
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvanced 96-microtiter plate based bioelectrochemical platform reveals molecular short cut of electron flow in cytochrome P450 enzyme†
Ronny
Frank
 a,
Christoph
Prönnecke
a,
Ronny
Azendorf
a,
Heinz-Georg
Jahnke
a,
Annette G.
Beck-Sickinger
b and
Andrea A.
Robitzki
*a
a,
Christoph
Prönnecke
a,
Ronny
Azendorf
a,
Heinz-Georg
Jahnke
a,
Annette G.
Beck-Sickinger
b and
Andrea A.
Robitzki
*a
aCentre for Biotechnology and Biomedicine, Molecular biological-biochemical Processing Technology, Leipzig University, Deutscher Platz 5, D-04103 Leipzig, Germany. E-mail: andrea.robitzki@bbz.uni-leipzig.de; Fax: +49 341 9731249; Tel: +49 341 9731241
bInstitute of Biochemistry, Leipzig University, Brüderstraße 34, D-04103 Leipzig, Germany
First published on 20th March 2020
Abstract
In bioelectrocatalysis, immobilised redox enzymes are activated in a bioelectronic interface without redox equivalents such as NADPH, thus enabling heterogeneous flow chemistry. The functional contact between enzyme and electrode requires a high degree of optimisation regarding choice of electrode material, electrode pre-treatment, enzyme immobilisation and reaction conditions. So far, however, there are no systems that can easily enable an optimisation procedure at a higher throughput. Here, we present an advanced platform with a vertical divided cell architecture in conjunction with a developed 96-multipotentiostat to be able to drive redox enzymes in 96 well microtiter plate based multielectrode arrays. This platform controls 96 independent three-electrode setups with arbitrary working electrode materials. We demonstrate its applicability in a mutation study of cytochrome P450 BM3 using indium tin oxide as electrode material and the 7-ethoxycoumarin product quantification assay. We show that the bioelectrocatalytic activity of P450 BM3 can be amplified when the cofactor FAD is erased from the enzyme by a single point mutation, so that FMN becomes the first electron entry point. Bioelectrocatalysis thus offers an approach to enzyme simplification as a remedy for the inherent instability of self-sufficient cytochrome P450 enzymes. In addition, we examined native and artificial enzyme activation with respect to ionic strength and buffer composition. The optimal conditions of the activation types differ substantially from each other and exhibit a new molecular facet in enzyme characteristics. In a proof-of-principle we demonstrate that the platform is also compatible with raw cell extracts, thus opening the door for random mutagenesis screenings.
1. Introduction
Enzymes are widely used as biocatalysts in the feed, food and technical industries.1–3 Their ability to catalyse specific chemical reactions under ambient conditions qualifies them especially with regard to green chemistry for the production of fine chemicals and pharmaceuticals. However, considering the available large variety of enzymes, only a minor part is suitable to be adapted for an industrial application. At present, enzymes from the group of hydrolases, which are used for the degradation of natural polymers, dominate in industrial biotechnology, while the group of oxidoreductases, which catalyse electron transfer reactions, are underrepresented.4 A decisive reason for this is the generally higher complexity of oxidoreductases, which can be seen, for example, in the dependence on redox cofactors and unstable and expensive redox mediators such as NADH or NADPH. There are several methods for circumventing native redox mediators, which are, however, primarily only suitable at a laboratory level.4 One of the promising techniques applicable to larger scale is direct electrode enzyme regeneration in which electrons are transferred directly to bound cofactors to operate the enzyme bioelectrocatalytically. Due to the necessary immobilisation on electrode surfaces, this method is directly compatible with flow reactor systems.Cytochrome P450 enzymes are a much-noticed subgroup of oxidoreductases. They are able to selectively derivatise thermodynamically indistinguishable and inert C–H bonds, which is hardly and sometimes not at all feasible using classical chemistry.5,6 However, in addition to redox mediator dependency, these enzymes are limited in terms of stability, solvent tolerance and often low activity to be used industrially.7,8 Thus, the performance and robustness of cytochrome P450 enzymes have to be improved for biotechnological use. Unfortunately, the simultaneous optimisation of qualitatively different enzyme characteristics has so far posed a major challenge,9 especially when bioelectrocatalytic product formation should be used as processing or evolutionary parameter.
Recently, we demonstrated the bioelectrocatalytic activation of cytochrome P450 BM3 immobilised on indium tin oxides electrodes.10 P450 BM3 is a 119 kDa class VIII self-sufficient bacterial flavocytochrome in which the cytochrome domain is naturally fused to a cytochrome-reductase domain that resembles the eukaryotic cytochrome P450 reductase.11 P450 BM3 stands out by one of the highest turnover numbers for ω-hydroxylation of fatty acids and epoxidation of arachidonic acid as well as for its simple recombinant expression.12 It is not only one of the best studied cytochrome P450 enzymes, but also a valuable resource for pharmaceutical purposes and research.13–15 From a mechanistic point of view, the native electron transfer in P450 BM3 is quite well understood.12,16–19 Electrons are transferred from NADPH to the non-covalent bound cofactor FAD and then forwarded to the heme cofactor by a shuttle subdomain containing FMN. Recently, we developed rationally designed P450 BM3 variants that showed significantly enhanced bioelectrocatalytic activity due to an introduced surface-affine peptide tag that improved enzyme orientation on the electrode surface.20 However, a major limitation for further quantitative investigation was the number of independent and parallel-addressable three-electrode setups to ensure equivalent test conditions for all samples. Therefore, to execute efficient laboratory-scale library screening and enzyme development strategies a considerably extended system of 96 parallel potentiostats is required. In view of the effort associated with a 96-multi-potentiostat system, alternatives to potentiostat driven systems that use the 96 well microtiter-plate format have been described. Typically this includes the mediated electron transfer, e.g. in case of cytochrome P450 BM3 using zinc dust as an electron source and cobalt sepulchrate as a mediator.21,22 The adjustment and variation of the electrochemical potentials for mechanistic studies is however not straightforward in these mediated systems. Described 96 well microtiter plate configurations that use potentiostats e.g. for direct electron transfer, have so far been limited in that they either work sequentially by multiplexing23 or only work with rare measuring methods in a two-instead of a typical three-electrode configuration.24,25 From a technological point-of-view, no 96-multi-potentiostat-multi-electrode-system is currently available for bioelectrocatalytic screening of immobilised redox enzymes that especially addresses the need for product quantification, combinability with optical methods and microtiter-plate based constraints.
This work introduces a fully parallelised 96 well microtiter plate based bioelectrochemical screening platform including a new developed 96-multi-potentiostat unit to investigate the bioelectrocatalytic activity of cytochrome P450 BM3 in conjunction with a photonic readout (Fig. 1). This platform marks a major improvement compared with our previous 12-potentiostat system, having not only eight times more potentiostats integrated in a portable unit but also a new 96 well electrode architecture. The new system is built on three separate modules that are simply stacked on each other to generate 96 completely independent three-electrode setups in vertically divided cell configuration. The first module is a modified standard 96 well microtiter plate with multi electrode array as bottom to provide 96 working electrode chambers. The second module is a 3D-printed unit that provides 96 partner chambers in the divided cell architecture and connects these by agarose bridges to the first module. The third module is the multi-potentiostat-controlled electrode main module through which counter and reference electrodes are supplied and to which the working electrodes are connected.
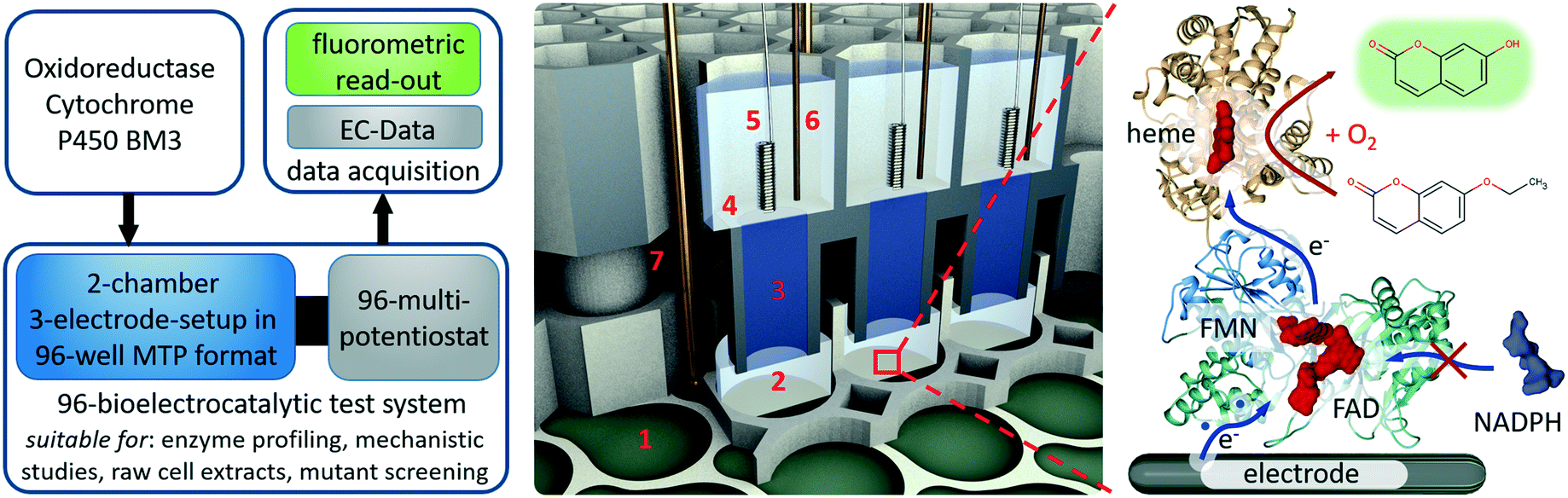 | ||
| Fig. 1 Concept of the developed 96 well bioelectrochemical analysis system. Left pipeline for testing new enzyme mutants and for profiling enzyme properties using the developed 96-bioelectrocatalytic system (MTP = microtiter plate, EC = electrochemical) middle design of the 96 well two-chamber three-electrode setup. The system is composed of three modules. Module 1: 1–2 = 96-microtiter plate with working electrode array (1) and the bottom chambers (2); module 2: 3–4 = 3d-printed unit to provide 96 agarose bridges (3) and 96 top chambers (4); module 3: 5–7 = the electrode main module carries the counter electrodes (5), reference electrodes (6) and the spring-loaded probes (7) to contact the working electrodes. Right bioelectrocatalysis: modelled structure of P450 BM3 showing the cofactors and substrate turnover for fluorometric product quantification (for illustration only). P450 BM3 catalyzes hydroxylation reactions under consumption of oxygen. | ||
In order to show the applicability of the developed platform to understand the bioelectrocatalytic process of redox enzymes, we investigated flavine-deficient mutants of the cytochrome P450 BM3 to find the electron entry point. Since the artificial enzyme activation can differ significantly from the native process, we demonstrate the suitability of the platform for mapping relevant reaction conditions. In addition, we demonstrate in a proof-of-concept the applicability for library screening purposes through compatibility with raw cell extracts.
2. Experimental section
2.1. Fabrication of 96 well multi-electrode array
Indium tin oxide working electrodes were produced via lift-off technique in a cleanroom (class 1000) using borosilicate plates with dimensions of 113.5 × 75 × 1 mm3 (Goettgens Industriearmaturen; Germany). Glass plates were cleaned with piranha solution, structured with positive photo resist AR-P 3510 (Allresist, Germany) and sputtered with 300–500 nm indium tin oxide (Sindlhauser Materials GmbH, Germany) in a CREAMET 500 (Creavac, Germany). Indium tin oxide electrodes were annealed at 500 °C for 10 min. The sheet resistance was in the range of 19–38 Ω sq−1 (more details can be found in the ESI,† Fig. S1).The prepared electrode array was bonded to the bottom of a milled black polypropylene microtiter plate (Nunc™ MicroWell™, Thermo Fisher, Germany) using epoxy resin EPO-TEK 302-3M (Epoxy Technologies, Germany) in a self-built bonding device (Fig. S2†).
On the day of use, the 96 well indium tin oxide array was cleaned with a RCA-1 solution of H2O2(30%)/NH3(25%)/H2O (v/v/v 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5) for 15 min at room temperature, afterwards 10 min in 1 M hydrochloric acid and another 5 min in H2O/H2O2/NH3 solution. ITO electrodes were stored in ultrapure water until use to avoid recontamination.
:1:5) for 15 min at room temperature, afterwards 10 min in 1 M hydrochloric acid and another 5 min in H2O/H2O2/NH3 solution. ITO electrodes were stored in ultrapure water until use to avoid recontamination.
Morphology of the ITO electrodes was investigated by atomic force microscopy using a JPK Nanowizard III (Bruker) in intermittent-contact mode (Fig. S3†). The mean RMS surface roughness of ITO electrodes was 4.6–4.7 nm.
2.2. Design and fabrication of the 288-electrode main module
The body of the electrode main module was designed in Inventor Professional 2016 and produced by additive manufacturing using an Ultimaker 3 extended with the Ultimaker PLA. Counter electrodes were made by coiling platinum wire (d = 0.3 mm, L = 5 cm, Chempur GmbH, Germany), which was soldered to a contact pin. Reference electrodes were custom-made Ag/AgCl electrodes generated from a silver wire (d = 0.8 mm, L = 2.45 cm) by galvanisation in potassium chloride. In order to contact the 96 working electrodes, spring loaded probes of a length of 45 mm and a hub of 4 mm were soldered on a double contact pin. In order to precisely position and align the spring-loaded probes to the microtiter plate, 4 × 4 well PLA stabiliser were inserted, which were also produced by additive manufacturing. The main module is connected via LSHM plugs (Samtec) to the 96-multi-potentiostat.2.3. Design and fabrication of the 96 well top chamber module
The top chamber module establishes 96 top chambers for the counter and reference electrodes, and connects these via an agarose cylinder to the 96 bottom chambers given by the microtiter plate. The structure was designed in Inventor Professional 2016 and 3d-printed vertically on a Lulzbot Mini using natural PLA (eSun). Each cylinder has an ellipsoid base (major axis 4.1 mm, minor axis 3.3 mm) and a length of 10 mm.2.4. Development of the 96-multi-potentiostat
The De-Ford-type potentiostat circuit was used as starting point. COM-board development and potentiostat circuit simulations were performed with PSPICE (Cadence Design Systems) and Mulisim (National Instruments). PCB layouts were built in EAGLE (Cadsoft), fabricated by Multi Circuit Boards and equipped by Sinus Messtechnik GmbH. Electronic components (e.g. microcontroller, AD/DA-converter, operational amplifier) were purchased from Mouser Electronics Inc., RS Components GmbH and Digi-Key Electronics. The chassis of the multi-potentiostat was designed in Inventor Professional 2016 (Autodesk) and produced by CNC Future Technics GmbH.2.5. Experimental procedure of the bioelectrocatalytic assay
Cloning and expression of P450 BM3 variants in E. coli is described in the ESI.† To avoid batch variability between single experiments, raw cell pellets from one expression were aliquoted to 0.3 g units and stored at −80 °C. In order to prepare the enzyme solution for bioelectrocatalytic activity testing, these cell pellets aliquots were thawed on ice and resuspended in 800 μL PBS (pH 7.2) with addition of protease inhibitor cocktail (Roche). The cells were disrupted by sonication (3 × 60 s, 30 s pause interval) with a UP50H sonicator (Dr. Hielscher GmbH, 0.5 cycle, 40% amplitude) on ice and centrifuged for 10 min at 16000 × g (Biofuge pico, Heraeus). The supernatant was further cleaned using a 0.45 μm PES filter (TPP). Thus prepared raw cell extract was always kept on ice and only used on the day of preparation. Purified P450 BM3 was generated from raw cell extract by IMAC using the C-terminal His-tag as described in the SI. Protein concentration of potentially active enzyme was assessed by CO-assay.26
Agarose bridges, which connect each chamber pair, were prepared in one step by immersion of the 96 well top chamber module in 4% agarose with 0.15 M KCl solution. After cure, the bottom of the bridges were placed in 0.15 M KCl solution of a 96-microtiter plate and 40 μL of 0.15 M KCl solution were sited on top of the agarose bridges to avoid shrinking during storage time. In order to maximize the time of electrolyte concentration stability in the reaction chamber, the 1 M KCl solution in the top chamber was added just in time, when the testing plate was ready for measuring.
The purified P450 BM3 was immobilised on the ITO electrode array from 60 μL of a 500 nM solution to which 2 μM FAD and 2 μM FMN were added. Immobilisation was performed by adsorption at 4 °C for 30 min. Stocks of the raw cell extracts were diluted by factor of 50 or higher to immobilise the unpurified P450 BM3. Afterwards, each well of the 96-microtiter plate was washed four times with 100 μL PBS (+2 μM FAD, +2 μM FMN). In case of testing different buffer compositions, washing was performed two times with PBS and two times with the buffer of interest. The top chambers were filled with 1 M KCl solution and the washing solution of the bottom chamber was replaced by 200 μL reaction solution, containing additional 1 mM 7-ethoxycoumarin (Alfa Aesar, Germany) and 1% DMSO. The modules were assembled and the reaction was started at −0.8 V for 30 min. After the reaction time, the setup was disassembled and 180 μL of the reaction solution were pH-shifted in 120 μL of 1.5 M glycine buffer (pH 10.2) to measure the fluorescence at Ex: 380/10 nm/Em: 465/35 nm (TECAN, F200).27 Procedure to assess enzyme activity with NADPH is described in the ESI.†
2.6. Software for potentiostat control
In order to control all 96 potentiostat modules, a configuration interface was generated in LabView 2010, in which each potentiostat module can be individually programmed and electrochemical data can be recorded and presented.2.7. Statistics
Graphpad Prism 5 was used for all statistical analysis. Presented graphs are given as mean ± SEM unless stated otherwise. Each experiment consisted of at least three replicates and sum of experiments is given as “n”. Significance of mean difference was analyzed by two-way ANOVA and Bonferroni post hoc test, considering *, P < 0.05 as significant, **, P < 0.01 very significant and ***, P < 0.001 extremely significant.3. Results and discussion
3.1. Concept and structure of the 96 well bioelectrocatalytic platform
Enzyme activation by direct electron transfer is artificial in its nature. Thus optimization of reaction conditions can severely differ regarding its results from the native reaction pathway. Given that, it is pivotal to adapt enzyme features directly at the site of operation. For direct electron transfer, this approach is typically limited by the number of available potentiostats and a manageable parallel setup assembly process (e.g. concerning space requirements, cable harnesses, surface pretreatments etc.).Here, we designed a 96 well divided cell three-electrode setup in conjunction with a 96-multi-potentiostat in order to be able to assess the bioelectrocatalytic activity of immobilised cytochrome P450 BM3 mutants in 96 well microtiter plate format (Fig. 1). The platform can be used to evaluate P450 BM3 mutants from raw cells extracts for library screening purposes or to profile single P450 BM3 mutants regarding e.g. pH, buffer concentration, additives and activity at certain electrochemical potentials. While the 96-multi-potentiostat drives the enzymes and records electrochemical data, the product turnover is measured fluorometrically using the 7-ethoxycoumarin/7-hydroxycoumarin (7-EC/7-HC) end-point assay, although also other fluorescent substrates such as benzyloxyresorufin can be applied. Unfortunately, the activity of the cytochrome P450 BM3 enzyme cannot be directly determined from the current signal of the multi-potentiostat. Due to the oxygen reduction reaction, which occurs concomitantly to the enzyme activation during cathodic polarization, the influence of oxygen on the measurement signal is too high.28 However, since oxygen is required for the enzymatic reaction, it cannot be omitted either. This is the reason we chose product quantification by fluorescence to determine the bioelectrocatalytic activity. Thus, we are measuring the functional electron transfer, which leads to product formation, as opposed to an electrochemical signal that could originate from free cofactor.29 Given that fluorescence is quite specific in terms of excitation and emission spectra, there are no cross-sensitivities to be concerned of. Due to the fact that the product quantification is performed fluorometrically, the counter electrode had to be spatially separated from the working electrode to a top chamber as otherwise the enzymatic product would be destroyed because of oxidative potentials during bioelectrocatalysis. Since all wells of the microtiter plate carry a working electrode filling the entire bottom, we provided the top chamber in a vertical construction. This way we avoided interconnection of two adjacent wells in the microtiter plate, which would result in only 48 three-electrode setups. For this purpose we designed a top chamber module that contained separate chambers for all 96 counter electrodes and connections to the working electrode chambers. These connections enable us to use agarose as a replaceable salt bridge and convection barrier.
The working electrode array consists of 96 pear-shape planar electrodes (Fig. 2A1). Each working electrode includes a section for enzyme immobilisation of the size of 44 mm2 and a section for electrical contacting. The section for enzyme immobilisation takes up the entire bottom of a chamber to achieve maximum sensitivity. Due to the short electrical path length between these two sections also semi-conductive materials such as indium tin oxide can be used. Furthermore, given the planar structure of the working electrodes, they can be produced on arbitrary substrates with techniques such as printing, sputtering or chemical vapour deposition. After bonding to an adapted 96 well microtiter plate, the electrode surface can further be modified dependent on the application of interest. The required 96 well microtiter plates allow electrical contacting of the working electrodes by providing a passage for spring-loaded probes between the adjacent chambers (Fig. 2A2). Although it is in principle possible to use additive manufactured microtiter plates e.g. from natural PLA, we generally observed higher variances in the bioelectrocatalytic assay than with adapted commercial 96 well microtiter plates. This is probably caused by the rougher surface structure due to the fused filament fabrication process, resulting in unequal washing efficiencies during the enzyme immobilisation in contrast to the high quality surface of microtiter plates produced by injection molding.
 | ||
| Fig. 2 96-Microtiter plate based bioelectrochemical analysis system. A1 geometry of single working electrodes (i: area for electrical contacting, ii: electrochemical active area). A2 geometry of the 96-microtiter plate (arrow indicates gap necessary for electrical contacting). B1 and B2 model of the 96-top chamber module. C1 and C2 overview and detail image of the electrode main module. D1–D3 assembly of the system. In D1, module 1 (microtiter plate with multi electrode array) is placed in the stacking device. In D2, module 2 is placed on top and vertically positioned by four nuts. In D3, module 3 is inserted (shown without cables). D4 system in working state incl. splash guard. D5 system in explosive presentation. E1–E3 detail images of the assembly, the agarose bridges and the spring loaded probes contacting transparent ITO electrodes. | ||
In order to generate top chambers for the counter electrodes, we produced a reusable 96 well top chamber module by additive manufacturing (Fig. 2B). A single top chamber has a volume of 400 μL and includes an open cylindrical structure on the bottom which is freshly filled with agarose for an experiment. The agarose cylinders have an average length of 8 mm and can easily be ejected after an experiment (Fig. S4†). The top chamber module also allows passage of the spring loaded probes.
In order to perform an assay in a 96 well microtiter plate, 96 three-electrode setups are realised from a single electrode main module, which is comprised of the 96 counter and 96 reference electrodes as well as 96 spring loaded probes that contact the working electrodes in the microtiter plate (Fig. 2C). These 288 units are plugged to sockets in the electrode main module and can easily be replaced. All sockets are soldered to a printed circuit board, which allows the connecting of all electrodes with the 96-multi-potentiostat (Fig. 2D5). In order to reduce the degrees of freedom of the long spring contact probes, additive manufactured alignment stabilizers were used in six 4 × 4 matrices.
The complete system is assembled by stacking the modules on each other, starting with the bottom chamber module (96 well microtiter plate with the multi electrode array), the top chamber module and finally the electrode main module (Fig. 2D1–D3). In a stacking device, screws help to guide the modules, while nuts are used to define the final positions and to generate the pressure of the spring contact probes on the working electrodes (Fig. 2D4). The printed circuit board translates all electrode positions into four LSHM plugs, which are connected by flexible flat band cables to the multi-potentiostat. The assembly is completed in approx. one minute. Although the insertion of the contact pins seems tricky, little training is required due to the low tolerances of the device.
The multi-potentiostat consists of 96 De-Ford type potentiostat modules30 arranged in two clusters of 48 modules on two communication boards, which are referred to as COM-boards (Fig. 3A and B). Each COM-board is connected via a USB port to a PC. The main purpose of a single COM-board is to transfer configuration settings to and to receive data from each of the 48 potentiostat modules attached to it (Fig. S5A†). Each module is assigned a specification number in a protocol and its data is sent to the user interface during a measurement in a constant stream to display live data. In addition, the COM-board and its firmware were developed to communicate with other types of measurement modules such as impedance analysers or analog data converters in the future.
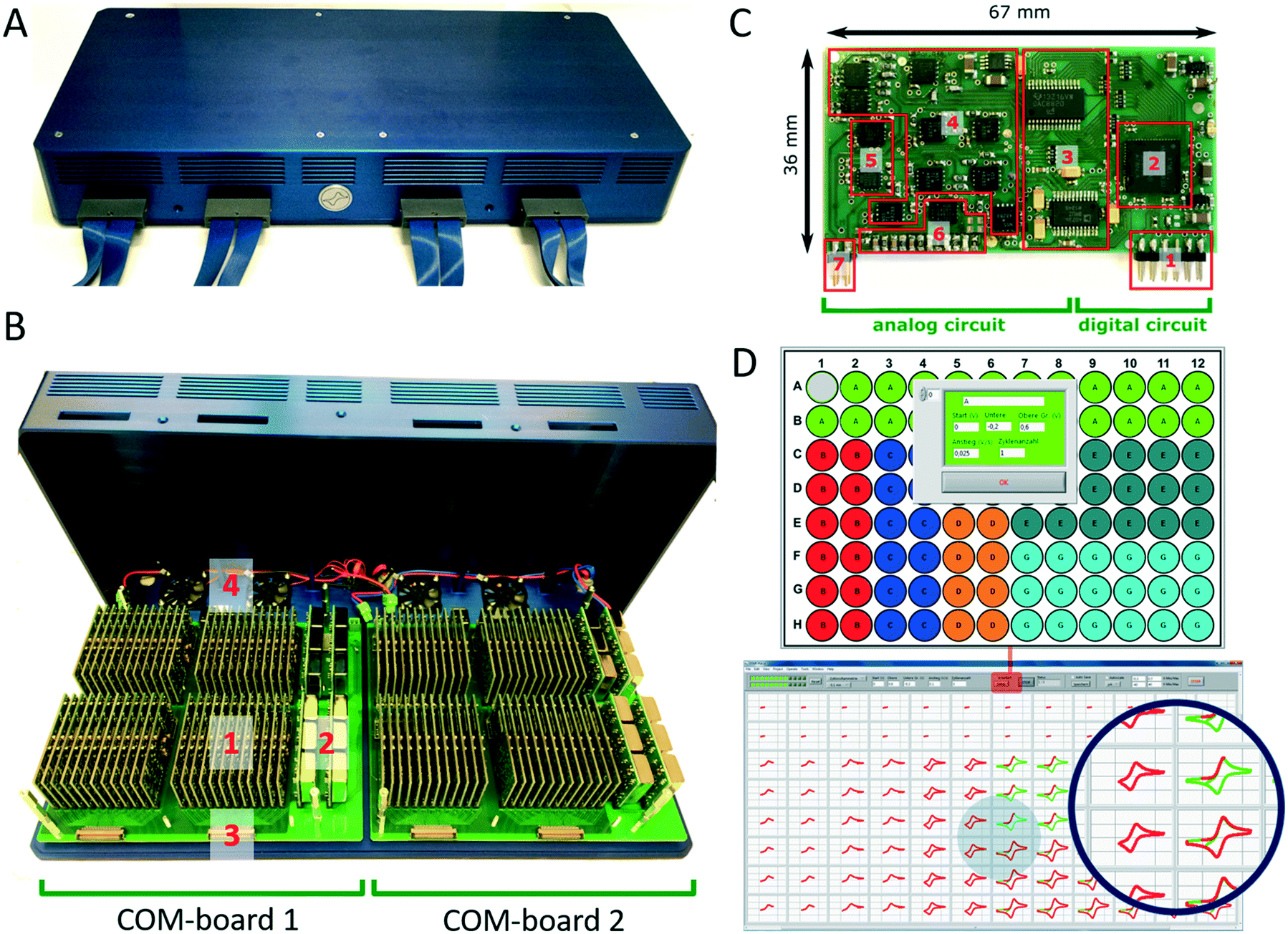 | ||
| Fig. 3 96-Multi-potentiostat unit to control the bioelectrocatalytic analysis system. A 96-multi-potentiostat unit with closed chassis, (B) inner structure consists of two COM-boards carrying (1) the potentiostat modules, (2) current transformer, (3) and LSHM plugs (3). Chassis includes active air cooling by 4 fans (4). C single potentiostat module, functional blocks: (1) connector pins to the COM-board for the SPI-data-interface as well as digital and analog power supply, (2) microcontroller for data-processing, (3) digital–analog converter to generate the excitation signal and analog–digital-converter to convert the measuring signal, (4) amplifier stages of the potentiostat circuit, (5) switches to choose the electrochemical method and to generate defined signals at unused amplifiers to avoid oscillation, (6) multiplexer to adjust the current ranges, (7) connector pins for the potentiostat electrodes. D software interface to configure each potentiostat module arranged analogous to the ANSI 96 standard format and data presentation during a cyclic voltammetric measurement using 0.2 mM ferri/ferrocyanide for scan rates of 25 mV s−1 to 800 mV s−1. | ||
The development of modules instead of a single board design with 96 potentiostat channels offers the option of simple upscaling, which is currently only limited by the number of USB ports available on the analysis computer and its computing power. The modular construction also allows replacement of broken units or to insert upgraded modules with additional functionality. Each module of the multi-potentiostat consist of a analog and digital circuit (Fig. 3C) and provides a voltage range of ±10 V with a resolution of 0.3 mV and current ranges from ±10 mA down to ±1 μA with a minimal resolution of 0.12 nA. A condensed drawing of the potentiostat circuit is shown in Fig. S5B.† Each module is able to perform staircase cyclic voltammetry and constant potential as well as open cell potential measurements. For techniques such as cyclic voltammetry, each module is capable to deliver a scan rate of up to 10 V s−1 at a sampling rate of up to 200 Hz. To demonstrate the functionality of the developed potentiostat modules we compared cyclic voltammetry and constant potential measurements performed in the bioelectrochemical platform with those of a commercial high-precision potentiostat (Fig. S6†).
The multi-potentiostat is controlled from a self-built program written in LabView. The graphical interface enables configuration of each potentiostat module and includes options for constant potential, cyclic voltammetry and open cell potential measurements (Fig. 3D). Once an electrochemical method has been selected for all potentiostats, each module can be configured individually with regard to the parameters of the method. Derived electrochemical data from the measurement are presented in real-time, which is shown in Fig. 3D for different scan rates in a cyclic voltammetry measurement.
3.2. Considerations concerning a vertical two-chamber setup
The vertical structure generally carries the risk that if the agarose barrier fails, the liquid cannot be held in the top chamber and short-circuits working electrodes of various potentiostat modules. However, it has been found that this case did not occur, which can be explained by the large interaction surface of the inner cylinder wall of the top chamber module and the agarose due to the high surface roughness produced by the additive manufacturing process.Since the agarose acts as a convection barrier, the question arises how long it would take for liquids in the bottom and top chamber to mix. We analysed this by impedance spectroscopy using different potassium chloride concentrations. Independent of agarose concentrations between 2.5% and 5%, mixing starts after 45 min (Fig. S7D†), which offers a measurement window with no cross-contamination between these two chambers taking place. This is also in accordance with a FEM simulation of diffusion (Fig. S8†).
This has also a direct consequence regarding the reference electrode, which is located in the top chamber in addition to the counter electrode. While this makes the system practical to handle, it can also prevent released ions, e.g. Ag(I) of the silver/silver chloride electrode, from contaminating the reaction solution in the bottom chamber. This might otherwise be a problem, as the interaction of silver ions with sulfhydryl or thioether groups can disrupt the function of sensitive enzymes.31,32 The localization of the Ag/AgCl reference electrode in the top chamber also allows the chloride concentration to be adjusted to fine-tune the reference potential. However, homogeneity of the reference electrodes is quite high with a maximal deviation of ±5 mV from the mean (Fig. S9†). In the other direction, the reference electrode is neither contaminated nor compromised, as it would be in the bottom chamber e.g. by residual protein, other electrolytes (e.g. potassium nitrate) or additives.
Apart from these advantages, however, a greater distance of approx. 12–14 mm between the reference electrode and the working electrode has to be accepted. This is accompanied by an increased ohmic drop depending on the electrolyte concentration in the bottom chamber, the agarose bridge and the top chamber (Fig. S7A†). To estimate the extent, the total solution resistance was impedimetrically measured for different electrolyte concentrations. In the normal operating state for bioelectrocatalytic measurements (meaning KCl concentration in bottom chamber and in agarose bridge is 0.15 M and in top chamber 1 M) this results in a resistance of 350 Ω (Table S1†). Given that the current during a bioelectrocatalytic measurement with indium tin oxide electrodes has always been below 10 μA (typically in the range of 2–5 μA) at a working potential of −0.8 V (Fig. S7C†), the ohmic drop would be 3.5 mV. This value increases slightly to 5.8 mV if the top chamber would be also filled with 0.15 M KCl solution, whether 2.5% or 5% agarose is used. In each case, the deviation from the target value is quite small for most applications, and certainly negligible for mutant screening purposes. There is also no significant change of the cell potential, when the potassium chloride concentration is 150 mM in the bottom chamber and 1 M in the top chamber, compared to an equilibrated state at 150 mM (Fig. S10†).
3.3. Investigation of the bioelectrocatalytic activity of P450 BM3
Recently, we showed a cytochrome P450 BM3 variant for a directed immobilisation on indium tin oxide using a surface-binding peptide.20 This enzyme variant was significantly more bioelectrocatalytically active than the untagged variant. However, there were certain drawbacks alongside. On the one hand the surface-binding tag blocked the NADPH binding site of the cytochrome P450 reductase domain, when not bound to the target surface, resulting in less activity with NADPH in solution. On the other hand, the enzyme variant was purified by FLAG affinity chromatography, which is suitable for sensing and not for high throughput purposes compared with other purification systems.33,34 In order to circumvent both aspects we found a new variant in which a hexahistidine tag is introduced between the C-terminus and the surface-binding tag. This new variant, which we refer to as BM3 FT6H and still has the three mutations A74G/F87V/L188Q, could be easily expressed and purified and showed almost complete restored activity in solution (Fig. S11, Table S2†), meaning the surface-binding tag does no longer block the NADPH entry site. Thus, native and artificial activation can be better compared.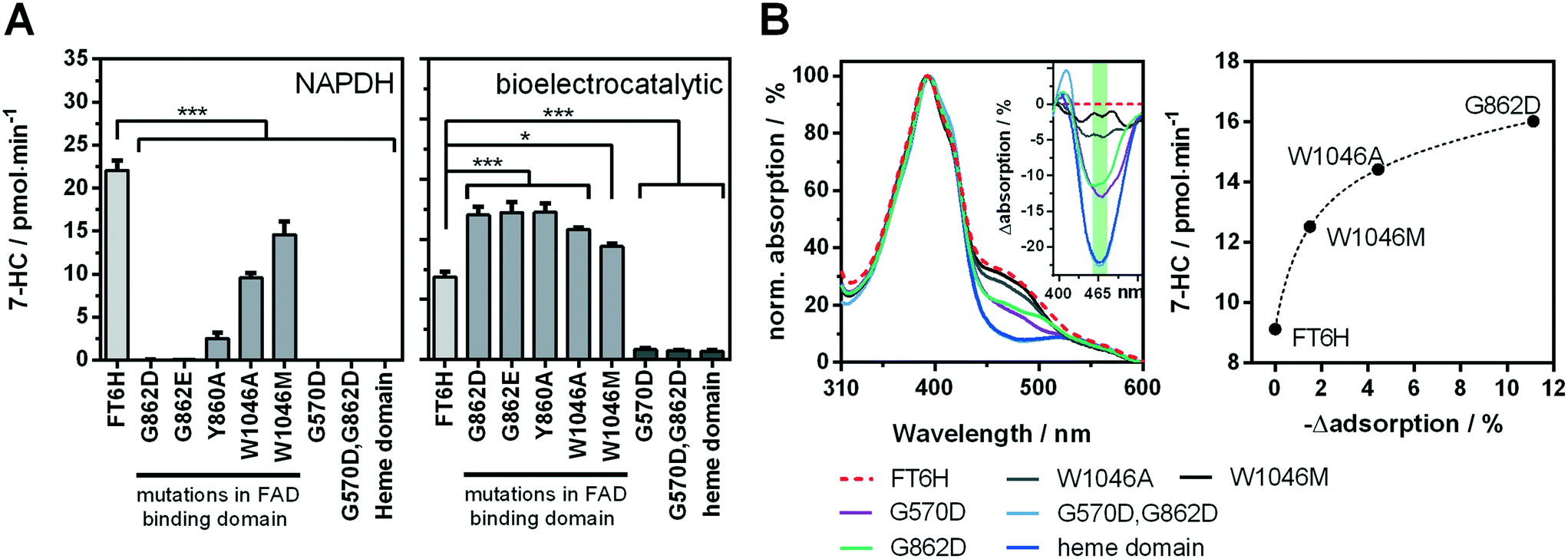 | ||
| Fig. 4 Analysis of the cofactor-dependence of the bioelectrocatalytic process. A enzyme activity of FT6H variants with mutations affecting the flavin-binding domains in solution with NADPH (left) and under bioelectrocatalytic conditions (right). B UV-vis spectra of FT6H variants in presence of the substrate octylbenzene sulfonate (left) and the difference spectra (insert). Correlation of FAD-signal (green area in difference spectra, mean value of 455–480 nm) with bioelectrocatalytic activity (right). Black dashed line: Non-linear regression (one-site total binding). For additional difference spectra see Fig. S12.† | ||
Low change in UV/vis spectra was observed for W1046M and medium change for W1046A, indicating only partial loss or at least altered FAD binding. Surprisingly, we found that loss of FAD led to a bioelectrocatalytic activity increase of up to 80% (Fig. 4A) that correlated inversely with the FAD signal in the UV/vis spectra. Loss of FMN by the G570D mutation led to inactivation of the enzyme, whether a concomitant G862D mutation, to delete FAD, was present or not. These results demonstrate for the first time that FMN but not FAD is essential for the bioelectrocatalytic process of cytochrome P450 BM3 on indium tin oxide, and strengthens the hypothesis that direct electron transfer takes place under consideration of previous findings.10,20 Although we can conclude that FMN is an electron entry point, we cannot exclude the same for FAD. To explain the activity increase in absence of FAD, it could be argued that an intermediate electron transfer step is eliminated, making the overall process more efficient. However, it is acknowledged that electron transfer within the reductase domain is not rate-limiting for the substrate turnover.38,39 Moreover, we performed a potential series from −0.3 V to −1.0 V for the initial FT6H variant and the G862D and Y860A mutant (Fig. S13†). Maximal activity is reached at −0.8 V whether FAD is present or not. This absent of a positive shift in case of the absence of FAD, could be interpreted that FMN is always the electron entry point, taking into account the redox potential order of the native pathway.16 A further explanation lies in the peculiarity of P450 BM3 of having an inactive “over-reduced” state in which FMN and FAD exist as quinone and semiquinone, respectively.39 By deleting FAD, this state might be no longer possible, leading to a prolonged catalysis. Although this finding might suggest an increase in efficiency by truncating the FAD domain, new cryo-EM data show that dimerization in BM3 takes place at the reductase domain.40 Thus, the FAD binding domain forms a central core for the active dimer structure and its absence is consistent with the limitations of such truncated variants.41
It will be interesting to see, whether other electrode materials perform even better than indium tin oxide e.g. due to a different over potential regarding flavin reduction. However first results with titanium and titanium nitride electrodes demonstrate the superiority of indium tin oxide for the tested mutant FT6H G862D (Fig. S14†), which is also a question of enzyme orientation and electrode/cofactor distance.
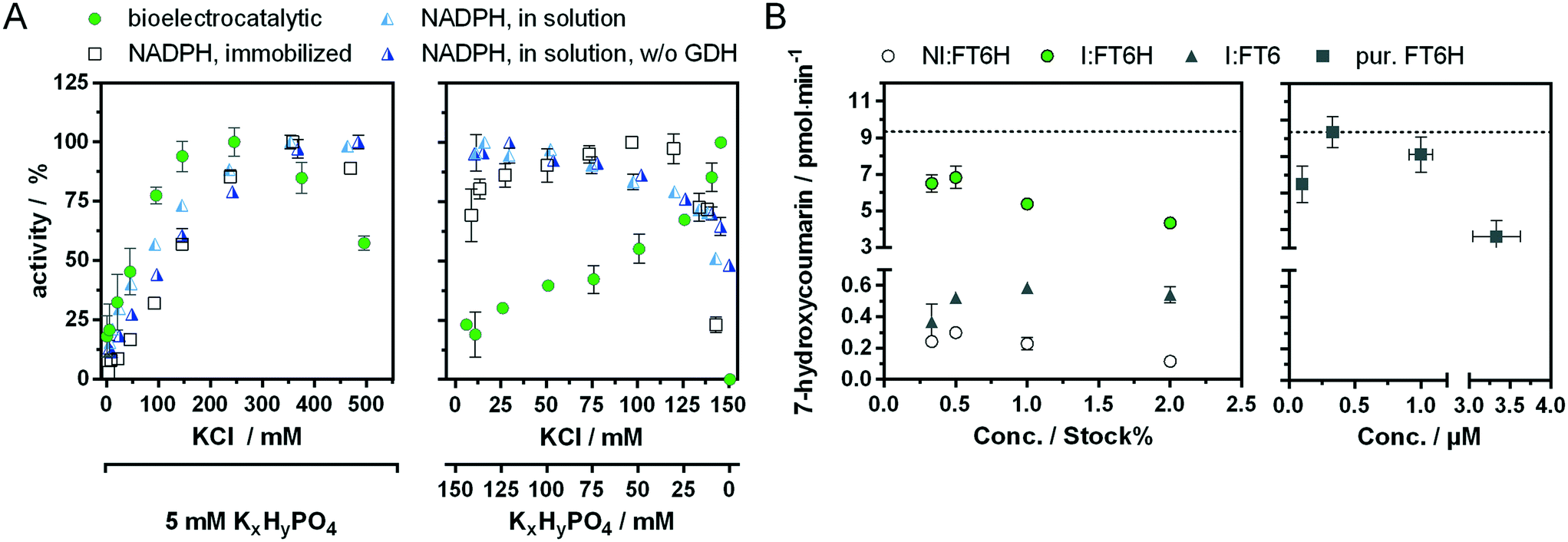 | ||
| Fig. 5 Mapping of bioelectrocatalytic activity of P450 BM3 in the developed analysis system. A activity of P450 BM3 FT6H in a potassium chloride concentration series (left) and at different ratios of potassium chloride and hydrogenphosphate (right) compared with the activation of the immobilised enzyme by NADPH. The pH of all solutions was 7.2. w/o GDH: without the NADPH regeneration system (consisting of glucose and glucose-dehydrogenase). B BM3 immobilised from raw cell extract (left) compared to the purified enzyme (right) at different immobilisation concentrations (BM3 variants include mutations A74G/F87V/L188Q; FT6H = Flag-BM3-H6-(RTHRK)3, FT6 = Flag-BM3-H6; NI = not induced, I = induced with IPTG). | ||
It is interesting to note that at very low hydrogenphosphate concentrations, the change of 10 mM to 5 mM results in a stronger increase in bioelectrocatalytic activity (+15%) than would have been expected from the simultaneous minimal change of 140 to 145 mM in potassium chloride concentration (Fig. S15†). This suggests that hydrogenphosphate might inhibit the bioelectrocatalytical process. To see whether this effect is unique to hydrogenphosphate, it will be interesting to test more electrolytes of singly and doubly charged ions and compare those activity profiles e.g. with the Hofmeister series.
4. Conclusion
This work has introduced a fully parallelised 96 well microtiter plate based bioelectrochemical screening platform with a 96-multi-potentiostat capable of assessing the bioelectrocatalytic activity of cytochrome P450 BM3 by product quantification in conjunction with a photonic readout. The benefit of a multi-potentiostat lies especially in the manifold possibilities for enzyme activation. Not only constant potential activation is possible, but also activation by potential pulses, switching potentials or by a dynamic potential range as in cyclic voltammetry. The applicability of the developed platform was demonstrated for mechanistic studies, mapping of optimal reactions conditions and compatibility with raw cell extracts and thus efficient mutant screening strategies. Compared to our previous 12-potentiostat system, it was possible for the first time to build a fully parallel 96-microtiter plate system in which also semiconducting materials can be used without any problems due to the short conduction paths. In addition, the high number of parallel reaction rooms allows effective screening of variable reaction parameters in a single experiment.The separation of the working and counter electrode into different cells aligned vertically in 96-microtiter plate format for activity testing with fluorescent substrates is ideal to achieve the largest possible surface of a planar working electrode and hence highest possible sensitivity without additional surface roughening. Enzyme variants with improved bioelectrocatalytic activity originating from this system will elicit a new round of miniaturisation, since less surface area is required for further advancement. Smaller working electrodes make room to shrink the total height of the system down to a common microtiter plate and would allow the direct integration of the electrode system into a fluorescence reader for simultaneous electrochemical and optical real-time monitoring.
As derived from the developed analysis system, the reduction of enzyme complexity by deletion of the FAD binding in P450 BM3, which leads to an increased product turnover, reveals the potential of bioelectrocatalytic enzyme activation. New reaction pathways will dismiss domains from some of their natural functions and make them highly mutable. Instead of truncating these domains, we reason they will be best treated by assigning them new functions that are specific and beneficial for the bioelectronic interface such as pH-stability, reversible surface attachment and oxidative stress tolerance. Thus, this platform can be a valuable tool to study and improve redox enzymes in bioelectronic interfaces, e.g. for flow reactor systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Leibniz Collaborative Excellence (Grant No. K67/2017) and by the Saxon Ministry of Science and the Fine Arts (SMWK, project OlEnz, Grant No. 100316512) and is tax-supported on the basis of the budget approved by the members of the parliament of the Free State of Saxony. We thank the CeLecta GmbH for providing the original BM3 containing plasmid.References
- S. Jemli, D. Ayadi-Zouari, H. B. Hlima and S. Bejar, Biocatalysts: application and engineering for industrial purposes, Crit. Rev. Biotechnol., 2016, 36(2), 246–258 CrossRef CAS PubMed.
- J. M. Choi, S. S. Han and H. S. Kim, Industrial applications of enzyme biocatalysis: Current status and future aspects, Biotechnol. Adv., 2015, 33(7), 1443–1454 CrossRef CAS PubMed.
- R. Singh, M. Kumar, A. Mittal and P. K. Mehta, Microbial enzymes: industrial progress in 21st century, 3 Biotech., 2016, 6(2), 174 CrossRef PubMed.
- R. Wichmann and D. Vasic-Racki, Cofactor regeneration at the lab scale, Technology Transfer in Biotechnology: From Lab to Industry to Production, 2005, vol. 92, pp. 225–260 Search PubMed.
- P. R. Schreiner and A. A. Fokin, Selective alkane C-H-bond functionalizations utilizing oxidative single-electron transfer and organocatalysis, Chem. Rec., 2004, 3(5), 247–257 CrossRef CAS PubMed.
- Y. Qin, L. H. Zhu and S. Z. Luo, Organocatalysis in Inert C-H Bond Functionalization, Chem. Rev., 2017, 117(13), 9433–9520 CrossRef CAS PubMed.
- E. O'Reilly, V. Kohler, S. L. Flitsch and N. J. Turner, Cytochromes P450 as useful biocatalysts: addressing the limitations, Chem. Commun., 2011, 47(9), 2490–2501 RSC.
- S. T. Jung, R. Lauchli and F. H. Arnold, Cytochrome P450: taming a wild type enzyme, Curr. Opin. Biotechnol., 2011, 22(6), 809–817 CrossRef CAS PubMed.
- J. D. Bloom, S. T. Labthavikul, C. R. Otey and F. H. Arnold, Protein stability promotes evolvability, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(15), 5869 CrossRef CAS PubMed.
- R. Frank, M. Klenner, R. Azendorf, M. Bartz, H.-G. Jahnke and A. A. Robitzki, Novel 96-well quantitative bioelectrocatalytic analysis platform reveals highly efficient direct electrode regeneration of cytochrome P450 BM3 on indium tin oxide, Biosens. Bioelectron., 2017, 93, 322–329 CrossRef CAS PubMed.
- A. Gutierrez, A. Grunau, M. Paine, A. W. Munro, C. R. Wolf and G. C. K. Roberts, et al. Electron transfer in human cytochrome P450 reductase, Biochem. Soc. Trans., 2003, 31(3), 497 CrossRef CAS PubMed.
- H. M. Girvan, A. J. Dunford, R. Neeli, I. S. Ekanem, T. N. Waltham and M. G. Joyce, et al. Flavocytochrome P450 BM3 mutant W1046A is a NADH-dependent fatty acid hydroxylase: Implications for the mechanism of electron transfer in the P450 BM3 dimer, Arch. Biochem. Biophys., 2011, 507(1), 75–85 CrossRef CAS PubMed.
- D. J. Cook, J. D. Finnigan, K. Cook, G. W. Black and S. J. Charnock, Cytochromes P450: History, Classes, Catalytic Mechanism, and Industrial Application, Adv. Protein Chem. Struct. Biol., 2016, 105, 105–126 CAS.
- B. M. van Vugt-Lussenburg, E. Stjernschantz, J. Lastdrager, C. Oostenbrink, N. P. Vermeulen and J. N. Commandeur, Identification of critical residues in novel drug metabolizing mutants of cytochrome P450 BM3 using random mutagenesis, J. Med. Chem., 2007, 50(3), 455–461 CrossRef CAS PubMed.
- L. L. Wong, P450(BM3) on steroids: the Swiss Army knife P450 enzyme just gets better, ChemBioChem, 2011, 12(17), 2537–2539 CrossRef CAS PubMed.
- S. N. Daff, S. K. Chapman, R. A. Holt, S. Govindaraj, T. L. Poulos and A. W. Munro, Redox Control of the Catalytic Cycle of Flavocytochrome P-450 BM3, Biochemistry, 1997, 36(45), 13816–13823 CrossRef CAS PubMed.
- A. W. Munro, D. G. Leys, K. J. McLean, K. R. Marshall, T. W. B. Ost and S. Daff, et al. P450 BM3: the very model of a modern flavocytochrome, Trends Biochem. Sci., 2002, 27(5), 250–257 CrossRef CAS PubMed.
- I. F. Sevrioukova, H. Li, H. Zhang, J. A. Peterson and T. L. Poulos, Structure of a cytochrome P450–redox partner electron-transfer complex, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(5), 1863 CrossRef CAS PubMed.
- R. Verma, U. Schwaneberg and D. Roccatano, Insight into the redox partner interaction mechanism in cytochrome P450BM-3 using molecular dynamics simulations, Biopolymers, 2014, 101(3), 197–209 CrossRef CAS PubMed.
- S. Zernia, R. Frank, R. H.-J. Weiße, H.-G. Jahnke, K. Bellmann-Sickert and A. Prager, et al. Surface-Binding Peptide Facilitates Electricity-Driven NADPH-Free Cytochrome P450 Catalysis, ChemCatChem, 2018, 10(3), 525–530 CrossRef CAS.
- U. Schwaneberg, C. Schmidt-Dannert, J. Schmitt and R. D. Schmid, A continuous spectrophotometric assay for P450 BM-3, a fatty acid hydroxylating enzyme, and its mutant F87A, Anal. Biochem., 1999, 269(2), 359–366 CrossRef CAS PubMed.
- J. Nazor, S. Dannenmann, R. O. Adjei, Y. B. Fordjour, I. T. Ghampson and M. Blanusa, et al. Laboratory evolution of P450 BM3 for mediated electron transfer yielding an activity-improved and reductase-independent variant, Protein Eng., Des. Sel., 2008, 21(1), 29–35 CrossRef CAS PubMed.
- M. M. P. S. Neves, M. B. González-García, D. Hernández-Santos and P. Fanjul-Bolado, Screen-Printed Electrochemical 96-Well Plate: a High-Throughput Platform for Multiple Analytical Applications, Electroanalysis, 2014, 26(12), 2764–2772 CrossRef CAS.
- S. Abdellaoui, A. Noiriel, R. Henkens, C. Bonaventura, L. J. Blum and B. Doumeche, A 96-well electrochemical method for the screening of enzymatic activities, Anal. Chem., 2013, 85(7), 3690–3697 CrossRef CAS PubMed.
- A. Ezquerra, J. C. Vidal, L. Bonel and J. R. Castillo, A validated multi-channel electrochemical immunoassay for rapid fumonisin B1 determination in cereal samples, Anal. Methods, 2015, 7(9), 3742–3749 RSC.
- T. Omura and R. Sato, The Carbon Monoxide-Binding Pigment of Liver Microsomes. I. Evidence for Its Hemoprotein Nature, J. Biol. Chem., 1964, 239, 2370–2378 CAS.
- A. Aitio, A simple and sensitive assay of 7-ethoxycoumarin deethylation, Anal. Biochem., 1978, 85(2), 488–491 CrossRef CAS PubMed.
- M. Choi, K. Jo and H. Yang, Effect of Different Pretreatments on Indium-Tin Oxide Electrodes, Bull. Korean Chem. Soc., 2013, 34(2), 421–425 CrossRef CAS.
- R. D. Milton and S. D. Minteer, Direct enzymatic bioelectrocatalysis: differentiating between myth and reality, J. R. Soc., Interface, 2017, 14(131), 20170253 CrossRef PubMed.
- P. T. Kissinger and W. R. Heineman, Laboratory techniques in electroanalytical chemistry, CRC Press, Boca Raton, 2nd edn, revised and expanded, 2018 Search PubMed.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Antimicrobial activity of metals: mechanisms, molecular targets and applications, Nat. Rev. Microbiol., 2013, 11(6), 371–384 CrossRef CAS PubMed.
- L. Mazzei, M. Cianci, A. Gonzalez Vara and S. Ciurli, The structure of urease inactivated by Ag(i): a new paradigm for enzyme inhibition by heavy metals, Dalton Trans., 2018, 47(25), 8240–8247 RSC.
- D. S. Waugh, Making the most of affinity tags, Trends Biotechnol., 2005, 23(6), 316–320 CrossRef CAS PubMed.
- M. E. Kimple, A. L. Brill and R. L. Pasker, Overview of affinity tags for protein purification, Curr. Protoc. Protein Sci., 2013, 73, 9.9 Search PubMed : Unit-9.
- M. L. Klein and A. J. Fulco, Critical residues involved in FMN binding and catalytic activity in cytochrome P450BM-3, J. Biol. Chem., 1993, 268(10), 7553–7561 CAS.
- S. Eiben, L. Kaysser, S. Maurer, K. Kühnel, V. B. Urlacher and R. D. Schmid, Preparative use of isolated CYP102 monooxygenases—A critical appraisal, J. Biotechnol., 2006, 124(4), 662–669 CrossRef CAS PubMed.
- A. Gutierrez, O. Doehr, M. Paine, C. R. Wolf, N. S. Scrutton and G. C. K. Roberts, Trp-676 Facilitates Nicotinamide Coenzyme Exchange in the Reductive Half-Reaction of Human Cytochrome P450 Reductase: Properties of the Soluble W676H and W676A Mutant Reductases, Biochemistry, 2000, 39(51), 15990–15999 CrossRef CAS PubMed.
- M. L. Kelin and A. J. Fulco, The interaction of cytochrome c and the heme domain of cytochrome P-450BM-3 with the reductase domain of cytochrome P-450BM-3, Biochim. Biophys. Acta, Gen. Subj., 1994, 1201(2), 245–250 CrossRef.
- C. J. Whitehouse, S. G. Bell and L. L. Wong, P450(BM3) (CYP102A1): connecting the dots, Chem. Soc. Rev., 2012, 41(3), 1218–1260 RSC.
- H. Zhang, A. L. Yokom, S. Cheng, M. Su, P. F. Hollenberg and D. R. Southworth, et al. The full-length cytochrome P450 enzyme CYP102A1 dimerizes at its reductase domains and has flexible heme domains for efficient catalysis, J. Biol. Chem., 2018, 293(20), 7727–7736 CrossRef CAS PubMed.
- A. J. Ruff, Advances in directed monooxygenase evolution : from diversity generation and flow cytometry screening to tailor-made monooxygenases, PhD Thesis, RWTH Aachen University, Aachen, 2012 Search PubMed.
- J. Petrovic, R. A. Clark, H. Yue, D. H. Waldeck and E. F. Bowden, Impact of surface immobilization and solution ionic strength on the formal potential of immobilized cytochrome C, Langmuir, 2005, 21(14), 6308–6316 CrossRef CAS PubMed.
- F. Baneyx, Recombinant protein expression in Escherichia coli, Curr. Opin. Biotechnol., 1999, 10(5), 411–421 CrossRef CAS.
- P. Déjardin, Proteins at Solid-Liquid Interfaces, Springer-Verlag, 1st edn, 2006, p. 345 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc01220f |
| This journal is © The Royal Society of Chemistry 2020 |