
Electrochemical intramolecular C–H/N–H functionalization for the synthesis of isoxazolidine-fused isoquinolin-1(2H)-ones†
Lin-Bao
Zhang
 ,
Rui-Sen
Geng
,
Zi-Chen
Wang
,
Guang-Yi
Ren
,
Li-Rong
Wen
* and
Ming
Li
*
,
Rui-Sen
Geng
,
Zi-Chen
Wang
,
Guang-Yi
Ren
,
Li-Rong
Wen
* and
Ming
Li
*
State Key Laboratory Base of Eco-Chemical Engineering, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: wenlirong@qust.edu.cn; liming928@qust.edu.cn
First published on 26th November 2019
Abstract
A general and practical protocol for the construction of isoxazolidine-fused isoquinolin-1(2H)-ones has been described by electrochemical-oxidation-induced intramolecular annulation via amidyl radicals. In an undivided cell, isoquinolinones could be easily generated from various available amides bearing CONHOR groups under metal-free, additive-free and external oxidant-free conditions. Moreover, this transformation proceeded smoothly by using cheap 95% ethanol as the green solvent and could be extended to the gram scale.
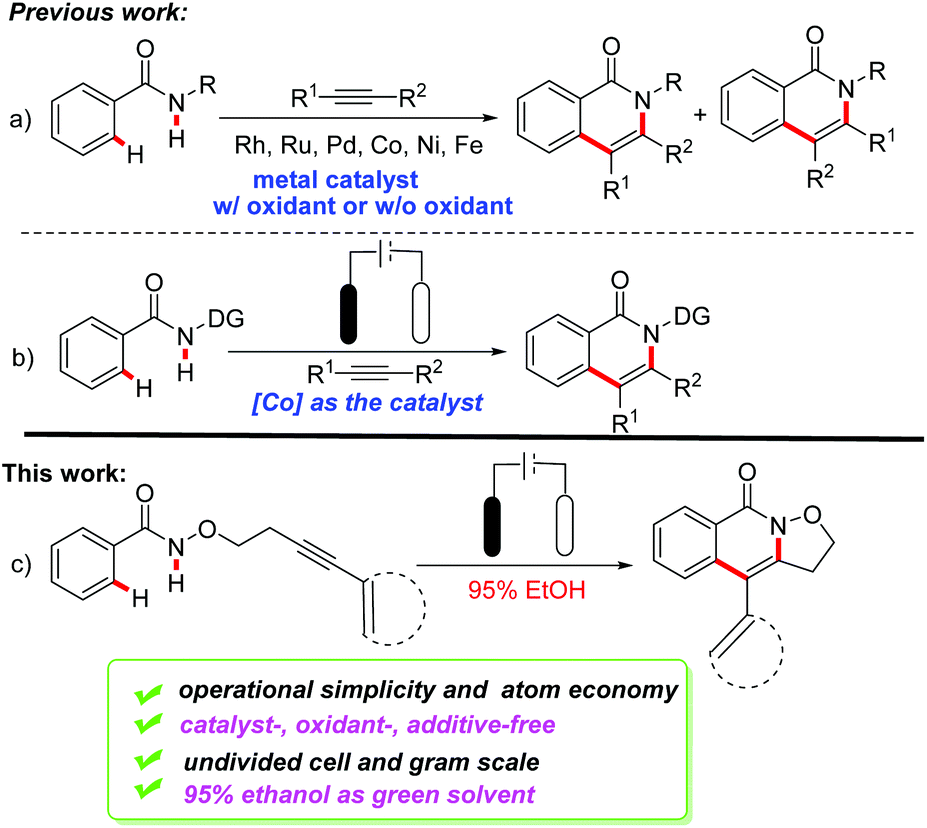
Isoquinolinones are essential frameworks in biologically active natural products, and widely exist in pharmaceutical products.1 Hence the development of their synthetic strategies based on green, sufficient, and sustainable methodologies has drawn great attention. Recently, transition-metal-catalyzed C–H/N–H functionalization of benzamides with alkynes has emerged as a powerful and step-economical strategy (Scheme 1a).2 Among them, the direct synthesis of such scaffolds via noble-metal-catalyzed C–H bond functionalization has been achieved by Rh,2a–i Ru,2j–n and Pd2o–q catalysts. Soon after, Co-, Ni-, Fe- and Cu-catalyzed C–H/N–H bond activation was reported by Daugulis, Chatani Nakamura, and Han.3 Although the reported catalytic systems afforded good yields and eliminated the need for prefunctionalized substrates, these approaches suffered from several limitations, such as the low control of regioselectivity, the use of toxic solvents and the requirement for oxidants and metal catalysts. As a consequence, there is an urgent demand for developing a green and sustainable protocol for the synthesis of isoquinolinones from available building blocks without utilizing a metal catalyst or an oxidant.
 | ||
| Scheme 1 The synthesis of isoquinolinones via C–H/N–H functionalization. | ||
N-Centered radicals (NCRs) are versatile intermediates for the synthesis of N-containing compounds.4 The established approaches for the NCRs required either prefunctionalization of the N atom or the use of strong oxidants. Recent developments have employed photoredox catalysis as a reliable tool for the generation of N-radicals through a mild process.5 Generally, the generation of amidyl radicals is somewhat limited by virtue of the fact that (1) their precursors are difficult to obtain; (2) the homolysis of a strong amidyl N–H bond was difficult due to the high energy barrier (N–H BDFE ≈ 99 kcal mol−1);6 (3) the harsh conditions preclude the compatibility of many functional groups. Recently, Knowles7 and Xu8a–c respectively reported the generation of amidyl radicals using photoredox and electrochemical approaches. Moreover, Xu also reported an electrochemical method for the generation of iminyl radicals under metal-free reaction conditions.8d,e
Electrochemical synthesis has attracted great attention and considerable development has been seen.9 C–H bond functionalization induced by electrochemical-oxidation provided an environmentally-benign strategy in contrast to the traditional synthetic protocol.10 Various heterocyclic compounds have been reported in succession through a range of electrochemical annulations.11 In particular, the groups of Ackermann12a,b and Lei12c have respectively disclosed a similar electrochemistry enabled C–H/N–H functionalization with the assistance of cobalt catalysis in 2018 (Scheme 1b). We have recently developed an AgI-mediated dearomative annulation of indoles via amidyl radicals using CONHOR directing groups.13a Following the electrochemical synthetic strategy, herein we develop an efficient, green and practical electrochemical intramolecular annulation for the synthesis of isoxazolidine-fused isoquinolin-1(2H)-ones through metal-free direct electrolysis. The present work differs from Xu's work as follows: (1) an amidyl radical was generated instead of the iminyl radical shown in Xu's work; (2) two N-heterocyclic motifs were obtained through the radical cascade reactions, in which the C–C bond and C–N bond were formed in one step while benzimidazoles were afforded via C–H/N–H cross-coupling in Xu's work.8d,e It is worth mentioning that the reaction is conducted in 95% ethanol, which is one of the recommended green and cheap solvents (Scheme 1c).14
Initially, N-((4-phenylbut-3-yn-1-yl)oxy)benzamide 1a was chosen as the model substrate for the optimization investigation. The desired annulation product 2a was obtained in 93% yield, when the reaction system was carried out under 2.0 mA constant current in the presence of 1.0 equivalent of n-Bu4NBF4 in refluxing 95% ethanol using an undivided cell for 4 h (Table 1, entry 1). Increasing the current of the cell afforded lower yields probably due to the decomposition of the starting material and the product under high current conditions (Table 1, entries 2 and 3). Replacing the carbon felt electrodes with other carbon or platinum electrodes resulted in decreased yields (Table 1, entries 4–6). As for solvent, MeCN and MeOH could promote the transformation but exhibited relatively lower efficiency compared with EtOH (Table 1, entries 7–9). In particular, only a trace amount of product was obtained in the absence of n-Bu4NBF4 (Table 1, entry 10). The control experiments indicated that it was as necessary as conducting the electrolysis under a nitrogen atmosphere and electricity was essential for the reaction (Table 1, entries 11 and 12).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), n-Bu4NBF4 (0.1 mmol), 95% EtOH (5.0 mL) in an undivided cell with a carbon felt anode and cathode (2.0 cm × 1.0 cm × 0.5 cm), constant current = 2.0 mA, nitrogen, reflux, 4 h, (3 F). b Isolated yield of 2a. n.r = no reaction. | ||
| 1 | None | 93 |
| 2 | 5 mA | 62 |
| 3 | 3 mA | 75 |
| 4 | Pt(+)|Pt(−) | 40 |
| 5 | C(+)|C(−) | 63 |
| 6 | C(+)|Pt(−) | 65 |
| 7 | MeCN as the solvent | 81 |
| 8 | MeOH as the solvent | 85 |
| 9 | EtOH as the solvent | 90 |
| 10 | Without an electrolyte | Trace |
| 11 | Under air | n.r |
| 12 | No electric current | n.r |

With the optimized conditions in hand, we investigated the substrate scope of alkyne-tethered N-alkoxyamides (Table 2). The tolerance of the functional group at the amide moiety was first explored and the corresponding products were obtained in 35%–99% yields. para-Substituted benzamides with electron-withdrawing and electron-donating groups were well compatible in the electrochemical C–H/N–H activation with good to excellent yields. For meta-substituted benzamides, the reaction dominantly took place at the less hindered position of aromatic rings with excellent yields (2k and 2k′; 2l and 2l′). The ortho-substituted functional groups of amides have not shown an obvious inhibitory effect on the transformation, providing the desired products 2m–2p in moderate to good yields. The reaction of ditrisubstituted amides proceeded smoothly to give the corresponding products (2q and 2r) in good yields. In particular, the protocol was also appropriate for the piperonyl amide 1s, furnishing a separable mixture at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.7 ratio with an almost quantitative yield. The naphthyl amide 1t successfully reacted at the 2-position to deliver product 2t in 95% yield.
:0.7 ratio with an almost quantitative yield. The naphthyl amide 1t successfully reacted at the 2-position to deliver product 2t in 95% yield.

Moreover, amide 1aq bearing the easily oxidizable functional group SMe was also tolerated under electrochemical conditions. Notably, the valuable electrophilic halogen groups, such as aryl bromide or aryl iodine were tolerated, providing the possibility of further transformation.
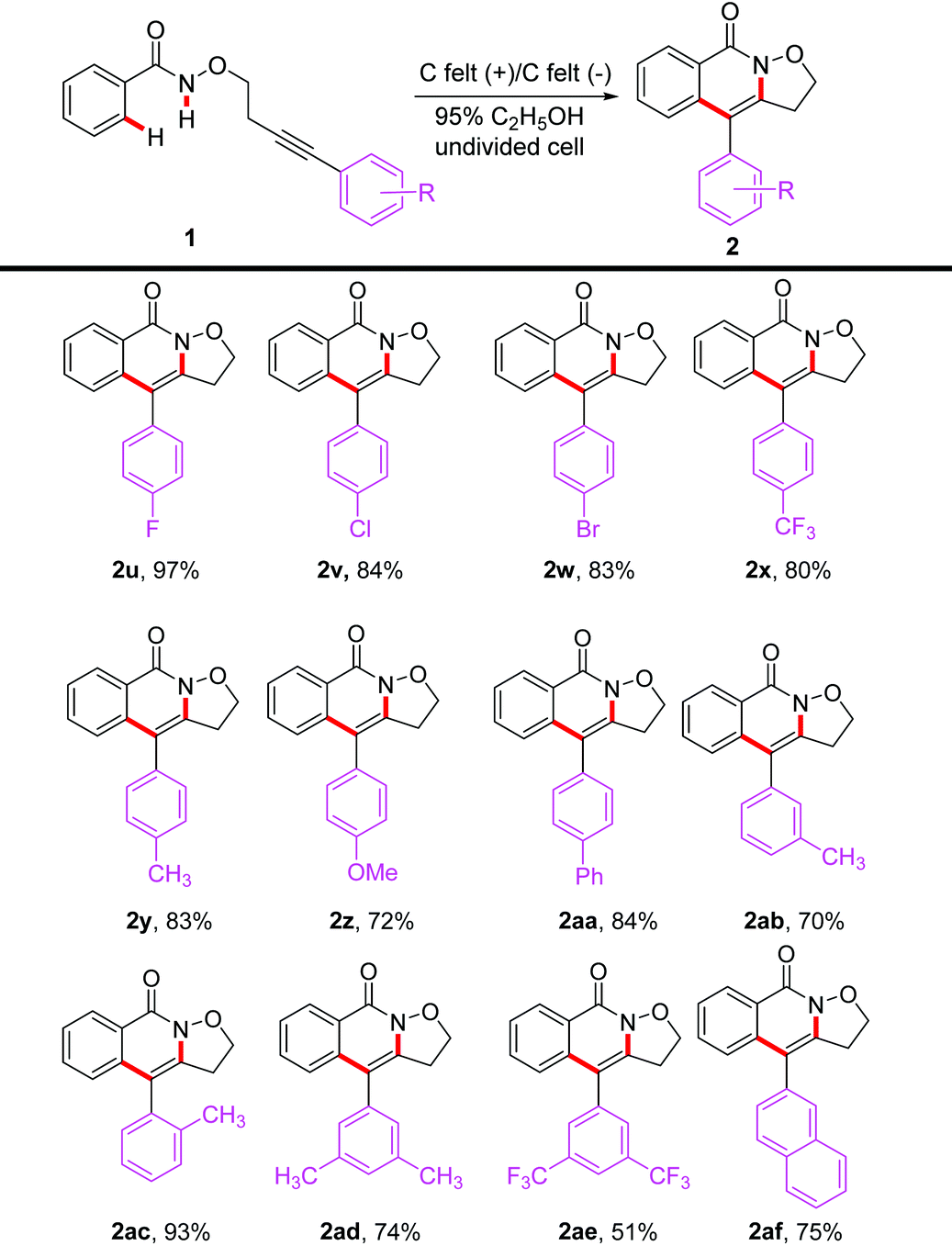
Next, we studied the compatibility of the alkyne moieties (Table 3) and found that the reaction of N-alkoxybenzamides bearing various substituents including halogen, methyl, methoxy, tert-butyl, trifluoromethyl, phenyl, and naphthyl on the aromatic ring of the alkyne moiety proceeded smoothly under the optimal reaction conditions with an excellent reaction efficiency. Generally, the electron-withdrawing groups at the para-position of the phenyl ring had a better performance in contrast to the electron-donating substituents (2u–2xvs.2y–2aa). The amide substrates (1ab and 1ac) with methyl at the meta- or ortho-position of phenyl were also good candidates for the transformation. The annulation strategy was also compatible with ditrisubstituted phenyl alkyne tethered N-alkoxybenzamides, as shown in the cases of 2ad and 2ae. 2-Naphthyl alkynyl tethered N-alkoxyamide could undergo the transformation well, providing 2af in 75% yield.
Subsequently, other conjugated substituents on the alkyne moiety were also investigated in the intramolecular electrochemical C–H/N–H transformation and the results are presented in Table 4. The ene-ynebenzamides, which are obtained via the Sonogashira coupling reaction, could proceed successfully under electrolytic conditions by adjusting the current to 0.5 mA, furnishing the corresponding products 2ag–2al in 58%–78% yields. Notably, the fluorinated alkenyl-substituted product 2al could be obtained in an acceptable yield in this electrochemical reaction. In addition, the sensitive alkene moiety of the products was left intact under electrochemical-oxidation reaction conditions. Moreover, with regard to diyne-containing benzamide, the electrochemical reaction could proceed well to furnish 2am in 72% yield. In addition, benzamides bearing three carbon atoms between the oxygen atom and the alkyne were also investigated under 2 mA electrolytic conditions. Gratifyingly, the representative oxazinane-fused isoquinolin-1(2H)-ones 2an–2ap were smoothly obtained in 82%–87% yields.

In order to demonstrate the practicability of the electrochemical strategy, a gram-scale reaction of alkyne-tethered benzamide 1a was performed on the 5 mmol scale (Scheme 2). The desired product 2a was finally obtained in 86% yield through prolonging the reaction time to 120 h, which showed the potential application of the electrochemical annulation strategy.
 | ||
| Scheme 2 Gram scale experiments. | ||
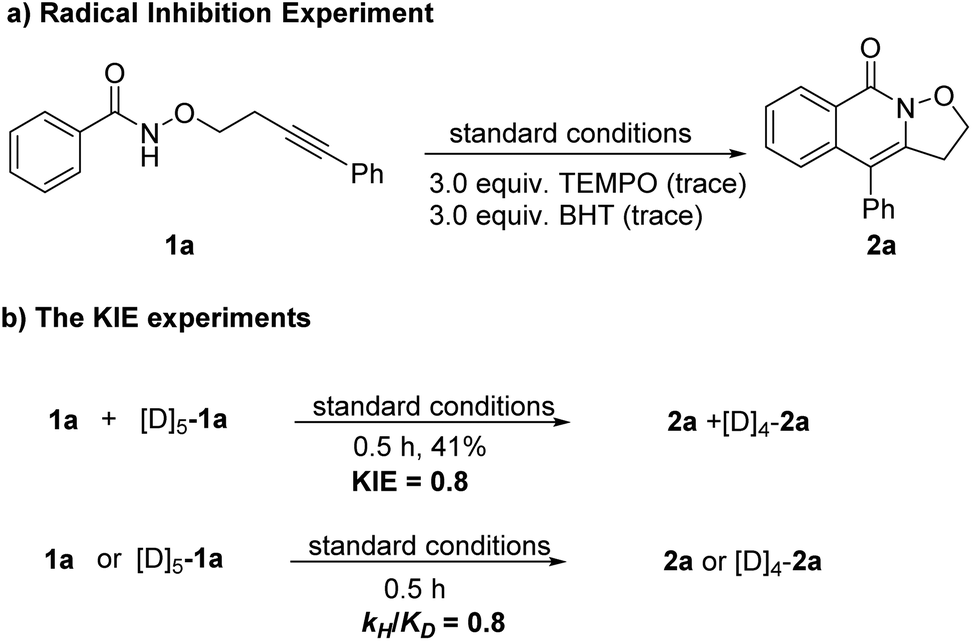
To gain insight into this reaction mechanism, a series of control experiments were carried out (Scheme 3). The addition of a radical scavenger, namely 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylhydroxytoluene (BHT) to the reaction system completely inhibited the transformation, indicating that a radical process might be involved (Scheme 3a). The lack of a KIE (∼0.8) was observed in the competition reaction of 1a and [D5]-1a in one vessel under the standard reaction conditions. Moreover, a similar result was obtained from two parallel reactions (Scheme 3b).
 | ||
| Scheme 3 Control experiments. | ||
The cyclic voltammetry experiments were carried out in EtOH (Fig. 1). A significant oxidation peak at 1.5 V of the background was observed, which suggested that ethanol might be oxidized at about 1.5 V. The oxidation potential of 1a and 1ag is about 0.8 V, which is much lower than that of ethanol. As a consequence, amides 1 were oxidized through anodic oxidation under the electrochemical conditions, while the reduction of ethanol forms ethoxide EtO- and H2 on the cathode surface.
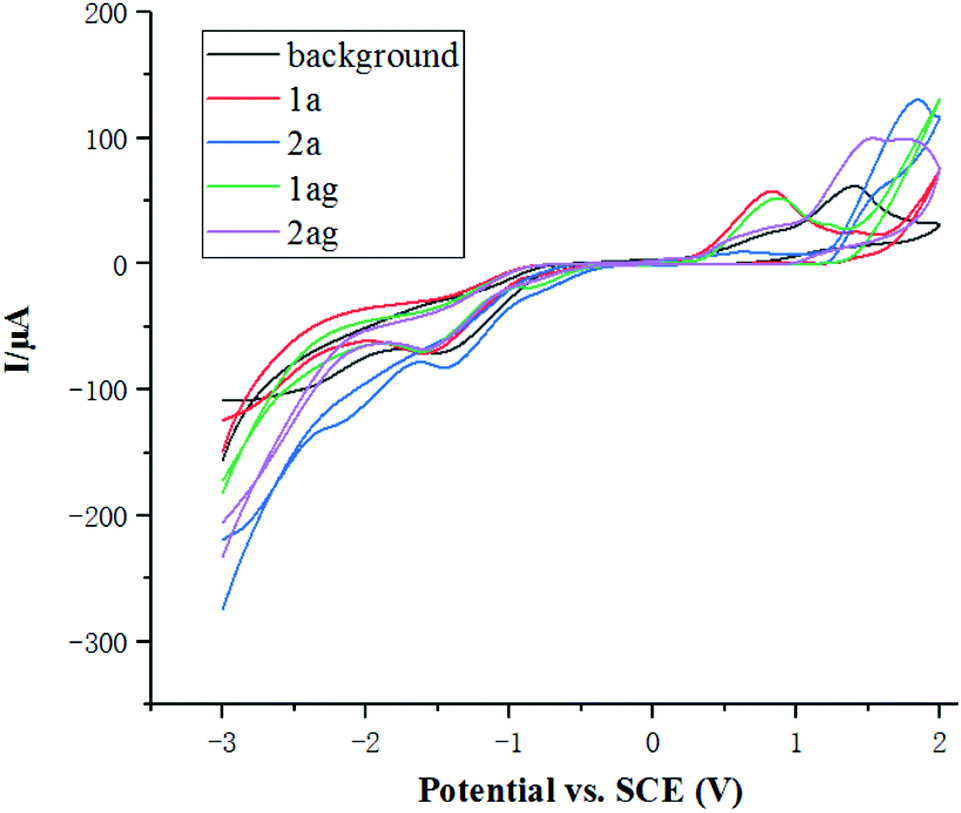 | ||
| Fig. 1 Cyclic voltammograms of related compounds in 0.02 M nBu4NPF6/EtOH using a glassy carbon electrode as the working electrode, and Pt wire and SCE as counter and reference electrodes, respectively, at 100 mV s−1 (scan rate: background): nBu4NPF6 (0.02 M). (1a): 1a (0.02 M), nBu4NPF6 (0.02 M). (2a): 2a (0.02 M), nBu4NPF6 (0.02 M). (1ag): 1ag (0.02 M), nBu4NPF6 (0.02 M). (2ag): 2ag (0.02 M), nBu4NPF6 (0.02 M). | ||
Based on the control experiments, DFT calculations (see the ESI†) and previous reports,8 a plausible mechanism is proposed in Scheme 4. Firstly, the cathodic reduction of ethanol forms ethoxide EtO-, which deprotonates substrate 1a to generate the anion A, subsequently, single-electron-transfer (SET) oxidation of A occurs to produce N-centered radical B through anode oxidation. B participates in the 5-exo-dig annulation to give the vinyl radical C, which proceeds to a second annulation to afford the delocalized radical D. Finally, the desired product 2a is formed through the rearomatization of D by electron oxidation.
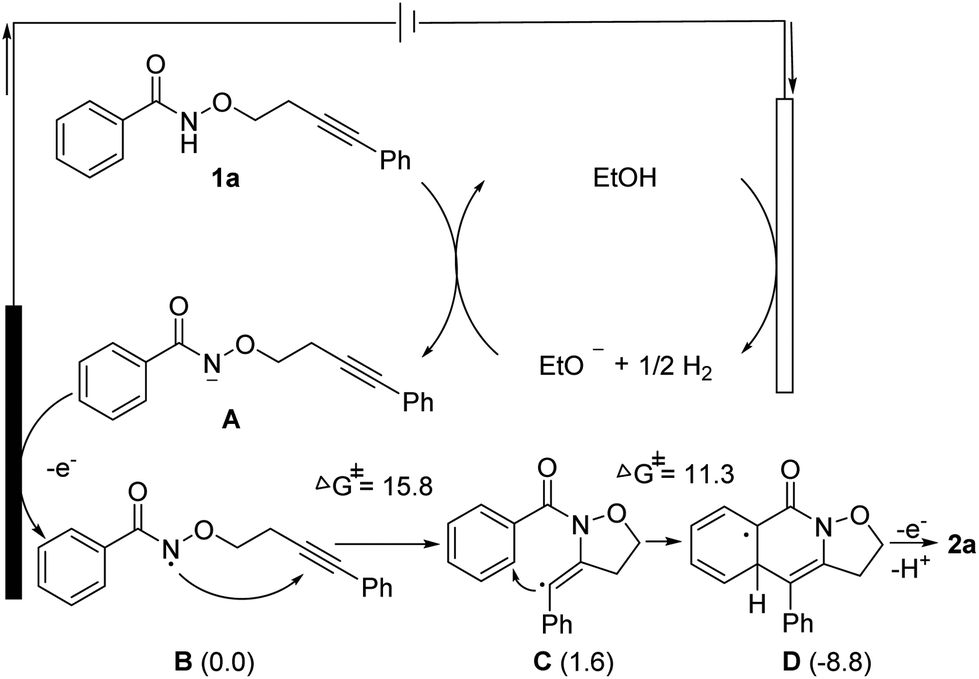 | ||
| Scheme 4 Proposed mechanism. The Gibbs free energies (kcal mol−1), shown within parentheses, are those relative to B and were calculated using DFT. | ||
Conclusions
In conclusion, we have demonstrated electrochemical-oxidation-induced intramolecular annulation for the synthesis of isoxazolidine-fused isoquinolin-1(2H)-ones via amidyl radicals. In an undivided cell, isoquinolinones could be easily obtained from various available amides bearing CONHOR groups by using 95% ethanol as the green solvent. This dehydrogenative protocol for the construction of isoquinolinone derivatives exhibits a broad substrate scope and operational simplicity under metal-free and external-oxidant-free reaction conditions. In addition, this reaction proceeded under mild conditions and could be performed on the gram scale. Further efforts to develop more electrochemical-oxidation transformations are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21801152 and 21572110) and the Natural Science Foundation of Shandong Province (ZR2019BB005 and ZR2019MB010).Notes and references
-
(a) V. A. Glushkov and Y. V. Shklyaev, Chem. Heterocycl. Compd., 2001, 37, 663 CrossRef CAS
; (b) G. R. Pettit, Y. Meng, D. L. Herald, K. A. N. Graham, R. K. Pettit and D. L. J. Doubek, Nat. Prod., 2003, 66, 1065 CrossRef CAS PubMed
-
(a) N. Quiñones, A. Seoane, R. García-Fandiño, J. L. Mascareñas and M. Gulías, Chem. Sci., 2013, 4, 2874 RSC
-
(a) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef CAS
-
(a) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069 RSC
-
(a) L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607 CrossRef CAS PubMed
- J. P. Cheng and Y. Y. Zhao, Tetrahedron, 1993, 49, 5267 CrossRef CAS
-
(a) D. C. Miller, G. J. Choi, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 13492 CrossRef CAS
-
(a) L. Zhu, P. Xiong, Z. Y. Mao, Y. H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226 CrossRef CAS
-
(a) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS PubMed
-
(a) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769 CrossRef CAS
-
(a) X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370 CrossRef CAS
-
(a) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383 CrossRef CAS PubMed
-
(a) L.-B. Zhang, M.-H. Zhu, S.-F. Ni, L.-R. Wen and M. Li, ACS Catal., 2019, 9, 1680 CrossRef CAS
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, A. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03290h |
| This journal is © The Royal Society of Chemistry 2020 |