
Selective electrochemical CO2 conversion to multicarbon alcohols on highly efficient N-doped porous carbon-supported Cu catalysts†
Hyunsu
Han
a,
Yuseong
Noh
a,
Yoongon
Kim
a,
Seongmin
Park
a,
Wongeun
Yoon
a,
Daehee
Jang
a,
Sung Mook
Choi
b and
Won Bae
Kim
 *a
*a
aDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: kimwb@postech.ac.kr; Fax: +82-54-279-5528; Tel: +82-54-279-2397
bMaterials Center for Energy Department, Surface Technology Division, Korea Institute of Materials Science, Changwon, 642831, Republic of Korea
First published on 7th October 2019
Abstract
The selective and efficient electrocatalytic transformation of carbon dioxide (CO2) to multicarbon alcohols (e.g., C2H5OH and C3H7OH) is a challenge in renewable and sustainable energy research. Herein, a series of hybrid catalysts consisting of Cu nanoparticles supported on N-doped porous carbon (Cu/NPC) were prepared. It was demonstrated that the selectivity for C2H5OH or C3H7OH could be tuned by introducing N-doped porous carbon materials as cocatalysts with different pyridinic N contents, which could in situ produce a reactive CO intermediate from CO2. By varying the pyridinic N content, highly selective production of multicarbon alcohols was achieved using the Cu/NPC hybrid catalysts with a high faradaic efficiency for one pot production of multicarbon alcohols up to 73.3% at −1.05 V (vs. RHE). The faradaic efficiency for C2H5OH and C3H7OH was 64.6% and 8.7%, respectively. The pyridinic N species were likely the CO-producing sites and together with Cu catalytic sites acted cooperatively to produce C2H5OH and C3H7OH via a two-site mechanism for efficient CO2 reduction to multicarbon alcohols. These findings provide novel guidance for the rational design of electrocatalysts and for tuning the catalytic activity and selectivity for multicarbon alcohol production from CO2.
1. Introduction
The growing use of fossil fuels for energy leads to the accumulation of CO2, a major greenhouse gas, which causes serious climate problems that may eventually harm human society.1,2 As a potential remedy, CO2 can be electrochemically converted to value-added products using renewable electricity, enabling both renewable energy storage and carbon recycling. Electrochemical CO2 reduction can be powered by renewable energy sources and operated under ambient conditions with selective formation of products by controlling the reaction conditions.3,4Significant efforts have been made to develop efficient electrocatalysts that can selectively convert CO2 in aqueous solutions over the competing hydrogen evolution reaction (HER) since the groundbreaking study by Hori et al.5,6 Major progress in electrocatalytic CO2 reduction has resulted in the production of gaseous carbon products including CO, CH4, and C2H4, as well as liquid-phase products such as HCOO−.7–10 However, the production of liquid-phase alcohols with multiple carbons (i.e., two or more carbons) is highly desired, as it may enable the synthesis of sustainable fuels that have high energy densities (21 MJ l−1 for C2H5OH, 27 MJ l−1 for C3H7OH) suitable for long-range and heavy freight transportation. In addition, the current market values of C2H5OH and C3H7OH make the process commercially attractive.11 Unfortunately, the production of multicarbon alcohols via direct electrocatalytic CO2 reduction remains challenging and is not economically viable due to the low catalytic activity and limited selectivity.12,13 Therefore, improving the catalytic activity and selectivity toward products with greater economic value is an attractive technological target.
Among electrocatalysts for CO2 reduction, Cu and Cu-based materials are the most promising for the conversion of CO2 to multicarbon alcohols via multiple electron transfer reactions.14–16 During CO2 reduction, the adsorbed CO intermediate (*CO) is formed on a catalyst and is then further reduced and coupled to form multicarbon alcohols.17 However, insufficient surface coverage of *CO remains a challenge, preventing efficient and productive multicarbon alcohol generation.18 Although several methods are available to tune the reaction pathway for selective production of multicarbon alcohols, overcoming the previously mentioned limitations remains an issue. Li et al. demonstrated that Cu nanoparticles can catalyze direct CO reduction in 0.1 M KOH with faradaic efficiencies of up to 57% for the production of C2H5OH, CH3COO−, and C3H7OH.19 Recently, the so-called “CO-insertion mechanism” in electrochemical CO2 reduction reactions was proposed by several groups. For example, Ren et al. reported that C2H5OH can be produced using CuxZn catalysts, in which metallic Zn weakly binds to CO, producing an in situ source of mobile CO. The desorbed CO can subsequently diffuse to neighboring Cu sites to react with another C1 intermediate, generating multicarbon alcohols.20 Lum et al. showed that CO generated by a nearby CO-producing catalyst could be transferred to Cu, thus increasing the production of oxygenates (C2H5OH and C3H7OH).21 Furthermore, first-principles calculations have shown that all known products from CO2 reduction on Cu catalysts, except formate, involve CO as the primary intermediate.22–24 Thus, it is reasonable to expect that changes in the local CO concentration in the vicinity of the catalyst could affect product selectivity. Based on the above discussion, a feasible strategy to overcome the lack of CO solubility and insufficient CO coverage on the catalytic sites is to introduce a cocatalyst to facilitate CO formation in situ during electrocatalytic CO2 reduction. Thus, a catalyst system can be prepared by coupling two sites, where one site can efficiently reduce CO2 to CO, which then diffuses to a second site where it reacts to form longer carbon chain species via C–C coupling. For the CO-producing site, N-doped carbon materials have exhibited appealing catalytic performance for CO2 reduction to CO with high selectivity. Theoretical and experimental studies revealed that pyridinic N species in N-doped carbon materials are efficient catalysts for CO production.25–28
The design of catalysts with high activity for CO2 reduction and high selectivity toward the formation of multicarbon alcohols could be produced using a combination of pyridinic N as a CO-producing site and Cu nanoparticles as a catalytic site where the latter could provide additional opportunities for C–C coupling between *C1 or *C2 intermediates and *CO. Therefore, highly dispersed Cu nanoparticles supported on N-doped porous carbon materials (Cu/NPC) were designed with different contents of pyridinic N species (e.g., Cu/NPC-700, Cu/NPC-800 and Cu/NPC-900; where the number is the pyrolysis temperature in °C for NPC synthesis). The results indicate that the selective CO2 reduction to multicarbon alcohols (e.g., C2H5OH and C3H7OH) can be tuned by varying the pyridinic N content in the Cu/NPC hybrid catalysts. Furthermore, the introduction of N-doped porous carbon materials affected the size and electronic structure of the Cu catalyst and contributed toward efficient gas transport for improved accessibility to pyridinic N where CO2 is adsorbed and reduced to CO. Supported by the results presented herein and previous studies, a two-site mechanism (pyridinic N as a CO-producing site and Cu as a catalytic site) was proposed to rationalize the efficient CO2 reduction to C2H5OH and C3H7OH over the Cu/NPC hybrid catalysts. To the best of our knowledge, this is the first report of the introduction of N-doped porous carbon materials as a cocatalyst to catalyze CO formation in situ during electrocatalytic CO2 reduction for the generation of multicarbon alcohols.
2. Experimental section
2.1 Chemicals and materials
Pyrrole (C4H5N, Acros Organics), ammonium persulfate ((NH4)2S2O8, Acros Organics), sodium citrate tribasic dihydrate (C6H5Na3O7·2H2O, Sigma Aldrich), sulfuric acid (H2SO4, Sigma Aldrich), acetone (C3H6O, Sigma Aldrich), Triton X-100 (C14H22O(C2H4O)n, Sigma Aldrich), ethylene glycol (C2H6O2, JUNSEI Chemical Co.), and copper nitrate hydrate (Cu(NO3)2·xH2O, Alfa Aesar) were purchased from the specified suppliers. Deionized water (DI, 18 MΩ cm, Millipore) was used for all of the experimental steps.2.2 Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) containing Triton X-100 (0.15 M) by using ultrasonic treatment for 0.5 h, followed by addition of the pyrrole monomer (2.2 mmol). Then 10 mL of distilled water that contained ammonium persulfate (1.3 mmol) was added dropwise into the suspension with continuous stirring for 4 h. The obtained black powder was filtered and washed with distilled water, followed by drying in a freezing dryer. Finally, polypyrrole coated PC (PPyPC) powder samples were heat-treated under an Ar atmosphere at different temperatures of 700, 800 and 900 °C for 2 h. The final products were denoted as NPC-700, NPC-800 and NPC-900, respectively.
:1) containing Triton X-100 (0.15 M) by using ultrasonic treatment for 0.5 h, followed by addition of the pyrrole monomer (2.2 mmol). Then 10 mL of distilled water that contained ammonium persulfate (1.3 mmol) was added dropwise into the suspension with continuous stirring for 4 h. The obtained black powder was filtered and washed with distilled water, followed by drying in a freezing dryer. Finally, polypyrrole coated PC (PPyPC) powder samples were heat-treated under an Ar atmosphere at different temperatures of 700, 800 and 900 °C for 2 h. The final products were denoted as NPC-700, NPC-800 and NPC-900, respectively.
2.3 Physicochemical characterization
The morphology and metal particle size of samples were investigated using scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, as collected on a JEOL-JSN7500F instrument operated at 5 kV and a JEOL-JEM1011 instrument operated at 200 kV, respectively. X-ray diffraction (XRD) analysis data were collected on a Bruker-Advanced D8 diffractometer using Cu Kα (λ = 1.5418 Å) radiation operated at 40 kV and 100 mA. The Brunauer–Emmett–Teller (BET) surface area of samples was measured on a Quadrasorb SI (Quantachrome) by using N2 adsorption/desorption isotherms at −196 °C; the pore size distribution curve was obtained from the Barrett–Joyner–Halenda (BJH) and Horvath–Kawazoe (HK) model. The CO2 adsorption capacity was measured on a BELSORP-mini (MicrotracBEL). The content of Cu in the samples was measured by using an inductively coupled plasma atomic emission (ICP) spectrometer (Optima 7300DV). A high-performance X-ray photoelectron spectrometer (HP-XPS, VG multilab 2000) was used in order to obtain the XPS spectra of the surface elements. Fourier transform infrared (FT-IR) spectroscopy was carried out on a Frontier (PerkinElmer) with powder samples in a KBr pellet form. The XANES spectra of the Cu K-edge were obtained on the 7D beamline of the Pohang Accelerator Laboratory (PLS-II), in which ATHENA software was used for the XANES analysis. CO temperature-programmed desorption (CO-TPD) experiments were performed at 40 °C by injecting a gas mixture of 4% CO/He, and the desorbed CO was detected by using a mass spectrometer while the temperature was increased from 40 to 240 °C at a heating rate of 5 °C min−1. Before the CO-TPD process, the samples were pre-treated at 200 °C with a gas flow of He at 30 mL min−1 for 2 h. CO2 temperature-programmed desorption (CO2-TPD) experiments were carried out by injecting CO2 gas for 1 h at 50 °C, followed by a He rinsing step for 1 h. The desorbed CO2 was analyzed with a TCD detector while the temperature was increased from 50 to 300 °C at a heating rate of 10 °C min−1. Before the CO2-TPD experiment, the samples were pre-treated at 300 °C with a gas flow of He at 30 mL min−1 for 1 h.2.4 Electrochemical measurements
The electrochemical measurements were performed in an electrochemical reactor (H-type cell), composed of three electrodes, which were separated by Nafion 212 into two compartments (Fig. S1, ESI†). A graphite plate (geometric size, 1 cm × 1.5 cm) was used as the working electrode. The catalyst loading in the working electrode was 2 mg cm−2. Pt coil and an Ag/AgCl electrode were used as the counter and reference electrodes, respectively. The potential was converted to a RHE using the following equation:| E (vs. RHE) = E (vs. Ag/AgCl) + 0.209 V + 0.0591 × pH |
Linear sweep voltammograms (LSVs) were recorded in CO2-saturated 0.2 M KHCO3 aqueous solution at a scan rate of 20 mV s−1. Electrochemical reduction of CO2 was performed in 0.2 M KHCO3 aqueous solution, which was stirred at a constant rate of 200 rpm, and CO2 was purged for at least 0.5 h before the experiments. During the electrolysis, CO2 was purged continuously into the cathodic compartment at a rate of 20 mL min−1. The IR drop was compensated using the current interrupt mode. The gas and liquid products from the electrochemical CO2 reduction were detected and analyzed by using a gas chromatograph (GC, Agilent 6890) and a high performance liquid chromatograph (HPLC, Waters e2695), respectively. Electrochemical impedance spectroscopy (EIS) was performed at −1.05 V (vs. RHE) in CO2-saturated 0.2 M KHCO3 aqueous solution.
3. Results and discussion
The carbon atoms in sodium citrate are crucial for the formation of carbon frameworks, while the sodium species contribute to the generation of pores. This template-free one-step process for the fabrication of porous carbon materials involves a simple pyrolysis process without several complex steps that use large amounts of corrosive chemical agents (i.e. KOH, NaOH, ZnCl2, and phosphoric acid).29 The prepared porous carbon (PC) was used as a precursor for the fabrication of N-doped porous carbon hybrid catalysts with deposition of Cu nanoparticles. To obtain various N-doped porous carbons, polypyrrole-coated porous carbon (PPyPC) materials were heat treated at 700 °C, 800 °C, and 900 °C, denoted as NPC-700, NPC-800, and NPC-900, respectively. Herein, polypyrrole was selected as the N source due to its facile and low-cost synthesis, as well as sufficient nitrogen content.30 Finally, the N-doped porous carbons were impregnated with Cu nanoparticles (Cu/NPC-700, -800, and -900) via microwave-assisted polyol reduction.31 A schematic of the Cu/NPC hybrid catalyst fabrication is shown in Scheme 1. The physicochemical characteristics of the prepared Cu/NPC catalysts and their application in electrochemical CO2 reduction will be discussed in detail. | ||
| Scheme 1 Schematic illustration of the overall fabrication process for Cu/NPC hybrid catalysts. | ||
The microstructure and surface morphology of the prepared samples were investigated via SEM analysis. The PC exhibited a well-developed hierarchical porous structure with different types of open pores in multidirectional channels (Fig. 1a). Because of the polymerization process, polypyrrole coated PC (PPyPC) exhibited a relatively smooth surface without the porous structure of PC (Fig. S2, ESI†), indicating that the polypyrrole was preferentially formed as a uniform layer on the PC. After pyrolysis at different temperatures (700 °C, 800 °C, and 900 °C), the NPC materials maintained their original PC structures without notable changes (Fig. 1b–d), containing a myriad of macropores 50–500 nm in size. To further investigate the pore structure of the prepared carbon materials, N2 adsorption/desorption isotherms of the four samples (PC, NPC-700, NPC-800, and NPC-900) were analyzed (Fig. S3, ESI†). The qualitative behavior of the isotherms clearly illustrated that all carbon materials had a similar pore structure (Fig. S3a, ESI†). Moreover, the relevant pore size analysis in Fig. S3b (ESI†) showed that all samples exhibited a uniform mesopore distribution, particularly from 2–20 nm, according to the Barrett–Joyner–Halenda (BJH) characterization. In addition, analysis of the N2 desorption isotherm by the Horvath–Kawazoe (HK) model showed uniformly distributed micropores with sizes centered at approximately 0.4 nm (Fig. S3c, ESI†). The textural properties are summarized in Table S1 (ESI†) with BET surface areas and porosity. It has been widely reported that the porous structure in electrodes plays an important role in the overall CO2 reduction reaction.32–34 Therefore, the well-developed hierarchical porous structure should provide efficient gas transportation channels for improved accessibility to the N-doped sites.33,35
 | ||
| Fig. 1 SEM image of (a) PC, (b) NPC-700, (c) NPC-800 and (d) NPC-900; the inset shows the high-magnitude SEM image. | ||
The nature of the N species on the prepared sample surfaces was analyzed by XPS measurements. From the peak deconvolution of XPS N 1s spectra shown in Fig. S4 (ESI†), PPyPC showed a major peak at 400.0 eV, which was attributed to a neutral pyrrolic N (–N–), and a minor peak at 400.8 eV corresponding to a positively charged nitrogen (–N+–).36 During pyrolysis, PPyPC was gradually transformed to N-doped carbonaceous materials, whereas most pyrrolic N in PPyPC was converted into various N species, yielding the N-doped carbon compounds. Thus, the NPC-700, NPC-800, and NPC-900 samples showed two major peaks at approximately 398.3 and 401.1 eV, which were assigned to pyridinic N and graphitic N, respectively, and minor peaks at 400.2 and 403.2 eV which were ascribed to pyrrolic N and oxidized N, respectively (Fig. 2a).37 The total N contents with different N species in the NPC materials are summarized in Fig. 2b, suggesting that the pyrolysis temperature is highly correlated with the transformation behavior of the N species. With the increase of the pyrolysis temperature from 700 to 800 °C, the content of pyrrolic N is decreased whereas the contents of pyridinic N and graphitic N are increased due to the reconstruction of the N-doped carbon structure along with transformations of the N species. However, both the pyrrolic N and pyridinic N contents showed a decrease at temperatures greater than 800 °C with a simultaneous increase of the graphitic N content (Fig. 2b), which can be ascribed to the fact that the pyrrolic N and pyridinic N are not thermally stable compared to the graphitic N.38,39 In addition, with increasing pyrolysis temperature from 700 to 900 °C, the total N content in the NPC materials decreased from 11.2 to 9.6 at%, which was attributed to the removal of relatively unstable C–N bonds.35 The chemical features of PC, PPyPC, and NPC-800 were further investigated using FTIR spectroscopy, as shown in Fig. S5 (ESI†), to examine the functional groups at characteristic wavenumbers.40 PPyPC showed distinct peaks at 1209 and 926 cm−1, assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+–C and C–N+, respectively, and a peak at 1040 cm−1 corresponding to the in-plane C–H vibrations. Notably, the successful incorporation of N atoms into the PC can be monitored by the disappearance of the characteristic peaks of polypyrrole in NPC-800. The PC exhibited a sharp peak at 1615 cm−1 corresponding to the CC group, and NPC-800 showed the same peaks for CC/CN groups at 1625 cm−1. From Fig. 2c, the CO2 uptake at 25 °C of the prepared samples followed the order: NPC-800 (2.26 mmol g−1) > NPC-700 (1.62 mmol g−1) > NPC-900 (1.20 mmol g−1) > PC (0.45 mmol g−1). In terms of the CO2 adsorption capacity, micropores and pyridinic N are considered to be important factors.41,42 However, because the micropore volume of all carbon materials was similar for the materials prepared herein, the difference in CO2 adsorption capacity was attributed to the pyridinic N content. Therefore, the PC without pyridinic N showed a lower CO2 adsorption capacity than the NPC materials (Table S2, ESI†). In addition, the hierarchical porosities likely facilitated gas transportation for improved accessibility to the pyridinic N site where CO2 is adsorbed.33,43
N+–C and C–N+, respectively, and a peak at 1040 cm−1 corresponding to the in-plane C–H vibrations. Notably, the successful incorporation of N atoms into the PC can be monitored by the disappearance of the characteristic peaks of polypyrrole in NPC-800. The PC exhibited a sharp peak at 1615 cm−1 corresponding to the CC group, and NPC-800 showed the same peaks for CC/CN groups at 1625 cm−1. From Fig. 2c, the CO2 uptake at 25 °C of the prepared samples followed the order: NPC-800 (2.26 mmol g−1) > NPC-700 (1.62 mmol g−1) > NPC-900 (1.20 mmol g−1) > PC (0.45 mmol g−1). In terms of the CO2 adsorption capacity, micropores and pyridinic N are considered to be important factors.41,42 However, because the micropore volume of all carbon materials was similar for the materials prepared herein, the difference in CO2 adsorption capacity was attributed to the pyridinic N content. Therefore, the PC without pyridinic N showed a lower CO2 adsorption capacity than the NPC materials (Table S2, ESI†). In addition, the hierarchical porosities likely facilitated gas transportation for improved accessibility to the pyridinic N site where CO2 is adsorbed.33,43
 | ||
| Fig. 2 (a) High resolution N 1s spectra of NPC-700, NPC-800 and NPC-900. (b) Summary of N atomic contents and relative concentrations. (c) CO2 uptake isotherms at 25 °C. | ||
TEM images with size distribution histograms of the Cu nanoparticles deposited on the PC and NPC materials are shown in Fig. 3. The three Cu/NPC hybrid catalysts exhibited significantly smaller particle sizes than Cu/PC. From the size distribution histograms, the average Cu particle sizes of Cu/PC, Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900 were approximately 12.1, 5.3, 5.2, and 5.4 nm, respectively, as estimated using 200 randomly selected particles from the TEM images. Despite using the same synthesis method, the difference in the Cu nanoparticle size between the PC and NPC materials arises from the N-doped sites in the NPC materials that facilitate the nucleation/growth of Cu.44,45 The crystallographic data of the prepared samples were obtained via XRD, as shown in Fig. 4. The broad width and weak intensity peak centered at approximately 23.5° for the (002) plane of graphitic carbons (Fig. 4a) suggests a low degree of graphitization.35 The presence of metallic Cu on the NPC materials (Cu/PC, Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900) was confirmed by the three strong Cu diffraction peaks at approximately 43°, 50°, and 74°, corresponding to the (111), (200) and (220) planes of the fcc crystal structure of metallic Cu. Meanwhile, the full width at half maximum (FWHM) of the Cu peaks of the Cu/NPC materials was relatively smaller than that observed for the Cu/PC sample (Fig. 4b), indicating smaller Cu nanoparticles with a higher degree of dispersion on the NPC materials.44 The actual amounts of Cu on the PC and NPC materials were determined quantitatively using ICP spectrometry. The nominal Cu metal loading was fixed at 20 wt% and the actual Cu contents in the Cu/PC, Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900 samples were ca. 21.6, 19.7, 19.8, and 20.2 wt%, respectively.
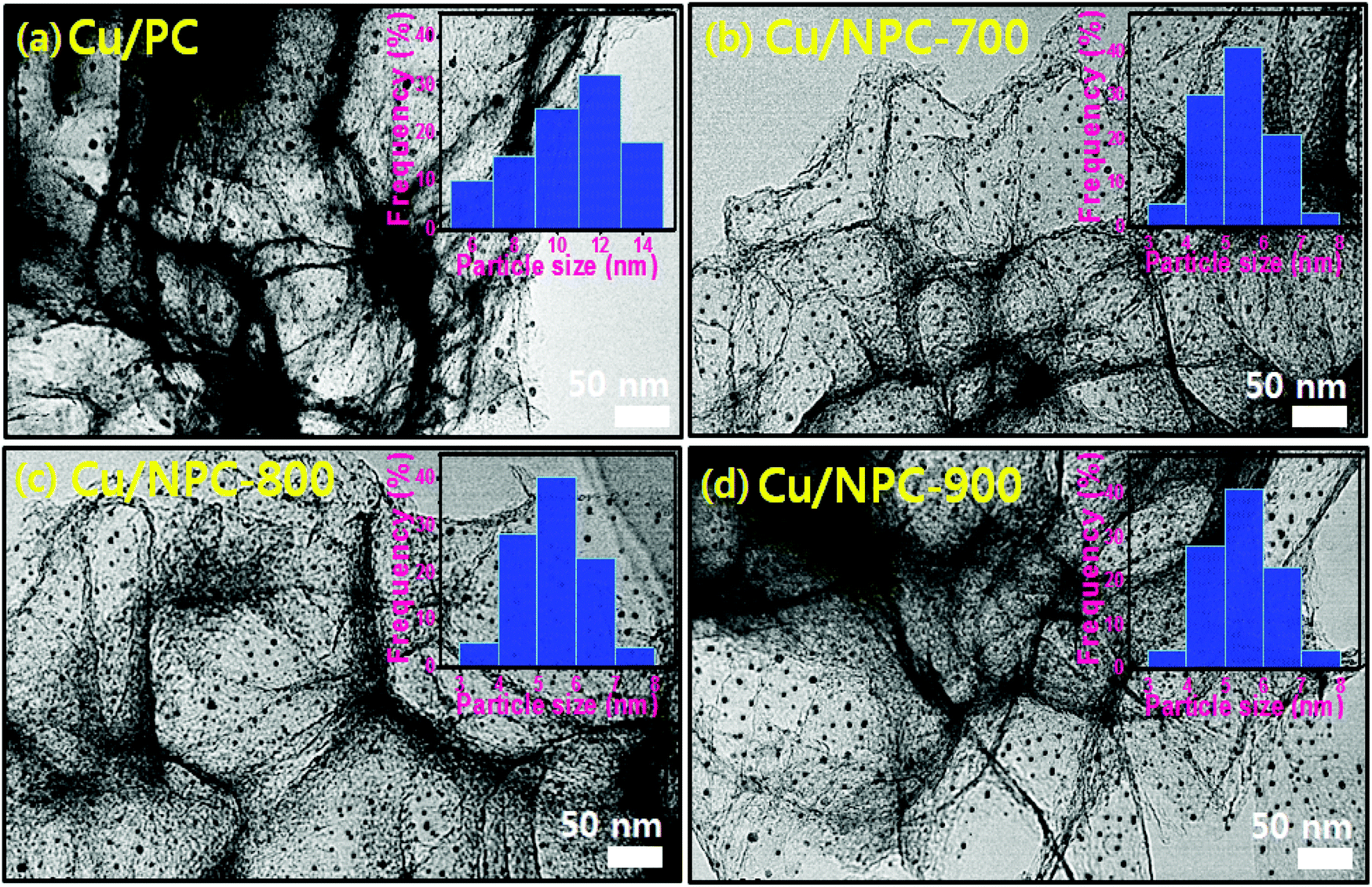 | ||
| Fig. 3 TEM images with particle size distribution of (a) Cu/PC, (b) Cu/NPC-700 (c) Cu/NPC-800 and (d) Cu/NPC-900 hybrid catalysts. | ||
 | ||
| Fig. 4 XRD patterns of (a) PC, NPC-700, NPC-800 and NPC-900 and (b) Cu/PC and Cu/NPC-700, Cu/NPC-800 and Cu/NPC-900 hybrid catalysts. | ||
Fig. S6a–c (ESI†) show the peak deconvolution of XPS N 1s spectra of Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900. Four types of N species were also detected: pyridinic, pyrrolic, graphitic, and oxidized N. In addition, even after the Cu nanoparticles were decorated on the NPC material, the pyridinic N content was still the highest for the Cu/NPC-800 sample (Fig. S6d and Table S3, ESI†). However, the Cu nanoparticles on the NPC materials induced changes in the N 1s spectra. The significant decrease in the relative graphitic N content suggested that most of the Cu nanoparticles were anchored on graphitic N sites. This is consistent with the results that showed that the NPC materials promoted the formation of Cu nanoparticles with smaller sizes and better dispersity due to their graphitic N species content with a relatively high electron density.46–48 The slight increase in the relative percentage of pyridinic, pyrrolic, and oxidized N suggests that these N species are not associated with Cu nucleation and growth. The Cu 2p3/2 XPS spectra of Cu/PC, Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900 are shown in Fig. 5a. In all samples, the Cu 2p3/2 spectra were fitted with a major peak at 932.4 ± 0.3 eV associated with (Cu0 + Cu+).49 Compared to that of Cu/PC, the binding energies of Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900 shifted to higher binding energies by 0.4–0.5 eV. This shift toward higher energy indicated that more electron transfer from the Cu metal to NPC materials occurred, which further strengthened the electronic interaction.44 The Cu average particle sizes, Cu contents, and analytical results from the Cu 2p3/2 spectra of all the prepared catalysts are summarized in Table S4 (ESI†).
 | ||
| Fig. 5 (a) High resolution Cu 2p3/2 spectra of Cu/PC, Cu/NPC-700, Cu/NPC-800 and Cu/NPC-900 hybrid catalysts. (b) Cu K edge XANES spectra of reference samples, Cu/PC and Cu/NPC hybrid catalysts with (c) comparison in detail. | ||
Because XANES is sensitive to the chemical valence state of the probed atoms, detailed local structural properties of the prepared samples were further investigated at the Cu K-edge. The normalized XANES spectra of commercial Cu2O, CuO, Cu metal foil, and the prepared hybrid catalysts are shown in Fig. 5b. It is clear that the Cu K-edge XANES spectra of the Cu/NPC hybrid catalysts were similar to those of the Cu foil, indicating that the Cu nanoparticles on NPC-700, NPC-800, and NPC-900 mainly exist in a metallic state. The strong peak above the edge energy position (Fig. 5c) is closely related to the empty d states of Cu and the white line (WL) intensity can be used to measure changes in the local electronic structure of Cu.50 Compared to the WL intensity of Cu/PC, the increased intensity of Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900 was attributed to increased density of empty Cu d states due to electron transfer to the NPC materials.50 This phenomenon can be explained based on the previous XPS analysis results. The strong electronic interaction involving electron transfer between the Cu nanoparticles and NPC materials changes the electronic structure of Cu, which influenced the CO adsorption and C–C coupling during electrocatalytic CO2 reduction.51 Additionally, to quantify the relative composition of Cu species in the Cu/PC and Cu/NPC hybrid catalysts, linear combination fitting with the XANES spectra was performed (Fig. S7 and Table S5, ESI†). All samples contained mainly metallic Cu rather than the oxidized Cu+ or Cu2+. This result agrees with the XRD analysis, where Cu0 was identified and no oxide species were detected. However, the proportions of Cu+ and Cu2+ in the Cu/NPC hybrid catalysts were relatively higher than those of Cu/PC, suggesting that the smaller metal nanoparticles were more easily oxidized on the surface of the catalysts than the larger nanoparticles. In addition, the relative compositions of Cu species were similar in all the Cu/NPC hybrid catalysts.
As shown in Fig. 6, electrochemical CO2 reduction over the prepared samples was first estimated by LSV measurements in a CO2-saturated 0.2 M KHCO3 aqueous solution at a scan rate of 20 mV s−1. It should be noted that the onset potentials of the Cu/NPC hybrid catalysts (Cu/NPC-700, Cu/NPC-800, and Cu/NPC-900) were more positive than those of the other samples. In addition, the Cu/NPC hybrid catalysts exhibited the largest reductive current densities over the entire potential range, suggesting that the integration of Cu nanoparticles and N-doped porous carbon materials facilitated the efficient reduction of CO2 in the electrolyte solution. Chronoamperometry measurements were performed from −0.6 to −1.1 V (vs. RHE) in CO2-saturated 0.2 M KHCO3 aqueous solution using a H-type cell to further analyze the CO2 electroreduction products. The faradaic efficiencies for various liquid/gas products as a function of applied potential on the PC, NPC-800, Cu/PC, and Cu/NPC-800 samples are shown in Fig. 7. The PC alone showed almost no catalytic activity for CO2 reduction and instead the hydrogen evolution reaction (HER) was dominant because of incapability for catalyzing CO2 electroreduction (Fig. 7a and Table S6, ESI†). For NPC-800, CO was observed as the major gas product and the faradaic efficiency of CO formation increased from −0.6 to −1.0 V (vs. RHE), reaching a maximum (61.8%) at −1.0 V (Fig. 7b and Table S6, ESI†). The same trend was observed for the NPC-700 and NPC-900 catalysts (Fig. S8 and Table S6, ESI†). As previously reported, pyridinic N is the most preferable and selective site for the CO production from CO2 as it exhibits relatively weak binding energy for *CO and facilitates CO desorption.25–28,52 As shown in Fig. S9 (ESI†), the change in CO2 adsorption capacity as a function of pyridinic N content matched with the maximum faradaic efficiency for CO. In contrast to the NPC materials, Cu/PC showed a very broad product distribution comprising C2H4 and C2H5OH. In the applied potential range, Cu/PC produced C2H4 with a maximum faradaic efficiency of 30.2% at −1.0 V (vs. RHE) and C2H5OH with a maximum faradaic efficiency of 14.3% at −0.9 V (vs. RHE). Furthermore, trace C3H7OH was detected (Fig. 7c and Table S7, ESI†). Interestingly, all three Cu/NPC hybrid catalysts showed enhanced selectivities toward multicarbon alcohols (C2H5OH and C3H7OH) compared to Cu/PC (Fig. 7d, Fig. S10 and Table S7, ESI†). The maximum faradaic efficiencies for C2H5OH and C3H7OH were 64.6% and 8.7%, respectively, at −1.05 V (vs. RHE) for Cu/NPC-800. The partial current densities for the production of C2H5OH and C3H7OH (jC2H5OH and jC3H7OH) were analyzed to compare the performances of the Cu/PC and Cu/NPC hybrid catalysts (Fig. 8a and b). The observed trend was similar to that described for the faradaic efficiencies of C2H5OH and C3H7OH using the same catalysts. The values of jC2H5OH and jC3H7OH clearly indicate the superior catalytic activity of Cu/NPC-800 for multicarbon alcohol production over the entire potential range. In addition, among the Cu/NPC hybrid catalysts, Cu/NPC-800 showed the highest total current density at −1.05 V (vs. RHE), indicating the maximum faradaic efficiency for multicarbon alcohol generation (Fig. 8c). In Table S8 (ESI†), this novel class of hybrid catalysts, which is selective for multicarbon alcohols (C2H5OH and C3H7OH) over other C2–C3 products, was compared with the previously reported Cu-based catalysts.
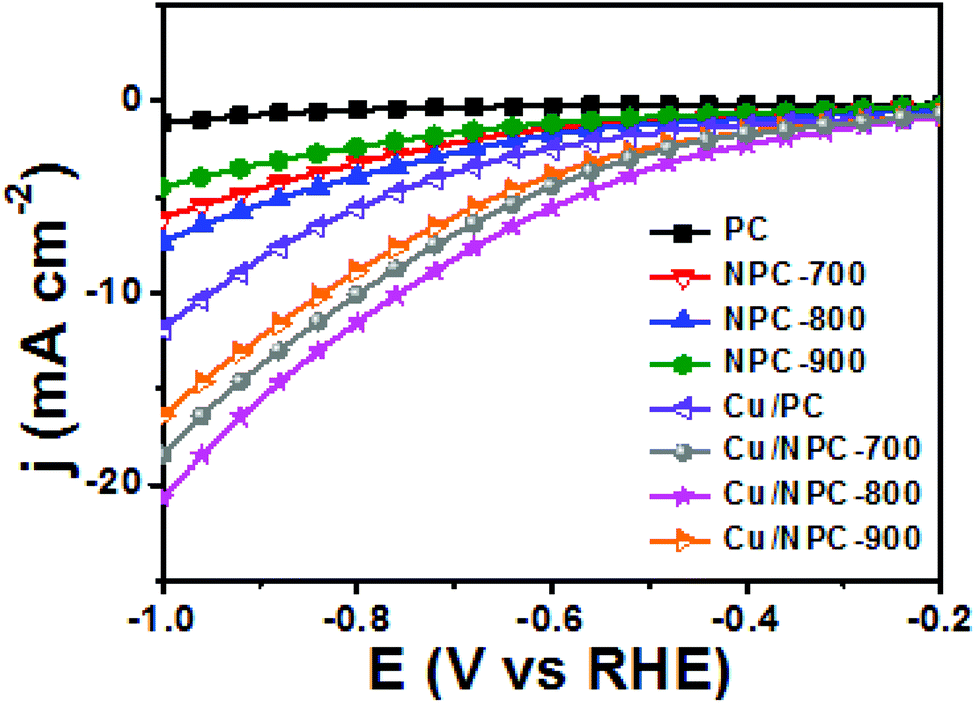 | ||
| Fig. 6 LSV curves of PC, NPC materials and Cu/NPC hybrid catalysts in CO2-saturated 0.2 M KHCO3 aqueous solution. | ||
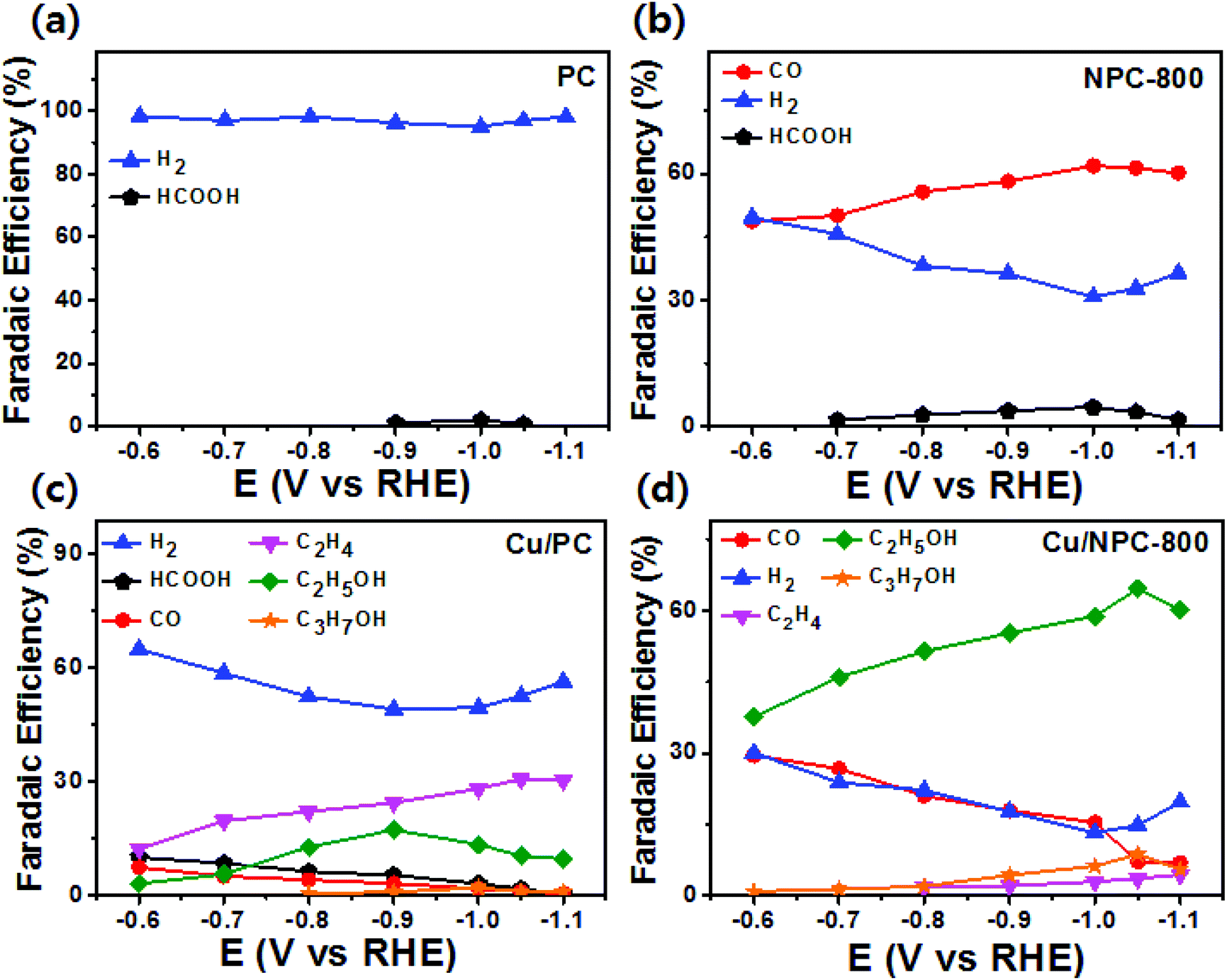 | ||
| Fig. 7 Faradaic efficiencies of liquid and gaseous products for CO2 reduction in CO2-saturated 0.2 M KHCO3 aqueous solution on (a) PC, (b) NPC-800, (c) Cu/PC, and (d) Cu/NPC-800 hybrid catalysts. | ||
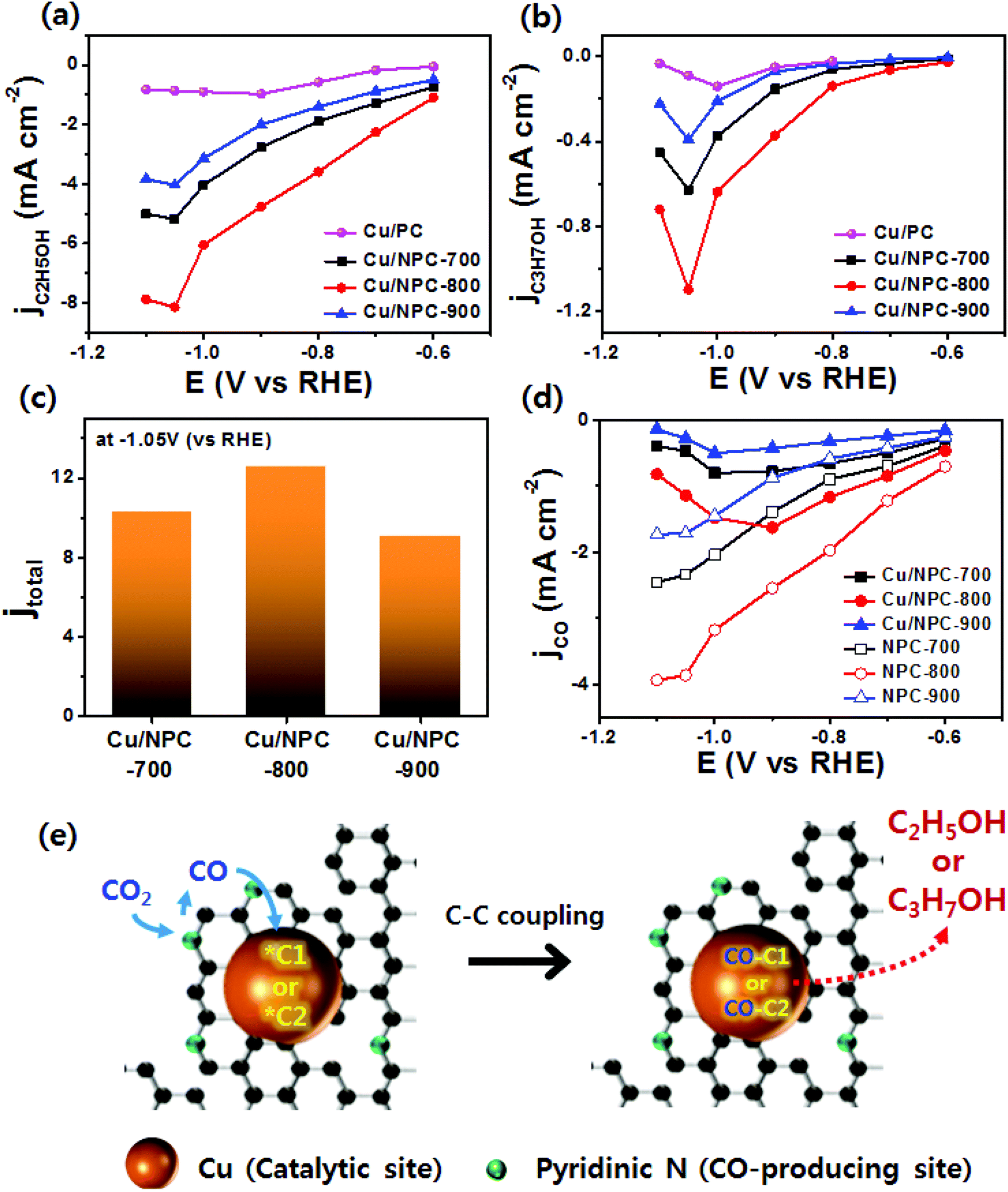 | ||
| Fig. 8 Partial current densities of (a) C2H5OH and (b) C3H7OH production on Cu/NPC hybrid catalysts. (c) Total current densities measured at −1.05 V (vs. RHE). (d) Partial current densities of CO on NPC materials and Cu/NPC hybrid catalysts. (e) Proposed two-site mechanism (via CO-insertion route) scheme for electrocatalytic CO2 reduction to multicarbon alcohols on Cu/NPC hybrid catalysts, indicating the CO-producing site (pyridinic N) and catalytic site (Cu nanoparticle). | ||
The calculated faradaic efficiencies of the major reaction products over Cu/PC and Cu/NPC hybrid catalysts showed interesting selectivity for multicarbon alcohols. In the low negative potential region, CO production was significantly increased over the Cu/NPC hybrid catalysts. The faradaic efficiency of CO decreased monotonically with increasingly negative applied potential, whereas C2H5OH and C3H7OH production began and increased in the potential region, indicating that the *CO intermediate was intimately connected to C2H5OH and C3H7OH formation.20,53,54 The aforementioned trends in C2H5OH and C3H7OH formation over the Cu/NPC hybrid catalysts suggest that the pyridinic N and Cu nanoparticles acted cooperatively to promote C2H5OH and C3H7OH formation. This cooperative catalysis was demonstrated by analyzing the faradaic efficiency of CO over these catalysts (Fig. 7d and Fig. S10a, b, ESI†). Otherwise, it could be anticipated that increasing faradaic efficiency of CO on the Cu/NPC hybrid catalysts as larger negative potentials were applied would be achieved due to the production of CO from pyridinic N (Fig. 7d and Fig. S10a, b, ESI†). Therefore, it was assumed that gaseous CO formed at the pyridinic N sites may further react to yield C2H5OH and C3H7OH. This speculation is corroborated by the negative shift in maximum faradaic efficiencies of C2H5OH and C3H7OH production by 150 and 50 mV over the Cu/NPC hybrid catalysts compared to those achieved using Cu/PC. Importantly, most of the CO produced at the pyridinic N sites of the NPC materials diffused toward Cu sites for further reaction, as indicated by the low CO partial current densities (Fig. 8d).
To understand the key factors influencing the catalytic performance for CO2 reduction, the analytical and experimental results of prepared samples were compared (Tables S1–S5, ESI†). It was found that textural properties of NPC materials were similar to each other. Furthermore, the effects of the particle size, loading amount, composition and electronic properties of Cu nanoparticles were also negligible because they were similar for all Cu/NPC hybrid catalysts. In contrast, the pyridinic N content in the Cu/NPC hybrid catalysts has a critical effect on producing multicarbon alcohols, as shown by the fact that plenty of pyridinic N species in Cu/NPC-800 hybrid catalysts could directly contribute to the improvement of the catalytic activity and selectivity toward production of multicarbon alcohols from CO2 reduction. In addition, the Cu catalysts supported on different carbon materials might explain why the selective production of multicarbon alcohols was enhanced on the Cu/NPC hybrid catalysts. It seems that the introduction of N-doped carbon materials is an important factor for the selective production of multicarbon alcohols because they not only affect the size and electronic structure of the Cu, but also promote the adsorption of CO2 as well as CO production. The different catalytic performances of Cu/PC and Cu/NPC hybrid catalysts could be further supported through the EIS measurements. As shown in Fig. S11 (ESI†), the Nyquist plots revealed that the Cu/NPC-800 exhibited a smaller semicircle radius compared with that of Cu/PC, indicating accelerated charge transfer. This is associated with a lower activation energy55 for the CO2 reduction reaction on Cu/NPC-800, as determined by LSV measurements. These results suggest that the Cu/NPC-800 hybrid catalysts were highly active for CO2 reduction due to the introduction of N-doped porous carbon materials.
Moreover, previous studies showed that the differences in CO binding energy are responsible for generating different products.56,57 To investigate CO adsorption capabilities, CO-TPD measurement and voltammetric experiments were performed and the results are shown in Fig. S12 (ESI†). To clearly identify the desorbed CO signal, the mass-to-charge ratio (m/z) was set to 28 to detect the desorbed CO signal. The CO desorption signal appeared at the higher temperature for all the Cu/NPC hybrid catalysts as compared to the Cu/PC, suggesting the higher binding energies of CO over the Cu/NPC hybrid catalysts (Fig. S12a, ESI†). Furthermore, to investigate the CO adsorption strength under actual experimental conditions, voltammetric measurements were performed in a CO-saturated 0.2 M KHCO3 aqueous solution with an applied potential of −0.12 V (vs. RHE) for 120 s (Fig. S12b, ESI†). This applied potential was chosen for a maximum amount of adsorbed CO before the occurrence of any electrochemical reactions. With the addition of N2 gas after 120 s, the cathodic current density decrease was much slower for Cu/NPC-800 than that for Cu/PC. The CO-TPD and voltammetric results showed that the enhanced CO adsorption strength was caused by changes in the electronic properties of Cu,51 as indicated by the XPS and XANES spectra. In addition, the larger surface area of Cu nanoparticles with ∼5 nm sizes could lead to a higher chance of offering optimized geometries for CO adsorption.18 Thus, from these results, we could assume that change in CO adsorption capability on the Cu/NPC hybrid catalysts might tune the energetics of intermediate binding at the Cu site, affecting selectivity toward multicarbon alcohols.54,58,59 This might explain why the production of multicarbon alcohols was favored over C2H4 on all Cu/NPC hybrid catalysts, whereas C2H4 was selectively produced over multicarbon alcohols on Cu/PC, although more information regarding the selectivity determining steps is necessary for mechanistic determination. To examine the adsorption behavior of CO2 over Cu/NPC-700, Cu/NPC-800 and Cu/NPC-900, the CO2-TPD measurements were performed as shown in Fig. S13a (ESI†). The first desorption peak could originate from physisorbed/chemisorbed CO2, whereas the second peak was related to strongly chemisorbed CO2.60 The CO2-TPD peak areas for all Cu/NPC hybrid catalysts followed the order: Cu/NPC-800 > Cu/NPC-700 > Cu/NPC-900 (Fig. S13b, ESI†). These results suggested a correlation between the CO2 adsorption capacity and content of pyridinic N (Table S3 and Fig. S6d, S13, ESI†).
Generally, C2H4 is a favorable product of Cu-based catalysts over C2H5OH and C3H7OH via CO2 reduction.12,13 However, the product selectivity for multicarbon alcohols with a higher CO concentration was observed with other catalysts,20,21,53 suggesting that a CO atmosphere is crucial for controlling product selectivity. Therefore, to assess the role of the CO concentration, the direct CO electroreduction on Cu nanoparticles (Cu NPs), Cu/PC, and Cu/NPC hybrid catalysts was performed at −1.05 V (vs. RHE) in 0.2 M KHCO3 aqueous solution (Fig. S14, ESI†). As a control, Cu NPs prepared using the same synthetic method without carbon supports were prepared, for which the morphology and crystallographic information are provided in Fig. S15 (ESI†). It should be noted that the relative ratio of faradaic efficiencies of multicarbon alcohols to C2H4 significantly increased over the Cu nanoparticles and Cu/PC, whereas the Cu/NPC hybrid catalysts did not show substantial changes compared to CO2 reduction (Fig. S14, ESI†). These results indicated that the favorable product could be changed from C2H4 to multicarbon alcohols by tuning the CO concentration, indicating that pyridinic N as the CO-producing site significantly affected the production of C2H5OH and C3H7OH during electrocatalytic CO2 reduction. Thus, the CO population in accordance with the pyridinic N content of the Cu/NPC hybrid catalysts is a crucial factor for the selectivity toward multicarbon alcohols. Notably, the Cu/NPC hybrid catalysts showed a similar selectivity trend for C2H5OH and C3H7OH as a function of applied potential (Table S7, ESI†), suggesting that these alcohols may share common intermediates along their reaction pathways.54 Based on the results presented herein and the recent literature, a two-site mechanism was proposed to rationalize the action of the Cu nanoparticles and pyridinic N species in N-doped porous carbon for the electrocatalysis of CO2 to form multicarbon alcohols (Fig. 8e). The incoming CO2 molecules bind to pyridinic N sites and are reduced to CO,25–28 which adsorbs weakly at the pyridinic N sites, where it can later desorb. These steps are in agreement with product analysis (Fig. 7b, S8, and S9, ESI†). The desorbed CO from the pyridinic N sites subsequently diffuses toward the Cu nanoparticles, adsorbing onto the Cu surface where it can react with *C1 or *C2 intermediates.20,61–67 These intermediates can be further converted to either C2H5OH or C3H7OH via C–C coupling. In this mechanism, the cooperative catalytic effect of the Cu nanoparticles and pyridinic N species is crucial for the production of C2H5OH and C3H7OH. As summarized in Fig. 9, it clearly demonstrates that the pyridinic N facilitates the transformation of CO2 to CO near the catalytically active Cu site, changing the product selectivity toward oxygenated products.53,64,68,69 This also explains why the faradaic efficiencies of multicarbon alcohols (C2H5OH and C3H7OH) increased while the faradaic efficiencies of H2 and C2H4 decreased as a function of pyridinic N species content. Furthermore, the larger surface area of the small Cu nanoparticles (∼5 nm) with modified electronic properties enhanced the adsorption of CO, promoting C–C coupling (between *C1 or *C2 intermediates and *CO) for multicarbon alcohol production.
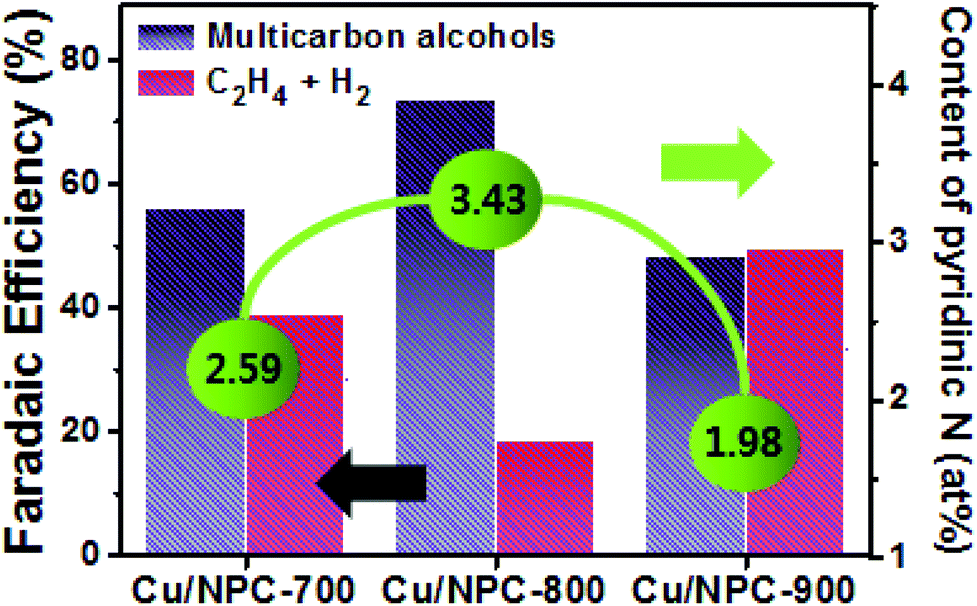 | ||
| Fig. 9 Schematic of the relationship between the contents of pyridinic N in Cu/NPC hybrid catalysts and the corresponding faradaic efficiencies of multicarbon alcohols, C2H4 and H2 in CO2-saturated 0.2 M KHCO3 aqueous solution at −1.05 V (vs. RHE). | ||
The cooperative catalytic effect was further examined for CO2 reduction using different Cu metal loadings of 10 and 30 wt% on the Cu/NPC-800 hybrid catalysts. The associated physicochemical characterization is provided in Fig. S16 and S17 (ESI†) and summarized in Table S9 (ESI†). Compared to Cu/NPC-800 with 20 wt% Cu loading, these catalysts exhibited smaller faradaic efficiencies toward multicarbon alcohols at −1.05 V (vs. RHE). For Cu10/NPC-800, the amount of Cu catalyst seems to be insufficient, but the catalytic activity toward CO formation is significantly increased with a larger pyridinic N species content in NPC-800 (Fig. S18a and Table S10, ESI†). In contrast, Cu30/NPC-800 showed increased faradaic efficiency for C2H4, but decreased efficiencies for C2H5OH and C3H7OH (Fig. S18b and Table S10, ESI†). This is likely due to overloading of the Cu nanoparticles. To maximize the cooperative catalytic effect, it is necessary for the Cu nanoparticles to stay close to or around the pyridinic N sites in the N-doped porous carbon structure. Thus, excessive loading of Cu may restrict the CO-producing ability of pyridinic N sites and attenuate the cooperative catalysis. Therefore, effective exposure of pyridinic N to the reactant and proper spacing with Cu nanoparticles are crucial for the CO2 reduction catalysis to form multicarbon alcohols.
Another crucial factor for the design of an ideal CO2 reduction catalyst is stability. Therefore, the stability test of Cu/NPC-800 hybrid catalysts was investigated at a constant applied potential of −1.05 V (vs. RHE) in CO2-saturated 0.2 M KHCO3 aqueous solution. As shown in Fig. 10, the Cu/NPC-800 revealed a quite stable performance in current density during 10 h. After the stability test, the catalyst was re-used in another CO2 reduction test in a fresh 0.2 M KHCO3 electrolyte under the same applied potential to estimate the faradaic efficiency for C2H5OH and C3H7OH. Total faradaic efficiencies of multicarbon alcohols were shown up to 71.1%, which is comparable to the initial value of 73.3% that was measured before the stability test. This result implied that the Cu/NPC-800 hybrid catalysts are relatively stable during the CO2 reduction reaction without significant performance loss.
 | ||
| Fig. 10 Chronoamperometric measurements at a constant potential of −1.05 V (vs. RHE) in CO2-saturated 0.2 M KHCO3 aqueous solution. | ||
4. Conclusions
In summary, a series of Cu/NPC hybrid catalysts were prepared with different pyridinic N contents for the selective production of multicarbon alcohols via electrocatalytic CO2 reduction. The production of total multicarbon alcohols was maximized for the Cu/NPC-800 hybrid catalysts with 20 wt% Cu loading at −1.05 V (vs. RHE), achieving a total faradaic efficiency of 73.3% for C2H5OH and C3H7OH. The Cu/NPC-800 hybrid catalysts were also catalytically stable for the production of C2H5OH and C3H7OH for 10 h. We discovered that the selective production of multicarbon alcohols on the Cu/NPC hybrid catalysts was significantly influenced by the pyridinic N content in the N-doped porous carbon materials. High contents of pyridinic N facilitated CO2 adsorption, and thus produced CO which efficiently diffused to neighboring Cu sites. Furthermore, suitable sized Cu nanoparticles with a modified electronic structure effectively catalyzed the production of *C1 or *C2 intermediates which subsequently reacted with CO. The strategy developed herein for cooperative catalysis between Cu nanoparticles and pyridinic N provides novel insights toward achieving the selective production of multicarbon alcohols via electroreduction of CO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Basic Science Research Program (NRF-2019R1A2C2088174) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT, and by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (20172010106300), and also by the Fundamental Research Program of the Korean Institute of Materials Science (Grant PNK6130).References
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242–3248 CrossRef PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- Z.-L. Wang, C. Li and Y. Yamauchi, Nano Today, 2016, 11, 373–391 CrossRef CAS.
- Y. Hori, Bull. Chem. Soc. Jpn., 1982, 55, 660–665 CrossRef CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Shin, Chem. Lett., 1986, 15, 897–898 CrossRef.
- L. Jin, B. Liu, P. Wang, H. Yao, L. A. Achola, P. Kerns, A. Lopes, Y. Yang, J. Ho, A. Moewes, Y. Pei and J. He, Nanoscale, 2018, 10, 14678–14686 RSC.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- X. Zheng, J. Han, Y. Fu, Y. Deng, Y. Liu, Y. Yang, T. Wang and L. Zhang, Nano Energy, 2018, 48, 93–100 CrossRef CAS.
- K. J. P. Schouten, F. Calle-Vallejo and M. Koper, Angew. Chem., Int. Ed., 2014, 53, 10858–10860 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- W. Zhu, K. Zhao, S. Liu, M. Liu, F. Peng, P. An, B. Qin, H. Zhou, H. Li and Z. He, J. Energy Chem., 2019, 37, 176–182 CrossRef.
- S. Shen, X. Peng, L. Song, Y. Qiu, C. Li, L. Zhou, J. He, J. Ren, X. Liu and J. Luo, Small, 2019, 15, 1902229 CrossRef PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019 DOI:10.1038/s41563-019-0445-x.
- Z. Gu, H. Shen, L. Shang, X. Lv, L. Quan and G. Zheng, Small Methods, 2018, 2, 1800121 CrossRef.
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. D. Luna, C.-T. Dinh, T.-T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Z. Finfrock, L. Ma, S.-H. Hsieh, Y.-S. Liu, G. A. Botton, W.-F. Pong, X. Du, J. Guo, T.-K. Sham, E. H. Sargent and D. Sinton, Nat. Commun., 2018, 9, 4614 CrossRef PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Adv. Funct. Mater., 2018, 28, 1800499 CrossRef.
- M. Sevilla and A. B. Fuertes, J. Mater. Chem. A, 2013, 1, 13738–13741 RSC.
- X. Yuan, X.-L. Ding, C.-Y. Wang and Z.-F. Ma, Energy Environ. Sci., 2013, 6, 1105–1124 RSC.
- W. X. Chen, J. Y. Lee and Z. Liu, Chem. Commun., 2002, 2588–2589 RSC.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- X. Sun, L. Lu, Q. Zhu, C. Wu, D. Yang, C. Chen and B. Han, Angew. Chem., Int. Ed., 2018, 57, 2427–2431 CrossRef CAS.
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef.
- H. Han, Y. Noh, Y. Kim, W. S. Jung, S. Park and W. B. Kim, Nanoscale, 2019, 11, 2423–2433 RSC.
- H. Kim, Y. Kim, Y. Noh, S. Lee, J. Sung and W. B. Kim, ChemCatChem, 2017, 9, 1503–1510 CrossRef CAS.
- Z. Lin, G. H. Waller, Y. Liu, M. Liu and C.-P. Wong, Nano Energy, 2013, 2, 241–248 CrossRef CAS.
- H. J. Cui, H. M. Yu, J. F. Zheng, Z. J. Wang, Y. Y. Zhu, S. P. Jia, J. Jia and Z. P. Zhu, Nanoscale, 2016, 8, 2795–2803 RSC.
- Z. Lin, G. Waller, Y. Liu, M. Liu and C.-P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS.
- H. Han, Y. Noh, Y. Kim, V. S. K. Yadav, S. Park, W. Yoon, S. Lee and W. B. Kim, ChemistrySelect, 2017, 2, 6260–6268 CrossRef CAS.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360–6368 CrossRef CAS.
- G. P. Hao, W.-C. Li, D. Qian and A.-H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS.
- H. Yang, Y. Wu, Q. Lin, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Angew. Chem., 2018, 130, 15702–15706 CrossRef.
- R. Shi, J. Zhao, S. Liu, W. Sun, H. Li, P. Hao, Z. Li and J. Ren, Carbon, 2018, 130, 185–195 CrossRef CAS.
- M. V. Morales, E. Asedegbega-Nieto, B. Bachiller-Baeza and A. Guerrero-Ruiz, Carbon, 2016, 102, 426–436 CrossRef CAS.
- Y.-X. Yu, Phys. Chem. Chem. Phys., 2013, 15, 16819–16827 RSC.
- C. Ma, X. Shao and D. Cao, J. Mater. Chem., 2012, 22, 8911–8915 RSC.
- Z.-J. Jiang and Z. Jiang, Sci. Rep., 2016, 6, 27081 CrossRef CAS.
- P. Mondal, A. Sinha, N. Salam, A. S. Roy, N. R. Jana and S. M. Islam, RSC Adv., 2013, 3, 5615–5623 RSC.
- R. Shi, M. Ren, H. Li, J. Zhao, S. Liu, Z. Li and J. Ren, Mol. Catal., 2018, 445, 257–268 CrossRef CAS.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, C.-W. Huang, C.-S. Tan, Y. Li, P. D. Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. D. Luna, P. L. Hsieh, T. Burdyny, H. H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. D. Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2019, 2, 86–93 CrossRef CAS.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. Mckeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS.
- T. A. Maark and B. R. K. Nanda, J. Phys. Chem. C, 2017, 121, 4496–4504 CrossRef.
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS.
- P. Yang, H. Zhuzhang, R. Wang, W. Lin and X. Wang, Angew. Chem., 2019, 131, 1146–1149 CrossRef.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- I. Ledezma-Yanez, E. P. Gallent, M. T. M. Koper and F. Calle-Vallejo, Catal. Today, 2016, 262, 90–94 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. Van der Han, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- H. Xiao, T. Cheng and W. A. Goddard, J. Am. Chem. Soc., 2016, 139, 130–136 CrossRef.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, J. Catal., 2013, 308, 237–249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03088c |
| This journal is © The Royal Society of Chemistry 2020 |