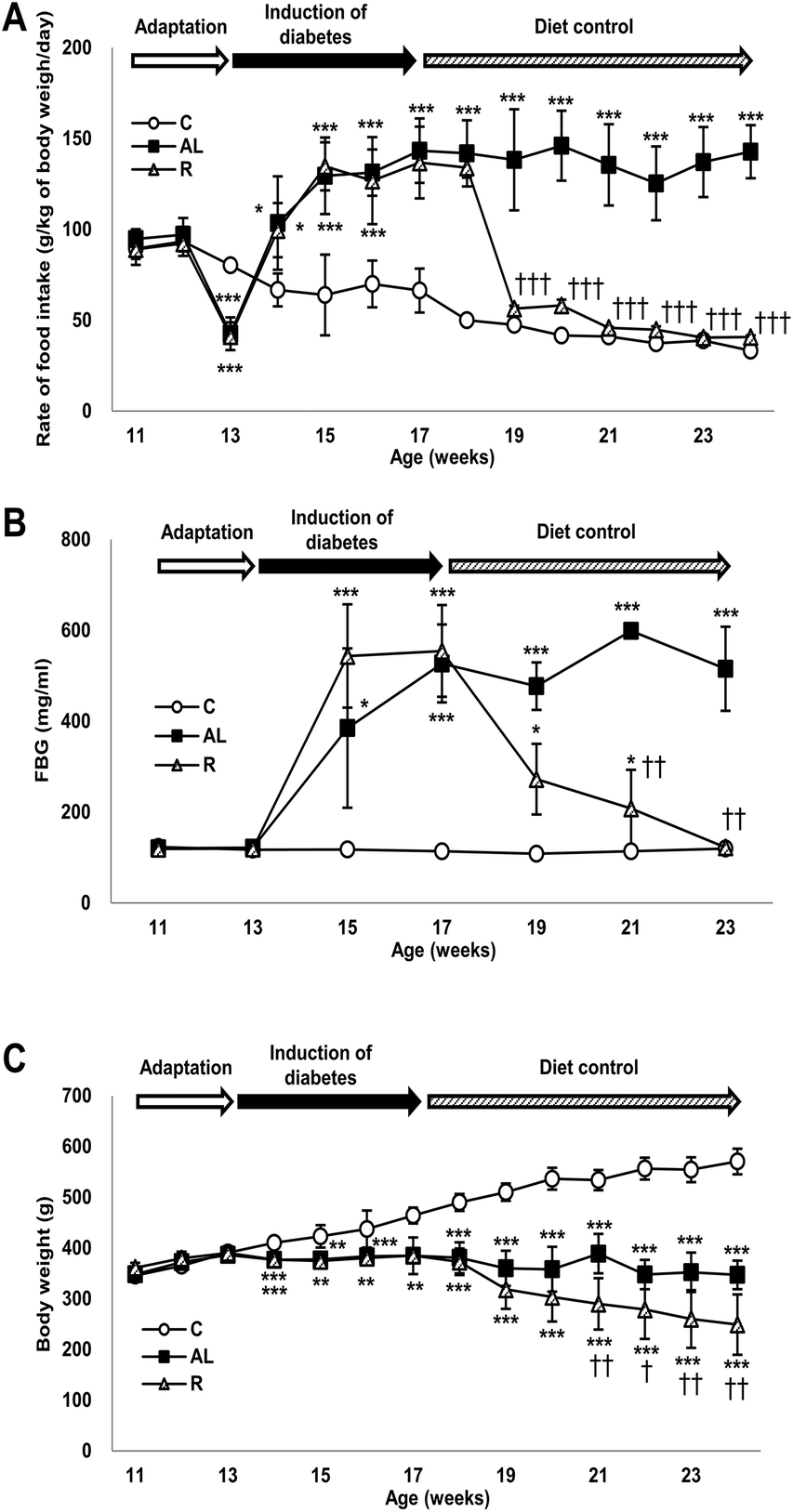 | ||
| Fig. 1 Sequential changes in (A) daily food intake (g per kg body weight per day), (B) fasting blood glucose (FBG) levels, and (C) body weights. Data are presented as means ± SD and were analyzed with one-way ANOVA followed by Tukey's post hoc test. C: sham-operated rats fed ad libitum (n = 5); AL: pancreatectomized diabetic rats fed ad libitum (n = 6); R: pancreatectomized diabetic rats fed controlled diet to achieve euglycaemia during the diet control period (n = 5). *P < 0.001, **P < 0.05, and *P < 0.01 versus C; †P < 0.01, ††P < 0.05, and †††P < 0.001 versus AL. | ||
Results
Control of the food intake rate
Fig. 1A shows changes in the rate of food intake (g per kg of body weight per day) of all groups throughout the study. The food intake rate showed significant interactions between dietary treatment and duration (F = 240.303, P < 0.001). The rate of food intake in the sham-control C group continuously decreased during the entire study; however, the rate of food intake in the Px groups increased significantly during the induction period of diabetes compared with that in the C group. The rate of food intake in the hyperglycemic Px group fed ad libitum (AL group) continuously increased for 4 weeks after surgery and then maintained until the end of the study. During the diet control period, the rate of food intake in the euglycaemic Px group fed with calorie-restriction diet (R group) decreased as in the C group. The food intake rates of the C and R groups were 1/3 of that of the AL group and were not significantly different during the diet control period.Changes in fasting blood glucose levels
Fig. 1B shows changes in the FBG levels of all groups throughout the study, in which we found a significant time × treatment interaction (F = 34.786, P < 0.001). The C group maintained euglycaemia throughout the study. However, the FBG levels of the Px groups increased significantly and showed hyperglycaemia during the induction period of diabetes compared with those of the C group (approximately 4.2-fold). During the diet control period, the FBG levels of the R group decreased similarly to those of the C group, while the AL group maintained hyperglycaemia until the end of the study.Changes in body weight
Fig. 1C shows changes in the body weight (g) of all groups throughout the study and there was a significant time × treatment interaction (F = 60.418, P < 0.001). The body weight of the C group increased continuously throughout the study; however, the body weight of the AL group was maintained after surgery until the end of the study. The body weight of the R group was maintained during the induction period of diabetes but decreased continuously and significantly during the diet control period compared with that of the AL group.Fasting plasma insulin and C-peptide levels
To confirm that we generated insulin deficiency in the Px groups, we measured the plasma insulin (μU mL−1) and C-peptide (nmol L−1) levels. Fig. 2 shows the mean plasma insulin (A) and C-peptide (B) levels. The mean plasma insulin levels of the Px groups, including the AL group and R group, were 7.9% and 4.4% of that of the C group, respectively. There was also a significant difference (F = 36.509, P < 0.001 by one-way ANOVA). The mean plasma C-peptide levels in the Px groups, including the AL group and R group, were 19.1% and 8.7% of that in the C group, respectively (P < 0.001 for all).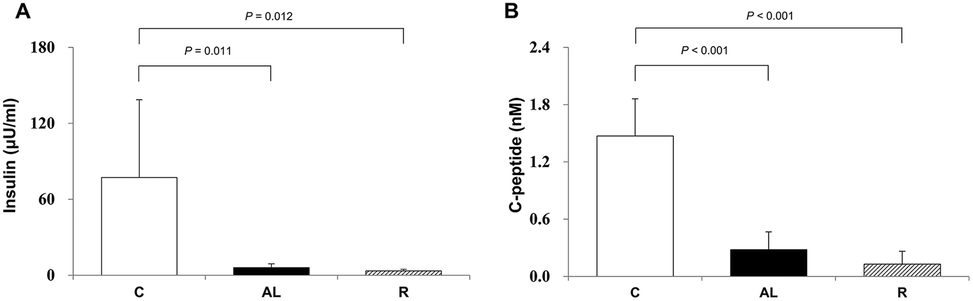 | ||
| Fig. 2 Endogenous plasma insulin and C-peptide levels. Data are presented as means ± SD and were analyzed with one-way ANOVA followed by Tukey's post hoc test. C: sham-operated rats fed ad libitum (n = 5); AL: pancreatectomized diabetic rats fed ad libitum (n = 6); R: pancreatectomized diabetic rats fed diet control to achieve euglycaemia during the diet control period (n = 5). | ||
Induction of AMPK activation and tau hyperphosphorylation in the hippocampus
To confirm that the diet control to achieve euglycaemia induced AMPK activation and tau hyperphosphorylation in the hippocampus of insulin-deficient DM rats, we investigated the phosphorylation of AMPK and tau phosphorylation at Ser199/202 and Ser396 (Fig. 3). In the AMPK phosphorylation, there was a significant difference among the groups (F = 2386.885, P < 0.001). AMPK phosphorylation in the R group was 3.1-fold higher than that in the C group (P < 0.001) and 2.5-fold higher than that in the AL group (P < 0.001). However, there was no significant difference in the levels of AMPK phosphorylation between the C and AL groups. The levels of tau phosphorylation at Ser199/202 of the Px groups (the AL and R groups) were 2.4- and 5.0-fold higher, respectively, than that of the C group (C vs. AL, P = 0.036; C vs. R, P < 0.001). The level of tau phosphorylation at Ser199/202 in the R group was 2.1-fold higher than that in AL (AL vs. R, P = 0.002). The levels of tau phosphorylation at Ser396 of the AL and R groups were also 2.2- and 3.1-fold higher, respectively, than that of the C group (C vs. AL, P = 0.018; C vs. R, P = 0.001). Furthermore, the level of tau phosphorylation at Ser396 of the R group was 1.4-fold higher than that of the AL group (AL vs. R, P = 0.047).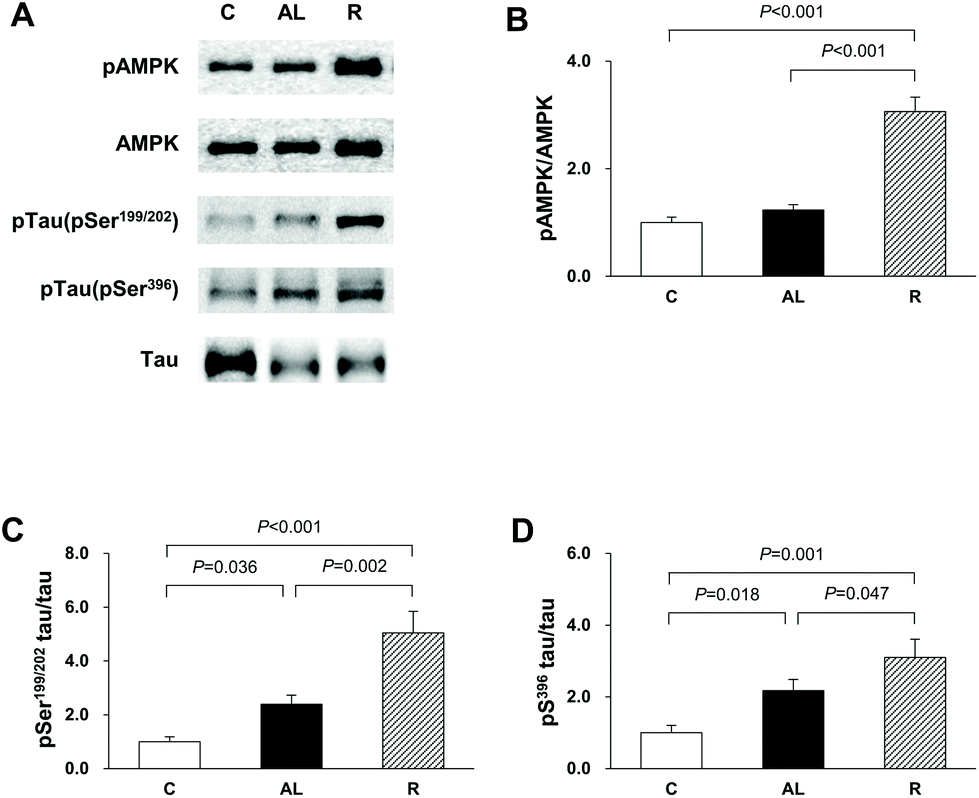 | ||
| Fig. 3 (A) Western blot analyses. (B) The ratio of pAMPK to AMPK. The ratio of (C) ptau at Ser199/202, and (D) ptau at Ser396 to tau. The blots shown are representative of triplicate experiments. Data are presented as means ± SD and were analyzed with one-way ANOVA followed by Tukey's post hoc test. C: Sham-operated rats fed ad libitum (n = 5); AL: pancreatectomized diabetic rats fed ad libitum (n = 6); R: pancreatectomized diabetic rats fed diet control to achieve euglycaemia during the diet control period (n = 5). | ||
Akt, GSK-3β, and PP2A activities in the hippocampus
To determine the mechanism for the significantly higher tau phosphorylation in the hippocampus of euglycaemic insulin-deficient DM rats, we examined the Akt and GSK-3β phosphorylation levels (Fig. 4). Akt phosphorylation in the AL group was over 2-fold higher than that in the C or R group (AL vs. C, P = 0.003; AL vs. R, P = 0.002). However, there was no significant difference between the C and R groups. The levels of GSK-3β phosphorylation at Ser9, an inhibitory modification of GSK-3β by Akt, in the AL and R groups were 2.6- and 2.0-fold higher, respectively, than that in the C group (C vs. AL, P < 0.001; C vs. R, P = 0.002). However, the level of the R group significantly decreased by 76.4% compared to that of the AL group (AL vs. R, P = 0.016).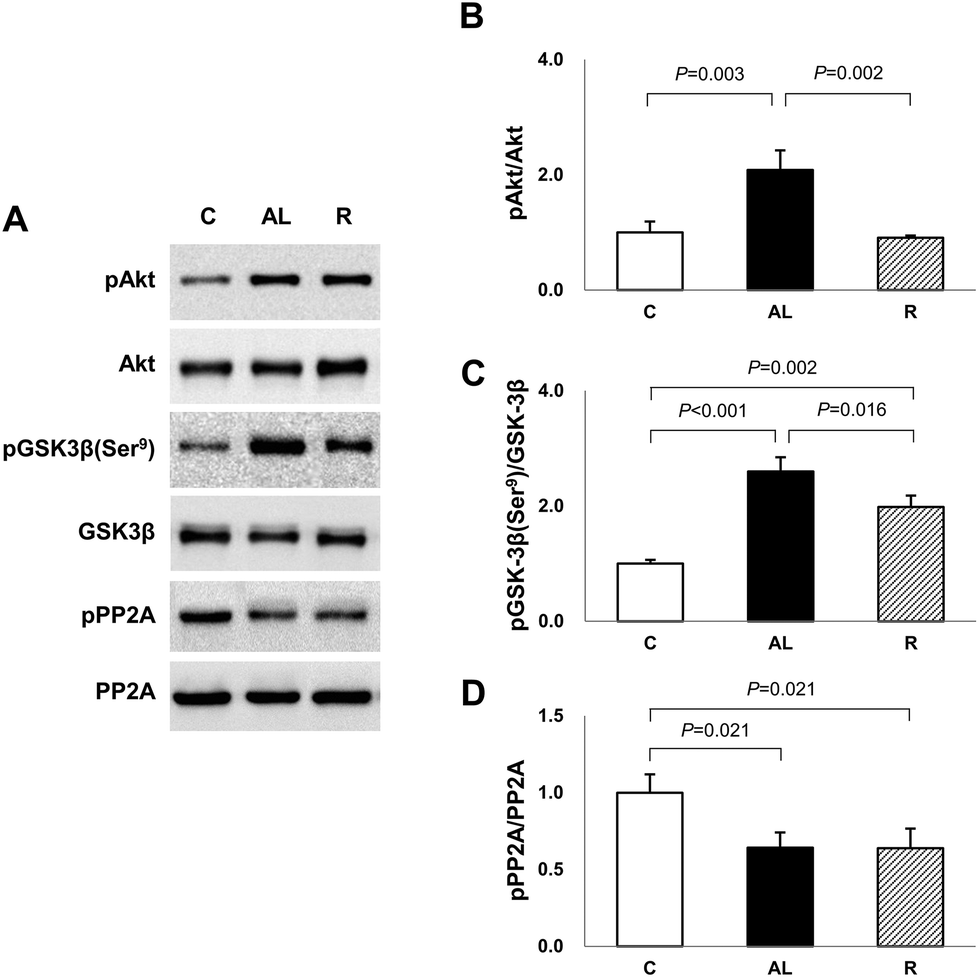 | ||
| Fig. 4 (A) Western blot analyses. (B) The ratio of pAkt to Akt. (C) The ratio of pGSK-3β at Ser9 to GSK-3β. (D) The ratio of pPP2A to PP2A. The blots shown are representative of triplicate experiments. Data are presented as means ± SD and were analyzed with one-way ANOVA followed by Tukey's post hoc test. C: Sham-operated rats fed ad libitum (n = 5); AL: pancreatectomized diabetic rats fed ad libitum (n = 6); R: pancreatectomized diabetic rats fed diet control to achieve euglycaemia during the diet control period (n = 5). | ||
To further explain the mechanism of the increased AMPK activation and tau phosphorylation in the hippocampus of euglycaemic insulin-deficient DM rats, although their GSK-3β activity was inhibited compared to that of the C group, we investigated PP2A activity (Fig. 4). The levels of PP2A phosphorylation of Px groups decreased by 64% compared to that of the C group (C vs. AL or R, P = 0.021 for all). However, there was no significant difference between the Px groups.
Histopathology of the hippocampal CA1 region
To determine whether diet control to achieve euglycaemia induced tau hyperphosphorylation, we investigated the AD pathology in the CA1 region of the hippocampus (Fig. 5). CV staining showed that the cell layer of the AL group was thinner than that of the C group, however, the cell layer of the R group was the thinnest among all groups. Furthermore, immunohistochemistry (IHC) staining against phospho-tau showed that the density of phospho-tau was higher in the AL group than that in the C group (P = 0.022), but the density was the highest in the R group (P < 0.001 vs. C; P = 0.003 vs. AL, shown in Fig. 5C).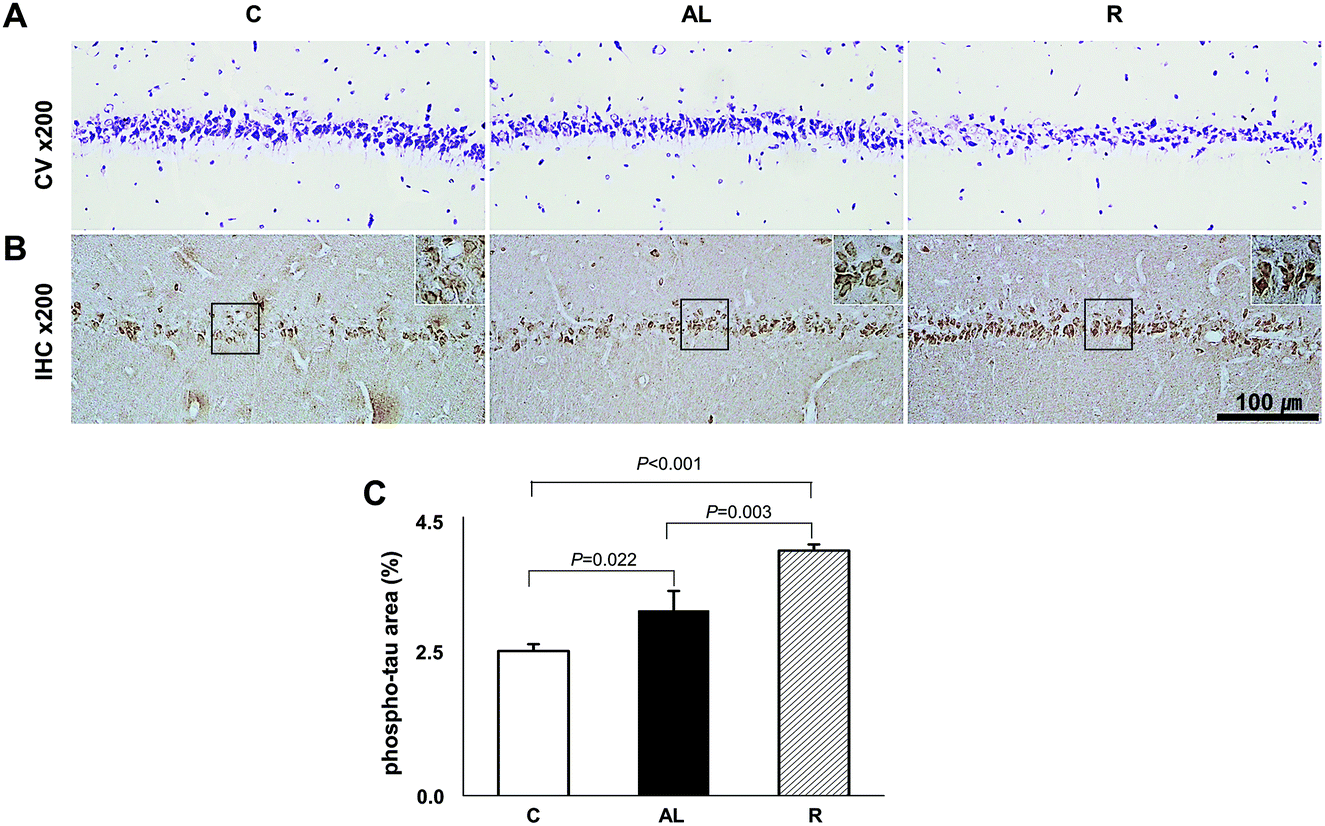 | ||
| Fig. 5 Hippocampus CA1 region histology. (A) Crystal violet staining. (B) Immunohistochemistry against phospho-Tau. (C) The comparison of density area (%) of phospho-Tau. See Fig. S1 in the ESI.† We examined the histopathology of the hippocampus by hematoxylin and eosin (H&E) staining including dentate gyrus (DG) and the CA3 region as well as the CA1 region. C: Sham-operated rats fed ad libitum (n = 3); AL: pancreatectomized diabetic rats fed ad libitum (n = 3); R: pancreatectomized diabetic rats fed diet control to achieve euglycaemia during the diet control period (n = 3). | ||
Discussion
In this study, we demonstrated that a diet control to achieve euglycaemia induced AMPK activation and tau hyperphosphorylation in the hippocampus of insulin-deficient DM rats. We suggest that diet control to achieve euglycaemia has a dangerous effect on maintaining energy metabolism and structural integrity, although achieving euglycaemia is a major therapeutic target in DM.AMPK is a cellular sensor that regulates the activity of various metabolic enzymes to maintain energy homeostasis. AMPK is a heterotrimeric Ser/Thr protein kinase composed of a catalytic α subunit and two regulatory β and γ subunits. This kinase is activated via Thr172 phosphorylation in the activation loop of the catalytic α2 subunit during energy stress by different upstream kinases.22 AMPK is expressed in most mammalian tissues and cell types, including cerebral neurons, and is thought to play an important role in regulating energy homeostasis.23
In this study, we found that AMPK phosphorylation was higher in the R group than in the other groups, although the FBG level of the R group did not differ from that of the C group. We also observed that the levels of phospho Ser199/202 and Ser396 tau proteins were higher in the hippocampus of the euglycaemic insulin-deficient R group, in which the level of phospho AMPK was the highest, than those of the hyperglycemic insulin-deficient AL group (Fig. 3). Although the level of tau phosphorylation was higher in the hyperglycemic AL group than in the C group, the level was the highest in the R group, where the level of AMPK phosphorylation was higher in the R group than in the AL group. Recently, many studies have reported that AMPK plays a key role in tau phosphorylation and directly regulates various tau phosphorylation sites.16,24,25 We suggest that the diet control to achieve euglycaemia during insulin deficiency may aggravate AD, because the significantly high AMPK activity following calorie-restriction may lead to tau hyperphosphorylation in the hippocampus of insulin-deficient DM animals.
As Akt-GSK3β is known to be a typical upstream pathway involved in the regulation of tauopathy in the development of AD, we also investigated whether diet control to achieve euglycaemia affected Akt-GSK3β signaling. We confirmed that the phosphorylation of Akt and GSK3β Ser9 in the AL group, which was maintained with hyperglycaemia following ad libitum diet during insulin deficiency, was greater than that in the C group. However, the phosphorylation of GSK3β Ser9 in the R group was also greater than that in the C group, although the level of Akt phosphorylation in the R group did not differ compared to that in the C group (Fig. 4). GSK3β is phosphorylated at Ser9 by activated Akt, which is activated during hyperglycaemia in the brain,26 and our present study demonstrated the significantly high Akt-GSK3β activity in the hippocampus of the hyperglycemic AL group. However, it was unclear why GSK3β activity was inhibited in the hippocampus of the euglycaemic insulin-deficient R group, where Akt was not activated as compared to that of the AL group. To explain this, we investigated the activity of PP2A, which is a kinase inhibitor. PP2A is a soluble protein mainly in the cytoplasm, but it is also found in the nucleus, mitochondria, cytoskeleton, and cell membrane. PP2A has multiple roles in cell cycle regulation, cell morphology, and signal transduction by dephosphorylating different substrates and critically regulating the integrity of the cytoskeleton.27 PP2A is also known to directly dephosphorylate various serine sites of tau, including Ser199/202 and Ser28,396 and to regulate the activation of GSK3β via dephosphorylation at Ser9.29 In addition, decreased PP2A activity and decreased phosphorylation of GSK3β at Ser9 in the cortex and hippocampus of streptozotocin-injected DM mice were observed compared with those in normal mice.30 Our study showed that PP2A activity decreased in the hippocampus of Px rats compared with that in the C group rats. Therefore, we speculate that the increase in the phosphorylation of GSK3β at Ser9 despite the decrease in Akt phosphorylation in the R group can be explained by PP2A, which was dephosphorylated and then may not dephosphorylate pGSK3β at Ser9 in the R group, resulting in the maintenance of the level of GSK3β Ser9 phosphate.
PP2A is also known to regulate the phosphorylation of the AMPK α-subunit at Thr172.20 Although the exact pathway between AMPK and PP2A was unclear in this study, we speculate that an insulin-deficient DM condition and diet control to achieve euglycaemia dephosphorylate PP2A and affect the phosphorylation of AMPK and GSK3β; therefore, this protein seems to be involved in tau hyperphosphorylation.
Based on these findings, we suggest that GSK3β activation may not be involved in tau hyperphosphorylation in the hippocampus of insulin-deficient DM rats, for the level of PP2A phosphorylation decreased in our DM rats, by which GSK3β was maintained inactive while tau was hyperphosphorylated compared to the C group. In addition, since the diet control to achieve euglycaemia during insulin deficiency has a starvation-like effect, this diet control induced a significant tau hyperphosphorylation via activated AMPK together with the decreased PP2A activity in the hippocampus of the euglycemic insulin-deficient R group compared with that of the hyperglycemic AL group. Biguanide metformin has been reported to have a beneficial effect on tau hyperphosphorylation via activated AMPK and PP2A in vivo and in vitro.31 However, they did not use DM model animals but used murine primary neurons from wild-type and human tau transgenic mice for their experiment. Therefore, we propose that the significantly high AMPK activation in insulin-deficient DM condition has a harmful effect on tau hyperphosphorylation in the hippocampus. Fig. 6 briefly shows the protein signal transduction that occurred in the insulin-deficient hippocampus in this study.
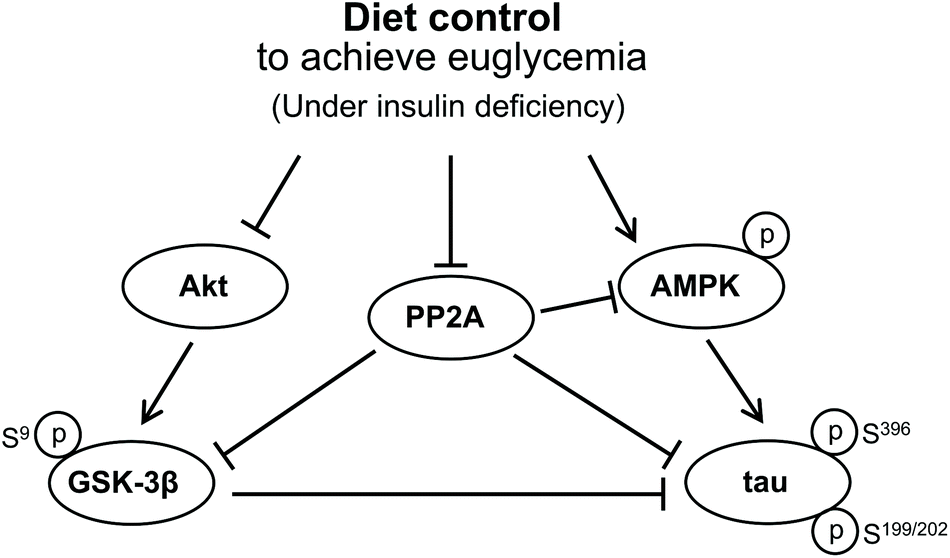 | ||
| Fig. 6 Signal transduction that concerned the tau hyperphosphorylation in the brain under an insulin deficiency condition. | ||
Conclusion
We suggest that diet control to achieve euglycaemia may increase AMPK activity and decrease PP2A activity, thus playing a key role in tau hyperphosphorylation in the hippocampus of insulin-deficient DM. Although euglycaemia can be achieved, a diet control and/or combination of AMPK-activating oral anti-diabetic agent treatment should be considered carefully in insulin-deficient DM patients because of the possibility of AD development through tau hyperphosphorylation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Konkuk University in 2015.References
- I. Grundke-Iqbal, K. Iqbal, Y. C. Tung, M. Quinlan, H. M. Wisniewski and L. I. Binder, Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology, Proc. Natl. Acad. Sci. U. S. A., 1986, 8, 4913–4917 CrossRef PubMed.
- T. Hartmann, S. C. Bieger, B. Brühl, P. J. Tienari, N. Ida, D. Allsop, G. W. Roberts, C. L. Masters, C. G. Dotti, K. Unisicker and K. Beyreuther, Distinct sites of intracellular production for Alzheimer's disease a beta40/42 amyloid peptides, Nat. Med., 1997, 3, 1016–1020 CrossRef CAS PubMed.
- T. L. Spires-Jones, W. H. Stoothoff, A. de Calignon, P. B. Jones and B. T. Hyman, Tau pathophysiology in neurodegeneration: A tangled issue, Trends Neurosci., 2009, 32, 150–159 CrossRef CAS PubMed.
- P. A. Maher and D. R. Schubert, Metabolic links between diabetes and Alzheimer's disease, Expert Rev. Neurother., 2009, 9, 617–630 CrossRef CAS PubMed.
- G. J. Biessels, S. Staekenborg, E. Brunner, C. Brayne and P. Scheltens, Risk of dementia in diabetes mellitus: A systematic review, Lancet Neurol., 2006, 5, 64–67 CrossRef PubMed.
- Z. Arvanitakis, R. S. Wilson, J. L. Bienias, D. A. Evans and D. A. Bennett, Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function, Arch. Neurol., 2004, 61, 661–666 CrossRef PubMed.
- C. G. Jolivalt, C. A. Lee, K. K. Beiswenger, J. L. Smith, M. Orlov, M. A. Torrance and E. Masliah, Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: Parallels with Alzheimer's disease and correction by insulin, J. Neurosci. Res., 2008, 86, 3265–3274 CrossRef CAS PubMed.
- Z. Qu, Z. Jiao, X. Sun, Y. Zhao, J. Ren and G. Xu, Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain, Brain Res., 2011, 1383, 300–306 CrossRef CAS PubMed.
- I. M. Stratton, A. I. Adler, H. A. Neil, D. R. Matthews, S. E. Manley, C. A. Cull, D. Hadden, R. C. Turner and R. R. Holman, Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study, Br. Med. J., 2000, 321, 405–412 CrossRef CAS PubMed.
- C. J. Currie, J. R. Peters, A. Tynan, M. Evans, R. J. Heine, O. L. Bracco, T. Zagar and C. D. Poole, Survival as a function of HbA(1c) in people with type 2 diabetes: A retrospective cohort study, Lancet, 2010, 375, 481–489 CrossRef CAS.
- J. MacLeod, M. J. Franz, D. Handu, E. Gradwell, C. Brown, A. Evert, A. Reppert and M. Robinson, Academy of nutrition and dietetics nutrition practice guideline for type 1 and type 2 diabetes in adults: Nutrition intervention evidence reviews and recommendations, J. Acad. Nutr. Diet., 2017, 117, 1637–1658 CrossRef.
- American Diabetes Association, 4. Lifestyle management: Standards of medical care in diabetes-2018, Diabetes Care, 2018, 41, S38–SS50 CrossRef PubMed.
- J. H. Lee, J. H. Lee, M. Jin, S. D. Han, G. R. Chon, I. H. Kim, S. Kim, S. B. Choi and Y. H. Noh, Diet control to achieve euglycaemia induces significant loss of heart and liver weight via increased autophagy compared with ad libitum diet in diabetic rats, Exp. Mol. Med., 2014, 46, e111 CrossRef CAS PubMed.
- J. H. Lee, S. B. Choi, M. Jin, J. H. Lee, S. D. Han, H. Bae, I. Lim and Y. H. Noh, Euglycaemia in diabetic rats leads to reduced liver weight via increased autophagy and apoptosis through increased ampk and caspase-3 and decreased mtor activities, J. Diabetes Res., 2015, 2015, 497431 Search PubMed.
- C. Thornton, N. J. Bright, M. Sastre, P. J. Muckett and D. Carling, Amp-activated protein kinase (ampk) is a tau kinase, activated in response to amyloid beta-peptide exposure, Biochem. J., 2011, 434, 503–512 CrossRef CAS PubMed.
- V. Vingtdeux, P. Davies, D. W. Dickson and P. Marambaud, AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer's disease and other tauopathies, Acta Neuropathol., 2011, 121, 337–349 CrossRef CAS PubMed.
- S. E. Kahn, S. Zarika, K. M. Utzschneider and R. L. Hull, The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality, Diabetologia, 2009, 52, 1003–1012 CrossRef CAS.
- D. Tripathy, M. Carlsson, P. Almgren, B. Isomaa, M. R. Taskinen, T. Tuomi and L. C. Groop, Insulin secretion and insulin sensitivity in relation to glucose tolerance: lessons from the Botnia Study, Diabetes, 2000, 49, 975–980 CrossRef CAS PubMed.
- M. E. Lean, W. S. Leslie, A. C. Barnes, N. Brosnahan, G. Thom, L. McCombie, C. Peters, S. Zhyzhneuskaya, A. Al-Mrabeh, K. G. Hollingsworth, A. M. Rodrigues, L. Rehackova, A. J. Adamson, F. F. Sniehotta, J. C. Mathers, H. M. Ross, Y. McIlvenna, R. Stefanetti, M. Trenell, P. Welsh, S. Kean, I. Ford, A. McConnachie, N. Sattar and R. Taylor, Primary care-led weight management for remission of type 2 diabetes (DiRECT): an open-label, cluster-randomised trial, Lancet, 2018, 391, 541–551 CrossRef.
- S. P. Davies, N. R. Helps, P. T. Cohen and D. G. Hardie, 5′-amp inhibits dephosphorylation, as well as promoting phosphorylation, of the amp-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2c alpha and native bovine protein phosphatase-2ac, FEBS Lett., 1995, 377, 421–425 CrossRef CAS.
- J. M. Sontag and E. Sontag, Protein phosphatase 2A dysfunction in Alzheimer's disease, Front. Mol. Neurosci., 2014, 7, 16 Search PubMed.
- S. M. Jeon, Regulation and function of ampk in physiology and diseases, Exp. Mol. Med., 2016, 48, e245 CrossRef CAS.
- G. V. Ronnett, S. Ramamurthy, A. M. Kleman, L. E. Landree and S. Aja, AMPK in the brain: Its roles in energy balance and neuroprotection, J. Neurochem., 2009, 109, 17–23 CrossRef CAS PubMed.
- A. Salminen, K. Kaarniranta, A. Haapasalo, H. Soininen and M. Hiltunen, AMP-activated protein kinase: A potential player in Alzheimer's disease, J. Neurochem., 2011, 118, 460–474 CrossRef CAS.
- Z. Cai, L. J. Yan, K. Li, S. H. Quazi and B. Zhao, Roles of AMP-activated protein kinase in Alzheimer's disease, NeuroMol. Med., 2012, 14, 1–14 CrossRef CAS.
- B. Clodfelder-Miller, P. De Sarno, A. A. Zmijewska, L. Song and R. S. Jope, Physiological and pathological changes in glucose regulate brain Akt and glycogen synthase kinase-3, J. Biol. Chem., 2005, 280, 39723–39731 CrossRef CAS.
- V. Janssens and J. Goris, Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling, Biochem. J., 2001, 353, 417–439 CrossRef CAS.
- Q. Tian and J. Wang, Role of serine/threonine protein phosphatase in Alzheimer's disease, Neurosignals, 2005, 11, 262–269 CrossRef.
- X. W. Zhou, B. Winblad, Z. Guan and J. J. Pei, Interactions between glycogen synthase kinase 3beta, protein kinase B, and protein phosphatase 2A in tau phosphorylation in mouse N2a neuroblastoma cells, J. Alzheimer's Dis., 2009, 17, 929–937 CAS.
- B. J. Clodfelder-Miller, A. A. Zmijewska, G. V. Johnson and R. S. Jope, Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency, Diabetes, 2006, 55, 3320–3325 CrossRef CAS PubMed.
- E. Kickstein, S. Krauss, P. Thornhill, D. Rutschow, R. Zeller, J. Sharkey, R. Williamson, M. Fuchs, A. Köhler, H. Glossmann, R. Schneider, C. Sutherland and S. Schweiger, Biguanide metformin acts on tau phosphorylation via mtor/protein phosphatase 2A (pp2a) signaling, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21830–21835 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo00709a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |