
Ginsenoside Rk1 induces apoptosis and downregulates the expression of PD-L1 by targeting the NF-κB pathway in lung adenocarcinoma†
Manling
Hu‡
abc,
Jing
Yang‡
abc,
Linlin
Qu
abc,
Xuqian
Deng
abc,
Zhiguang
Duan
abc,
Rongzhan
Fu
abc,
Lihua
Liang
abc and
Daidi
Fan
 *abc
*abc
aShaanxi Key Laboratory of Degradable Biomedical Materials, School of Chemical Engineering, Northwest University, Taibai North Road 229, Xi'an, Shaanxi 710069, China. E-mail: fandaidi@nwu.edu.cn; Fax: +86-29-88305118; Tel: +86-29-88305118
bShaanxi R&D Center of Biomaterials and Fermentation Engineering, School of Chemical Engineering, Northwest University, Taibai North Road 229, Xi'an, Shaanxi 710069, China
cBiotech & Biomed Research Institute, Northwest University, Taibai North Road 229, Xi'an, Shaanxi 710069, China
First published on 29th November 2019
Abstract
Ginsenoside Rk1 is a substance derived from ginseng and exhibits various activities such as anti-diabetic, anti-inflammatory and anti-cancer effects; however, its anti-tumor effect and target signaling mechanism in lung adenocarcinoma are not well understood. Here, we show that Rk1, a natural drug product, can function as an antitumor modulator that induces apoptosis in lung adenocarcinoma cells by inhibiting NF-κB transcription and triggering cell cycle arrest. Mechanistically, Rk1 suppressed the proliferation and clonal formation of two lung adenocarcinoma cell lines (A549 and PC9) in vitro and caused G1 phase cell arrest. In the A549 xenograft model, Rk1 significantly inhibited tumor growth and had few toxic side effects on normal organs. Western blotting results showed that Rk1 increased the protein expression of Bax, cleaved caspase-3, -8, and -9, and PARP, decreased the expression of Bcl-2 and blocked the NF-κB signaling pathway. Furthermore, ginsenoside Rk1 also reduced the high expression of PD-L1 in lung adenocarcinoma cells by inhibiting NF-κB signaling. These data revealed a previously unreported antitumor mechanism of Rk1, providing new ideas and an experimental basis for further study of the mechanism of action of Rk1 in lung adenocarcinoma.
1. Introduction
Lung cancer is one of the most common malignancies worldwide and the main cause of cancer death in recent years, with the highest morbidity and mortality in the world.1 CA journal estimated that there will be 2.1 million new cases of lung cancer and 1.8 million lung cancer deaths, accounting for 18.4% of all cancer deaths.2 Among them, lung adenocarcinoma is the most common histopathological type of lung cancer, with an incidence of approximately 40%,3 and is characterized by a low early diagnosis rate, low surgical resection rate, low postoperative survival rate, unsatisfactory chemotherapy effect and poor prognosis.4 Although the development of medicine and surgery has increased the overall survival rate of patients, the long-term prognosis is still poor due to the high rate of metastasis of lung adenocarcinoma, which reduces the efficacy of therapeutics for a number of patients.5 Therefore, there is an urgent need to develop a new therapeutic strategy to effectively treat this malignant disease to meet the needs of patients.Nuclear factor-κB (NF-κB) is a protein transcription factor involved in many complex biological processes such as cell proliferation, apoptosis, invasion and angiogenesis.6 According to the classical NF-κB pathway cascade, phosphorylated IKK (IκB kinase) further phosphorylates IκB, a cytoplasmic inhibitor of the NF-κB complex. Upon phosphorylation, IκB is ubiquitinated and degraded in a proteasome-dependent manner, resulting in the release and nuclear translocation of the NF-κB complex.7 Inactivation of NF-κB can reduce tumor progression and enhance the efficacy of therapeutic agents in lung cancer in vitro and in vivo.6 Therefore, targeting NF-κB is considered a promising strategy for the treatment of lung cancer. The PD-1/PD-L1 axis protects tumor cells from immune-mediated surveillance and plays a key role in the tumor microenvironment.8 PD-L1 is highly expressed on the cell surface of various human cancers, such as ovarian cancer, hepatocellular carcinoma, malignant melanoma, and non-small cell lung cancer.9 It has been reported that PD-L1 inhibitors inhibit the occurrence of cancer. Therefore, PD-L1 is a new target for treating lung adenocarcinoma cells.
Natural products, especially Chinese herbal medicines, have attracted extensive attention due to their outstanding efficacy and low toxicity in cancer drug development processes.10 Ginsenoside, the main active ingredient of ginseng, has been proved to show an excellent pharmacological activity, including anti-tumor effects.11 Among them, ginsenoside Rk1 is a rare ginsenoside extracted from black ginseng,12 which shows a variety of biopharmacological activities including anti-diabetic,13 anti-tumor,14 anti-inflammatory,15 and anti-platelet aggregation activities.16 Intriguingly, recent studies have highlighted that ginsenoside Rk1 is a promising antitumor agent. Mechanistically, its antitumor effect has been ascribed to direct targeting of autophagy, apoptosis, and cell cycle arrest.17 However, whether ginsenoside Rk1 plays a role in blocking NF-κB signaling against cancer remains unexplored. Therefore, in the present study, we investigated the anti-tumor effects of Rk1 in lung adenocarcinoma to elucidate the contribution of this phytochemical. The molecular mechanism of Rk1 may involve promoting intrinsic and extrinsic apoptosis and inhibiting the high protein expression of PD-L1 by suppressing the NF-κB signaling pathway in lung adenocarcinoma cells. Furthermore, we studied the effectiveness of Rk1 in lung adenocarcinoma using an A549-xenograft nude mouse model to estimate the potential of Rk1 as a promising new therapeutic lead molecule against lung adenocarcinoma.
2. Materials and methods
2.1 Chemicals and reagents
Ginsenoside Rk1 (Fig. 1A) (purity ≥99%) was purchased from Puruifa Technology Development Co., Ltd (Chengdu, China). Fetal bovine serum (FBS), Roswell Park Memorial Institute (RPMI) medium, penicillin and streptomycin were purchased from GIBCO (Grand Island, NY, USA). Z-VAD-FMK, JSH-23, and ceruletide were purchased from Med ChemExpress (NJ, USA). Primary antibodies against Bax, Bcl-2, PD-L1, p65, and IκBα were obtained from Proteintech Group Inc. (Chicago, IL, USA). Primary antibodies against p-p65, p-IκBα, p-IKKα, cleaved caspase-3, -8, and -9 and PARP were purchased from Abcam (MA, USA). Primary antibodies against p53, p21, cyclin D1, CDK4, IKKα, β-actin, and GAPDH were obtained from Cell Signaling Technology (Danvers, MA, USA). Gefitinib powders (purity ≥99%) were bought from Aladdin (Shanghai, China).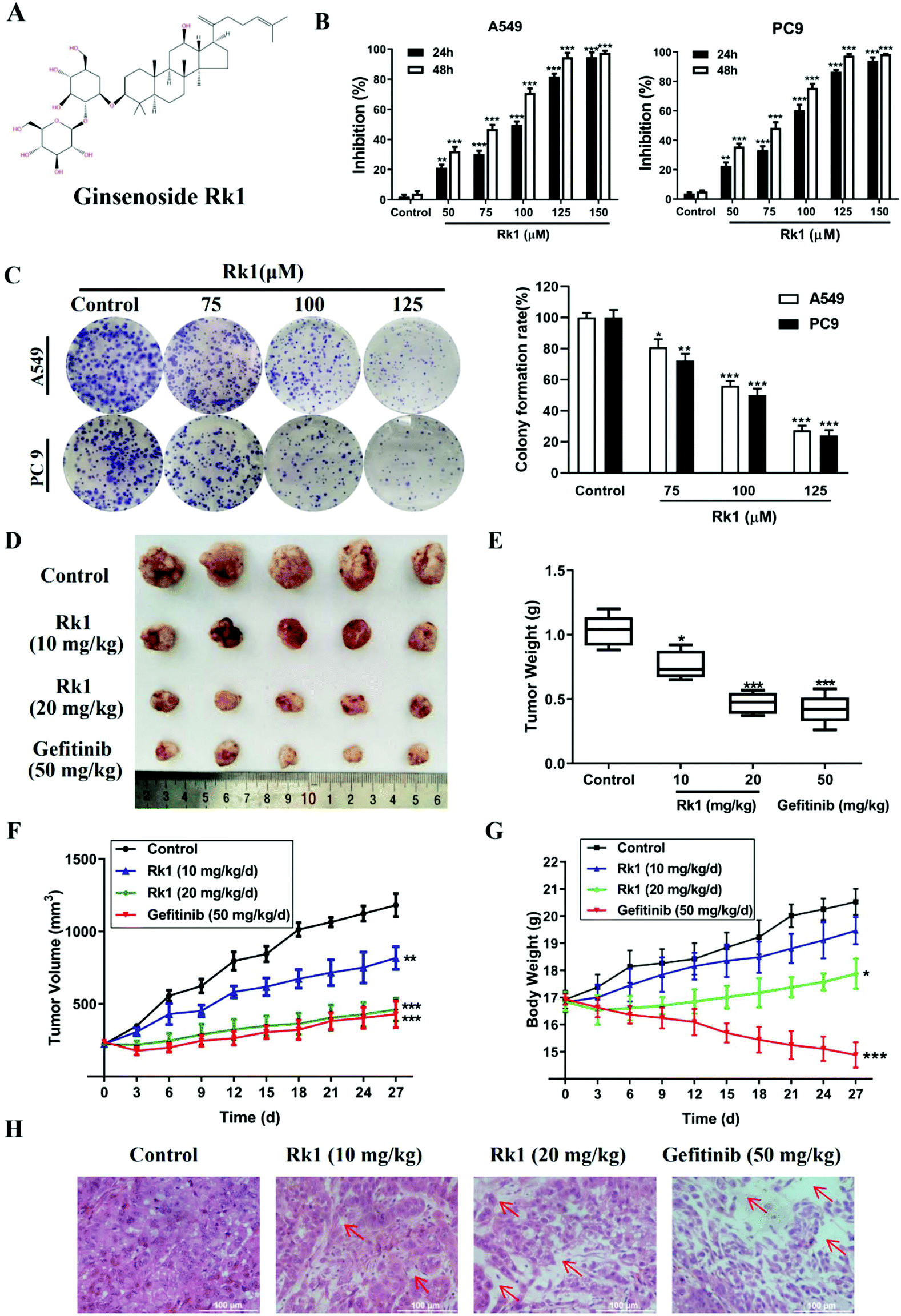 | ||
| Fig. 1 Ginsenoside Rk1 significantly inhibited the proliferation of lung adenocarcinoma cells in vitro and in vivo. (A) The chemical structure of ginsenoside Rk1. (B) The cell viability was measured via the MTT assay after Rk1 treatment. (C) A549 and PC9 cells were treated with Rk1, and colony formation assay was accomplished. (D) Representative pictures of A549-xenograft tumors from the control group, Rk1-treated groups and Gefitinib-treated group. (E) Tumor weight of tumor-bearing nude mice at the 28th day. (F) Tumor volume and (G) body weight of mice were measured every three days. (H) H&E staining of tumors in each group. Scale bars = 100 μm. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
2.2 Cell culture
The lung adenocarcinoma cell lines A549 and PC9 and the human normal lung epithelial cell Bease-2b were purchased from the American Type Culture Collection (ATCC, Maryland, USA). Both cell lines were cultured in RPMI-1640 medium, supplemented with 10% FBS and 1% penicillin and streptomycin. All cells were cultured at 37 °C in a humidified incubator containing 5% CO2.2.3 MTT assay
A549 and PC9 cells were plated in 96-well plates with 1 × 104 cells per well. After incubation for 24 h, different concentrations of Rk1 (0–200 μM) were added to each well, and control cells were incubated with culture media containing 0.1% DMSO. After incubation with Rk1 for 24 and 48 h, 50 μL of 5 mg mL−1 MTT was added, and the cells were incubated at 37 °C for another 2–4 h. Then, MTT was aspirated, 150 μL of DMSO was added, and the formazan was fully dissolved. The absorbance (A) was measured at 490 nm using an ELx800 automated microplate reader (BioTek Instruments, Inc.). IC50 was taken as the concentration that caused 50% inhibition of cell viability and was calculated by the logit method.2.4 Colony formation assay
A549 and PC9 cells were plated in 6-well plates at a density of 800 cells per well. The cells were treated with 0.1% DMSO or different doses of Rk1 (75, 100, and 125 μM). After 24 h, the medium was replenished and subsequently changed once every 3 days without treatment. After 2 weeks, the medium was removed, and the cells were washed twice with phosphate-buffered saline (PBS). Then, the cells were fixed with methanol for 10 min; the colonies were stained with Giemsa stain for 5 min and scored under a microscope (Nikon, Tokyo, Japan).2.5 Cell cycle assay
To determine the effect of Rk1 on the cell cycle phase, propidium iodide (PI) staining was performed and the cells were analyzed with a flow cytometer (Becton Dickson, CA). In brief, A549 and PC9 cells were plated in 6-well plates and treated with 0.1% DMSO or different concentrations of Rk1 at 75, 100 and 125 μM for 24 h. Then, the cells were collected, fixed with 75% ice-cold ethanol at 4 °C overnight, and stained with 100 μg mL−1 RNase A and 50 μg mL−1 PI. The fluorescence per nucleus was measured via flow cytometry.2.6 Hoechst 33342 staining assay
Cell morphological changes were evaluated with Hoechst 33342 staining. Briefly, A549 and PC9 cells were exposed to Rk1 at 100 μM for 24 h, washed twice with pre-cooled PBS and incubated with 10 μg mL−1 of Hoechst 33342 (Solarbio, Beijing, China) for 15 min. The cell morphological characteristics were observed under an inverted fluorescence microscope using UV illumination.2.7 Quantification of apoptosis using the Annexin V/PI staining assay
A549 and PC9 cells were seeded onto 6-well plates at a density of 2 × 105 cells per well. After treatment with Rk1 for 24 h, the cells were harvested, washed twice with PBS and stained with Annexin V-FITC and PI for 15 min at room temperature. Apoptosis was determined via flow cytometry. According to the manufacturer's instructions, PI single-stained cells are dead cells, Annexin V single-stained cells are apoptotic cells, and Annexin V/PI double-stained cells are early and late apoptotic cells, and the apoptotic ratio was calculated by adding the percentage of early and late apoptotic cells.2.8 Transmission electron microscopy (TEM)
A549 and PC9 cells were treated with 0.1% DMSO or 100 μM Rk1 for 24 h. The cells were digested with trypsin and centrifuged. After washing with PBS, the cells were fixed overnight at 4 °C with 2.5% glutaraldehyde and 2% osmium acid. Gradient ethanol dehydration, poly(diallyl terephthalate) immersion, embedding, polymerization, ultrathin sectioning and staining were performed to observe the ultrastructure of the cells under a TEM (JEM-1230, Japan Electron Optics Laboratory, Japan).2.9 Western blotting analysis
A549 and PC9 cells were cultured and treated as described previously. The cells were lysed with RIPA buffer containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1% phosphatase inhibitor cocktail (Solarbio, Beijing, China) on ice for more than 30 min. Then, the cell lysates were centrifuged for 20 min at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C, and protein concentrations were determined using a BCA protein assay kit (Solarbio, Beijing, China). Equal amounts of proteins were resolved via SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. Subsequently, the PVDF membranes were blocked with 5% skimmed milk at room temperature for 2 h and incubated with the corresponding antibodies at 4 °C overnight. The next day, the membranes were washed five times with 1× TBST and incubated using horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Then, the protein bands were imaged by using an ECL system (PerkinElmer, Waltham, MA, USA). Information on antibodies for western blotting was provided in Table S1.†
000 rpm at 4 °C, and protein concentrations were determined using a BCA protein assay kit (Solarbio, Beijing, China). Equal amounts of proteins were resolved via SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. Subsequently, the PVDF membranes were blocked with 5% skimmed milk at room temperature for 2 h and incubated with the corresponding antibodies at 4 °C overnight. The next day, the membranes were washed five times with 1× TBST and incubated using horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Then, the protein bands were imaged by using an ECL system (PerkinElmer, Waltham, MA, USA). Information on antibodies for western blotting was provided in Table S1.†
2.10 Human lung cancer xenograft nude mouse model
To explore the anti-tumor effect of ginsenoside Rk1 in lung adenocarcinoma in vivo, a tumor-bearing nude mouse model was established. Four-week-old female BALB/c athymic nude mice (14 ± 2 g) were obtained from SJA Lab Animal Co., Ltd (Hunan, China). The mice were fed under sterile conditions. After adaption for one week, A549 cells (1 × 107 cells per mouse) were inoculated into the left forelimb pits of the mice. When the tumor volumes of the mice reached 150 mm3 in one week, the mice were randomly divided into 6 groups (n = 5): normal group, normal + 20 mg kg−1 d−1 Rk1 group (intragastric, i.g.), control group, 50 mg kg−1 d−1 gefitinib group18,19 (intragastric, i.g.), 10 mg kg−1 d−1 Rk1 group and 20 mg kg−1 d−1 Rk1 group (intragastric, i.g.). The body weights and tumor sizes of the mice were measured every three days, and the tumor volumes were calculated as follows: tumor volume = (length × width2)/2. After treatment for 28 days, the mice were euthanized, and the tumors and primary organs were taken out, weighed, and stored at −80 °C for future analysis. All experiments were conducted in accordance with the Animal Ethics Regulations of the People's Republic of China and the Animal Ethics Guidelines of the People's Republic of China and were approved by the Animal Ethics Committee of Northwest University (NWU-AWC-20190604M).2.11 Histopathology and immunohistochemistry
Histopathology and immunohistochemistry were performed as previously described.20 In brief, the tumor tissues and major organs, including the heart, liver, spleen, lung and kidney, were embedded in paraffin, and then, 5 μm sections were stained with hematoxylin and eosin (H&E). Tumor tissues were immunostained for caspase-3, Bax, Bcl-2, IKKα, p-IKKα, IκBα, p-IκBα, p65, p-p65, and PD-L1. The images were captured via a microscope (Nikon, Tokyo, Japan).2.12 Hemogram assay and measurement of biochemical parameters
Blood samples were obtained from each group on the 28th day. Peripheral blood was drawn from the orbital venous plexus and collected in tubes containing ethylenediaminetetraacetic acid to detect hematological parameters including the white blood cell (WBC) count, lymphocyte (LYM) count and granulocyte (GRAN) count in each sample using an automatic hematology analyzer (HC2200, Meiyilinm, China).Serum samples were collected from each group on the 28th day and were used to analyze the liver function parameters ALT and AST and the renal function parameters urea, UA and creatinine using commercial kits according to the manufacturer's instructions (Shanghai Enzyme Biotechnology Co., Ltd, Shanghai, China).
2.13 Statistical analysis
Statistical analysis was performed using SPSS version 19.0 software (SPSS Inc., IL, USA). The experimental data are represented as the mean ± standard deviation (SD) and were analyzed with one-way analysis of variance (ANOVA). For all experiments, a value of p < 0.05 was considered statistically significant.3. Results
3.1 Rk1 inhibited lung adenocarcinoma growth in vivo and in vitro
To investigate the effect of Rk1 on the proliferation of lung adenocarcinoma cells and normal lung epithelial cells, the cell viability of A549, PC9, and Bease-2b cells was determined by MTT assay. As shown in Fig S1,† when the Rk1 concentration reached 150 μM, the cell survival rate was still 85% at 48 h, so Rk1 was substantially non-toxic to normal lung epithelial cells. For lung adenocarcinoma cells, the results showed that Rk1 markedly reduced cell viability in a time- and dose-dependent manner. In particular, the cell proliferation inhibition rate reached 94.52 ± 3.09% after treatment with 125 μM Rk1 for 48 h in A549 cells, and similar results were observed in PC9 cells (97.51 ± 1.09%). In addition, the half maximal inhibitory concentration (IC50) of Rk1 was 69.25 μM in A549 cells and 66.12 μM in PC9 cells after treatment with Rk1 for 48 h (Fig. 1B). Furthermore, in the colony formation assay, Rk1 significantly inhibited the colony formation of A549 and PC9 cells, and the results showed that Rk1 inhibited lung adenocarcinoma for a long period of time (Fig. 1C). In conclusion, our results indicate that Rk1 can significantly decrease the proliferation of lung adenocarcinoma cells in vitro.To further illustrate the anti-tumor effect of Rk1 in lung adenocarcinoma, a lung adenocarcinoma xenograft nude mouse model was established with A549 cells. As shown in Fig. 1D and E, after treatment with Rk1, the tumor sizes of the tumor-bearing nude mice were dramatically restrained (p < 0.05) compared to the control group. In addition, compared with the tumor volume in the control group, the tumor volume inhibition rates of Rk1 and gefitinib on tumor volume were 30.83%, 60.87%, and 63.74%, and there was no significant difference between the effects in the Rk1 high dose group and the gefitinib group (p > 0.05) (Fig. 1F). Moreover, on the 28th day of administration, the average body weight of the model control group (20.26 ± 0.78 g) was similar to that of the 10 mg kg−1 Rk1 group (19.11 ± 0.49 g), and there was no significant difference between the two groups (p > 0.05). The average body weight of the mice in the 20 mg kg−1 Rk1 group was 17.58 ± 0.68 g, but the average weight of the mice in the gefitinib positive control group observably decreased to 15.11 ± 0.78 g (Fig. 1G) (p < 0.05). H&E staining of tumor tissues was shown in Fig. 1H. The tumor cells in the control group were closely arranged, the staining was deep, and the intercellular substance was small. The tumor cells in the Rk1 and the gefitinib groups were loosely arranged, with lighter staining, partial degeneration and necrosis. Taken together, these results indicated that Rk1 blocked the growth of human lung adenocarcinoma in vivo, which was consistent with the results of in vitro studies.
3.2 Toxicity evaluation of Rk1 in murine tumor xenograft models
To evaluate the toxicity of Rk1 in the xenograft nude mice, we first measured the levels of immune cells in the peripheral blood of each group. The results showed that there was no significant difference in the number of WBC, LYM and GRAN in the peripheral blood between the normal group, the normal + 20 mg kg−1 Rk1 group, the control group, the low dose Rk1 group and the high dose Rk1 group (p > 0.05; Fig. 2A). However, the number of these immune cells in the gefitinib group was obviously decreased compared to that in the normal group (p < 0.05; Fig. 2A), indicating that gefitinib might destroy the immune system of the nude mice, while Rk1 caused no damage to the immune system. In addition, we investigated the effect of Rk1 on hepatic and renal functions. As shown in Fig. 2B and C, administration of Rk1 did not result in notable changes in liver function parameters (ALT and AST) or renal function parameters (urea, uric acid and creatinine) in the mice compared to those in the normal group. Therefore, Rk1 did not cause liver or kidney toxicity. However, the liver function parameters of the gefitinib group were significantly elevated, suggesting that gefitinib had certain side effects on liver function (Fig. 2C). Moreover, H&E staining showed no obvious pathological changes in the physiological appearance of vital organs in the normal group, normal + Rk1 (20 mg kg−1) group, control group and Rk1 treatment group, while organs in the gefitinib group showed vacuolization around hepatocyte nuclei, alveolar structure collapse, alveolar septum widening, and renal structural disorder (Fig. 2D). Together, these results demonstrate that Rk1 has no effect on the normal functions of the vital organs and immune cells in vivo.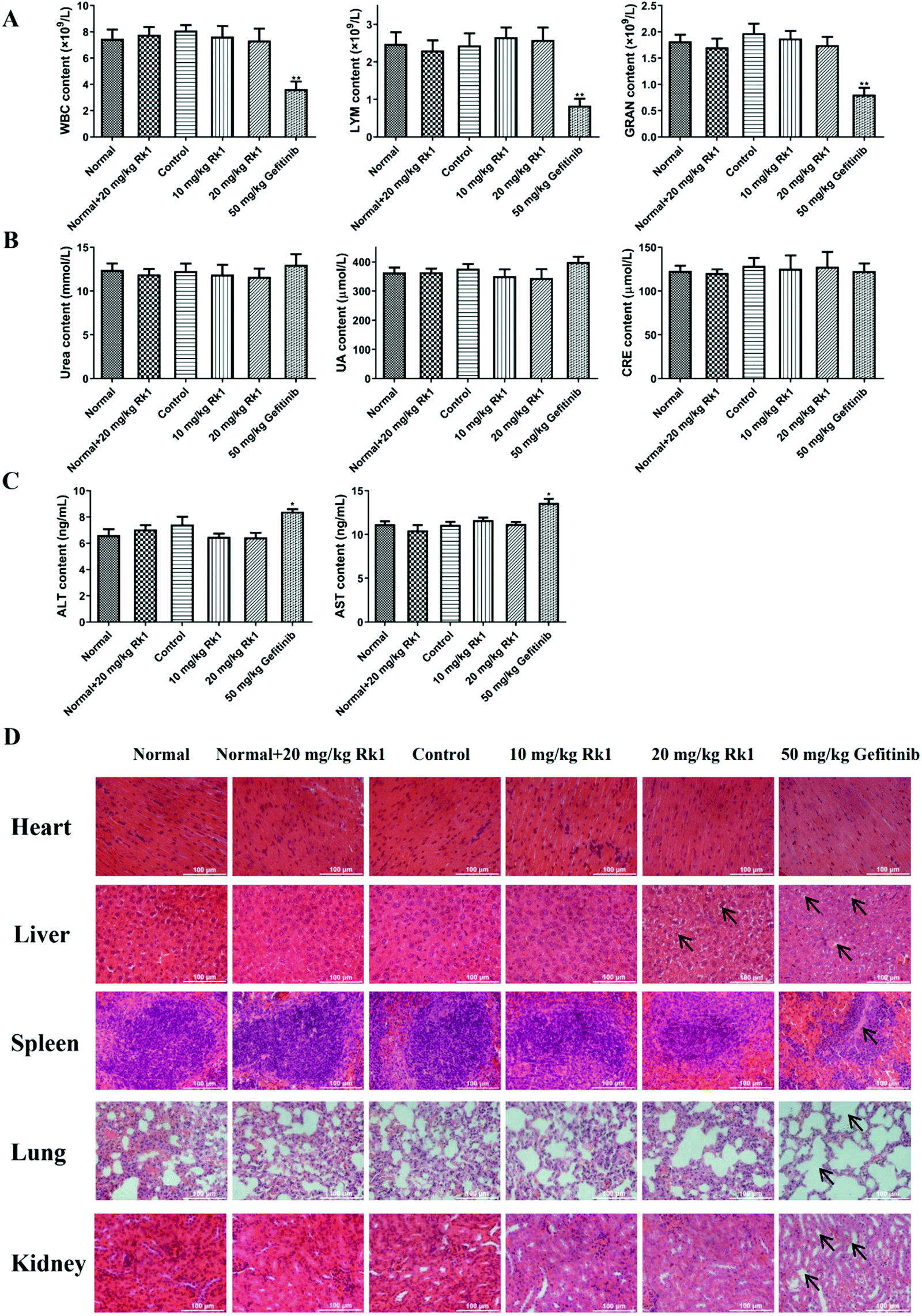 | ||
| Fig. 2 Effect of ginsenoside Rk1 on major organs and pivotal indexes in tumor-bearing nude mice. (A) The number of white blood cells (WBC), lymphocytes (LYM) and granulocytes (GRAN) in peripheral blood of each group was analyzed by using the Automatic Hematology Analyzer. The levels of (B) renal function parameters (urea, UA and CRE) and (C) liver function parameters (ALT and AST) in the serum from each group. (D) H&E staining of the main organs in each group including the heart, liver, spleen, lungs and kidneys. Scale bars = 100 μm. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
3.3 Rk1 induced cell cycle arrest in the G1 phase
To verify the effect of Rk1 on the cell cycle of lung adenocarcinoma cells, we used flow cytometry and western blotting analysis. As shown in Fig. 3A, Rk1 induced G1 phase arrest in lung adenocarcinoma cells. In comparison with the control group, after treatment with 125 μM Rk1, the number of cells in the G1 phase increased in both A549 and PC9 cells, while the number of cells in the S phase and G2 phase decreased. Specifically, the proportion of G1 phase cells in A549 cells increased from 57.49% to 75.60%, the number of cells in the S phase decreased from 15.62% to 3.39%, and the number of cells in the G2 phase decreased from 26.89% to 21.01%. Similarly, the proportion of PC9 cells in G1, S and G2 phases varied from 42.35%, 34.06% and 23.59% to 93.15%, 4.61% and 2.24%, respectively. Therefore, these results indicate that Rk1 induced G1 phase arrest in two lung adenocarcinoma cell lines.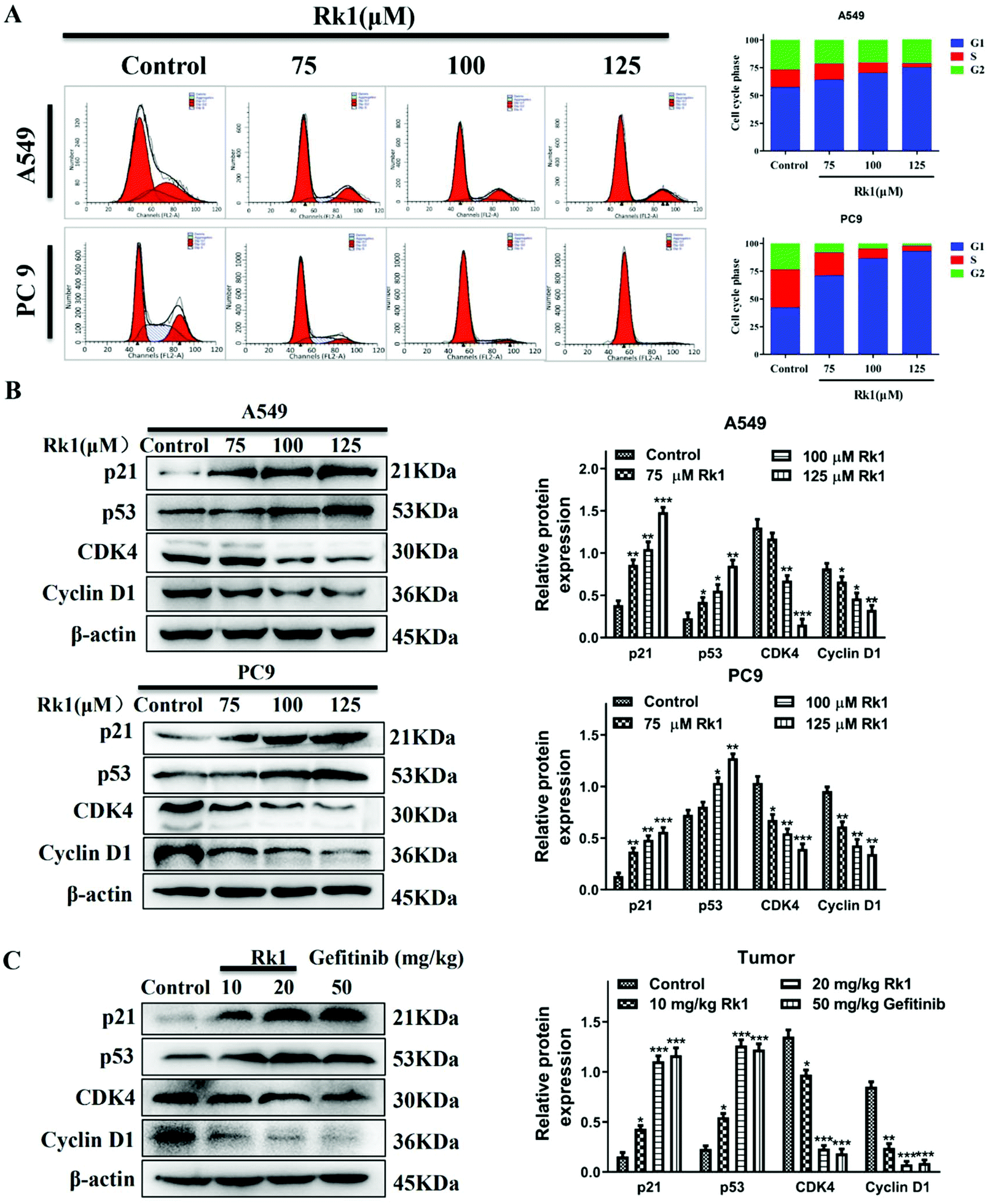 | ||
| Fig. 3 The effect of ginsenoside Rk1 on the cycle of lung adenocarcinoma cells in vitro and in vivo. (A) The cell cycle distribution was detected via flow cytometry. Western blotting was used to detect the expression of G1 phase-related proteins in (B) lung adenocarcinoma cells (A549 and PC9 cells) and (C) each group of tumor tissues. β-Actin was used as an endogenous reference. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
To further investigate whether Rk1 induced G1 arrest in lung adenocarcinoma cells, we analyzed the expression of G1 phase-associated proteins by western blotting after Rk1 treatment of A549 and PC9 cells and tumor tissues. The results are shown in Fig. 3B, and the protein expression levels of p21 and p53 were markedly increased (p < 0.05), while those of CDK4 and cyclin D1 were dramatically decreased in a dose-dependent manner by Rk1 treatment in A549 and PC9 cells (p < 0.05) compared to the control group. Moreover, similar results were detected in tumor tissues, and there was no significant difference between the effects in the Rk1 high dose and the gefitinib groups (Fig. 3C). In conclusion, these results demonstrate that Rk1 cause cell death by inducing G1 phase arrest in A549 and PC9 cells.
3.4 Rk1 induced cell apoptosis by a caspase-dependent pathway
To analyze whether Rk1 causes apoptosis of lung adenocarcinoma cells in vitro and in vivo, Hoechst 33342 staining, TEM and flow cytometry assays were performed. Hoechst 33342 staining showed that the cells of the control group showed a light blue color, and the cell morphology did not change and the state was good. However, the 100 μM Rk1 treated cells exhibited a bright blue state. And the number of cells in the Rk1 group was significantly reduced compared with those of the control group (Fig. 4A). Apoptotic features such as membrane shrinkage, nuclear fragmentation, chromatin margination and vacuolation were observed by TEM in A549 and PC9 cells after treatment with 100 μM Rk1 for 24 h (Fig. 4B) compared to the control group. Moreover, in Fig. 4C, the upper left quadrant indicates necrotic cells, the lower left quadrant indicates normal cells, the upper right quadrant indicates late apoptotic cells, and the lower right quadrant indicates early apoptotic cells. Annexin V/PI double staining results showed that the apoptosis rate of A549 and PC9 cells was enhanced to 66.50% and 76.24% after 125 μM Rk1 treatment. Consequently, these results indicate that Rk1 can cause apoptotic cell death in lung adenocarcinoma cells.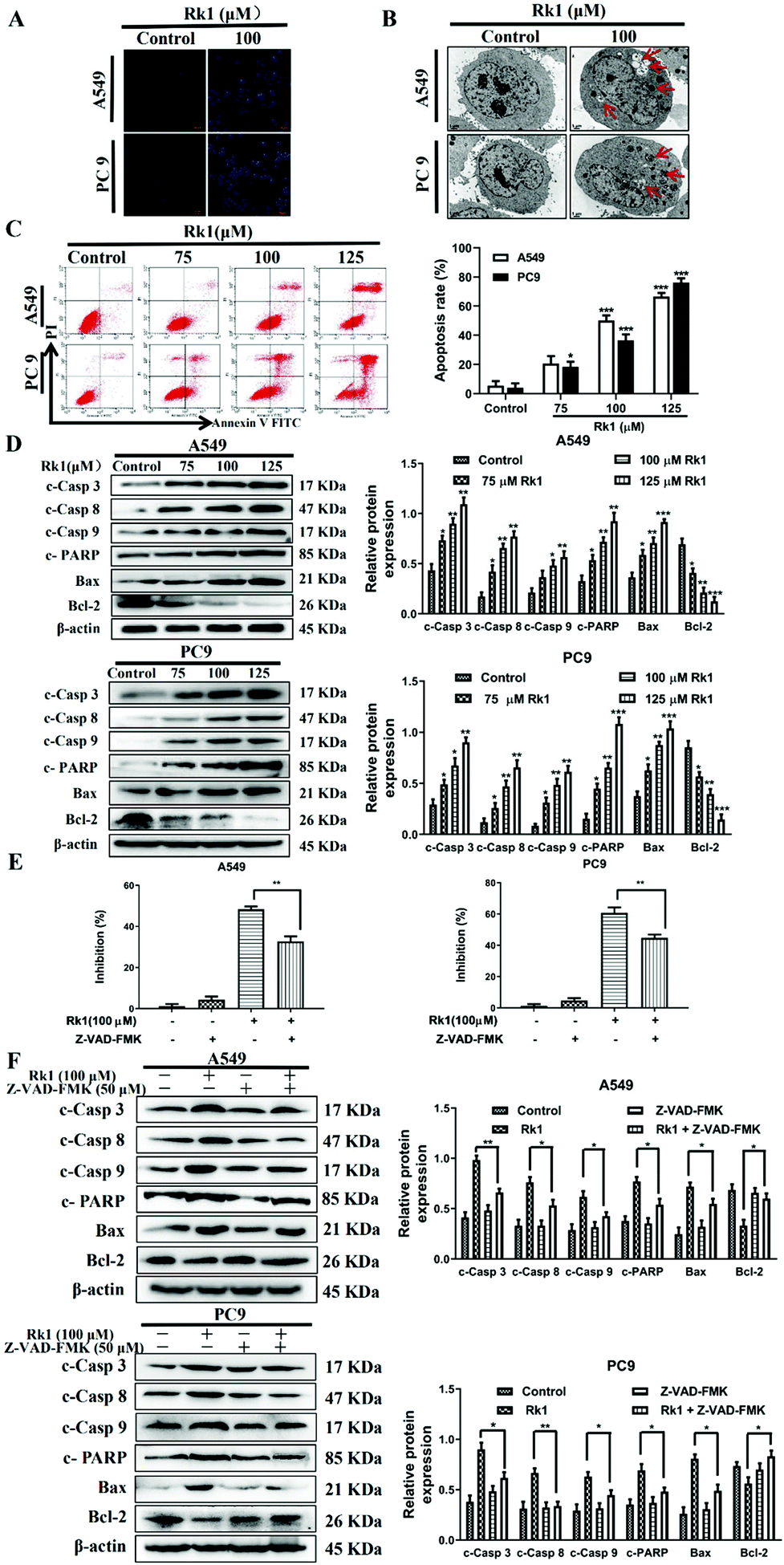 | ||
| Fig. 4 Ginsenoside Rk1 induced apoptosis of lung adenocarcinoma cells in vitro. (A) Hoechst 33342 staining. Scale bar = 100 μm. (B) Apoptosis was measured by flow cytometry after Annexin V/PI staining. (C) Ultrastructure of apoptotic cells was observed via TEM. Scale bar = 1 μm. (D) The expression levels of apoptosis-related proteins in A549 and PC9 cells were detected by western blotting. (E) Cells were pretreated with 50 μM Z-VAD-FMK for 2 h followed by co-treatment with Rk1 for another 24 h, and then processed for the MTT assay. (F) Cells were pretreated with 50 μM Z-VAD-FMK for 2 h followed by co-treatment with Rk1 for another 24 h, and then expression of apoptosis-related proteins was detected by western blotting. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
Furthermore, to validate the apoptosis signaling pathway that is induced by Rk1 in lung adenocarcinoma cells, apoptosis-related protein expression was measured. As shown in Fig. 4D, Rk1 up-regulated the expression of Bax, cleaved caspase-3, -8, and -9 and PARP, and down-regulated the expression of Bcl-2 in A549 and PC9 cells (p < 0.05) compared to the control group. In addition, similar results were detected in lung adenocarcinoma tumor tissues, and there was no dramatic difference between the high-dose Rk1 group and the gefitinib group (Fig. 5A). Immunohistochemical analysis of tumor tissues showed that the positive expression areas of cleaved caspase-3 and Bax in the Rk1 and gefitinib groups were higher than those in the control group, while Bcl-2 was lower than that in the control group (p < 0.05) (Fig. 5B). To conclude, these findings indicate that Rk1 induces caspase-dependent apoptosis in vitro and in vivo through both extrinsic and intrinsic pathways.
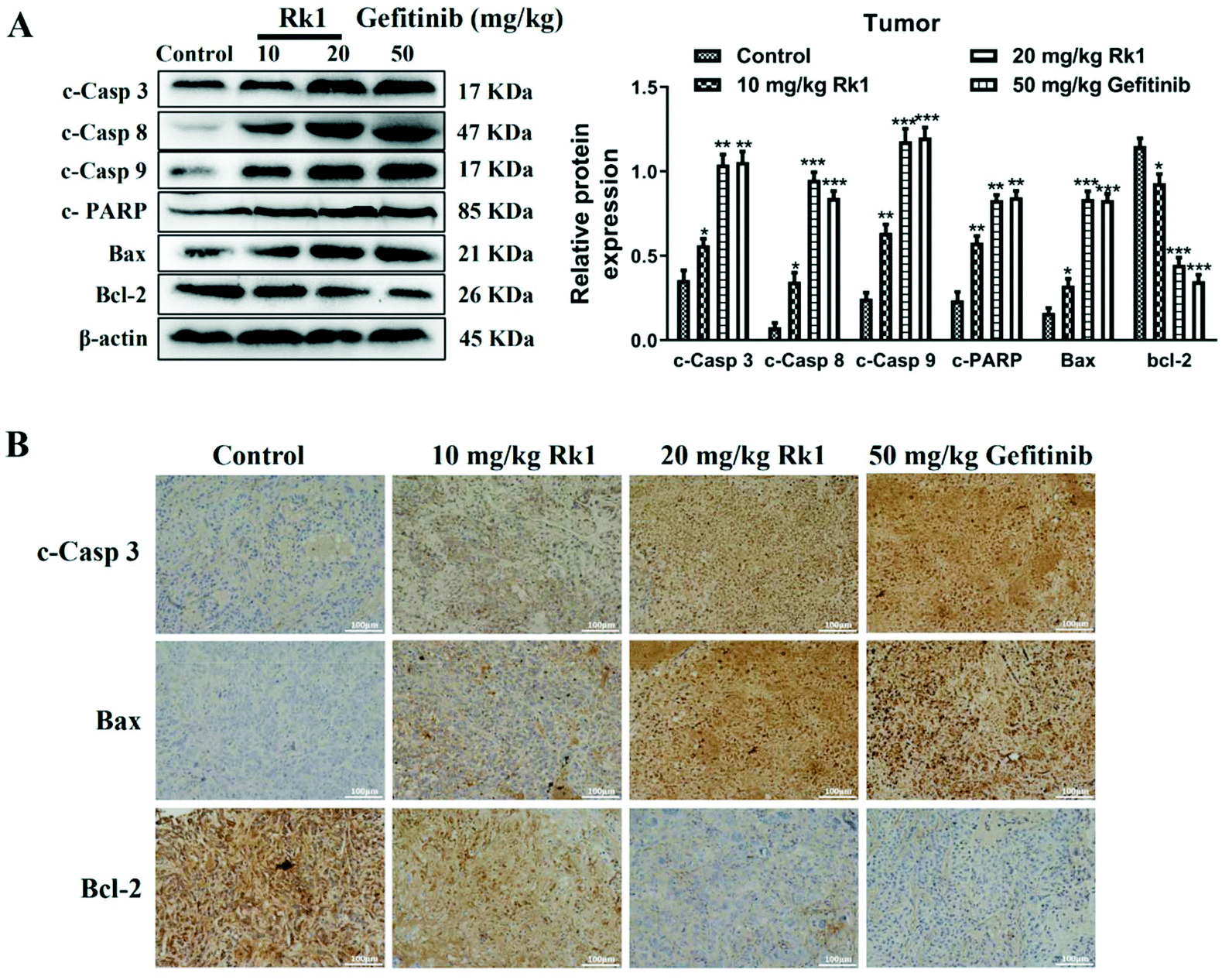 | ||
| Fig. 5 Ginsenoside Rk1 induced apoptosis of lung adenocarcinoma in vivo. (A) The expression of apoptosis-related proteins was detected by western blotting in tumor tissues per group. (B) Immunohistochemical staining was performed to measure the expression of apoptosis-associated proteins in tumor tissues from each group. Scale bars = 100 μm. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
Furthermore, to further verify these findings, we studied the effect of Z-VAD-FMK (a pan-caspase inhibitor) on the survival rate of lung adenocarcinoma cells and the expression of related proteins. After pretreatment with 50 μM Z-VAD-FMK for 2 h, MTT assay showed that Z-VAD-FMK attenuated Rk1-induced cell death (Fig. 4E). In addition, Z-VAD-FMK partially eliminated cleaved caspase-3, -8, and -9, PARP and Bax protein levels and increased the expression of Bcl-2 compared with those of Rk1 alone (p < 0.05, Fig. 4F). Taken together, these results further demonstrate that Rk1 promotes lung adenocarcinoma cell apoptosis via a caspase-dependent pathway.
3.5 Rk1 induced apoptosis of lung adenocarcinoma cells via the NF-κB signaling pathway
To explore whether Rk1 induces apoptosis in A549 and PC9 cells through the NF-κB pathway, we detected the protein levels of the NF-κB pathway by western blotting. Moreover, compared with the control group, A549 and PC9 cells treated with various concentrations of Rk1 attenuated the expression of phosphorylated IKKα, phosphorylated IκBα, p65 and phosphorylated p65, and led to a certain decrease in IKKα expression but largely increased the expression of IκBα compared with those of the control group (Fig. 6A). Similar results were found in tumor tissues (Fig. 6B). These findings suggest that Rk1 inhibits cell survival by mediating the NF-κB pathway. Immunohistochemical analysis showed that Rk1 and gefitinib reduced the positive expression of NF-κB-related protein, which was consistent with the western blotting results (Fig. 6C). To verify the key role of NF-κB in this process, 30 μM JSH-23 (an NF-κB inhibitor) and 1 μM CER (ceruletide, an NF-κB activator) were pretreated for 2 h, and then were co-treated with 100 μM Rk1, respectively. First, the MTT results showed that CER partially abolished the inhibition of growth of Rk1 on lung adenocarcinoma cells, while JSH further enhanced the effect of Rk1 (Fig. 7A). Then, as shown in Fig. 7B, compared with the effects of 100 μM Rk1 alone, pretreatment with JSH-23 further down-regulated the protein expression of IKKα, p-IKKα, p-IκBα, p65 and p-p65, and up-regulated IκBα protein levels, while CER treatment partially reversed the Rk1-induced expression in A549 cells. Furthermore, JSH-23 further promoted the expression of cleaved caspase-3 and -8 and Bax and suppressed Bcl-2 levels in A549 cells, while pretreatment with CER showed the opposite results (Fig. 7B). Consequently, these results indicate that Rk1 induces apoptosis of lung adenocarcinoma cells via the NF-κB signaling pathway.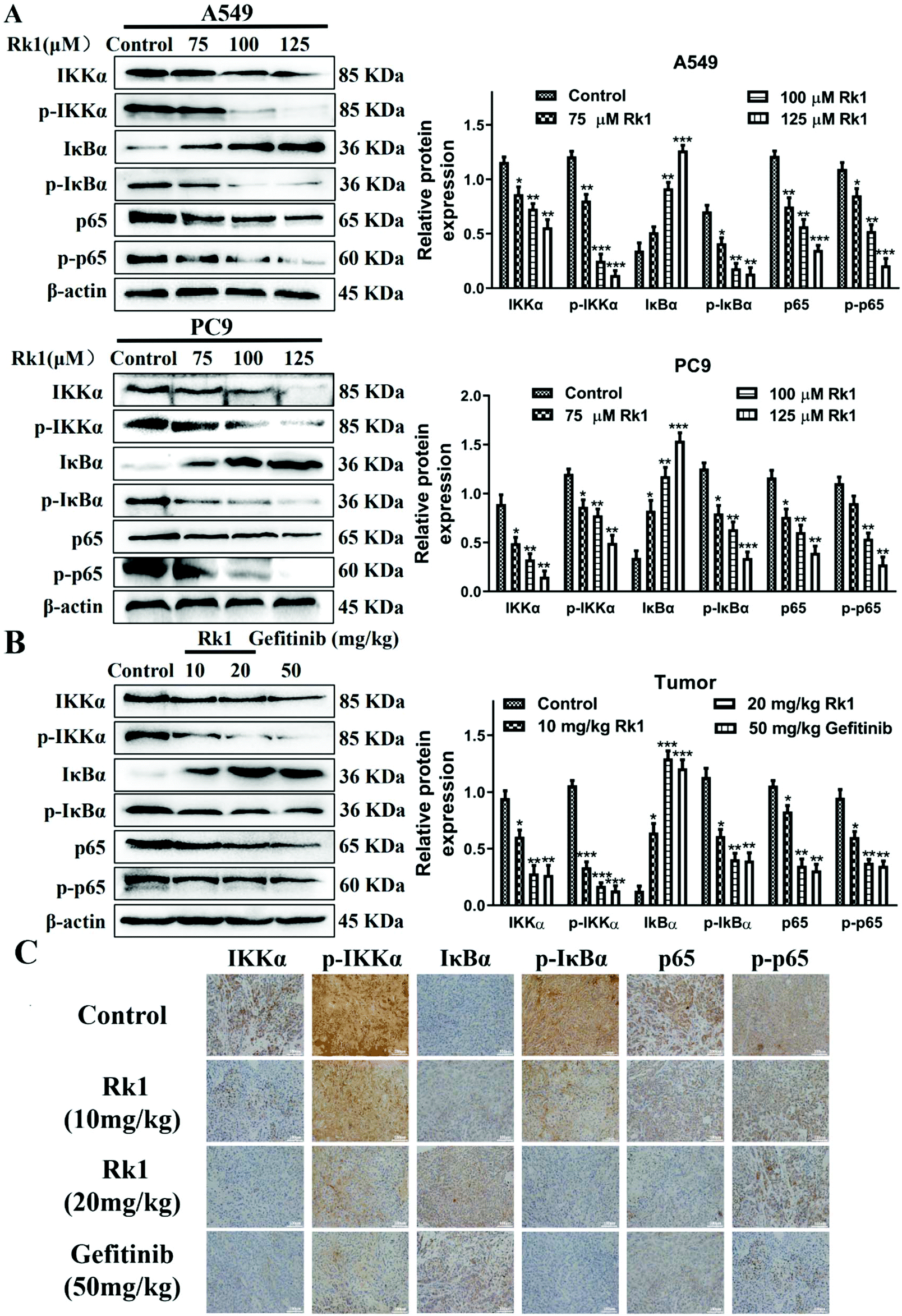 | ||
| Fig. 6 Effects of ginsenoside Rk1 on the NF-κB signaling pathway in lung adenocarcinoma cells in vitro and in vivo. Western blotting was performed to measure the expression of IKKα, p-IKKα, IκBα, p-IκBα, p65 and p-p65 in (A) A549 and PC9 cells and (B) tumor tissues. (C) The expression of NF-κB-associated proteins in tumor tissues from each group were detected via immunohistochemical staining. Scale bars = 100 μm. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
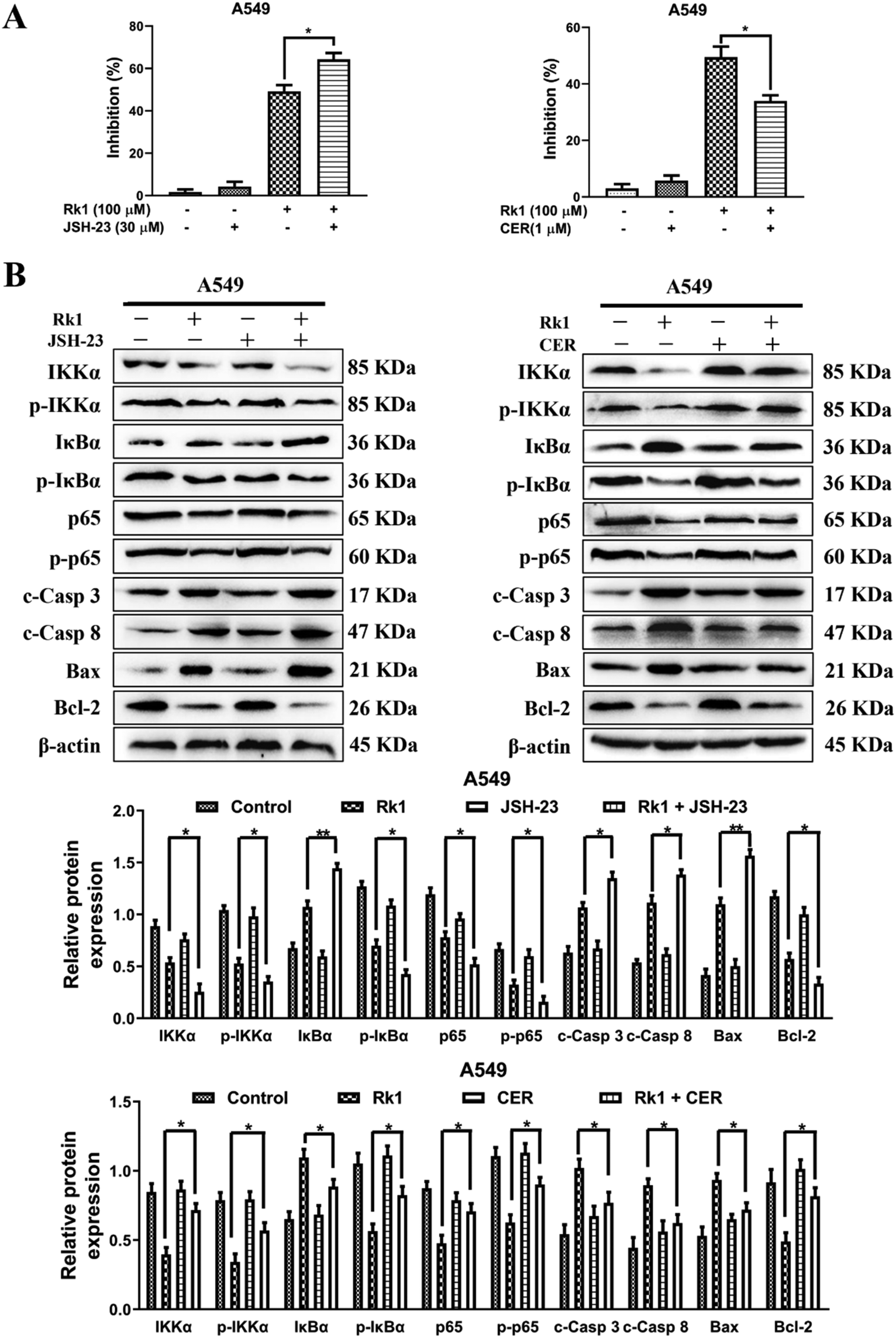 | ||
| Fig. 7 Rk1 induced apoptosis through blocking the NF-κB signaling pathway in lung adenocarcinoma cells. (A) Cells were pretreated with 30 μM JSH-23 or 1 μM CER for 2 h followed by co-treatment with Rk1 for another 24 h, and then processed for the MTT assay. (B) Cells were pretreated with 30 μM JSH-23 or 1 μM CER for 2 h followed by co-treatment with Rk1 for another 24 h, and then expression of NF-κB and apoptosis-related proteins was detected by western blotting. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
3.6. Rk1 reduced PD-L1 expression by down-regulating NF-κB
To explore whether Rk1 regulates the activity of PD-L1 in lung adenocarcinoma cells, the expression of PD-L1 in cells was analyzed by western blotting. Human lung adenocarcinoma cells were treated with different concentrations of Rk1 for 24 h, and the PD-L1 protein expression levels were gradually augmented in a dose-dependent manner (p < 0.05) compared to the control group. A similar result was detected in tumor tissues (Fig. 8A). Immunohistochemistry also showed that Rk1 down-regulated the strong positive expression of PD-L1 in tumor cells (Fig. 8B). Next, we examined whether the decrease in PD-L1 expression caused by Rk1 was affected by NF-κB. As shown in Fig. 8C, CER partly eliminated the Rk1-mediated protein expression inhibition of PD-L1, while JSH-23 further reduced the expression of PD-L1 in A549 and PC9 cells. These findings suggest that Rk1 regulates the expression of PD-L1 by down-regulating NF-κB activity. A schematic diagram of the mechanism by which Rk1 acts against lung adenocarcinoma was shown in Fig. 9.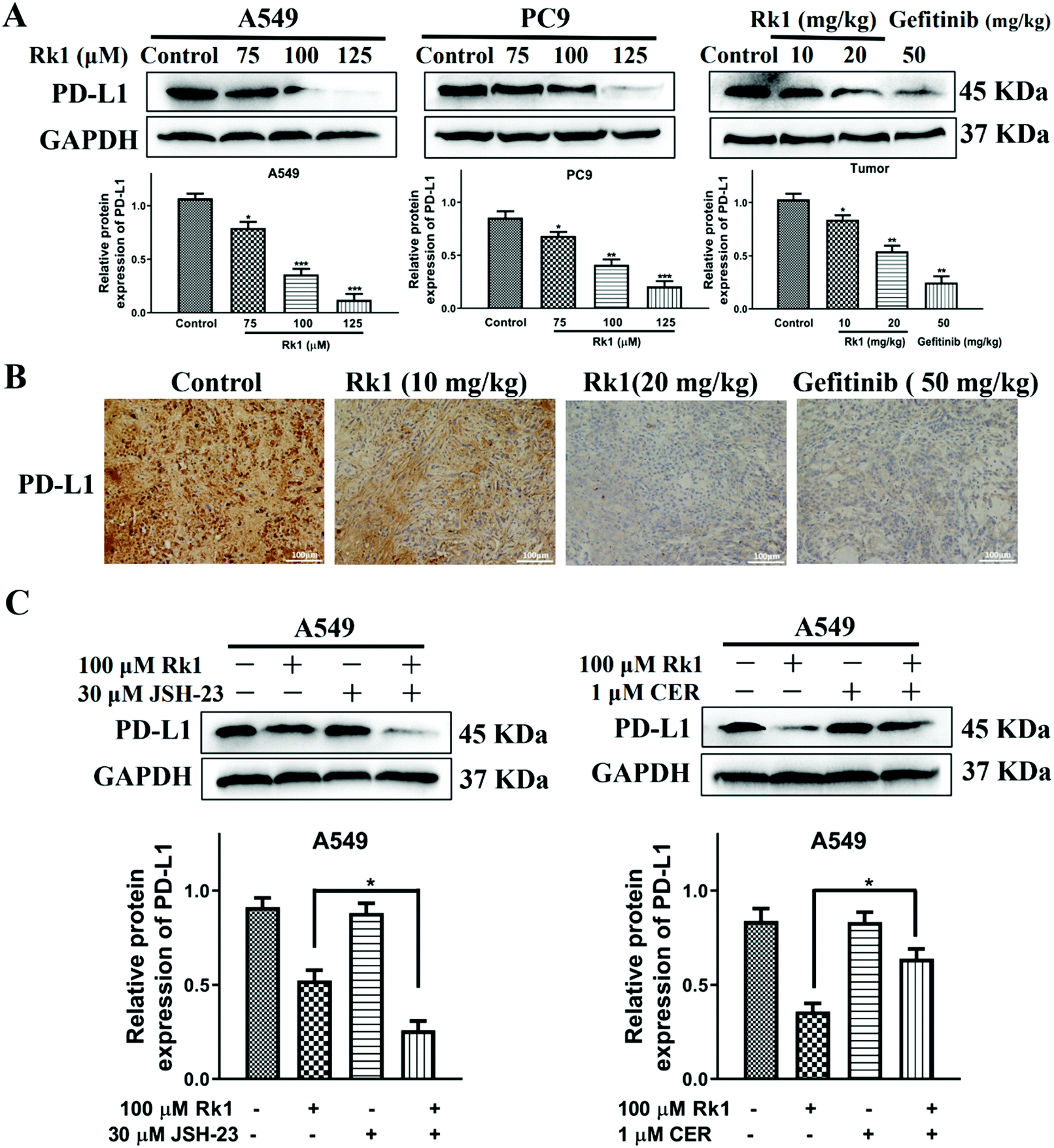 | ||
| Fig. 8 The effects of Rk1 on the expression of PD-L1 in lung adenocarcinoma cells in vitro and in vivo. (A) and (B) The basic PD-L1 expression in A549 and PC9 cells and tumor tissues via western blotting and immunohistochemical staining. Scale bars = 100 μm. (C) The expression of PD-L1 was analyzed by western blotting after pretreatment with JSH-23 or ceruletide. Data are presented as means ± SD of three independent experiments, *p < 0.05, **p < 0.01 and ***p < 0.001 compared with the control. | ||
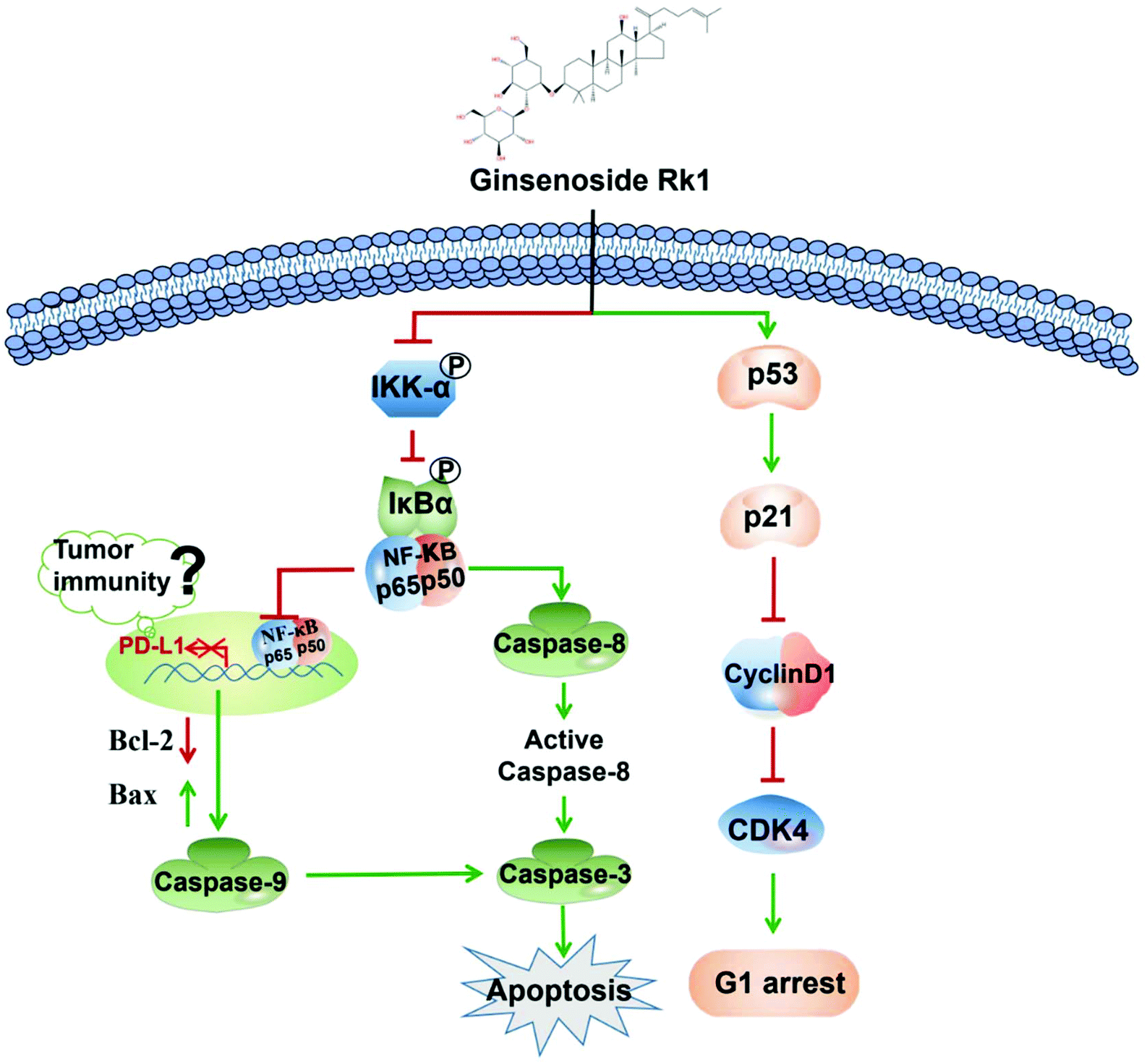 | ||
| Fig. 9 Proposed molecular mechanism of the anti-lung adenocarcinoma cancer activity of Rk1. | ||
4. Discussion
The discovery of natural drugs might provide new methods for the treatment of lung adenocarcinoma, which remains one of the most threatening malignancies in the world despite great progress in its treatment.21 Ginsenoside Rk1 is the main active ingredient extracted from black ginseng. Recent studies have shown that Rk1 exhibits strong anti-breast cancer activity through the ROS-mediated PTEN/PI3K/Akt/mTOR pathway in vivo and in vitro22 and induces neuroblastoma cell apoptosis through loss of mitochondrial membrane potential and activation of caspases.23 However, the antineoplastic effect of Rk1 in lung adenocarcinoma and its mechanism are still unclear. Therefore, in this study, we aimed to investigate the significant effects of ginsenoside Rk1 on the growth inhibition and cell apoptosis in lung adenocarcinoma cells (A549 and PC9) and a xenotransplantation model of lung adenocarcinoma. In the MTT and colony formation experiments, Rk1 reduced the activity of A549 and PC9 cells in vitro, especially with 150 μM Rk1 treatment, and the cell proliferation inhibition rate reached more than 90%. In the xenograft model, we found that 20 mg kg−1 Rk1 significantly inhibited tumor growth, and its tumor volume inhibition rate was 59.17%, which was not significantly different from that of the first-line drug gefitinib in lung adenocarcinoma; however, gefitinib caused severe weight loss in nude mice. These results demonstrate that Rk1 has a strong inhibitory proliferation effect in lung adenocarcinoma in vivo and in vitro.The first-line anticancer drug gefitinib, an aniline quinazoline small molecule compound, has long been used to treat advanced non-small cell lung cancer. However, gefitinib has side effects in the treatment of lung adenocarcinoma. Previous reports have proved that gefitinib can cause liver damage and lung injury.24 Consistently, our serum biochemical parameters and H&E staining of liver tissue treated with gefitinib showed liver damage and mild lung injury. In addition, gefitinib significantly decreased the number of immune cells, including WBC, LYM and GRAN, compared to those in the normal group, indicating that gefitinib was highly toxic. In contrast, blood test results showed that Rk1 did not reduce the number of immune cells, indicating that Rk1 caused no immune damage. H&E staining showed that Rk1 treatment did not affect the normal function of the heart, liver, spleen, lungs, or kidneys. Serum biochemical parameters showed that Rk1 did not damage liver or renal function. These results indicate that Rk1 exhibits few side effects in treating lung adenocarcinoma. Therefore, ginsenoside Rk1 may be a new anti-lung adenocarcinoma drug.
The cell cycle is the period from the end of one division of the proliferating cell to the beginning of the next division. In the past few years, it has been reported that cancer could be regarded as a disease of the cell cycle.25 The cell cycle is triggered by an independent control system. At the heart of this system is a series of protein kinases called cyclin-dependent kinases (CDKs), whose activity increases and decreases as the cells progress through the circulation.26 Inhibition of cell growth is typically accompanied by redistribution of the cell cycle.27 Therefore, regulation of the cell cycle has become a crucial approach to treating cancer. In the current study, the percentage of A549 and PC9 cells in the G1 phase increased after ginsenoside Rk1 treatment, while the percentage of cells in the S phase and G2 phase decreased. Consistent with our results, the curcumin derivative MHMM-41 induces an increase in the percentage of cells in the G1 phase, causing A549 cells to trigger G1 arrest.28 Therefore, Rk1 induced lung adenocarcinoma cell arrest in the G1 phase. More importantly, p53 is a transcription factor that plays a protective role by detecting DNA damage. Activation of p53 levels increases the response to DNA damage by blocking the cells in the G1 phase to repair DNA.29 In other words, when cells have DNA damaged, p53 stops the cell cycle by increasing p21 or induces apoptosis.30 In addition, CDKs with regulatory subunit cyclins control cell cycle progression. Specifically, cyclin D1 promotes cell migration from the quiescent to the mitotic phase by activating CDK4, which is a G1 phase-specific cyclin-dependent kinase.31 Fortunately, we found that Rk1 induced G1 arrest in lung adenocarcinoma cells by up-regulating p53 and p21 levels and decreasing the expression of CDK4 and cyclin D1.
Apoptosis is also a major strategy for tumor therapy. In this study, we detected an increase in A549 and PC9 cell apoptosis after Rk1 intervention via fluorescent staining and flow cytometry. Apoptosis is mediated by members of the caspase family of cysteine proteases and is activated by two major pathways: the intrinsic mitochondrial pathway and the extrinsic death receptor pathway.32 The Bcl-2 protein family is the key initiator of the mitochondrial apoptotic pathway, including the pro-apoptotic proteins Bax and Bad and the anti-apoptotic protein Bcl-2.33 Previous studies have shown that Bcl-2 maintains cell activity and prevents apoptosis through the mitochondrial pathway, while the pro-apoptotic protein Bax activates caspase-9, which induces apoptosis through an intrinsic pathway.34 Caspase-3 is an important protein involved in this pathway-mediated apoptosis, and can activate downstream caspases, leading to apoptosis.35 In addition, caspase-8 mediates the extrinsic pathway and plays a key role in apoptosis induced through the stimulation of death receptors such as Fas and TNFR.14 Our results showed that Rk1 significantly increased the expression of cleaved caspase-3, -8, and -9 and Bax, and decreased the level of Bcl-2. Importantly, Z-VAD-FMK partially abolished Rk1-induced apoptosis. Similar to our results, kaempferol promotes Bax expression while inhibiting Bcl-2 expression, and increases the ratios of cleaved caspase-9/caspase-9 and cleaved caspase-3/caspase-3. Apoptosis of lung adenocarcinoma cells is induced by mitochondria and caspase-dependent pathways.36 Therefore, these results demonstrate that Rk1 induces apoptosis in lung adenocarcinoma cells through both intrinsic and extrinsic pathways.
The NF-κB pathway has recently become a promising target for tumor therapy.37 NF-κB-induced genes are involved in anti-apoptotic effects, proliferation, angiogenesis, invasion and metastasis, and play an important regulatory role in lung cancer.38 Many studies have reported that NF-κB is overexpressed in various cancers such as breast cancer,39 pancreatic cancer,40 and colorectal cancer,41 and NF-κB overexpression further promotes tumor development. Activation of NF-κB is attributed to its phosphorylation and degradation of IκB that is bound to NF-κB, resulting in the translocation of NF-κB into the nucleus to promote cell survival.42 Therefore, we hypothesized that Rk1 induces apoptosis of lung adenocarcinoma cells by inhibiting the activation of NF-κB. In addition, the most common form of NF-κB is a heterodimer composed of p65 and p50. Fortunately, we detected that Rk1 inhibited NF-κB-p65 and its upstream IκBα phosphorylation. Moreover, it has been reported that when tumors occur, IκB kinase α (IKKα) migrates into the nucleus, promoting cancer cell metastasis, spreading and proliferation.43 In this study, we found that total and phosphorylated IKKα in lung adenocarcinoma cells and tumor tissues were down-regulated by Rk1 treatment, preventing IKKα from entering the nucleus, which ultimately inhibited cell survival. This evidence indicates that Rk1 regulates the NF-κB pathway to inhibit lung adenocarcinoma proliferation. Similarly, treatment with PP2A inhibitors induces a decrease in the phosphorylation of IKKα, resulting in a consistent abatement in total IκBα protein levels, thereby inhibiting the expression of p65 in pancreatic cancer.44 The role of Rk1 in regulating the NF-κB signaling pathway in lung adenocarcinoma and the effect of the NF-κB pathway on apoptosis were further evaluated. The NF-κB activator CER treatment partially abolished the Rk1-mediated expression of p-p65/p65 signaling and partially reversed the expression of caspase family proteins. Therefore, these results revealed that Rk1 induces apoptosis in lung adenocarcinoma cells by regulating NF-κB.
Finally, western blotting showed that the expression of PD-L1 was significantly reduced after Rk1 treatment. Moreover, knockdown of EZH2 results in the down-regulation of PD-L1 in lung cancer cells.45 Metformin inhibits the expression of PD-L1 protein by AMPK activation, thereby inhibiting the proliferation of endometrial cancer cells.46 These studies are similar to our results. In addition, the expression of PD-L1 in tumor cells is dependent on the activation of NF-κB.47 Similarly, our results showed that CER partially offsets the effect of Rk1 on PD-L1 levels. Recent studies have shown that cancer immunosuppression and immune escape play an important role in tumor progression. Immune checkpoint blockade is the most promising strategy among various immunotherapies.48 Moreover, the PD-L1/PD-1 pathway is the most critical immunological checkpoint in immunotherapy.49 Our study showed that Rk1 inhibited the expression of PD-L1 protein. Therefore, an interesting future study will be devoted to determine whether Rk1 can achieve anti-tumor activity by regulating immunity.
5. Conclusion
Ginsenoside Rk1 induced apoptosis and triggered cell cycle arrest to inhibit cell proliferation in human lung adenocarcinoma A549 and PC9 cells. NF-κB protein expression was down-regulated by Rk1, and we found that Rk1 promotes cell apoptosis and inhibits PD-L1 expression by inhibiting NF-κB signaling. This study shows the innovative regulatory mechanism of Rk1 in lung adenocarcinoma that contributes to the promotion of Rk1 as an antitumor drug. In addition, the safety evaluation by in vivo experiments provides a theoretical basis for clinical applications.Abbreviations
| NF-κB | Nuclear factor-κB |
| FBS | Fetal bovine serum |
| RPMI | Roswell Park Memorial Institute |
| PBS | Phosphate-buffered saline |
| PI | Propidium iodide |
| TEM | Transmission electron microscopy |
| PMSF | Phenylmethylsulfonyl fluoride |
| PVDF | Polyvinylidene fluoride |
| H&E | Hematoxylin and eosin |
| WBC | White blood cell |
| LYM | Lymphocyte |
| GRAN | Granulocytes |
| CDKs | Cyclin-dependent kinases |
| IKKα | IκB kinase α |
Conflicts of interest
No potential conflict of interest was reported by the authors.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21838009, 21808184, 21776227, 21576223, and 21576160) and the Shaanxi Key Laboratory of Degradable Biomedical Materials Program (16JS106).References
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2018, CA-Cancer J. Clin., 2018, 60(5), 277–300 Search PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA-Cancer J. Clin., 2018, 68(6), 394–424 CrossRef PubMed.
- W. D. Travis, E. Brambilla, M. Noguchi, A. G. Nicholson, K. R. Geisinger, Y. Yatabe, D. G. Beer, C. A. Powell, G. J. Riely and P. E. Van Schil, International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma, J. Thorac. Oncol., 2011, 6(2), 244–285 CrossRef PubMed.
- S. Wu, D. Wu, Y. Pan, H. Liu, Z. Shao and M. Wang, Correlation between EZH2 and CEP55 and lung adenocarcinoma prognosis, Pathol., Res. Pract., 2019, 215(2), 292–301 CrossRef CAS PubMed.
- Y. Wang, J. Piao, Q. Wang, X. Cui, Z. Meng, T. Jin and Z. Lin, Paip1 predicts poor prognosis and promotes tumor progression through AKT/GSK-3β pathway in lung adenocarcinoma, Hum. Pathol., 2019, 86, 233–242 CrossRef CAS PubMed.
- S. Heavey, P. Godwin, A.-M. Baird, M. P. Barr, K. Umezawa, S. Cuffe, S. P. Finn, K. J. O'Byrne and K. Gately, Strategic targeting of the PI3K–NFκB axis in cisplatin-resistant NSCLC, Cancer Biol. Ther., 2014, 15(10), 1367–1377 CrossRef CAS PubMed.
- I. Jun-Ichiro, G. Jin, A. Taishin and S. Kentaro, NF-kappaB activation in development and progression of cancer, Cancer Sci., 2007, 98(3), 268–274 CrossRef PubMed.
- J. He, Y. Hu, M. Hu and B. Li, Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer, Sci. Rep., 2015, 5, 13110 CrossRef CAS PubMed.
- Z. Jiang, Y. Yang, Y. Yang, Y. Zhang, Z. Yue, Z. Pan and X. Ren, Ginsenoside Rg3 attenuates cisplatin resistance in lung cancer by downregulating PD-L1 and resuming immune, Biomed. Pharmacother., 2017, 96, 378–383 CrossRef CAS PubMed.
- Z. Yuan, H. Jiang, X. Zhu, X. Liu and J. Li, Ginsenoside Rg3 promotes cytotoxicity of Paclitaxel through inhibiting NF-κB signaling and regulating Bax/Bcl-2 expression on triple-negative breast cancer, Biomed. Pharmacother., 2017, 89, 227–232 CrossRef CAS PubMed.
- J. H. Kim, M. Kim, S.-M. Yun, S. Lee, J. H. No, D. H. Suh, K. Kim and Y. B. Kim, Ginsenoside Rh2 induces apoptosis and inhibits epithelial-mesenchymal transition in HEC1A and Ishikawa endometrial cancer cells, Biomed. Pharmacother., 2017, 96, 871–876 CrossRef CAS PubMed.
- J.-H. Cho, H.-Y. Chun, J. S. Lee, J.-H. Lee, K. J. Cheong, Y.-S. Jung, T.-G. Woo, M.-H. Yoon, A.-Y. Oh and S.-M. Kang, Prevention effect of rare ginsenosides against stress-hormone induced MTOC amplification, Oncotarget, 2016, 7(23), 35144 CrossRef PubMed.
- S. P. Ponnuraj, F. Siraj, S. Kang, H. Y. Noh, J.-W. Min, Y.-J. Kim and D.-C. Yang, Amelioration of insulin resistance by Rk1+Rg5 complex under endoplasmic reticulum stress conditions, Pharmacogn. Res., 2014, 6(4), 292 CrossRef PubMed.
- Y.-J. Kim, H. C. Kwon, H. Ko, J. H. Park, H. Y. Kim, J.-H. Yoo and H. O. Yang, Anti-tumor activity of the ginsenoside Rk1 in human hepatocellular carcinoma cells through inhibition of telomerase activity and induction of apoptosis, Biol. Pharm. Bull., 2008, 31(5), 826–830 CrossRef CAS PubMed.
- Y. Qian, Z. Ke-Wu, M. Xiao-Li, Y. Jiang, T. Peng-Fei and W. Xue-Mei, Ginsenoside Rk1 suppresses pro-inflammatory responses in lipopolysaccharide-stimulated RAW264. 7 cells by inhibiting the Jak2/Stat3 pathway, Chin. J. Nat. Med., 2017, 15(10), 751–757 Search PubMed.
- H. K. Ju, J. G. Lee, M. K. Park, S.-J. Park, C. H. Lee, J. H. Park and S. W. Kwon, Metabolomic investigation of the anti-platelet aggregation activity of ginsenoside Rk1 reveals attenuated 12-HETE production, J. Proteome Res., 2012, 11(10), 4939–4946 CrossRef CAS PubMed.
- Y. Hong and D. Fan, Ginsenoside Rk1 induces cell cycle arrest and apoptosis in MDA-MB-231 triple negative breast cancer cells, Toxicology, 2019, 418, 22–31 CrossRef CAS PubMed.
- X. Pang, P. Yang, L. Wang, J. Cao, Y. Cheng, D. Sheng, X. Wan, Q. Guo, K. Qian and Q. Zhang, Human serum albumin nanoparticulate system with encapsulation of gefitinib for enhanced anti-tumor effects in non-small cell lung cancer, J. Drug Delivery Sci. Technol., 2019, 52, 997–1007 CrossRef CAS.
- J. Shou, L. You, J. Yao, J. Xie, J. Jing, Z. Jing, L. Jiang, X. Sui, H. Pan and W. Han, Cyclosporine A sensitizes human non-small cell lung cancer cells to gefitinib through inhibition of STAT3, Cancer Lett., 2016, 379(1), 124–133 CrossRef CAS PubMed.
- R. Kong, B. Sun, H. Jiang, S. Pan, H. Chen, S. Wang, G. W. Krissansen and X. Sun, Downregulation of nuclear factor-κB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer, Cancer Lett., 2010, 291(1), 90–98 CrossRef CAS PubMed.
- R. Sordella, D. W. Bell, D. A. Haber and J. Settleman, Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways, Science, 2004, 305(5687), 1163–1167 CrossRef CAS PubMed.
- Y. Hong and D. Fan, Ginsenoside Rk1 induces cell death through ROS-mediated PTEN/PI3K/Akt/mTOR signaling pathway in MCF-7 cells, J. Funct. Foods, 2019, 57, 255–265 CrossRef CAS.
- J.-M. Oh, J. Lee, W.-T. Im and S. Chun, Ginsenoside Rk1 Induces Apoptosis in Neuroblastoma Cells through Loss of Mitochondrial Membrane Potential and Activation of Caspases, Int. J. Mol. Sci., 2019, 20(5), 1213 CrossRef CAS PubMed.
- C. Bunchorntavakul and K. R. Reddy, Drug Hepatotoxicity: Newer Agents, Clin Liver Dis., 2017, 21(1), 115–134 CrossRef PubMed.
- S. Diaz-Moralli, M. Tarrado-Castellarnau, A. Miranda and M. Cascante, Targeting cell cycle regulation in cancer therapy, Pharmacol. Ther., 2013, 138(2), 255–271 CrossRef CAS PubMed.
- M. Zhang and H. Yang, Negative growth regulators of the cell cycle machinery and cancer, Pathophysiology, 2009, 16(4), 305–309 CrossRef CAS PubMed.
- Y. Zhang, S. Li, H. He, Q. Han, B. Wang and Y. Zhu, Influence of Tanshinone IIA on apoptosis of human esophageal carcinoma Eca-109 cells and its molecular mechanism, Thorac. Cancer, 2017, 8(4), 296–303 CrossRef CAS PubMed.
- G.-Z. Zhou, A.-F. Li, Y.-H. Sun and G.-C. Sun, A novel synthetic curcumin derivative MHMM-41 induces ROS-mediated apoptosis and migration blocking of human lung cancer cells A549, Biomed. Pharmacother., 2018, 103, 391–398 CrossRef CAS PubMed.
- R. A. Weinberg, Tumor suppressor genes, Science, 1991, 254(5035), 1138–1146 CrossRef CAS PubMed.
- S.-J. Kim and A. K. Kim, Anti-breast cancer activity of Fine Black ginseng (Panax ginseng Meyer) and ginsenoside Rg5, J. Ginseng Res., 2015, 39(2), 125–134 CrossRef CAS PubMed.
- N. Gopalakrishnan, M. Saravanakumar, P. Madankumar, M. Thiyagu and H. Devaraj, Colocalization of β-catenin with Notch intracellular domain in colon cancer: a possible role of Notch1 signaling in activation of CyclinD1-mediated cell proliferation, Mol. Cell. Biochem., 2014, 396(1–2), 281–293 CrossRef CAS PubMed.
- S. Wang, L. Fu, Y. Wu, H. Xiao, J. Wang and G. Sun, Influence of luteolin on the apoptosis of esophageal cancer Eca109 cells and its mechanism of action, Food Sci. Human Well., 2019, 415, 73–85 CrossRef PubMed.
- B.-h. Zhou, P.-p. Tan, L.-s. Jia, W.-p. Zhao, J.-c. Wang and H.-w. Wang, PI3K/AKT signaling pathway involvement in fluoride-induced apoptosis in C2C12 cells, Chemosphere, 2018, 199, 297–302 CrossRef CAS PubMed.
- B. G. Eimani, M. H. Sanati, M. Houshmand, M. Ataei, F. Akbarian and N. Shakhssalim, Expression and prognostic significance of bcl-2 and bax in the progression and clinical outcome of transitional bladder cell carcinoma, Cell J., 2014, 15(4), 356 CAS.
- M. Brentnall, L. Rodriguez-Menocal, R. L. De Guevara, E. Cepero and L. H. Boise, Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis, BMC Cell Biol., 2013, 14(1), 32 CrossRef CAS PubMed.
- X. Han, C.-F. Liu, N. Gao, J. Zhao and J. Xu, Kaempferol suppresses proliferation but increases apoptosis and autophagy by up-regulating microRNA-340 in human lung cancer cells, Biomed. Pharmacother., 2018, 108, 809–816 CrossRef CAS PubMed.
- M. Karin, Y. Yamamoto and Q. M. Wang, The IKK NF-κB system: a treasure trove for drug development, Nat. Rev. Drug Discovery, 2004, 3(1), 17 CrossRef CAS PubMed.
- J. Wang, L. Tian, M. N. Khan, L. Zhang, Q. Chen, Y. Zhao, Q. Yan, L. Fu and J. Liu, Ginsenoside Rg3 sensitizes hypoxic lung cancer cells to cisplatin via blocking of NF-κB mediated epithelial–mesenchymal transition and stemness, Cancer Lett., 2018, 415, 73–85 CrossRef CAS PubMed.
- D. K. Biswas, Q. Shi, S. Baily, I. Strickland, S. Ghosh, A. B. Pardee and J. D. Iglehart, NF-κB activation in human breast cancer specimens and its role in cell proliferation and apoptosis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(27), 10137–10142 CrossRef CAS PubMed.
- D.-G. Wu, P. Yu, J.-W. Li, P. Jiang, J. Sun, H.-Z. Wang, L.-D. Zhang, M.-B. Wen and P. Bie, Apigenin potentiates the growth inhibitory effects by IKK-β-mediated NF-κB activation in pancreatic cancer cells, Toxicol. Lett., 2014, 224(1), 157–164 CrossRef CAS PubMed.
- M. Patel, P. G. Horgan, D. C. McMillan and J. Edwards, NF-κB pathways in the development and progression of colorectal cancer, Transl. Res., 2018, 197, 43–56 CrossRef CAS PubMed.
- J.-N. Dai, Y. Zong, L.-M. Zhong, Y.-M. Li, W. Zhang, L.-G. Bian, Q.-L. Ai, Y.-D. Liu, J. Sun and D. Lu, Gastrodin inhibits expression of inducible NO synthase, cyclooxygenase-2 and proinflammatory cytokines in cultured LPS-stimulated microglia via MAPK pathways, PLoS One, 2011, 6(7), e21891 CrossRef CAS PubMed.
- J.-L. Luo, W. Tan, J. M. Ricono, O. Korchynskyi, M. Zhang, S. L. Gonias, D. A. Cheresh and M. Karin, Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing Maspin, Nature, 2007, 446(7136), 690 CrossRef CAS PubMed.
- W. Li, Z. Chen, Y. Zong, F. Gong, Y. Zhu, Y. Zhu, J. Lv, J. Zhang, L. Xie and Y. Sun, PP2A inhibitors induce apoptosis in pancreatic cancer cell line PANC-1 through persistent phosphorylation of IKKα and sustained activation of the NF-κB pathway, Cancer Lett., 2011, 304(2), 117–127 CrossRef CAS PubMed.
- Y. Zhao, X.-x. Wang, W. Wu, H. Long, J. Huang, Z. Wang, T. Li, S. Tang, B. Zhu and D. Chen, EZH2 regulates PD-L1 expression via HIF-1α in non-small cell lung cancer cells, Biochem. Biophys. Res. Commun., 2019, 517(2), 201–209 CrossRef CAS PubMed.
- J. Xue, L. Li, N. Li, F. Li, X. Qin, T. Li and M. Liu, Metformin suppresses cancer cell growth in endometrial carcinoma by inhibiting PD-L1, Eur. J. Pharmacol., 2019, 859, 172541 CrossRef CAS PubMed.
- K. Gowrishankar, D. Gunatilake, S. J. Gallagher, J. Tiffen, H. Rizos and P. Hersey, Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-κB, PLoS One, 2015, 10(4), e0123410 CrossRef PubMed.
- A. Ribas and J. D. Wolchok, Cancer immunotherapy using checkpoint blockade, Science, 2018, 359(6382), 1350–1355 CrossRef CAS PubMed.
- A. Constantinidou, C. Alifieris and D. T. Trafalis, Targeting programmed cell death-1 (PD-1) and ligand (PD-L1): a new era in cancer active immunotherapy, Pharmacol. Ther., 2018, 194, 84–106 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo02166c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |