
Efficient hydrodeoxygenation of sulfoxides into sulfides under mild conditions using heterogeneous cobalt–molybdenum catalysts†
Kaiyue
Yao‡
,
Ziliang
Yuan‡
,
Shiwei
Jin
*,
Quan
Chi
,
Bing
Liu
 *,
Renjie
Huang
and
Zehui
Zhang
*,
Renjie
Huang
and
Zehui
Zhang
Key Laboratory of Catalysis and Materials Sciences of Hubei, South-Central University for Nationalities, Wuhan, 430074, China. E-mail: liubing@mail.scuec.edu.cn; jinsw@mail.scuec.edu.cn; Fax: +86-27-67842572; Tel: +86-27-67842572
First published on 20th November 2019
Abstract
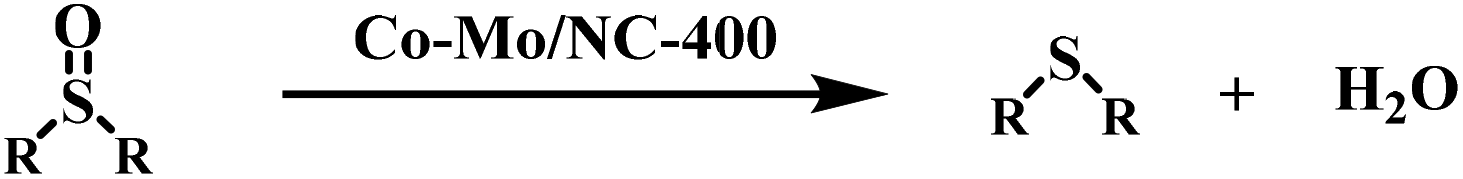
Nitrogen-doped carbon-supported cobalt–molybdenum bimetallic catalysts (abbreviated as Co–Mo/NC) are active for the hydrodeoxygenation of sulfoxides to sulfides under mild conditions (25–80 °C and 10 bar H2), which represents the first example of the use of heterogeneous non-noble metal catalysts for this transformation. MoO3 with Lewis acid sites assists the hydrodeoxygenation of sulfoxides into sulfides by hydrogen over cobalt nanoparticles.
The selective hydrodeoxygenation of sulfoxides into sulfides is one of the most important organic transformations in organic chemistry,1 which has attracted great interest in the fine chemical industry as well as in biochemistry. For example, sulfoxides are now widely utilized as chirons in numerous asymmetric transformations wherein the sulfoxide groups, after stereoselective introduction, are removed by deoxygenation to their corresponding sulfides followed by hydrodesulfurization.2 In the early days, the hydrodeoxygenation of sulfoxides was performed by the use of excess amounts of sacrificial agents such as metal hydrides and zinc metal,3–5 which produce large quantities of waste. There have also been some reports on metal-free deoxygenation of sulfoxides with halides, such as iodides in the presence of acids.6,7 However, these methods produce halide by-products, which are not environmentally friendly.
Currently, catalytic hydrodeoxygenation represents an environmentally friendly method. Compared with other reducing agents such as alcohols8 and silanes,9 hydrogen is a clean and environmentally friendly reducing agent because of high atom-efficiency with only the release of water. Homogeneous catalysts were earlier utilized for the hydrodeoxygenation of dimethyl sulfoxide,10,11 but it is difficult to recycle homogeneous catalysts and there is the purification of the products to be considered. Obviously, the use of heterogeneous catalysts can address the drawbacks associated with homogeneous catalytic systems. However, the deoxygenation of sulfoxides with H2 is unprecedented over heterogeneous catalysts.12 In 2014, Mitsudome and co-workers made a great contribution with the hydrodeoxygenation of sulfoxide over a Ru/TiO2 catalyst, which was reported to be active under atmospheric H2 pressure.13 Later, heterogeneous platinum catalysts (Pt–MoOX/TiO2 and Pt/V0.7Cr0.3OX) were developed for the hydrodeoxygenation of sulfoxides, which was performed at 50–150 °C under hydrogen pressure (1–7 bar).14,15 Although heterogeneous noble metal catalysts are effective for this transformation, the use of heterogeneous non-noble metal catalysts is highly attractive because of the lower cost.
To the best of our knowledge, no heterogeneous non-noble metal catalysts have been reported for this transformation. In recent years, nitrogen-doped carbon material-supported non-noble metal catalysts have received a great deal of attention in the replacement of noble metal catalysts. Our group recently discovered that nitrogen-doped carbon material-supported cobalt catalysts demonstrate excellent activity in hydrogenation and oxidation reactions.16,17 Therefore, we also tried to use nitrogen-doped carbon-supported cobalt catalysts for the hydrodeoxygenation of sulfoxides, but failed. Inspired by the fact that some metals such as Re, Mo and W are oxyphilic elements, nitrogen-doped carbon material-supported Co–Mo bimetallic catalysts were constructed, and found to be active for the selective hydrodeoxygenation of sulfoxides.
ZIF-67 as one of the most investigated metal–organic frameworks was used for the preparation of Co–Mo/NC-T catalysts, where T represents the reduction temperature. In order to introduce the oxyphilic element molybdenum, (NH4)6Mo7O24·4H2O was introduced into ZIF-67 via a double-solvent method, and the mixture was subjected to pyrolysis at different temperatures (see the Experimental details in the ESI†). Powder X-ray diffraction (PXRD) was performed to determine the crystal structure of the Co–Mo/NC-T catalysts. As shown in Fig. 1, three characteristic peaks at 2θ = 44.2°, 51.5° and 75.9° corresponding to metallic Co (111), (200), (220) reflections were observed for the Co–Mo/NC-T catalysts (PDF 15-0806).18 The average size of Co nanoparticles was estimated to be 9.8, 28.5 and 33.9 nm from the line broadening of the Co (111) reflection for the Co–Mo/NC-400, Co–Mo/NC-600 and Co–Mo/NC-800 catalysts, respectively. There were no XRD peaks of molybdenum species for the Co–Mo/NC-400 and Co–Mo/NC-600 catalysts, while characteristic peaks at 2θ = 34.5°, 38.1° and 39.5° were observed in the PXRD pattern of the Co–Mo/NC-800 catalyst, which were assigned to the (021), (200), and (121) reflections of Mo2C.19 According to the reported results,19 (NH4)6Mo7O24·4H2O was gradually changed into MoO3 from 300 °C to 500 °C and MoO2 from 500 °C to 700 °C, and then to Mo2C beyond 775 °C. No PXRD peaks of MoO3 or MoO2 were observed for the Co–Mo/NC-400 and Co–Mo/NC-600 catalysts, which was possibly due to the fact that MoO3 or MoO2 did not form crystal structures or the size of them was too small to be visible.
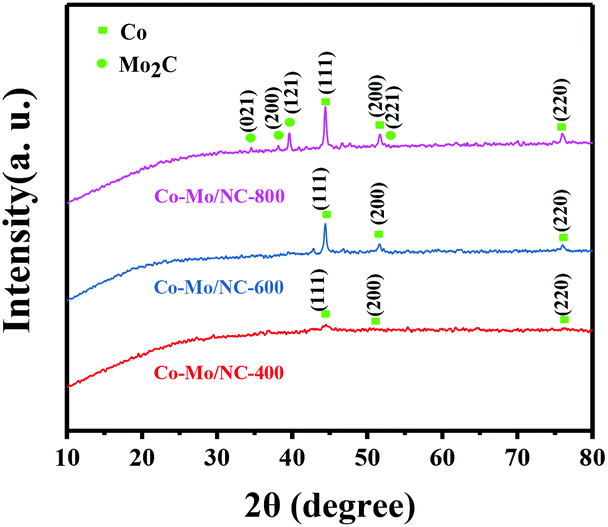 | ||
| Fig. 1 PXRD patterns of the Co–Mo/NC-T catalysts. | ||
As shown in Fig. S1,† two peaks at around 1347 and 1594 cm−1 were observed in the Raman spectra of the Co–Mo/NC-T catalysts, corresponding to the D and G bands, respectively. The G band represents the graphitic structure of the catalysts, while the D band can be attributed to the lattice defects.20 The intensity ratios of the D and G bands (ID/IG) were calculated to be 1.005, 0.979 and 0.904 for the Co–Mo/NC-400, Co–Mo/NC-600 and Co–Mo/NC-800 catalysts, and the decrease in the number of defects may result from the decrease in the number of nitrogen atoms in the Co–Mo/NC-T catalysts as a result of the increase in the reduction temperature.21
A transmission electron microscope (TEM) image (Fig. 2a) revealed that the Co nanoparticles were homogeneously dispersed on the surface of the Co–Mo/NC-400 catalyst with an average size of 10.1 nm in agreement with the XRD results. However, the growth of cobalt nanoparticles into larger particle size as well as aggregation were observed in the TEM images of the Co–Mo/NC-600 and Co–Mo/NC-800 catalysts. As shown in high-resolution TEM (HR-TEM) images (Fig. S2 and S3†), the lattice spacing measured for the crystalline plane was 0.20 nm (Fig. 2b), which corresponded to the d values of the (111) planes for metallic cobalt nanoparticles, while no lattices for the Mo species were found in the HR-TEM image of the Co–Mo/NC-400 catalyst.
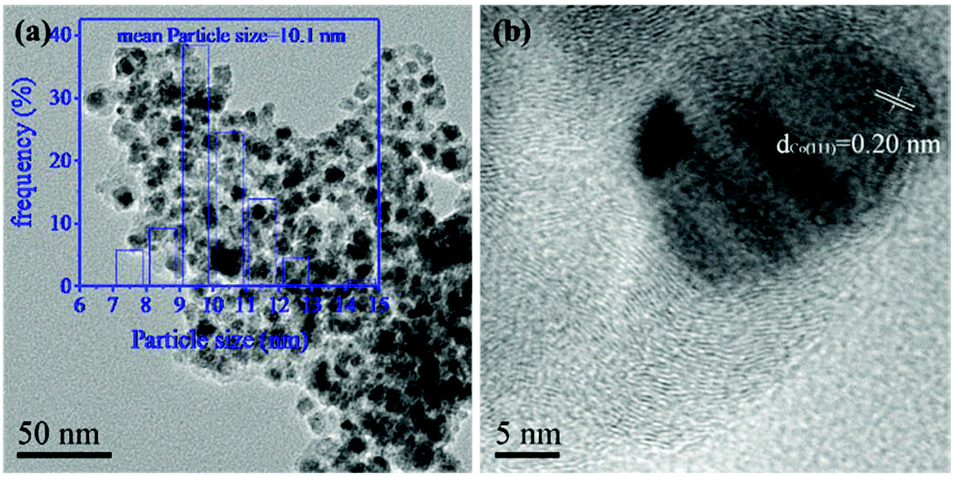 | ||
| Fig. 2 TEM image (a) and HR-TEM image (b) of the Co–Mo/NC-400 catalyst. | ||
X-ray photoelectron spectroscopy (XPS) was further used to study the valence states of the Co and Mo species in the Co–Mo/NC-T catalysts. Two peaks with binding energies at 781.4 and 796.6 eV were assigned to the Co 2p3/2 and Co 2p1/2 of Co2+, while two peaks with binding energies at 779.5 eV and 795.0 eV were attributed to the Co 2p3/2 and Co 2p1/2 of Co3+. Two peaks with binding energies at 785.7 and 802.4 eV corresponded to the Co2+ LMM auger peak in Co3O4.22 The presence of Co3O4 in the Co–Mo/NC-400 catalyst was due to the oxidation of surface metallic cobalt nanoparticles during storage in the air. Similarly, the surface of cobalt nanoparticles was also present in the oxidation states for Co–Mo/NC-600 and Co–Mo/NC-800 catalysts (Fig. S4†). The Mo 3d5/2 XPS peak could be deconvoluted into two peaks with binding energies of 231.6 and 232.2 eV, which are attributed to the Mo5+ and Mo6+ oxidation states for the Co–Mo/NC-400 catalyst (Fig. 3), respectively,23 and Mo6+ was the main oxidation state, corresponding to MoO3. Peaks with binding energies at 231.1, 232.4 and 227.6 eV were observed for the Mo 3d5/2 XPS peak for the Co–Mo/NC-600 catalyst, corresponding to the Mo5+, Mo6+ oxidation states and metallic Mo (Fig. S5†).23 Two peaks with binding energies at 232.4 and 228.6 eV were the characteristic Mo 3d5/2 XPS peaks of Mo6+ and Mo2C of the Co–Mo/NC-800 catalyst (Fig. S5†), and Mo2C was the main state in the Co–Mo/NC-800 catalyst, consistent with the PXRD results.
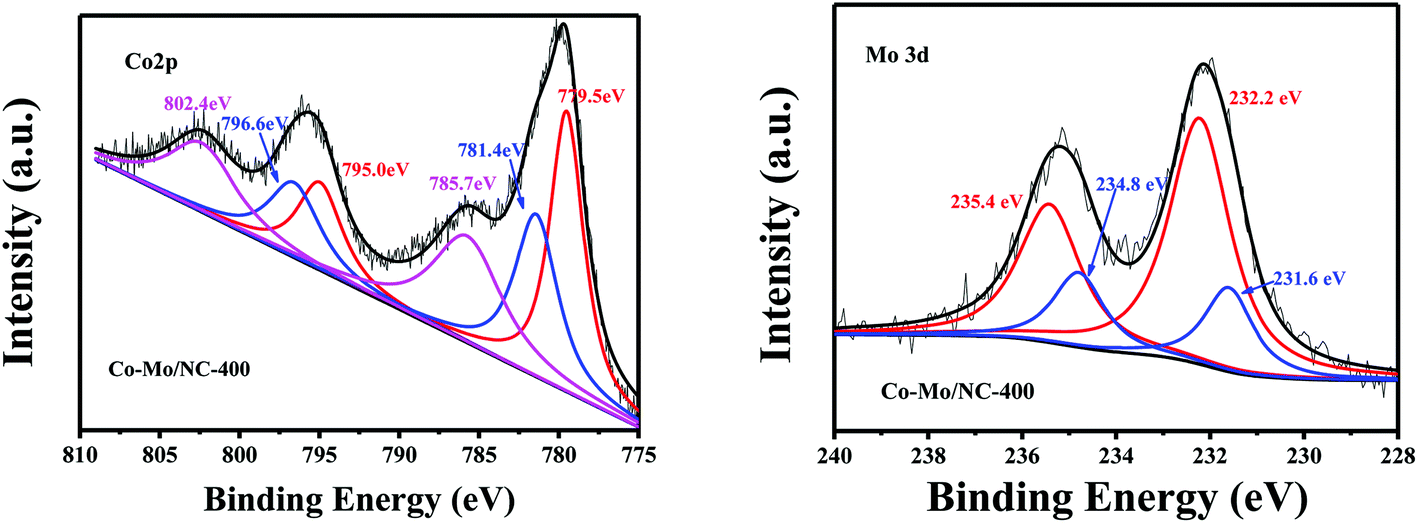 | ||
| Fig. 3 XPS spectra of Co 2p and Mo 3d in the Co–Mo/NC-400 catalyst. | ||
The hydrodeoxygenation of diphenyl sulfoxide into diphenyl sulfide was used as a model reaction to study the catalytic activity of the Co–Mo/NC-T catalysts (Table 1). Initially, the reaction was conducted in the presence of a Co/NC-400 catalyst, which was prepared by the same method as described for the Co–Mo/NC-400 catalyst without the introduction of Mo. No conversion of diphenyl sulfoxide was observed (Table 1, entry 1). In order to cleave the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, Mo as a common oxyphilic element was introduced into the nitrogen-doped carbon-supported Co catalysts to activate the SO bond. As expected, the Co–Mo/NC-400 catalyst could smoothly promote the hydrodeoxygenation of diphenyl sulfoxide into diphenyl sulfide, affording a conversion of 77% after 2 h at 80 °C under 10 bar H2 (Table 1, entry 2). However, the conversion of diphenyl sulfoxide sharply decreased to 24% over the Co–Mo/NC-600 catalyst (Table 1, entry 3), and no conversion was observed over the Co–Mo/NC-800 catalyst (Table 1, entry 4). The decrease in the catalytic activity of the Co–Mo/NC-T catalysts with an increase in the reduction temperature could be mainly due to the following two reasons. On the one hand, cobalt nanoparticles became larger and aggregated with an increase of the reduction temperature, which provided fewer active sites for the activation of H2. On the other hand, the Mo species in the Co–Mo/NC-T catalysts also played a crucial role in the hydrodeoxygenation of diphenyl sulfoxide. As characterized by XPS, MoO3 was the major component for the Co–Mo/NC-400 catalyst, while MoO2 was the major component for the Co–Mo/NC-600 catalyst. MoO3 with abundant oxygen vacancy sites (also called Lewis acid sites) was reported to be effective in combining with oxygen atoms in the SO bond,24 thus facilitating the hydrodeoxygenation of diphenyl sulfoxide. This was further confirmed that in the Co–Mo/NC-800 catalyst was completely inactive towards the hydrodeoxygenation of diphenyl sulfoxide, in which Mo species was Mo2C. A Co–V/NC-400 catalyst was also prepared with a similar method as for the Co–Mo/NV-400 catalyst, but it showed no catalytic activity for the hydrodeoxygenation of sulfoxides (Table 1, entry 6). These results suggested that vanadium oxides could not activate the substrates. We prepared an active carbon-supported Co–Mo catalyst by a similar method, and used it in the hydrodeoxygenation of sulfoxide. However, it showed no activity under the same reaction temperature of 80 °C (Table 1, entry 7). These results suggested that the nitrogen atoms in the carbon materials played important roles in the hydrodeoxygenation of sulfoxides. According to previous reports,17,26 nitrogen atoms might play the following roles in the catalyst activity: (a) nitrogen atoms act as Lewis base sites to promote the heterolytic cleavage of H2 in coordination with the Co nanoparticles; (b) the nitrogen atoms should have a strong interaction with Co nanoparticles to improve the catalytic activity of Co nanoparticles and to stabilize them.
O bond, Mo as a common oxyphilic element was introduced into the nitrogen-doped carbon-supported Co catalysts to activate the SO bond. As expected, the Co–Mo/NC-400 catalyst could smoothly promote the hydrodeoxygenation of diphenyl sulfoxide into diphenyl sulfide, affording a conversion of 77% after 2 h at 80 °C under 10 bar H2 (Table 1, entry 2). However, the conversion of diphenyl sulfoxide sharply decreased to 24% over the Co–Mo/NC-600 catalyst (Table 1, entry 3), and no conversion was observed over the Co–Mo/NC-800 catalyst (Table 1, entry 4). The decrease in the catalytic activity of the Co–Mo/NC-T catalysts with an increase in the reduction temperature could be mainly due to the following two reasons. On the one hand, cobalt nanoparticles became larger and aggregated with an increase of the reduction temperature, which provided fewer active sites for the activation of H2. On the other hand, the Mo species in the Co–Mo/NC-T catalysts also played a crucial role in the hydrodeoxygenation of diphenyl sulfoxide. As characterized by XPS, MoO3 was the major component for the Co–Mo/NC-400 catalyst, while MoO2 was the major component for the Co–Mo/NC-600 catalyst. MoO3 with abundant oxygen vacancy sites (also called Lewis acid sites) was reported to be effective in combining with oxygen atoms in the SO bond,24 thus facilitating the hydrodeoxygenation of diphenyl sulfoxide. This was further confirmed that in the Co–Mo/NC-800 catalyst was completely inactive towards the hydrodeoxygenation of diphenyl sulfoxide, in which Mo species was Mo2C. A Co–V/NC-400 catalyst was also prepared with a similar method as for the Co–Mo/NV-400 catalyst, but it showed no catalytic activity for the hydrodeoxygenation of sulfoxides (Table 1, entry 6). These results suggested that vanadium oxides could not activate the substrates. We prepared an active carbon-supported Co–Mo catalyst by a similar method, and used it in the hydrodeoxygenation of sulfoxide. However, it showed no activity under the same reaction temperature of 80 °C (Table 1, entry 7). These results suggested that the nitrogen atoms in the carbon materials played important roles in the hydrodeoxygenation of sulfoxides. According to previous reports,17,26 nitrogen atoms might play the following roles in the catalyst activity: (a) nitrogen atoms act as Lewis base sites to promote the heterolytic cleavage of H2 in coordination with the Co nanoparticles; (b) the nitrogen atoms should have a strong interaction with Co nanoparticles to improve the catalytic activity of Co nanoparticles and to stabilize them.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Conversion (%) | Sel. (%) |
| a Reaction conditions: Diphenyl sulfoxide (0.5 mmol), 1,4-dioxane (10 mL), H2 (10 bar), catalysts (20 mg), 80 °C. | ||||
| 1 | Co/NC-400 | 2 | 0 | — |
| 2 | Co–Mo/NC-400 | 2 | 77 | 100 |
| 3 | Co–Mo/NC-600 | 2 | 24 | 100 |
| 4 | Co–Mo/NC-800 | 2 | 0 | — |
| 5 | MoO3 | 2 | 0 | — |
| 6 | Co–V/NC-400 | 2 | 0 | — |
| 7 | Co–Mo/AC-400 | 2 | 0 | — |

According to the above results, we can conclude that metallic Co nanoparticles, nitrogen atoms and MoO3 played a synergistic role in the hydrodeoxygenation of diphenyl sulfoxide. MoO3 would activate the SO groups by combination with the oxygen atom in the SO bond. Metallic Co nanoparticles activated hydrogen molecules with the assistance of nitrogen atoms to generate the Co–H− hybrid and N–H+ species. The H+ in N–H+ was transferred to the oxygen atoms in SO bonds to generate –S–OH species, and the sulfur–oxygen bond was then cleaved by H− to give the target product.
The reaction conditions were then optimized over the Co–Mo/NC-400 catalyst. Firstly, the effect of the reaction solvents was studied. Generally, protic solvents with strong polarity as well as water gave high to quantitative conversions of diphenyl sulfoxide (Table S1, entries 1 vs. 4†). Particularly, it is interesting to note that the conversion of diphenyl sulfoxide in alcohols slightly increased in the order iso-PrOH, ethanol, and methanol. Moderate conversions of diphenyl sulfoxide were produced in N,N-dimethylformamide (DMF) and three other kinds of solvents with moderate polarity (Table S1, entries 5 vs. 8†). The reaction in hexane with non-polarity produced the lowest conversion of diphenyl sulfoxide of 45%. The excellent catalytic performance of the Co–Mo/NC-400 catalyst in water and alcohols could be for the following two reasons. On the one hand, the alcohol solvents and water should have the ability to activate the substrates or the intermediates via hydrogen-bonding interactions, similar to the hydrogenation of carbonyl groups in ethanol.25 On the other hand, solvents with strong polarity resulted in a high dispersion of diphenyl sulfoxide and the Co–Mo/NC-400 catalyst, as both of them were hydrophilic. The best results were obtained in methanol, which produced diphenyl sulfide with a 100% yield at 80 °C and 10 bar H2. In addition, the catalyst was active even at 25 °C and 10 bar, but it took 60 h to get a high yield (Table S1, entry 10†).
The excellent catalytic performance of the Co–Mo/NC-400 catalyst at 80 °C inspired us to carry out the deoxygenation of diphenyl sulfoxide under milder conditions. At a low temperature of 40 °C, it only produced 5% conversion of diphenyl sulfoxide after 2 h under 10 bar H2 (Fig. S6†). Then the conversion greatly increased with an increase in the reaction temperature from 50 to 80 °C. A quantitative conversion of diphenyl sulfoxide was observed at 80 °C after 2 h. Then the effect of hydrogen pressure on the hydrodeoxygenation of diphenyl sulfoxide was studied over the Co–Mo/NC-400 catalyst at 50 °C. As shown in Fig. S7,† the conversion of diphenyl sulfoxide increased with an increase of hydrogen pressure, especially from 1 to 10 bar. For example, the conversion of diphenyl sulfoxide greatly increased from 5% at 1 bar to 26% at 10 bar, and then slowly increased to 33% at 20 bar. The increase in hydrogen pressure resulted in an increase in the hydrogen concentration in the reaction solution, resulting in an increase in the reaction rate. Fig. S8† depicts the time course of the product distribution of the hydrodeoxygenation of diphenyl sulfoxide over the Co–Mo/NC-400 catalyst at 50 °C and 10 bar H2. The percentage of unreacted diphenyl sulfoxide gradually decreased during the reaction process, and the yield of diphenyl sulfide gradually increased on prolonging the reaction time. After 10 h, a full conversion of diphenyl sulfoxide was obtained at 50 °C and 10 bar H2 with a 100% selectivity of diphenyl sulfide. It is worth noting that the Co–Mo/NC-400 catalyst demonstrated comparable or even superior activity to the reported noble metal catalysts.14 For example, a long reaction time of 24 h was required to complete the conversion of diphenyl sulfoxide over a Pt–MoOX/TiO2 catalyst at 50 °C under 7 bar H2,14 while the reaction time to get full conversion of diphenyl sulfoxide was only 12 h over the Co–Mo/NC-400 catalyst at 50 °C under 10 bar H2.
Then, the scope of the as-prepared Co–Mo/NC-400 catalyst towards the hydrodeoxygenation of various sulfoxides was investigated. A number of aromatic, benzylic and aliphatic sulfoxides were smoothly converted into the corresponding sulfides with excellent yields (Table 2, entries 2–6). Compared with diphenyl sulfoxide, substituted diphenyl sulfoxides with large steric hindrance were less active, which required a longer reaction time to achieve high conversions (Table 2, entries 1–4). Meanwhile, a substituted diphenyl sulfoxide with electron-withdrawing groups showed lower activity than counterparts (Table 2, entry 2 vs. entries 3 and 4). Phenylalkyl sulfoxides demonstrated similar activity to diphenyl sulfoxides (Table 2, entry 2 vs. entries 3 and 4), which have small steric hindrance but low electronic density of the sulfoxide groups. Dialkyl sulfoxides were also successfully converted into the corresponding alkyl sulfides, which were performed at high reaction temperature of 80 °C, due to the low activity of the dialkyl sulfoxides with low electron density of the sulfoxide groups. Notably, the Co–Mo/NC-400 catalyst was effective for the selective dehydrogenation of sulfoxide substrates without affecting other reducible functional groups such as CC double bonds, Cl, and Br.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Temperature (°C) | Time (h) | Yield (%) |
| a Reaction conditions: Substrates (0.5 mmol), methanol (10 mL), H2 (10 bar), Co–Mo/NC-400 catalyst (20 mg). | ||||
| 1 |
|
50 | 10 | 100 |
| 2 |
|
50 | 15 | 100 |
| 3 |
|
50 | 18 | 100 |
| 4 |
|
50 | 20 | 100 |
| 5 |
|
50 | 24 | 100 |
| 6 |
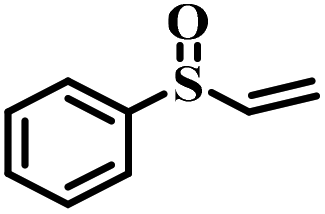
|
50 | 22 | 100 |
| 7 |
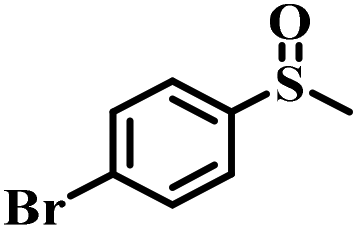
|
50 | 26 | 100 |
| 8 |
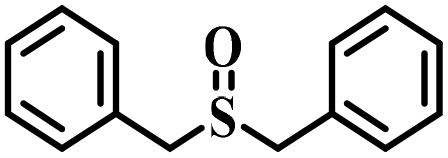
|
80 | 25 | 94 |
| 9 |
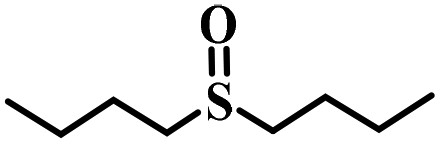
|
80 | 15 | 98 |
| 10 |
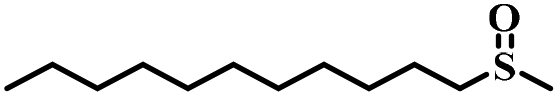
|
80 | 18 | 85 |
Finally, recycling experiments of the Co–Mo/NC-400 catalyst were conducted. As shown in Fig. S9,† the catalyst showed similar conversions of diphenyl sulfoxide of around 58% during 6 runs, and the selectivity of diphenyl sulfide remained at 100%. These results suggested that the Co–Mo/NC-400 catalyst showed a high stability. ICP-AES analysis of the solution after the first reaction showed that the Co and Mo contents in the solution were below the detection limits.
In conclusion, we have developed a new method for the use of heterogeneous non-noble metal catalysts for the catalytic hydrogenation of sulfoxides for the first time. Notably, the as-prepared Co–Mo/NC-400 catalyst exhibited an unprecedented activity under mild reaction conditions (25–80 °C and 10 bar H2) and greatly broadens the applicability to a wide range of sulfoxides. Control experiments demonstrate that metallic Co and Mo in the Co–Mo/NC-400 catalyst played a synergistic role in the hydrogenation of sulfoxides with H2. The activity of the as-prepared Co–Mo/NC-400 catalyst was comparable to or even higher than that of reported noble metal catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (no. 21203252) and Basic Scientific Research of Central Colleges, South-Central University for Nationalities (CZY 19005).Notes and references
- S. Sharma, D. Bhattacherjee and P. Das, Adv. Synth. Catal., 2018, 360, 2131–2137 CrossRef CAS
.
- R. Hille, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS PubMed
- D. J. Harrison, N. C. Tam, C. M. Vogels, R. F. Langler, R. T. Baker, A. Decken and S. A. Westcott, Tetrahedron Lett., 2004, 45, 8493–8496 CrossRef CAS
- J. Zhang, X. Gao, C. Zhang, J. Luan and D. Zhao, Synth. Commun., 2010, 40, 1794–1801 CrossRef CAS
- N. Iranpoor, H. Firouzabadi and H. R. Shaterian, J. Org. Chem., 2002, 67, 2826–2830 CrossRef CAS PubMed
- M. Madesclaire, Tetrahedron Lett., 1988, 44, 6537–6580 CrossRef CAS
- H. Firouzabadi and A. Jamalian, J. Sulfur Chem., 2008, 29, 53–97 CrossRef CAS
- Y. Mikami, A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. – Eur. J., 2011, 17, 1768–1772 CrossRef CAS PubMed
- Y. Takahashi, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Lett., 2014, 43, 420–422 CrossRef CAS
- S. C. A. Sousa, J. R. Bernardo, M. Wolff, B. Machura and A. C. Fernandes, Eur. J. Org. Chem., 2014, 1855–1859 CrossRef CAS
- M. Bagherzadeh and S. Ghazali-Esfahani, New J. Chem., 2012, 36, 971–976 RSC
- K. Ogura, M. Yamashita and G. Tsuchihashi, Synthesis, 1975, 6, 385–387 CrossRef
- T. Mitsudome, Y. Takahashi, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2014, 53, 8348–8351 CrossRef CAS
- A. S. Touchy, S. M. A. H. Siddiki, W. Onodera, K. Kon and K. Shimizu, Green Chem., 2016, 18, 2554–2560 RSC
- T. Uematsu, Y. Ogasawara, K. Suzuki, K. Yamaguchi and N. Mizuno, Catal. Sci. Technol., 2017, 7, 1912–1920 RSC
- S. Xu, P. Zhou, Z. H. Zhang, C. J. Yang, B. G. Zhang, K. J. Deng, S. Bottle and H. Y. Zhu, J. Am. Chem. Soc., 2017, 139, 14775–14782 CrossRef CAS PubMed
- P. Zhou, L. Jiang, F. Wang, K. J. Deng, K. L. Lv and Z. H. Zhang, Sci. Adv., 2017, 3, e1601945 CrossRef PubMed
- X. Dai, Z. Li, Y. Ma, M. Liu, K. Du, H. Su, H. Zhuo, L. Yu, H. Sun and X. Zhang, ACS. Appl. Mater. Interfaces, 2016, 8, 6439–6448 CrossRef CAS PubMed
- R. Ma, Y. Zhou, Y. Chen, P. Li and Q. Liu, Angew. Chem., Int. Ed., 2015, 54, 14723–14727 CrossRef CAS
- C. B. Lu, D. Tranca, J. Zhang, F. Rodríguez Hernández, Y. Z. Su, X. D. Zhuang, F. Zhang, G. Seifert and X. L. Feng, ACS Nano, 2017, 11, 3933–3942 CrossRef CAS
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS
- P. Zhou, C. L. Yu, L. Jiang, K. L. Lv and Z. H. Zhang, J. Catal., 2017, 352, 264–273 CrossRef CAS
- L. Lin, Z. K. Yang, Y. F. Jiang and A. W. Xu, ACS Catal., 2016, 6, 4449–4454 CrossRef CAS
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC
- R. Ouyang and D. E. Jiang, ACS Catal., 2015, 5, 6624–6629 CrossRef CAS
- S. G. Wang, P. Zhou, L. Jiang, Z. H. Zhang, K. J. Deng, Y. H. Zhang, Y. X. Zhao, J. L. Li, S. Bottle and H. Y. Zhu, J. Catal., 2018, 368, 207–216 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc02465d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |