
FSGHF3 and peptides, prepared from fish skin gelatin, exert a protective effect on DSS-induced colitis via the Nrf2 pathway†
Zhao
Deng
 a,
Chenbin
Cui
a,
Yanan
Wang
a,
Jiangjin
Ni
a,
Liufeng
Zheng
a,
Hong-Kui
Wei
*ab and
Jian
Peng
*ab
a,
Chenbin
Cui
a,
Yanan
Wang
a,
Jiangjin
Ni
a,
Liufeng
Zheng
a,
Hong-Kui
Wei
*ab and
Jian
Peng
*ab
aDepartment of Animal Nutrition and Feed Science, College of Animal Science and Technology, Huazhong Agricultural University, 430070, Wuhan, P. R. China. E-mail: weihongkui@mail.hzau.edu.cn; pengjian@mail.hzau.edu.cn; Tel: +86 027-87280122
bThe Cooperative Innovation Center for Sustainable Pig Production, Wuhan 430070, Hubei, China
First published on 3rd December 2019
Abstract
Inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), is a chronic inflammatory disease affecting the colon, and its incidence is rising worldwide. We previously found that fish skin gelatin hydrolysate fraction 3 (FSGHF3), isolated from fish skin gelatin hydrolysate, could exert antioxidant effects and maintain tight junctions in IPEC-J2 cells. Further HPLC-ESI-QqQ-MS results revealed that this fraction mainly included some peptides. Here, we aim to evaluate the effects of FSGHF3 and peptides in the mice model of dextran sodium sulfate (DSS)-induced colitis and LPS induced inflammation in IECs. The results show that FSGHF3 significantly ameliorates the clinical symptoms of DSS-induced colitis in mice, such as weight loss, disease activity index (DAI), colon shortening, spleen hypertrophy, histological scores, and MPO activity. FSGHF3 and peptide treatment inhibits pro-inflammatory cytokine production, leading to the maintenance of intestinal architecture in vivo and in vitro. Meanwhile, FSGHF3 and peptide treatment promotes antioxidant enzyme expression via activating Nrf2, which results in the removal of ROS and inhibition of NF-κB activation. Overall, our results suggest that FSGHF3 and peptides may be promising potential candidates for the alleviation of colitis.
1. Introduction
Inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), is a chronic relapsing inflammation of the gastrointestinal tract.1 CD is recognized by chronic intermittent inflammation usually involving the terminal ileum but can potentially affect any part of the gastrointestinal tract. UC is a chronic, relapsing inflammatory disease that most commonly affects the colon of adults and results in weight loss, diarrhea, rectal bleeding, and abdominal pain.2,3 Although the etiology and pathogenesis of IBD are complicated and remain poorly understood, environmental, genetic, and lifestyle factors, host immune system, intestinal microflora, and altered colonic barrier function are involved in the pathology of IBD.4–6In patients with IBD, the intestinal flora of the colon is disturbed and tight junctions are disrupted, allowing intestinal flora to enter the lamina propria. Intestinal epithelial cells and immune cells come into contact with a lot of bacteria, producing a large amount of ROS and pro-inflammatory factors, resulting in the activation of T cells which inflict further inflammatory damage on the epithelium and immune imbalance.7 Therefore, the inhibition of pro-inflammatory cytokines and ROS production, and maintenance of the integrity of the intestinal barrier may be important steps to treat IBD.7
The NF-κB signaling pathway plays a predominant role in IBD pathogenesis.8–12 Stimulation of the tumor necrosis factor receptor (TNFR) with bacterial lipopolysaccharides (LPS) results in NF-κB activation, thereby inducing the production of pro-inflammatory cytokines, such as TNF-α, interleukin-6 (IL-6), IL-8 and IL-12.12 To date, several in vitro and in vivo studies have indicated that anti-NF-κB therapies have successfully reduced pathology and morbidity in IBD.13,14
Nuclear factor-erythroid 2-related factor-2 (Nrf2) is a transcription factor that plays a critical role in cellular defenses against inflammation and oxidative stress through the induction of antioxidant enzymes.15 Once stimulated, Nrf2 translocates into the nucleus and then binds to the antioxidant response element (ARE), a cis-acting DNA regulatory element that activates the promoter regions of many gene encoding antioxidants, such as glutathione peroxidase (GPX), γ-glutamylcysteine ligase catalytic (GCLC) subunit, and glutamylcysteine ligase modulator (GCLM).16,17 Recently, several studies have focused on potent anti-inflammatory and antioxidant effects of Nrf2 in inflammatory diseases. In macrophages, Nrf2 inhibits inflammasome activation and ROS production.18 In IPEC-J2 cells, OEO (oregano essential oil) treatment induces SOD1 and GSH expression and alleviates oxidative stress through Nrf2 activation.19 Therefore, we assume that Nrf2 activation may be an effective way to treat intestinal inflammation.
Recently, numerous studies have reported that some nutrients, food components and bioactive peptides exert beneficial effects on IBD.20,21 For the treatment of colitis, some studies have demonstrated that some peptides, isolated from different gelatin sources, exert anti-inflammatory effects in vitro and in vivo.22–25 We previously found that fish skin gelatin hydrolysate fraction 3 (FSGHF3), isolated from fish skin gelatin hydrolysate, could alleviate oxidative stress and maintain the epithelial barrier through Nrf2 activation, and further HPLC-ESI-QqQ-MS results revealed that this fraction mainly included some peptides, such as Gly-Pro-Arg, Gly-Pro-Hyp, Leu-Hyp-Gly, and Hyp-Pro.26,27 However, the antioxidant and anti-inflammatory effects of FSGHF3 and peptides are not clear in vivo. The objectives of this study are to investigate the effects of FSGHF3 and peptides on dextran sodium sulfate (DSS) induced colitis. In the current study, we find that, through activating Nrf2, FSGHF3 and peptides up-regulate the expression of antioxidant enzymes and suppress ROS production, thereby inhibiting the inflammatory response and maintaining the intestinal barrier, resulting in the alleviation of DSS-induced colitis. Taken together, these findings may provide an effective nutrient peptide to ameliorate colitis.
2. Materials and methods
2.1 Reagents and antibodies
Synthetic peptides were purchased from Top-Peptide Biotechnology Co., Ltd (Top-Peptide, Shanghai, China). Dextran sodium sulfate (DSS, with a molecular weight of 36–50 kDa) was purchased from MP Biomedicals (Irvine, CA, USA). The protease inhibitor cocktail (C0001) was purchased from Target Mol (Topscience, Shanghai, China). Cell lysis buffer for western blot analysis (P0013) and BCA kit (P0011) were purchased from Beyotime (Shanghai, China). The TRIZOL reagent was purchased from Takara (Beijing, China). The first-strand cDNA synthesis kit was purchased from HLING (Shanghai, China). iTaq Universal SYBR Green Supermix was purchased from HLING (Shanghai, China).The antibodies against β-actin (AC026) were purchased from Abclonal (Wuhan, China). The antibodies against ZO-1 (AF5145) and occludin (DF7504) were purchased from Affinity (Cincinnati, USA). The antibody against Nrf2 (D1Z9C) was purchased from Cell Signaling Technology (Danvers, USA). The TNF-α ELISA kit (EMC102a) was purchased from NEOBIOSCIENCE (Shenzhen, China). MPO (A044-1-1), H2O2 (A064-1-1), GPX (A006-2-1) and GSH (A061-2-1) were purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China). PVDF membranes were purchased from Millipore Billerica (MA, USA).
2.2 Animals
Male C57BL/6 mice (5 weeks old) obtained from the Animal Experiment Center at the Huazhong Agricultural University (Wuhan, China) were used for the current study. The mice were housed under specific pathogen-free conditions in an air-conditioned room at 23 ± 2 °C. Food and water were supplied ad libitum. Animal welfare and experimental procedures were carried out based on the criteria outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978) and the related ethical regulations of Huazhong Agricultural University. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of Huazhong Agricultural University. All efforts were made to minimize animal suffering and to reduce the number of animals used.2.3 Establishment of a model of DSS-induced colitis in mice and its treatment
Acute colitis was induced by feeding mice with 3% (w/v) DSS, which was dissolved in drinking water, continuously for 7 days. The experiment was randomly divided into four groups: control group; DSS group; FSGHF3 (100 and 200 mg kg−1) + DSS. To assess the chemoprevention effect of FSGHF3 on DSS-induced acute colitis in C57BL/6 mice, the mice were treated with 3% DSS in their drinking water for 7 days to induce acute colitis. FSGHF3 was administered for 7 days before and during DSS treatment via oral gavage once per day. After 14 days, the mice were humanely euthanized, and colons were excised, measured, and sectioned for further analysis.2.4 Evaluation of colitis severity
We evaluated colitis severity based on body weight, colon length, and macroscopic and microscopic observations of the stool and colon, respectively. The disease activity index (DAI) score was determined by the method employed in previous studies, using five grades of weight change (0, no weight loss or gain; 1, 1–5% weight loss; 2, 5–10% weight loss; 3, 10–20% weight loss; and 4, more than 20% weight loss), stool consistency (0, normal; 1, mildly loose; 2, loose; 3, mild diarrhea; and 4, diarrhea), and stool bleeding (0, negative; 1, light bleeding; 2, mild bleeding; 3, severe bleeding; and 4, complete bleeding). Colon sections were prepared and stained with hematoxylin and eosin (H&E) according to the standard protocols. Histological scoring was performed using a previously described method.282.5 RNA isolation and quantitative real-time PCR
Total RNA was extracted using the TRIZOL reagent and transcribed into cDNA using a first-strand cDNA synthesis kit. Quantification of the mRNA level by quantitative real-time (qPCR) was performed on a real-time PCR system using iTaq Universal SYBR Green Supermix. The mean of the triplicate cycle thresholds (Ct) of the target gene was normalized to the mean of triplicate Ct of the reference β-actin and GAPDH gene using the formula “2−ΔΔCt”, which yielded relative gene expression level values. The primers used are listed in ESI, Table 1.†2.6 Western blot analysis
The cells were extracted with protein lysis buffer supplemented with a protease inhibitor cocktail. The protein concentration was determined using the BCA kit. Proteins (25–35 μg) were separated on a 10% polyacrylamide precast SDS gel followed by blotting on PVDF membranes.2.7 ELISA
TNFα in the serum was analyzed directly using an ELISA kit according to the manufacturer's instructions. For the determination of TNF-α concentrations in mice colon tissues, the tissues were homogenized in five volumes of ice-cold PBS containing 1 mM PMSF and 10 mM protease inhibitor cocktail and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min at 4 °C. The protein concentration was measured using the BCA kit, and TNF-α was detected using the ELISA kit.
000g for 10 min at 4 °C. The protein concentration was measured using the BCA kit, and TNF-α was detected using the ELISA kit.
2.8 MPO assay
Myeloperoxidase activity reflects the number and distribution of neutrophils in the tissues. The colon tissues were weighed and homogenized with phosphate buffered saline (PBS) [1:9 (w/v)]. The supernatants were collected. The activity of MPO was measured according to the manufacturer's instructions.
2.9 Serum antioxidant capacity and antioxidant enzymes
Hydrogen peroxide (H2O2), glutathione peroxidase (GPX), and reduced glutathione (GSH) in serum were analyzed directly using kits according to the manufacturer's instructions.2.10 HPLC-ESI-QqQ-MS/MS analysis
FSGHF3 samples were analyzed by LC-MS using a triple quadrupole mass spectrometer (6460 QqQ LC/MS system; Agilent Technologies, USA) coupled to an Agilent 1200 series HPLC system (Agilent Technologies, USA), and the analysis was performed using a previously described method.272.11 Statistical analysis
Data are presented as the mean ± standard error of the mean. Differences between group means were determined by one-way ANOVA using SAS 8.0 software. The Tukey post hoc multiple comparison test was performed to compare significant variations. Differences were considered significant at P < 0.05.3. Results
3.1 FSGHF3 attenuates weight loss and DAI in mice with DSS-induced colitis
In a previous study, we found that FSGHF3 derived from fish skin gelatin hydrolysate exerted anti-antioxidant activities and maintained the epithelial barrier in IPEC-J2 cells.26,27 To investigate the protective effect of FSGHF3 on intestinal inflammation, we established a model of DSS-induced colitis in mice by feeding C57BL/6 mice with 3% DSS mixed in drinking water for 7 days, and FSGHF3 (100, 200 mg kg−1) was administered for 14 days before and during DSS treatment via oral gavage once per day (Fig. 1A). The most frequent clinical index in colitis is weight loss and DAI. The mice treated only with DSS had a significant increase in weight loss and DAI scores compared with those of the control group (Fig. 1B and C). FSGHF3 treatment significantly attenuated DSS-induced weight loss and improved the DAI score (Fig. 1B and C). The results indicate that FSGHF3 treatment effectively alleviates DSS-induced colitis in mice.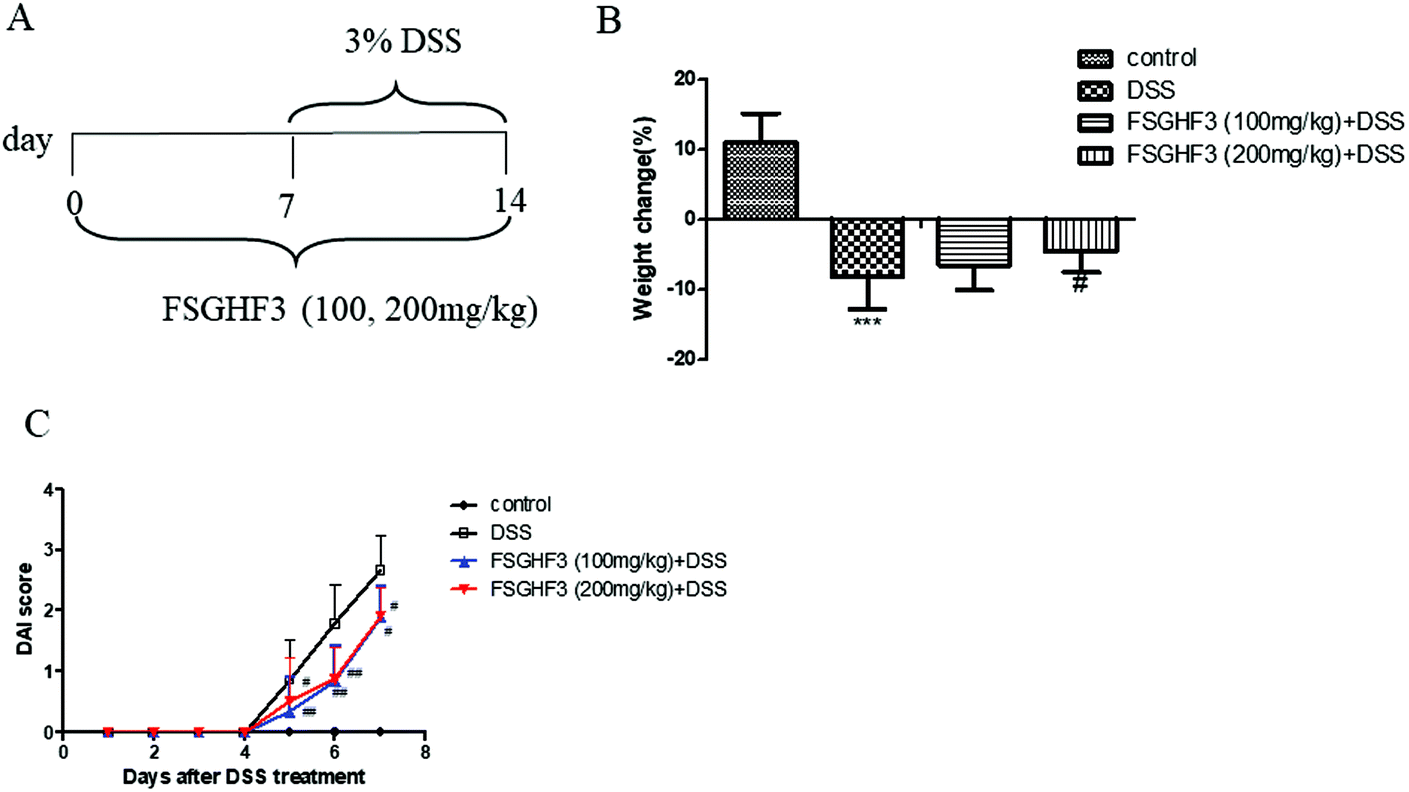 | ||
| Fig. 1 FSGHF3 attenuates weight loss and DAI in DSS-induced colitis mice. Mice were treated with 3% DSS in their drinking water for 7 days to induce acute colitis. FSGHF3 (100 and 200 mg kg−1) was administered for 7 days before and during DSS treatment via oral gavage once per day, and mice were sacrificed at day 14 after colitis induction. The experimental protocol with FSGHF3 in acute colitis model (A). Body weight changes during the experiment (B). Disease activity index (DAI) during the disease process (C). The results are expressed as mean ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. the control group on the same day; #p < 0.05, ##p < 0.01 and ###p < 0.001 vs. the DSS-treated group. | ||
3.2 FSGHF3 relieves DSS-induced colon shortening, histological changes, and spleen hypertrophy
It has been noted that DSS-induced colon shortening and histological changes are inversely linked with the severity of DSS-induced colitis in mice.29 Treatment with 3% DSS for 7 days caused colonic shortening, whereas this change was significantly ameliorated in FSGHF3 groups (Fig. 2A). Numerous studies report that the mice with DSS induced colitis develop symptoms of spleen hypertrophy.30,31 Compared with the control group mice, the DSS-induced colitis group mice showed spleen hypertrophy, and FSGHF3 treatment significantly alleviated spleen hypertrophy in mice with DSS-induced colitis (Fig. 2B). The severity of colonic inflammation was further evaluated by histopathological analysis. The DSS group exhibited distortion of crypts, loss of goblet cells, and severe mucosal damage. The administration of FSGHF3 improved these pathological changes (Fig. 2C). However, the number of goblet cells of colon tissues was significantly decreased in the DSS group compared with the control group, and FSGHF3 treatment dramatically increased the number of goblet cells compared to the DSS-induced colitis group (Fig. 2D). The results demonstrate that FSGHF3 could significantly reduce the level of colitis induced by DSS.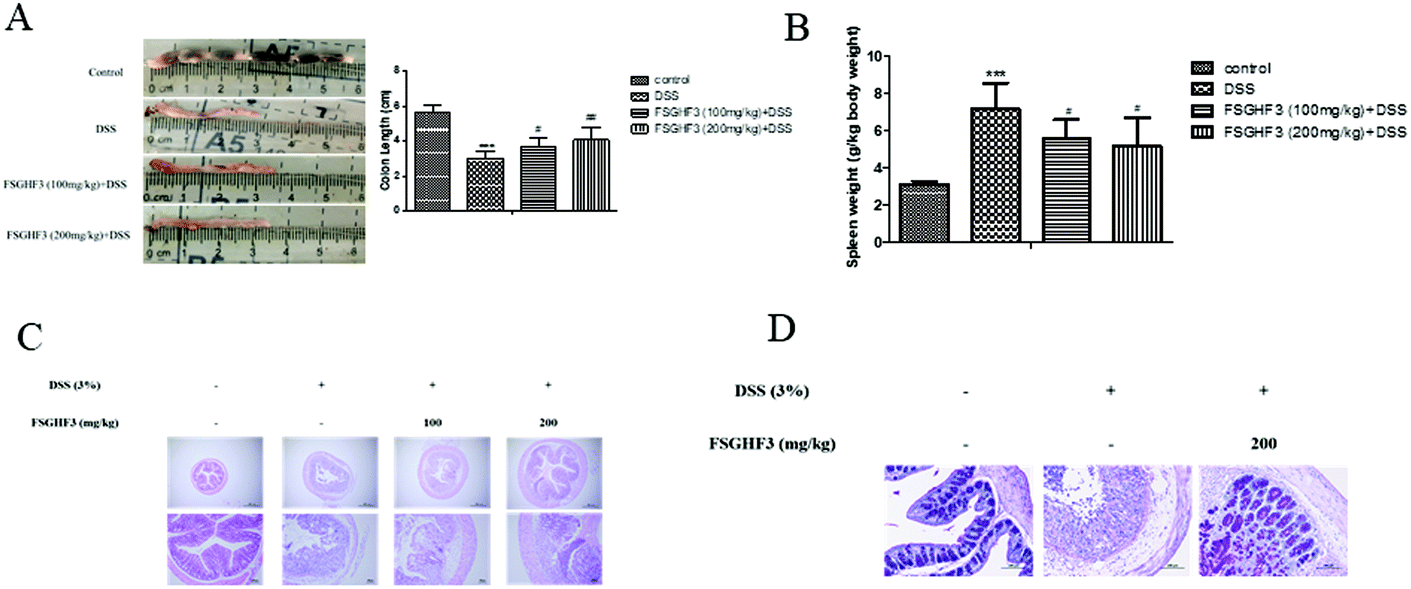 | ||
| Fig. 2 FSGHF3 relieves DSS-induced colon shortening, histological changes, and spleen hypertrophy. The lengths of colons of each group of mice were measured (A). Spleen weights were measured (B). The colons from each experimental group were processed for histological evaluation (hematoxylin–eosin staining, 40×, scale bar, 500 μm; 100×, scale bar, 100 μm), and histopathological scores of each group were determined (C). Representative images of the colon stained with PAS (D). The results are expressed as mean ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. the control group on the same day; #p < 0.05, ##p < 0.01 and ###p < 0.001 vs. the DSS-treated group. | ||
3.3 FSGHF3 and peptides contribute to the maintenance of tight junctions
Epithelial tight junction (TJ) proteins are especially important aspects of the mechanical barrier, preventing harmful substances from breaching the mucosal barrier, maintaining cellular integrity and permeability, and ensuring a relatively stable internal environment.32 ZO-1 and occludin are important epithelial TJ proteins. Thus, we determined the effect of FSGHF3 on epithelial TJ proteins (ZO-1 and occludin) in colon tissues, ZO-1 and occludin levels were decreased after DSS administration compared with the control group, while these changes were significantly ameliorated through pretreatment with FSGHF3 (Fig. 3A–C). These results suggest the importance of FSGHF3 for restoring the integrity of tight junctions.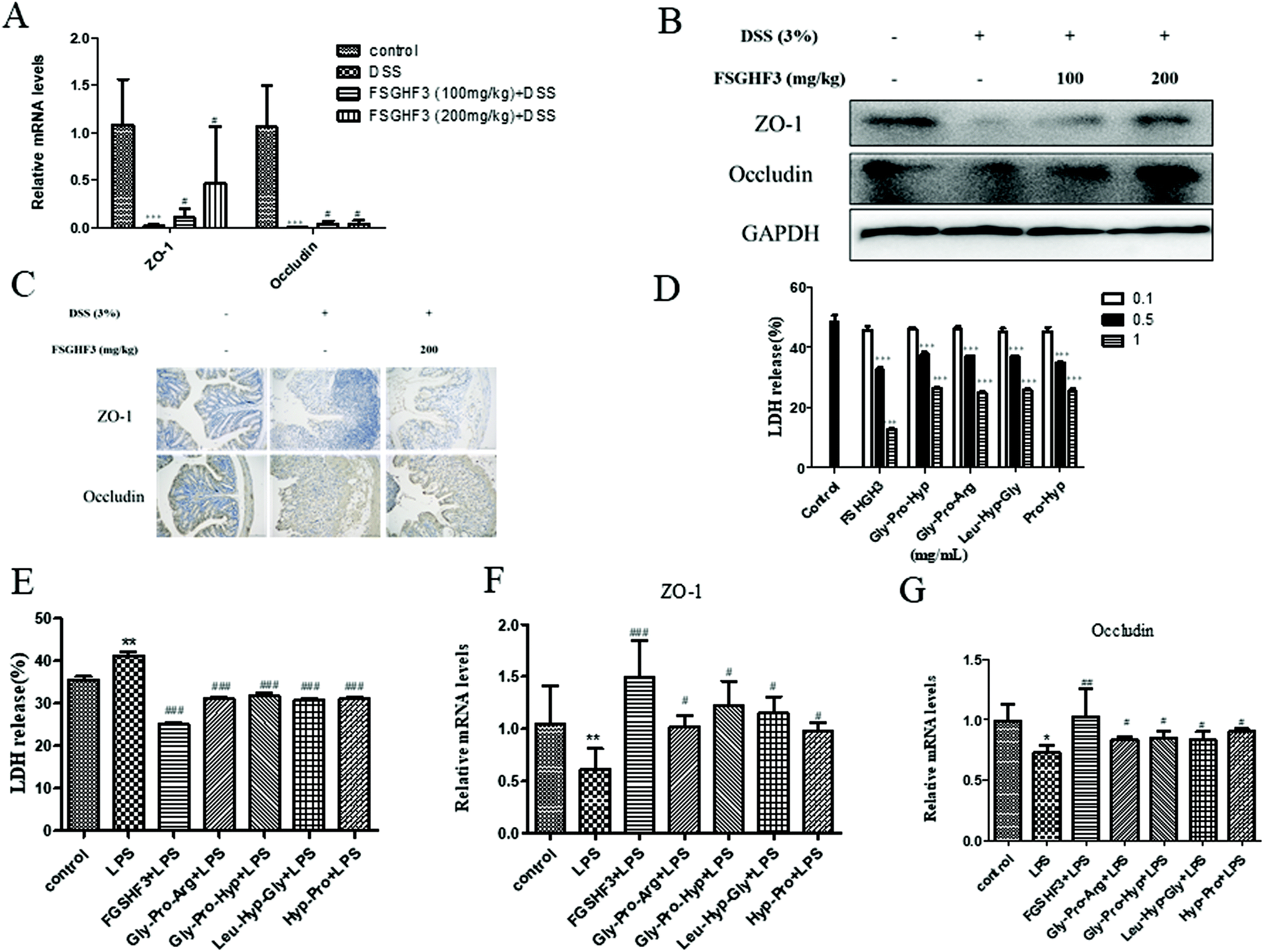 | ||
| Fig. 3 FSGHF3 and peptides contribute to the maintenance of tight junctions. The level of ZO-1 and occludin were analyzed by QPCR (A), western blotting (B), and immunohistochemical analysis (C). Cytotoxicity of IPEC-J2 cells treated with different concentrations of FSGHF3 or peptides for 12 hours was detected by LDH assay (D and E). IPEC-J2 cells pre-treated with different concentrations of FSGHF3 or peptides for 12 hours were exposed to LPS (2 μg ml−1) for 6 hours, and ZO-1 and occludin mRNA levels were detected by quantitative real-time PCR (F and G). The results are expressed as mean ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. the control group; #p < 0.05, ##p < 0.01 and ###p < 0.001 vs. the DSS-treated or LPS group. | ||
FSGHF3 was isolated from fish skin gelatin hydrolysate using ginger protease, and further HPLC-ESI-QqQ-MS analysis revealed that this fraction mainly includes some peptides, such as Gly-Pro-Arg, Gly-Pro-Hyp, Leu-Hyp-Gly, Hyp-Pro, etc. (Fig. S1†). Then, we evaluated whether FSGHF3 and peptides could exert anti-inflammatory effects and maintain tight junctions in vitro. Treatment with FSGHF3, Gly-Pro-Arg, Gly-Pro-Hyp, Leu-Hyp-Gly or Hyp-Pro alone did not cause any cytotoxic effects, which decreased the amount of LDH released in the IPEC-J2 cells (Fig. 3D). The protective effects of FSGHF3 and peptides against LPS induced cell membrane injury and tight junction breakdown were further explored. As expected, LPS stimulated a dramatic increase in LDH release and reduction in the expression of ZO-1 and occludin, whereas FSGHF3 or peptide treatment reduced LDH release and increased the level of ZO-1 and occludin (Fig. 3E–G). These findings suggest the importance of FSGHF3 and peptides for restoring the integrity of the tight junction and the cell membrane.
3.4 FSGHF3 and peptides suppress inflammatory response in vivo and in vitro
Several studies have demonstrated that MPO activity is an indicator of neutrophil infiltration and acute inflammation in DSS-induced colitis in mice.33 Therefore, we measured MPO activity to investigate the effect of FSGHF3 on mice with DSS-induced colitis. The MPO activity was significantly increased in the DSS-induced colitis group compared to the control group, and the administration of FSGHF3 significantly inhibited the activity of MPO in the colon of mice with DSS-induced colitis (Fig. 3A).The overproduction of pro-inflammatory cytokines is a hallmark of colon damage in the development of IBD.34 The anti-inflammatory effects of FSGHF3 on DSS-induced colitis were examined by measuring pro-inflammatory cytokines. Compared with the control group, the DSS-induced colitis group significantly increased the secretion of TNF-α in the serum. The secretion of TNF-α in the serum of the DSS-induced colitis group was significantly inhibited, when the mice were administered with FSGHF3 (Fig. 4B). Besides, to investigate the anti-inflammatory effect of FSGHF3, we determined the concentration of TNF-α in the colon tissue, and the results showed that FSGHF3 significantly inhibits the elevated expression of TNF-α (Fig. 4C). Furthermore, we determined the mRNA levels of TNF-α, IL-6, IL-8, IL-12 and IFN-γ in the colon tissue by qPCR, the levels of pro-inflammatory cytokines were significantly increased after DSS treatment, and the levels of pro-inflammatory cytokines in the colon tissue were dramatically reduced by FSGHF3 treatment (Fig. 4D).
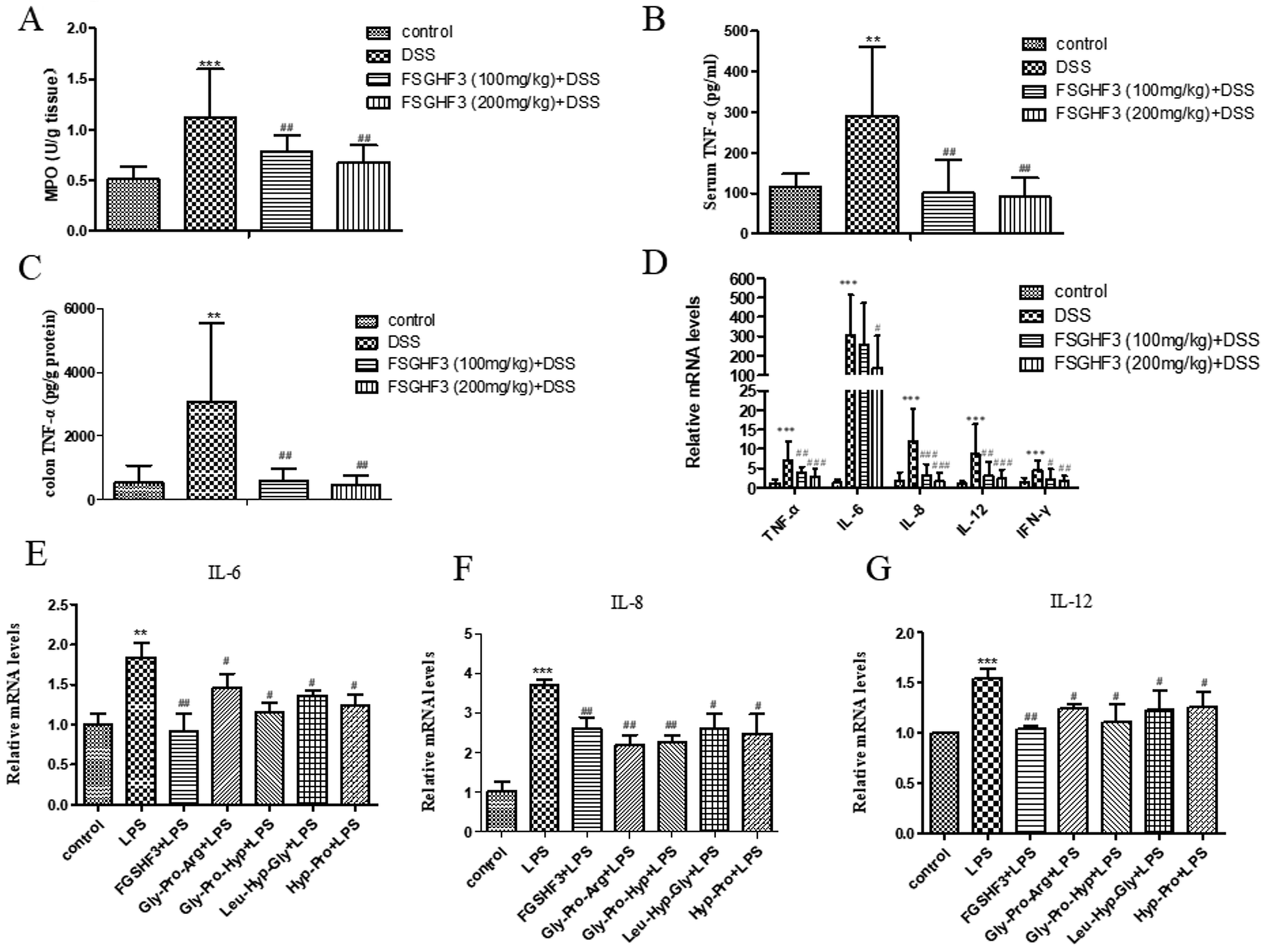 | ||
| Fig. 4 FSGHF3 and peptides suppress inflammatory response in vivo and in vitro. Myeloperoxidase (MPO) activity in the colonic tissues was detected (A). The production of inflammation-related cytokines TNF-α in serum (B). The production of inflammation-related cytokines TNF-α in colon tissues (C). TNF-α, IL-6, IL-8, IL-12 and IFN-γ mRNA levels in the colon tissues were measured by quantitative real-time PCR (D). IPEC-J2 cells pre-treated with different concentrations of FSGHF3 or peptides for 12 hours were exposed to LPS (2 μg ml−1) for 6 hours and IL-6, IL-8 and IL-12 mRNA levels were detected by quantitative real-time PCR (E–G). The results are expressed as mean ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. the control group on the same day; #p < 0.05, ##p < 0.01 and ###p < 0.001 vs. the DSS-treated group or LPS group. | ||
The protective effects of FSGHF3 and Gly-Pro-Arg, Gly-Pro-Hyp, Leu-Hyp-Gly or Hyp-Pro peptide on LPS-induced intestinal inflammation in IPEC-J2 cells were further explored. In the LPS induced inflammation model, FSGHF3 and peptide treatment significantly inhibited the production of the pro-inflammatory cytokines, including IL-6, IL-8, and IL-12 in the IPEC-J2 cells (Fig. 4E–G). The results demonstrate that FSGHF3 and peptides inhibit pro-inflammatory cytokine production in vivo and in vitro.
3.5 FSGHF3 and peptides inhibit the NF-κB pathway in vivo and in vitro
The NF-κB signaling pathway plays a predominant role in the pathogenesis of human inflammatory diseases, such as diabetes, IBD, and atherosclerosis.14 More importantly, NF-κB, as a transcriptional factor, regulates the transcription of some pro-inflammatory cytokines.12 To date, several in vitro and in vivo studies have reported that anti-NF-κB therapies have successfully reduced pathology and morbidity in IBD.13,14 Then, we analyzed p65 nuclear translocation by immunofluorescence, and the result showed that FSGHF3 administration alleviates DSS-induced p65 nuclear translocation (Fig. 5A). Next, we examined the effect of FSGHF3 and Gly-Pro-Arg, Gly-Pro-Hyp, Leu-Hyp-Gly or Hyp-Pro peptide on LPS-induced NF-κB activation in IPEC-J2 cells. We transfected the NF-κB reporter gene into IPEC-J2 cells, and found that FSGHF3 and peptide treatment alleviated LPS-induced NF-κB activation (Fig. 5B). These results demonstrate that FSGHF3 and peptides inhibit pro-inflammatory cytokine production by suppressing NF-κB activation.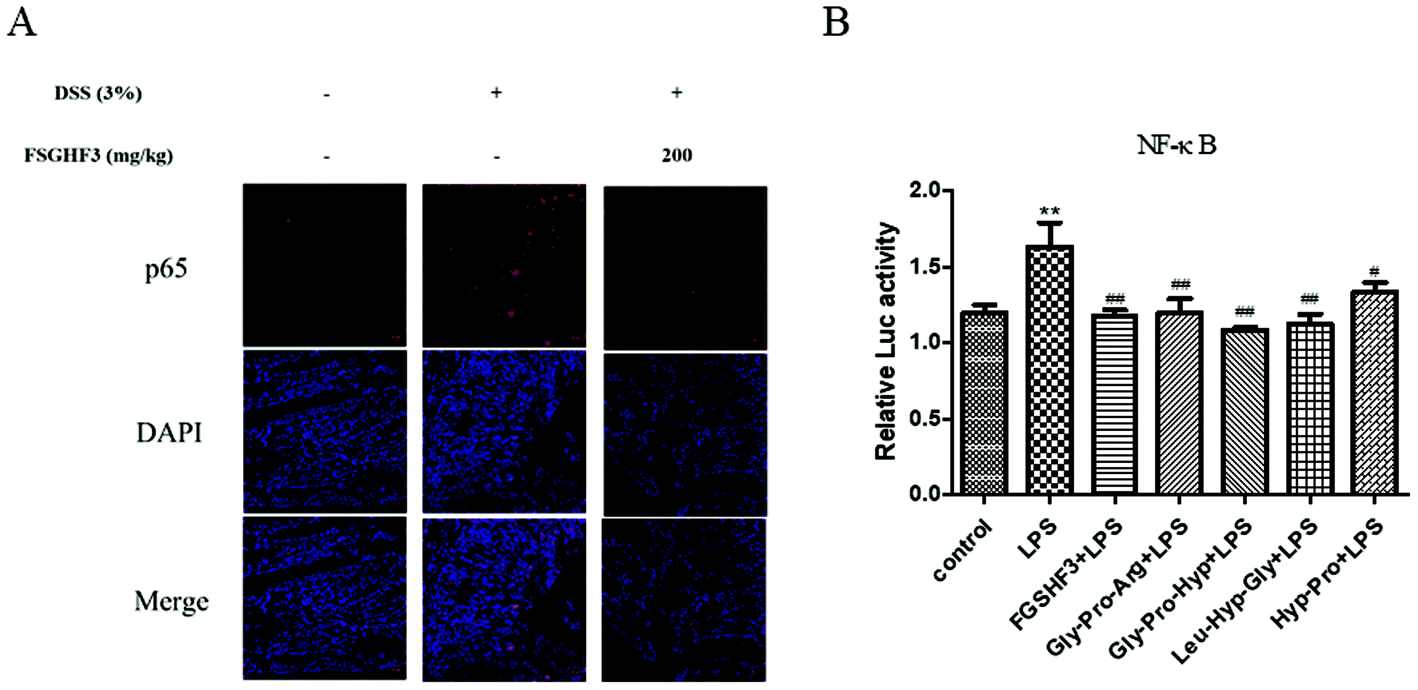 | ||
| Fig. 5 FSGHF3 and peptides inhibit the NF-κB pathway in vivo and in vitro. NF-κB p65, which entered the nucleus was detected by immunofluorescence (A). P65 (red) and DAPI (blue) scale bar, 20 μm. IPEC-J2 cells transfected with the PGL3-NF-κB reporter gene, then treated with different concentrations of FSGHF3 or peptides for 12 hours, were exposed to LPS (2 μg ml−1) for 6 hours and dual-luciferase activity of the reporter gene of NF-κB was measured (B). The results are expressed as mean ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. the control group on the same day; #p < 0.05, ##p < 0.01 and ###p < 0.001 vs. LPS group. | ||
3.6 FSGHF3 and peptides enhance antioxidant enzyme expression to inhibit oxidative stress in vivo and in vitro
Intestinal oxidative stress has been associated with the initiation and propagation of chronic intestinal pathologies such as IBD.35 Oxidative stress refers to excessive generation of ROS which causes cellular damage, and H2O2 is a kind of ROS.36 Meanwhile, ROS, as a signal molecule, activates several classical inflammatory signaling pathways such as the NF-κB pathway, leading to pro-inflammatory cytokine production in the intestinal tract, which further damages the intestinal barrier.37,38 In the current study, the production of H2O2 in the serum of the DSS-induced colitis group was significantly inhibited when the mice were administered with FSGHF3 (Fig. 6A). Antioxidant enzymes are the key driver of ROS elimination,39 then we determined antioxidant enzymes in the serum. We found that the administration of FSGHF3 markedly increases the level of GPX and GSH in the serum, compared with the DSS group (Fig. 6B and C). Furthermore, we analyzed the mRNA levels of antioxidant enzymes in the colon tissue by qPCR, compared with the control group mice, the mRNA levels for GCLC, GCLM, and GPX4 were significantly decreased in the DSS-induced colitis group mice, while the FSGHF3 treatment group showed significantly increased levels of GCLC, GCLM, and GPX4(Fig. 6D).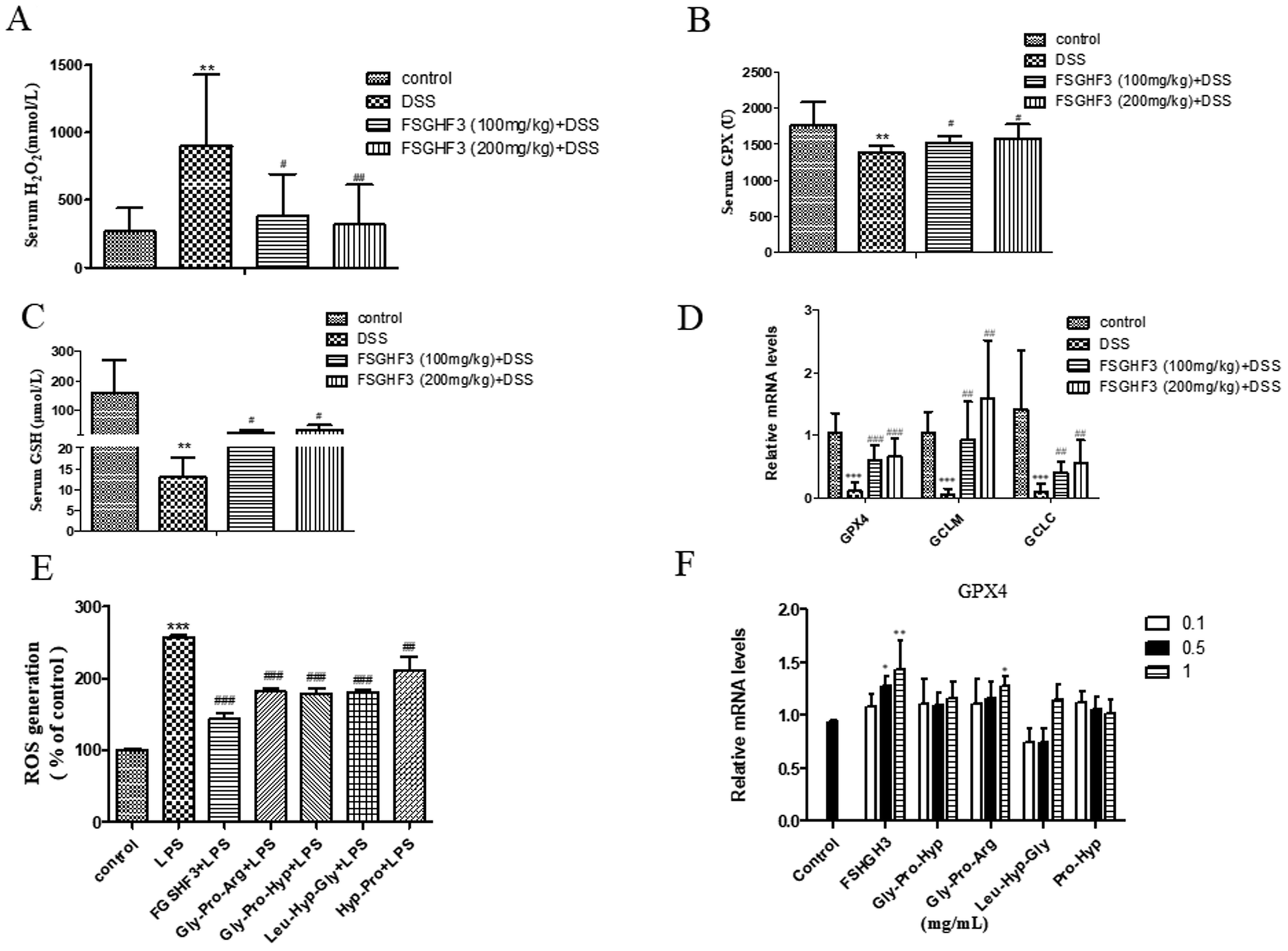 | ||
| Fig. 6 FSGHF3 inhibited oxidative stress in DSS-induced mice. The levels of H2O2, GPX and GSH were detected in the serum using kits (A–C). GPX4, GCLM and GCLC mRNA levels in the colon tissues were measured by quantitative real-time PCR (D). IPEC-J2 cells pre-treated with different concentrations of FSGHF3 or peptides for 12 hours were exposed to LPS (2 μg ml−1) for 6 hours, and ROS and GPX4 mRNA levels were detected (E and F). The results are expressed as mean ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. the control group on the same day; #p < 0.05, ##p < 0.01 and ###p < 0.001 vs. the DSS-treated group or LPS group. | ||
Next, in the IPEC-J2 cells, we found that FSGHF3 and peptides suppress ROS generation caused by LPS (Fig. 6E). FSGHF3 and peptide treatment alone enhanced the level of GPX4 and ameliorated LPS-induced inflammation, and also increased GPX4 expression (Fig. 6F). The results indicate that FSGHF3 and peptides drastically inhibited oxidative stress by increasing the level of antioxidant enzymes.
3.7 FSGHF3 and peptides enhance the level of Nrf2 in vivo and in vitro
Nrf2, as a classical transcription factor, plays a vital role in protecting various tissues against oxidative stress and inflammation.40 Then we determined the level of Nrf2 in colon tissues. As expected, treatment with FSGHF3 dramatically increased the level of Nrf2 (Fig. 7A and B). And in IPEC-J2 cells, we found that FSGHF3 and peptide treatment alone enhanced the level of Nrf2, and in LPS-induced inflammation, FSGHF3 and peptide treatment also increased Nrf2 expression (Fig. 6C and D). This indicates that FSGHF3 and peptides alleviate intestinal inflammation and oxidative stress by Nrf2 activation.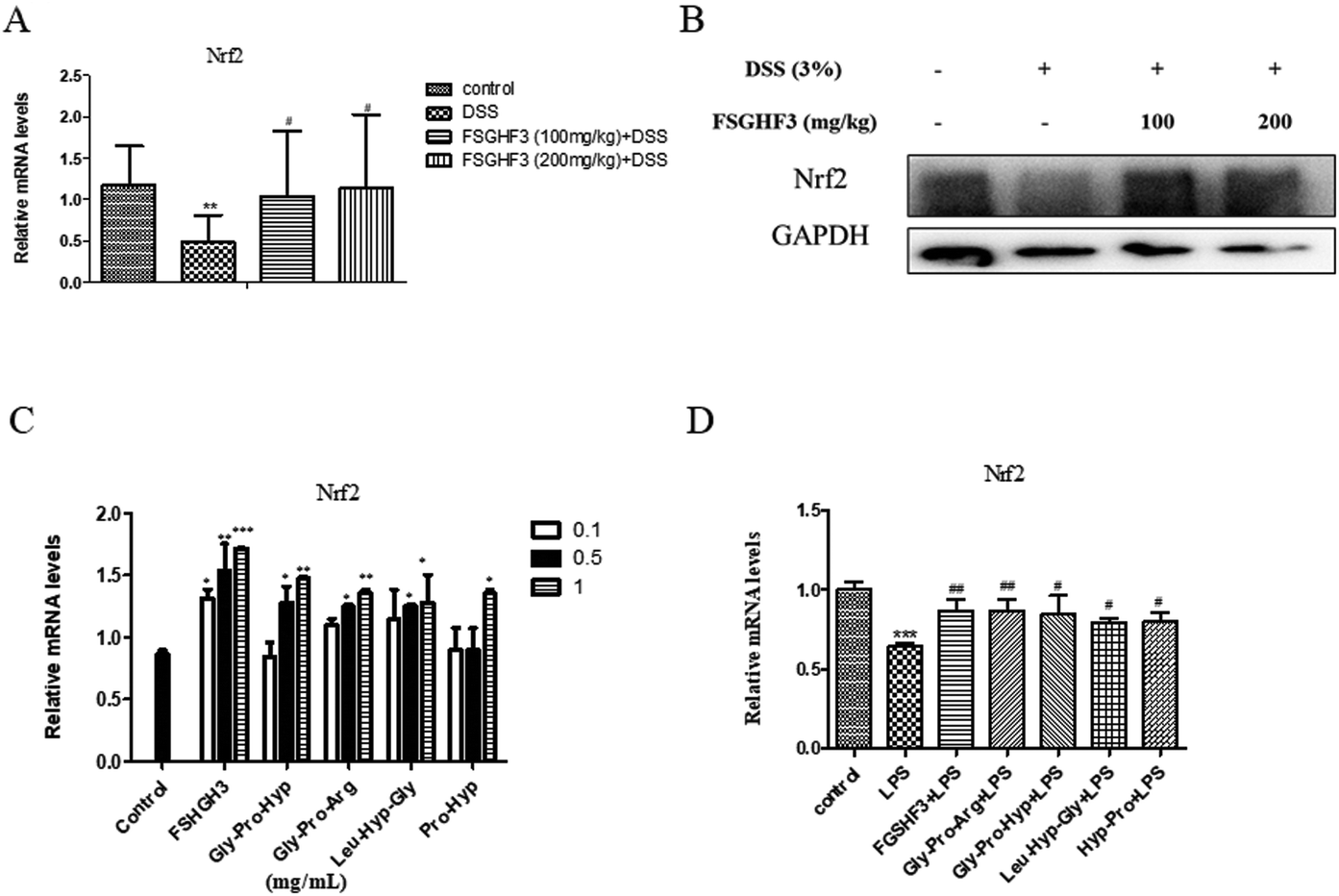 | ||
| Fig. 7 FSGHF3 enhanced the level of Nrf2 in the colon of DSS-induced mice. The level of Nrf2 was detected by qPCR (A), and western blotting (B) in colon tissues. IPEC-J2 cells pre-treated with different concentrations of FSGHF3 or peptides for 12 hours were exposed to LPS (2 μg ml−1) for 6 hours, and Nrf2 mRNA levels were detected (C and D). The results are expressed as mean ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. the control group on the same day; #p < 0.05, ##p < 0.01 and ###p < 0.001 vs. the DSS-treated group or LPS group. | ||
4. Discussion
The prevalence and incidence of IBD in the world is on the rise, which is affecting millions of people worldwide.41–43 In UC, the intestinal flora is disrupted, disrupting the tight junctions of epithelial cells, which allows microbes to migrate from the lumen into the lamina propria; immune cell and IECs engulf microbes, which activates the NF-κB pathway, ROS and pro-inflammatory cytokines are produced, which sets into motion a cycle of uncontrolled inflammation, which causes breakdown of the epithelium and immune dysregulation.7 In our study, we find that suppressing NF-κB activation, ROS and pro-inflammatory cytokine overproduction, and maintenance of the intestinal barrier, could be an effective way to alleviate colitis.Recently, many biologically active peptides, isolated from different gelatin sources, are reported to have antioxidant effects in vitro and in vivo.20 Previously, we found that FSGH and FSGHF3, isolated from fish skin gelatin hydrolysate using ginger protease, could exert antioxidative effects and maintain tight junctions in IPEC-J2.26,27 Further HPLC-ESI-QqQ-MS analysis revealed that this fraction mainly included some peptides, such as Gly-Pro-Arg, Gly-Pro-Hyp, Leu-Hyp-Gly, Hyp-Pro etc., which promoted ARE-luciferase activity, and exerted antioxidative effects.27 HPLC-ESI-QqQ-MS analysis showed that these peptides were Gly-Pro-Y and X-Hyp-Gly type peptides, and thus exhibited the highest antioxidant activity.27 These peptides significantly decrease the cytotoxicity and inhibit cytotoxicity induced by LPS in IEPC-J2 cells.
In the present study, in addition to the maintenance of the intestinal barrier, the inhibitory effects on inflammatory and oxidative stress are also observed in mice with DSS-induced colitis or LPS-induced intestinal inflammation pretreated with FSGHF3 and peptides. At present, the most traditional drugs for the treatment of IBD include salicylazosulfapyridine (SASP), immunosuppressive agents, and anti-TNF-α monoclonal antibodies.44,45 However, these drugs show limited efficacy and potential long-term toxicity.45 Instead, FSGHF3 and peptides are composed of L-amino acid residues, such as Gly, Pro, Ala, Arg, Ser, and Leu, which do not exert any cytotoxic effect in vitro.27 Therefore, this indicates that FSGHF3 and peptides, as nutrients, may support the treatment of intestinal inflammation.
Numerous studies reported that the intestinal barrier plays a key role in maintaining intestinal health.46,47 The structural abnormalities in tight junction proteins are the major cause of altered intestinal barrier in IBD.48 Tight junction proteins are capable of decreasing the permeability of intestinal mucosa and restraining foreign substances, such as LPS, from crossing the intestinal mucosa, which results in the suppression of inflammation.49,50 In the current study, we find that FSGHF3 and peptides exert a protective effect on barrier integrity by maintaining the expression of ZO-1 and occludin.
The NF-κB signaling pathway plays a predominant role in IBD pathogenesis.14 More importantly, NF-κB, as a transcriptional factor, regulates the transcription of some pro-inflammatory cytokines, such as TNF-α, IL-6, IL-8 and IL-12.12 To date, several in vitro and in vivo studies have indicated that anti-NF-κB therapies have successfully reduced pathology and morbidity in IBD.13,14 The overproduction of pro-inflammatory cytokines is a hallmark of colon damage in the development of IBD, and it is often associated with Th1, Th17, and Th2 cell-mediated secretion of pro-inflammatory cytokines such as IL-8, IL-12, and TNF-α.51 IL-8 is involved in inducing chemotaxis and activation of neutrophils52 and IL-12 is critical for the differentiation of Th1 cells and IFN-γ production, and enhancing cytotoxicity.53 In this study, FSGHF3 and peptides successfully suppress the increased production of pro-inflammatory cytokines by inhibiting NF-κB activation.
Oxidative stress has been recorded as a direct contributor to cellular damage, including cell death and membrane injury.54 More importantly, ROS, as a signal molecule, activates several classic inflammatory signaling pathways such as the NF-κB pathway, leading to pro-inflammatory cytokine production in the intestinal tract, which further damages the intestinal barrier.37,38 Therefore, it is essential to remove ROS to inhibit NF-κB activation and pro-inflammatory cytokine production, to maintain the intestinal barrier. Notably, cells have developed antioxidant defense systems to guard against oxidative stress.55 Therefore, increasing the expression of antioxidant enzymes could be considered as an effective target for therapies to prevent oxidative stress. Nrf2 is a redox-sensitive master regulatory transcription factor that plays a vital role in cellular defenses against oxidative stress through mediating the expression of a variety of antioxidant enzymes.55 Combined with previous studies,26,27 we suggest that FSGHF3 and peptides scavenge ROS by activating Nrf2 to up-regulate the expression of antioxidant enzymes.
In conclusion, our work explores a novel nutritional supplement for alleviating colitis. FSGHF3 and peptides enhance Nrf2 expression to up-regulate the expression of antioxidant enzymes, resulting in the removal of ROS and suppression of the NF-κB pathway. Ultimately, FSGHF3 and peptides alleviates intestinal inflammation and maintains the intestinal barrier. Therefore, our results suggested that FSGHF3 could potentially be used for the prevention of colitis.
Funding
This research was supported by the National Natural Science Foundation of China (31772609), the National key research and development program (No. 2018YFD0501003), the Hubei Provincial Creative Team Project of Agricultural Science and Technology (No. 2007-620), and the China Agriculture Research System (No. CARS-36).Conflicts of interest
The authors have declared that no conflicts of interest exist.Acknowledgements
We thank the Department of Animal Nutrition and Feed Science (Huazhong Agricultural University) for their support.References
- R. J. Xavier and D. K. Podolsky, Nature, 2007, 448, 427–434 CrossRef CAS PubMed.
- J. Torres, V. Billioud, D. B. Sachar, L. Peyrinbiroulet and J. F. Colombel, Inflamm. Bowel Dis., 2012, 18, 1356–1363 CrossRef PubMed.
- R. B. Michalčíková, K. Dryahina and P. Španěl, Int. J. Mass Spectrom., 2016, 396, 35–41 CrossRef.
- G. Bouma and W. Strober, Nat. Rev. Immunol., 2003, 3, 521–533 CrossRef CAS PubMed.
- T. T. Macdonald and G. Monteleone, Science, 2005, 307, 1920–1925 CrossRef CAS PubMed.
- J. R. Korzenik and D. K. Podolsky, Nat. Rev. Drug Discovery, 2006, 5, 197 CrossRef CAS PubMed.
- M. Eisenstein, Nature, 2018, 563, S34–S35 CrossRef CAS PubMed.
- Z. J. Chen, Immunol. Rev., 2012, 246, 95–106 CrossRef PubMed.
- M. S. Hayden and S. Ghosh, Cell, 2008, 132, 344–362 CrossRef CAS PubMed.
- M. Karin and E. Gallagher, Immunol. Rev., 2010, 228, 225–240 CrossRef PubMed.
- A. J. G. Schottelius and A. S. Baldwin Jr., Int. J. Colorectal Dis., 1999, 14, 18 CrossRef CAS PubMed.
- T. Lawrence, Cold Spring Harbor Perspect. Biol., 2009, 1, a001651 Search PubMed.
- J. H. Kang, S. Choi, J. E. Jang, P. Ramalingam, Y. T. Ko, S. Y. Kim and S. H. Oh, Food Funct., 2017, 8, 2865–2874 RSC.
- H. Okamoto, T. Yoshio, H. Kaneko and H. Yamanaka, Arthritis Rheum., 2010, 62, 82–92 CrossRef CAS PubMed.
- J. Kim, Y. N. Cha and Y. J. Surh, Mutat. Res., 2010, 690, 12–23 CrossRef CAS PubMed.
- M. M. Michael, I. Ken, Y. Masayuki and J. D. Hayes, J. Biol. Chem., 2003, 278, 21592–21600 CrossRef PubMed.
- H. Dreger and K. A. Westphal, Cardiovasc. Res., 2009, 83, 354–361 CrossRef CAS PubMed.
- X. Liu, X. Zhang, Y. Ding, W. Zhou, L. Tao, P. Lu, Y. Wang and R. Hu, Antioxid. Redox Signal., 2017, 26, 28–43 CrossRef CAS PubMed.
- Y. Zou, J. Wang, J. Peng and H. Wei, Oxid. Med. Cell. Longevity, 2016, 2016, 5987183 Search PubMed.
- M. Chalamaiah, W. Yu and J. Wu, Food Chem., 2018, 245, 205–222 CrossRef CAS PubMed.
- O. Martínez-Alvarez, S. Chamorro and A. Brenes, Food Res. Int., 2015, 73, 204–212 CrossRef.
- C. B. Ahn, Y. S. Cho and J. Y. Je, Food Chem., 2015, 168, 151–156 CrossRef CAS PubMed.
- S. Wada, K. Sato, R. Ohta, E. Wada, Y. Bou, M. Fujiwara, T. Kiyono, E. Y. Park, W. Aoi and T. Takagi, J. Agric. Food Chem., 2013, 61, 8807–8813 CrossRef CAS PubMed.
- J. Kovacs-Nolan, H. Zhang, M. Ibuki, T. Nakamori, K. Yoshiura, P. V. Turner, T. Matsui and Y. Mine, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 1753–1763 CrossRef CAS PubMed.
- G. Liu, W. Yan, S. Ding, H. Jiang, Y. Ma, H. Wang and J. Fang, Cell. Physiol. Biochem., 2018, 51, 441–451 CrossRef CAS PubMed.
- L. Zheng, H. Wei, H. Yu, Q. Xing, Y. Zou, Y. Zhou and J. Peng, J. Agric. Food Chem., 2018, 66, 11601–11611 CrossRef CAS PubMed.
- L. Zheng, H. Wei, H. Yu, Q. Xing, Y. Zou, Y. Zhou and J. Peng, J. Funct. Foods, 2018, 51, 104–112 CrossRef CAS.
- L. A. Dieleman, M. J. Palmen, H. Akol, E. Bloemena, A. S. Pena, S. G. Meuwissen and E. P. Van Rees, Clin. Exp. Immunol., 1998, 114, 385–391 CrossRef CAS PubMed.
- B. Chassaing, J. D. Aitken, M. Malleshappa and M. Vijay-Kumar, Curr. Protoc. Immunol., 2014, 104, Unit 15 25 Search PubMed.
- A. Satoko, N. Atsushi, M. Y. Mari, U. Mariko and M. Akira, BioFactors, 2012, 38, 226–233 CrossRef PubMed.
- M. Zhang, Y. Zhao, N. Wu, Y. Yao, M. Xu, H. Du and Y. Tu, Food Funct., 2018, 9, 6444–6454 RSC.
- E. J. Park, A. B. R. Thomson and M. T. Clandinin, J. Pediatr. Gastroenterol. Nutr., 2010, 50, 321 CrossRef CAS PubMed.
- F. Costa, M. G. Mumolo, L. Ceccarelli, M. Bellini, M. R. Romano, C. Sterpi, A. Ricchiuti, S. Marchi and M. Bottai, Gut, 2005, 54(3), 364–368 CrossRef CAS PubMed.
- D. C. Baumgart and W. J. Sandborn, Lancet, 2007, 369, 1641–1657 CrossRef CAS.
- A. Rezaie, R. D. Parker and M. Abdollahi, Dig. Dis Sci., 2007, 52, 2015–2021 CrossRef PubMed.
- M. Valko, D. Leibfritz, J. Moncol, M. T. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS PubMed.
- P. D. Ray, B. W. Huang and Y. Tsuji, Cell. Signalling, 2012, 24, 981–990 CrossRef CAS PubMed.
- M. Zhang, K. Sun, Y. Wu, Y. Yang, P. Tso and Z. Wu, Front. Immunol., 2017, 8, 942 CrossRef PubMed.
- J. Nordberg and E. S. Arner, Free Radicals Biol. Med., 2001, 31, 1287–1312 CrossRef CAS PubMed.
- T. O. Khor, M. T. Huang, A. Prawan, Y. Liu, X. Hao, S. Yu, W. K. Cheung, J. Y. Chan, B. S. Reddy and C. S. Yang, Cancer Prev. Res., 2008, 1, 187–191 CrossRef CAS PubMed.
- J. Cosnes, C. Gowerrousseau, P. Seksik and A. Cortot, Gastroenterology, 2011, 140, 1785–1794 CrossRef PubMed.
- K. Welcker, A. Martin, P. Kölle, M. Siebeck and M. Gross, Eur. J. Med.Res., 2004, 9, 456 CAS.
- I. Ordás, L. Eckmann, M. Talamini, D. C. Baumgart and W. J. Sandborn, Lancet, 2012, 380, 1606–1619 CrossRef.
- B. Bressler, J. K. Marshall, C. N. Bernstein, A. Bitton, J. Jones, G. I. Leontiadis, R. Panaccione, A. H. Steinhart, F. Tse and B. Feagan, Gastroenterology, 2015, 148, 1035–1058 CrossRef PubMed.
- M. F. Andréa, D. A. K. Queiroz, D. S. Juliana Celia, A. O. R. Pimentel and G. M. O. Fonseca, Redox Biol., 2015, 6, 617–639 CrossRef PubMed.
- M. A. Mcguckin, R. Eri, L. A. Simmsc, T. H. J. Florin and R. S. Dphil, Inflamm. Bowel Dis., 2010, 15, 100–113 CrossRef PubMed.
- M. Bashashati, H. R. Habibi, A. Keshavarzian, M. Schmulson and K. A. Sharkey, Curr. Drug Targets, 2015, 16(3), 199–208 CrossRef CAS PubMed.
- T. Kucharzik, S. V. Walsh, J. Chen, C. A. Parkos and A. Nusrat, Am. J. Pathol., 2001, 120, A187–A187 Search PubMed.
- T. Suzuki, Cell. Mol. Life Sci., 2013, 70, 631–659 CrossRef CAS PubMed.
- J. J. Jeong, K. A. Kim, S. E. Jang, J. Y. Woo, M. J. Han and D. H. Kim, PLoS One, 2015, 10, e0116533 CrossRef PubMed.
- W. Kori, Z. Li-Bo, K. Yoshitake and S. David, World J. Gastroenterol., 2014, 20, 6–21 CrossRef PubMed.
- N. Mukaida, Int. J. Hematol., 2000, 72, 391 CAS.
- M. H. Lexberg, A. Taubner, I. Albrecht, I. Lepenies, A. Richter, T. Kamradt, A. Radbruch and H. D. Chang, Eur. J. Immunol., 2010, 40, 3017–3027 CrossRef CAS PubMed.
- D. Babu, G. Leclercq, V. Goossens, Q. Remijsen, P. Vandenabeele, R. Motterlini and R. A. Lefebvre, Toxicol. Appl. Pharmacol., 2015, 288, 161–178 CrossRef CAS PubMed.
- T. W. Kensler, N. Wakabayashi and S. Biswal, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 89–116 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo02165e |
| This journal is © The Royal Society of Chemistry 2020 |