
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the charged nature and ion-exclusion mechanism of fluorine-enriched non-ionogenic polyamide derived thin film composite nanofiltration membranes†
Avishek
Pal
 ab,
T. K.
Dey
*a,
Kaustava
Bhattacharyya
c and
R. C.
Bindal
ab
ab,
T. K.
Dey
*a,
Kaustava
Bhattacharyya
c and
R. C.
Bindal
ab
aMembrane Development Section, Chemical Engineering Group, Bhabha Atomic Research Centre, Trombay, Mumbai – 400085, India. E-mail: tkdey@barc.gov.in
bHomi Bhabha National Institute, Anushakti Nagar, Mumbai – 400094, India
cChemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai – 400085, India
First published on 20th May 2020
Abstract
A thin film composite (TFC) nanofiltration (NF) membrane comprising a novel fluorine-enriched polyamide (FPA) as a skin layer polymer has been developed by in situ interfacial polycondensation between a cyclo-aliphatic diamine monomer, piperazine, and an acyclic fluorodiacyl chloride monomer, tetrafluorosuccinyl chloride. Instrumental investigations carried out by FTIR and XPS showed that the structure of FPA is devoid of intra- as well as inter-molecular H-bonds, which seems to be a rare finding in NF type membranes with decent performances. Stereoelectronic effects of the FPA structure, owing to the configurational mobility of polymer chains, electron-withdrawing ability of fluorine and lack of cohesive non-covalent type interactions, strongly influenced the surface morphology, topography and hydrophilicity of the TFC-NF membranes. The active skin layer of the membranes exhibited uniform charge-atmosphere in spite of not having any fixed charged groups in the polymer structure constituting it. Zeta potential measurement of the membranes revealed low values (−4.4 to −7 mV), indicating their low surface charge. However, a high Donnan-exclusion was observed towards a solute with multivalent anion (Na2SO4, solute rejection: 87–93%) as opposed to that with monovalent anion (NaCl, solute rejection: 23–26%), with a solvent flux as high as 54 L m−2 h−1 at 15 bar transmembrane pressure. The current class of TFC-NF membranes with unique physicochemical attributes thus provides a promising scope for a variety of nanofiltration applications.
1. Introduction
Separation, concentration and purification of molecular mixtures for different industrial applications are always associated with economic and environmental concerns. Nanofiltration, being a multi-dimensional membrane based separation process, has acquired immense importance for applications relevant to separation industries.1,2 This includes precise enrichment of high-value products in the food and pharmaceutical industries,3,4 production of high-quality water for communities and industries,5,6 removal or recovery of toxic or valuable heavy metal ions from industrial effluents,7 separation of toxic organic residues generated from the textile and dye manufacturing industries,8etc.Charged NF membranes, made out of polymers bearing fixed charged groups, fractionate mixed-electrolyte streams comprising ionic constituents of varying valances, utilizing their unique and promising feature of ion-selectivity through Donnan-exclusion.9,10 In the case of negatively or positively charged NF membranes, the skin layers’ polymeric structures carry stationary negatively or positively charged sites, respectively, and behave as electrically charged layers when they come in contact with aqueous electrolytic solutions.11–13 The pattern and extent of membrane–solute interactions, originating between the active skin layer of the membrane and the solvated ions of the feed solution, depend on the physicochemical attributes of the membrane and solution chemistry of the ionic constituents. Thus, during nanofiltration of an electrolytic solution, negatively charged NF membranes preferentially retain multivalent anions over monovalent anions owing to the stronger electrostatic repulsion (Donnan-exclusion) of the former. Conversely, it facilitates passage of multivalent cations more than monovalent cations.14
Discovery of high-performance polymeric materials is always of significant interest for sustainable growth of membrane-based separation processes. Therefore, investigation of the structure–property–performance relationship of novel polyamides has persistently drawn attention for the membrane industry. With advancement in research on prospective active layer formulations for TFC type membranes,15 various multifunctional acyl chloride monomers have been exploited hitherto, to tune the efficacy (optimum selectivity and water permeability) of membranes for applications involving a range of ionic feed solutions. These monomers tailor polymer structures of the membranes’ skin layers, wherein the residual functionalities mostly impart surface charge and influence the separation mechanism.16–20 Furthermore, amine monomers with ionogenic functionalities have also been investigated in fabricating the active skin layers of TFC membranes.21–24 In order to enhance the rejection of multivalent cations, the surface charge of a poly(piperazineamide)-based TFC type NF membrane was also modulated by using a multifunctional amine.25,26 In some studies, researchers even attempted to quench the acyl chloride groups with the aim that it would reduce the density of residual carboxyl groups of TFC membranes to increase the membranes’ surface hydrophobicity and mitigate fouling.27,28 In another study, the quenching of acyl chloride groups in fully aromatic TFC membranes was performed using amine, ammonia, and alcohol solutions.29
Fluorinated aromatic or semi-aromatic polyamides, derived using different fluorinated amines and ionogenic aromatic acyl chloride monomers, have been shown to exhibit Donnan-exclusion of ions for nanofiltration purposes.30–33 Developing a polyamide structure of low surface free energy by the use of a fluorinated polyamine has also become an important area of materials research for fouling-resistant membranes.30–33 Structural susceptibility of aromatic polyamides, being a major issue in membrane stability, is known to happen through electrophilic substitution, via N (amidic)-chlorination and subsequent aromatic ring-chlorination (Orton rearrangement).34,35 It is a serious concern with the polyamide class of polymers that they are poor chlorine-tolerant candidates under an oxidative environment. However, strong electron-withdrawing groups (e.g. –CF3) have introduced sufficient activity in the polyamide structure to sustain under an oxidative environment, by minimizing its inclination towards N-chlorination through amidic sites.34,35
Acknowledging the growing practical interest in fluorine-enriched polyamides, our study thus introduces an alternative fluoroacyl chloride monomer in fabricating NF membranes. Herein, the particulars of our effort enlighten the unique characteristics of a novel non-ionogenic polyamide structure as the skin layer of TFC-NF membranes. The active skin layers have been derived by in situ interfacial polycondensation of a cyclo-aliphatic diamine monomer, piperazine (PIP), with a diacyl chloride monomer having a fluoro-acyclic backbone, tetrafluorosuccinyl chloride (TFSC), on a porous polysulfone (Psf) base membrane, followed by conditioning of the nascent composite membranes under two different post-polymerization exposures. This work provides a facile in situ method to tailor the surface characteristics of polyamide-based TFC membranes, which could allow membrane manufacturers to avoid post-treatment fluorination strategies for specific applications.
2. Materials and methods
Polysulfone (Psf; M.W.: 30k Da), purchased from Solvay Speciality Polymers (India), N-methyl-2-pyrrolidone (NMP; purity: ≥ 99%), procured from Sigma-Aldrich, and polyvinyl pyrrolidone (PVP, K-30; M.W.: 40k Da), procured from SRL (India), were used to prepare the base membrane for the NF membranes. The nonwoven porous polyester fabric used as a support material for preparation of the base membrane had a density of 75 g m−2 and a thickness of 110 μm with an air permeability of 70 dm3 m−2 at 2 mbar. For evaluation of the molecular weight cut-off (MWCO) of the Psf base membrane, organic solutes, viz., poly(ethylene glycol) (PEG) of varying molecular weights, i.e., 12k, 20k and 35k Da, and poly(ethylene oxide) (PEO) of molecular weight 100k Da, were procured from Fluka (Germany) and Sigma-Aldrich, respectively. The cyclo–aliphatic diamine monomer PIP (purity: 99%) was procured from Sigma-Aldrich. The fluoroacyl chloride monomer TFSC (purity: 96%) was purchased from Chemos GmbH (Germany). Analytical grade n-hexane, magnesium chloride heptahydrate (MgCl2·7H2O), magnesium sulphate hexahydrate (MgSO4·6H2O), anhydrous sodium sulphate (Na2SO4), sodium chloride (NaCl) and potassium chloride (KCl) were procured from different companies in India. The organic probe-solvents, i.e., AR grade formamide (S D Fine-Chem Ltd, assay: 99%) and diiodomethane (Spectrochem, assay: 98%), were used as received for contact angle measurements. The conductivity of the Milli-Q ultra-pure water used in the experiments was below 2 μS cm−1.2.1 Fabrication of thin film composite nanofiltration membranes
An asymmetric flat sheet type Psf base membrane was prepared by a non-solvent induced phase inversion method. A polymer doping solution comprising 25 (w/vNMP)% Psf and 40 (w/wPsf)% PVP as a porogen was prepared in a hermetically sealed glass bottle through vigorous agitation for several hours, resulting in formation of a homogeneous viscous solution. After vacuum degassing to eliminate trapped air bubbles, the solution was cast over a nonwoven polyester support fabric using an automated casting machine operated at a steady casting shear, and maintained a uniform thickness of 100 μm. The cast film was immediately immersed in a precipitation bath containing ultra-pure water as a non-solvent, maintained at room temperature. To ensure the complete leaching of the porogen as well as the solvent from the membrane matrix, the membrane was rinsed several times in ample amounts of fresh water. The entire process of membrane casting was carried out under controlled atmospheric conditions by maintaining the room temperature at 24(±1) °C with a relative humidity of 30–35%.TFC-NF membranes were prepared through an in situ interfacial polymerization technique by using PIP as a diamine monomer and TFSC as a fluorine-bearing diacyl chloride monomer as presented in Fig. 1a. During the fabrication process (pictorially presented in Fig. S1, ESI†), a piece of porous Psf base membrane was superimposed on a flat sheet of Teflon, the skin surface facing outwards, and well-sealed with adhesive tape on all four sides. It was then immersed into an amine-bath containing a 4 (w/v)% aqueous solution of PIP and allowed to stay there for 4 min. After the soaking stage, the amine saturated base membrane was taken out of the amine-bath and squeezed through a pair of rubber rollers to remove the excess amine solution, ensuring a homogeneous distribution of the amine monomer on the skin surface of the base membrane. The as-treated membrane was then immersed in a reaction bath comprising an n-hexane solution containing 0.2 (w/v)% TFSC and kept dipped for a 60 s reaction period. A skin layer was thereby developed over the surface of the Psf base membrane following diffusion limited film growth. The composite membrane was then dried in air by conditioning under room atmospheric conditions, with a temperature of 25(±1) °C and relative humidity of 70–75%, for 12 h. The membrane specimen, denoted as TFC-NFA, was removed from the clamping unit and stored in a distilled water bath at room temperature.
 | ||
| Fig. 1 (a) Reaction scheme for in situ interfacial polymerization between PIP, as a diamine monomer, and TFSC, as a fluorine-bearing diacyl chloride monomer, to derive the skin layer polymer FPA of TFC-NF membranes; and (b) ATR-FTIR spectra of TFC-NFA and TFC-NFW over 600–4000 cm−1. | ||
Another route of preparation (illustrated in Fig. S1, ESI†) was adopted to fabricate the membrane employing the aforementioned combination and composition of monomers. Herein, a piece of porous Psf base membrane assembled with the clamping unit of a Teflon sheet was initially immersed in the amine-bath for 4 min, followed by removal of the excess solution from the surface of the base membrane. Then, the as-treated membrane was allowed to undergo in situ interfacial polycondensation by keeping it immersed in a reaction bath containing TFSC solution, for a period of 60 s. However, following a different way of the post-polymerization conditioning for the membrane, the nascent composite membrane was allowed to stand in room atmosphere (room temperature: 25(±1) °C and a relative humidity of 70–75%) for 4 min and subsequently annealed in a water-bath maintained at a temperature of 40(±1) °C for 10 min. The membrane was then taken out of the water-bath and conditioned under similar room atmospheric conditions for 12 h. The resulting membrane specimen, denoted as TFC-NFW, was stored in a distilled water bath at room temperature.
2.2 Physicochemical characterization of the TFC-NF membranes’ skin layers
The spectroscopic technique of Attenuated Total Reflectance (ATR)–FTIR was employed to investigate the structural aspect of the membranes’ skin layer polymer, which might be varied on their exposure to different post-polymerization conditioning environments. A Bruker Vertex 70 FTIR spectrometer equipped with an ATR accessory (ZnSe crystal, 45° angle of incidence and refractive index of 2.4) was employed for this purpose, where the membranes’ skin surfaces were kept face down on the ATR crystal element. The radiation penetration depth was 2 μm. All infrared spectra were recorded in absorbance mode over a wave number range 600–4000 cm−1 at 25 °C. For evaluation, 200 scans were taken with a spectral resolution of 2 cm−1.The surface chemical composition of the TFC–NF membranes was further investigated using an X-ray Photoelectron Spectroscopy (XPS) instrument, a DLD-150 electron analyzer (SPECS, Germany). It was equipped with non-monochromatized Mg-Kα X-rays (300 W, 1253.6 eV) as the excitation source. The spectrometer binding energy (BE) scale was calibrated with the Cu2p-3/2, Ag3d-5/2, and Au4f-7/2 photoelectron lines at 932.7, 368.3, and 83.95 eV, respectively. The spectral regions were scanned for several sweeps until a good signal-to-noise ratio was obtained, and then the survey scans were recorded in the range of 0–1400 eV at a 187.85 eV pass energy. The CasaXPS programme (Casa Software, UK) was used for data acquisition and analysis. A Shirley-type background was accurately subtracted from the signals. The binding energies were calculated with respect to the C–(C,H) component of the C1s peak of adventitious carbon fixed at 284.5 eV. The atomic concentration (in %) of each characteristic element (i.e., C, N, O and F) was determined from the recorded X-ray photoelectron survey-scan spectra, plotted as the intensity (number of counts per second) versus binding energy (BE). The appropriate relative sensitivity factor for each element was used from the Kratos library and the resulting atomic percent for each was measured. The recorded high resolution XPS spectra for the core levels of C1s and F1s were subjected to curve fitting using Gaussian functions to determine the BE values.
The topographical features of the membranes’ skin surfaces were characterised using an AFM instrument (NT-MDT SOLVER next; Russia) in semi-contact mode. Small pieces of the membrane, with an area of 1 cm2, were cut and glued onto a metal substrate. The rectangular cantilever (NSG 10, NT-MDT, Russia) used in scanning was made of Si3N4, with a spring constant of 11.8 N m−1, having a typical resonance frequency of 240 kHz and a nominal tip–apex radius of 10 nm with a high aspect ratio. The scanning was done on a 20 μm × 20 μm area of the membrane in air at room temperature, with a scanning frequency of 0.1 Hz. The scanned regions were flattened using a second-order polynomial to remove any curvatures and slopes from the image, and then the resulting best fit was extracted from it. Image acquisition and quantitative evaluation of surface roughness parameters by acquiring the three-dimensional (3D) topographies were performed using NOVA-P9 software. Surface roughness parameters of the membranes were calculated from the height profiles of the image in terms of the average roughness (Ra) and root mean square roughness (Rq).
The surface morphology of the TFC-NF membranes was characterized using a Scanning Electron Microscope (SEM), CamScan-CS3200LV (UK). The skin surfaces of the dry membranes (small strips: 2 mm2) were made electrically conducting through sputter coating with a gold–palladium alloy for 60 s employing a 30 mA current. Image acquisition was done at an acceleration voltage of 20 kV and 1000× magnification, operating in secondary electron mode.
Static contact angle measurements were conducted using the sessile drop method at ambient temperature. A contact angle measuring instrument (DSA 100 of KRÜSS Gmbh, Germany) was employed to assess the hydrophilicity of the skin layers of the TFC-NF membranes. The polar (γPolarSFE) and dispersive (γDispersiveSFE) components of the surface free energy (SFE) parameters were also determined. Three different wetting-solvents with known surface tension values, viz., water (γPS: 51.0 mN m−1 and γDS: 21.8 mN m−1), formamide (γPS: 19.0 mN m−1 and γDS: 29.0 mN m−1) and diiodomethane (γDS: 50.8 mN m−1), were used for this purpose. A sessile drop was slowly and steadily formed on the membrane surface by depositing 3 μl of the given solvents with a micro-syringe and the measurement was completed at the membrane–solvent–air interface with a residence time of 60 s. At least eight such measurements were done at different locations of each membrane surface and then averaged out to calculate the contact angle with their standard deviations using DSA 1 v 1.92 software. The Fowkes method was applied to calculate the SFE along with the polar and dispersive components of the membranes’ surfaces from the surface tension values of the probe-solvents and contact angle values of the membranes with respect to those probe-solvents.
The electrokinetic feature of the membranes was evaluated by employing a ZetaCAD electrokinetic analyser (CAD Inst., France). The streaming potential analyser consisting of a quartz-cell configuration was capable of holding two flat sheet membranes in such a way that the membranes remained detached by spacers and the skin layers faced each other creating a slit channel for a tangential flow of electrolytic solution across the membranes. An electrical potential difference was generated due to the flow of electrolyte solution (10−3 M KCl) in the two flow directions under a pressure gradient across the membrane and was measured by Ag/AgCl electrodes, equipped with the cell. The zeta potential of the membranes under such an environment was determined using the relationship between the measurable streaming potential (Vst) and the zeta potential (ζ), which is expressed by the Helmholtz–Smoluchowski equation (eqn (1))
 | (1) |
 | (2) |
The Debye-length (λDeb) was derived according to eqn (3)
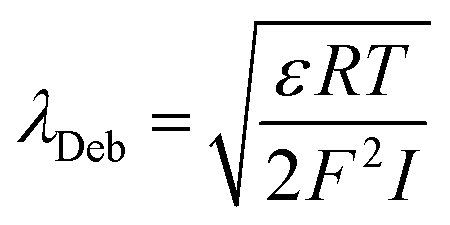 | (3) |
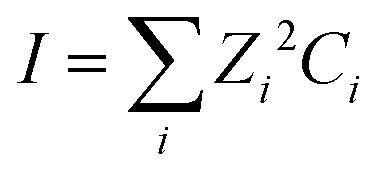 | (4) |
2.3 Performance evaluation of Psf and TFC-NF membranes
The molecular weight cut-off (MWCO) of the Psf base membrane was determined using the rejections of neutral organic solutes, viz., PEG (M.W. of 12k, 20k and 35k Da) and PEO (M.W. of 100k Da). Feed solutions of each solute, with a concentration of 200 ppm, were prepared by dissolving the requisite amounts of PEG or PEO in ultra-pure water and used for permeation experiments. The permeation study was carried out at a transmembrane pressure of 1 bar at room temperature. The concentrations of PEG and PEO in both the feed and product solutions were measured by analysing their total organic carbon (TOC) content, using a TOC analyser (ANATOC-II, SGE analytical science, Australia). The MWCO of the Psf membrane defines the molecular weight of a neutral solute for which the membrane delivers more than 90% rejection.The steady-state pure water permeability (L m−2 h−1; LMH) of the Psf base membrane was determined by measuring the permeate flow in water at 1 bar of transmembrane pressure, i.e., the volume (V, in L) collected during a time period (t, in h) through a membrane area (A, in m2). Under cross-flow permeation mode, a membrane with an effective membrane area of 14.4 cm2 was allowed to undergo hydraulic compaction for 2 h. Membranes were tested in triplicate to acquire reproducible performances.
Nanofiltration performances of the TFC-NF membranes were analysed using cross-flow permeation equipment (shown in Fig. S2, ESI†). Circular membrane specimens with an effective membrane area of 14.4 cm2 were properly placed in the test cell, wherein the membranes’ active skin surfaces faced the incoming feed solution. The entire experiment was carried out in recycle mode at a transmembrane pressure of 15 bar to maintain a steady feed concentration through recirculation of both concentrate and permeate streams into the feed reservoir. Isosmotic feed solutions comprising varying combinations of ionic species of different valences (viz., 0.03 M NaCl, 0.02 M Na2SO4, 0.02 M MgCl2 and 0.03 M MgSO4), maintained at 25(±2) °C, were employed for that purpose. Prior to the start of data accumulation, steady-state conditions were achieved for all the test experiments by operating for 4 h, allowing the membranes to get stabilized. The solute concentrations in the feed and permeate solutions were conductometrically determined using a digital conductivity meter (TESTRONIX 15, Microlab, Mumbai, India). For each category, the membrane specimens were investigated in triplicate and the steady-state results were accordingly documented as the mean along with the standard deviation.
The steady-state solvent flux was determined by direct measurement of the permeate flow, i.e., the volume (V, in L) collected during a time period (t, in h) through a membrane area (A, in m2) at a certain pressure, and expressed in terms of LMH. The percentage of solute rejection (SR) was determined by conductivity measurement of the respective solution and calculated as:
 | (5) |
3. Results and discussion
3.1 Structural aspects of FPA
The ATR-FTIR spectra (Fig. 1b) of the skin layers of the TFC-NF membranes, namely TFC-NFA and TFC-NFW, reflect the characteristic absorption bands pertaining to the synthesized FPA. The formation of the FPA-based thin skin layer of the TFC-NF membranes takes place by an in situ interfacial polycondensation route, through crossing over of the PIP monomers from the aqueous phase to the organic side having TFSC monomers, across the nascent interfacial polymer layer, in a diffusion controlled manner. The very strong bands appearing at 1668 and 1667 cm−1 are assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (νCO) in tertiary amide segments of the polymeric chains. The bands with lower intensities located at 1375 and 1373 cm−1 correspond to the C–N stretching vibration (νC–N) of tertiary amide segments of the polyamide structures.31,36,37 The peaks with higher intensities appearing around 1243 cm−1 and the shoulder peak appearing at 1166 cm−1 for both the membranes are assigned to the symmetric (νSym.) and asymmetric (νAsym.) stretching vibrations of C–F bonds, reflecting the >CF2 units of the fluoro-acyclic backbone of the polyamide, respectively.30,38,39 In secondary polyamides, the charged resonance structure of amide bond usually gets stabilized by the H-bonding at the CO sites, resulting in an increase in the C–N double bond character through a decrease in the CO bond order; however, the νC–N of tertiary amides does not show any such dependency on H-bonding.40 Notably, the absence of any absorption bands in the zone of 3200–3300 cm−1 indicates that the polyamide structure is free of amide N–H stretching, which straight away diminishes the possibility of any amidic H-bonding effect within the structure.30 In the FTIR spectra, the absence of the characteristic CO stretching vibration at 1720 cm−1 and strong absorption bands around 3000 cm−1, attributable to –OH stretching, substantiates that the polyamide structure is also exclusively devoid of pendant –COOH groups, and again reflects its inability in providing an H-bonding effect.27–29 In contrast, this kind of functional interference in the structural features is quite expected with the existing varying poly(piperazineamide) structures, wherein residual carboxyl groups appeared through trimesoyl chloride type monomers.25,27–29,31 The weak and broad absorption bands around 3000 cm−1 however showed the very low water content, because of the polymer structure's enrichment with fluorine.
O stretching vibration (νCO) in tertiary amide segments of the polymeric chains. The bands with lower intensities located at 1375 and 1373 cm−1 correspond to the C–N stretching vibration (νC–N) of tertiary amide segments of the polyamide structures.31,36,37 The peaks with higher intensities appearing around 1243 cm−1 and the shoulder peak appearing at 1166 cm−1 for both the membranes are assigned to the symmetric (νSym.) and asymmetric (νAsym.) stretching vibrations of C–F bonds, reflecting the >CF2 units of the fluoro-acyclic backbone of the polyamide, respectively.30,38,39 In secondary polyamides, the charged resonance structure of amide bond usually gets stabilized by the H-bonding at the CO sites, resulting in an increase in the C–N double bond character through a decrease in the CO bond order; however, the νC–N of tertiary amides does not show any such dependency on H-bonding.40 Notably, the absence of any absorption bands in the zone of 3200–3300 cm−1 indicates that the polyamide structure is free of amide N–H stretching, which straight away diminishes the possibility of any amidic H-bonding effect within the structure.30 In the FTIR spectra, the absence of the characteristic CO stretching vibration at 1720 cm−1 and strong absorption bands around 3000 cm−1, attributable to –OH stretching, substantiates that the polyamide structure is also exclusively devoid of pendant –COOH groups, and again reflects its inability in providing an H-bonding effect.27–29 In contrast, this kind of functional interference in the structural features is quite expected with the existing varying poly(piperazineamide) structures, wherein residual carboxyl groups appeared through trimesoyl chloride type monomers.25,27–29,31 The weak and broad absorption bands around 3000 cm−1 however showed the very low water content, because of the polymer structure's enrichment with fluorine.
The XPS survey-scan spectra of the membranes’ skin layers, reported in Fig. 2a for TFC-NFA and Fig. 2d for TFC-NFW, reveal their elemental compositions, particularly indicating the overall relative presence of the constituent elements (C, O, N and F) in the surfaces. Deconvolution of the high-resolution C-1s spectra of both TFC-NFA and TFC-NFW, presented in Fig. 2b and e, respectively, showed the appearance of peaks pertaining to the C–C, C–N, CO and C–F bonds at binding energies around 284.5, 285.7, 287.2 and 288.8 eV, respectively.30,32 It is noticed that the percentage area for the CO peak (Table 1) appears slightly higher in TFC-NFA (4%) than TFC-NFW (3.3%), whereas the percentage areas for the C–N and C–F peaks come substantially higher in TFC-NFW (5.8 and 2.5%) as compared to TFC-NFA (0.4 and 1.8%). This is further corroborated by the noteworthy difference in the relative intensity for the high-resolution F-1s spectra of TFC-NFA and TFC-NFW, as presented in Fig. 2c and f, respectively. These results thus suggest that the skin layers made with FPA are quite susceptible and structurally responsive to the exposure environment during their preparation. More specifically, the annealing in a water-bath seems to facilitate better enrichment with F in the skin layer of TFC-NFW, as is obvious by its lower value of the C/F ratio (Table 1) in comparison to TFC-NFA.
 | ||
| Fig. 2 XPS survey scan spectra (a and d); and high-resolution C-1s (b and e) and F-1s (c and f) spectra of membrane skin layers for TFC-NFA and TFC-NFW, respectively. | ||
| Membrane code | Percentage peak area | Atomic ratio | ||||
|---|---|---|---|---|---|---|
| C–C | C–N | CO |
C–F | C/F | C/O | |
| TFC-NFA | 93.8 | 0.4 | 4.0 | 1.8 | 52.1 | 23.5 |
| TFC-NFW | 88.4 | 5.8 | 3.3 | 2.5 | 35.4 | 26.8 |
It can be reasonably believed that the typical structure of the studied FPA permits only secondary non-covalent type interactions to happen between the polymeric chains. Interestingly, as discussed above, there is no spectral evidence for the presence of H-bonding in the structure of the polymer. So, it seems to be devoid of intra- as well as inter-molecular H-bonding, possibly because of the nonavailability of any amidic-H in the zone of the H-bonding distance. In the absence of such strong cohesive non-covalent interactions, there might be an interplay of other non-covalent types, which span from van der Waals to dipole–dipole interactions or n → π* electronic delocalization.41,42 With the similarity of the polyamide structure to those of proteins and peptides, the most probable n → π* electronic transition might happen through the delocalization (CO⋯CO) of a lone pair of electrons from a donor carbonyl group (nCO) to an antibonding orbital of a neighbouring acceptor carbonyl group (πCO*), subjected to having ideal spatial proximity.41–43 However, this electron delocalization reasonably gets attenuated in a polyamide structure having α-fluorocarbonyl segments, as occurred in both of our membranes, owing to the strong inductive electron withdrawing ability of α-F atoms, represented schematically in Fig. 3a. Another possible route (represented in Fig. 3b) of electronic delocalization could be through the donation of an F-lone pair (nF) to the π* orbital of the carbonyl group (πCO*) of the adjacent polymeric chains.43–46 However, the extent of such dipolar interaction (C–F⋯CO) through the πCO* and nF orbitals of the intrinsically polar CO and C–F units is likely to be dependent on the favourable spatial orientation (e.g. cis–gauche and gauche–gauche conformers) of the fluorinated-acyclic structure of the polyamide.43–46 It is assumed that the α-F atoms in FPA introduce molecular space between the polymeric chains, thereby mitigating strong cohesive non-covalent type interactions. Such attenuation of non-covalent interactions within polymer chains also encourages them to have unconstrained mobility or high configurational entropy, which concomitantly might have affected the micro-heterogeneity of the membrane skin layer polymer in both TFC-NFA and TFC-NFW.
 | ||
| Fig. 3 Structural representation of the skin layer polymer FPA, highlighted with the possible attenuated secondary non-covalent type interactions (a and b) and charge-transfer complex formation (c). | ||
3.2 Surface topography and morphology of FPA membranes
The surface topography was analysed for both TFC-NFA and TFC-NFW using AFM. Both 2D AFM images of the membranes’ skin surfaces, presented in Fig. 4a and c, show the formation of coalesced spherical domains for the structure of FPA, irrespective of their post-synthesis exposure to air or water. However, the average surface roughness (Ra) and root mean square roughness (Rq) appear distinctly higher for TFC-NFW (71.8 and 93.6 nm, Table 2) than TFC-NFA (31.1 and 40.4 nm, Table 2). This trend could be attributed to the respective spatial orientations of the polymeric chains of the membranes’ skin surfaces, wherein the influence of the post-synthesis exposure to water in the case of TFC-NFW turned out to be different, creating more randomized features, in comparison to the exposure to air. The features are more clearly observable in the corresponding 3D AFM images of the membranes, with well-defined peaks and valleys, presented in Fig. 4b and d for TFC-NFA and TFC-NFW, respectively. | ||
| Fig. 4 Skin surface topographies of FPA-based TFC-NF membranes, prepared by exposing them to two different environments during fabrication: (a and b) 2D and 3D AFM images of TFC-NFA and (c and d) 2D and 3D AFM images of TFC-NFW. | ||
| Membrane code | R a (nm) | R q (nm) | ζ (mV) | σ s (C m−2) × 10−3 |
|---|---|---|---|---|
| TFC-NFA | 31.1 | 40.4 | −4.4 | −3.1 |
| TFC-NFW | 71.8 | 93.6 | −7.0 | −4.9 |
The pattern of surface morphology as observed in the SEM images, presented in Fig. 5a for TFC-NFA and Fig. 5b for TFC-NFW, depicts the formation of coalesced and interspersed spherical domains in the microscale regime, which is consistent with their respective 2D AFM images as well. It is assumed that the amphiphilic network structure of FPA, consisting of hydrophilic amine or amide segments and fluorophilic chain segments, gets segregated at the surface of the Psf base membrane during in situ interfacial polycondensation, because of unfavourable enthalpic interactions between the two different phases of the polyamide polymer.47,48 Furthermore, such occurrence of phase separated morphology was most likely driven by the low surface energy fluorine-enriched segments.49,50 This confirms that the FPA structure gets strongly affected by its intrinsic stereoelectronic effects like configurational mobility of polymer chains, electron-withdrawing ability of F and structural hydrophilic–hydrophobic balance.48 The morphology of spherical domains also gets affected by the post-polymerization exposure, like air-drying for TFC-NFA and annealing in a water-bath for TFC-NFW; however, the variation is not visually resolvable on the scale of the current experiments.
 | ||
| Fig. 5 Skin surface morphology of FPA-based TFC-NF membranes, (a) TFC-NFA and (b) TFC-NFW, prepared by exposing them to two different environments during fabrication. | ||
3.3 Surface hydrophilicity and electrokinetic phenomena of FPA membranes
The hydrophilic characters of the membranes, evaluated by contact angle measurements and presented in Fig. 6a, reflect the intrinsic structural features of the membranes’ polymeric surfaces. The contact angle (θwater) values for TFC-NFA and TFC-NFW are 42° and 50°, respectively.30–32 They corroborate the trend of higher C–F population in TFC-NFW over TFC-NFA, as discussed through XPS analysis. As an index of intermolecular or interfacial attractive forces, the surface free energy (γ) is found to decrease from 59 mN m−1 for TFC-NFA to 54 mN m−1 for TFC-NFW (Fig. 6b).30–32 The polar contribution to the SFE (γPolar), attributed collectively to intrinsically polar Cδ+–Fδ− and amidic Cδ+–Oδ− as well as Cδ+–Nδ− bonds, is higher for TFC-NFA (15 mN m−1) than TFC-NFW (11 mN m−1). The higher value of γPolar for TFC-NFA as compared to that of TFC-NFW also indicates that the latter could exert greater hydrophobic forces towards polar solvents. The current class of FPA structure has the ability to form a charge-transfer complex under ideal spatial proximity.42,51 This is due to the interaction between the partially electron deficient N-site of the tertiary amide segments and the highly electronegative F-site of the >CF2 units, as represented schematically in Fig. 3c. The trend of γPolar indicates that the possibility of charge-transfer complex formation is higher in TFC-NFW over TFC-NFA, due to the favourable condition the polyamide experiences under water-exposure in the case of the former. Consequently, this slightly reduces the extent of polar interactions in the skin layer of TFC-NFW, owing to the lack of free polar sites, particularly through the interactive C–F and C–N bonds. With a change in the exposure environment from air (TFC-NFA) to hydrophilic (TFC-NFW), the FPA chains thus exploit their degree of freedom in the form of configurational mobility to acquire energetically favourable spatial orientations, and then strongly influence the hydrophilicity of the membranes. | ||
| Fig. 6 Hydrophilic characters (a) and surface free energy parameters (b) of TFC-NF membranes, TFC-NFA and TFC-NFW. | ||
Streaming potential measurements were conducted to understand the surface charge phenomena of the FPA membranes. Although the zeta potential (ζ) appears negative for both TFC-NFA (−4.4 mV) and TFC-NFW (−7.0 mV), the very low values of ζ (Table 2) substantiate that the current class of polyamide is inherently non-ionogenic in nature. The difference in the ζ values of these two membranes could reasonably be related to the extent of charge-transfer complex formation in TFC-NFA and TFC-NFW, as described before. Since the skin layer of TFC-NFW has fewer exposed free polar sites than TFC-NFA, the influence of the F-sites on their respective surface charge is predominantly higher in the case of the latter. With such low negative values of ζ, these polyamide-based NF membranes quite unlikely fall in the regime of negatively charged NF membranes. In comparison, in two different studies carried out by Y. Li et al.30,31 on polyamide NF membranes prepared using fluorinated polyamine and trimesoyl chloride as a monomeric combination, the reported values of ζ for the membranes were distinctly more negative (Table 3, −41.3 mV30 and −20.5 mV31). The origin of such surface charge character of the membranes was proposed to be due to the predominant contribution of pendant carboxyl groups, dissociated on hydrolysis of the residual acid chloride, which is obviously in sharp contrast to our cases.27–29 Hence, it is evidently clear that the fluorine-enriched structure of our polyamide class offers an ambience of negative surface charge, ostensibly due to the overwhelming presence of F atoms and the polarity that they bring to the polymer network, even without having any localized fixed charged moieties like in other charged polyamides.23,24,30–32 We assume that this might be responsible for making the membranes’ charge character apparently more uniform in nature.
3.4 Nanofiltration performances of FPA-based TFC-NF membranes
The nanofiltration performances of the TFC-NF membranes, evaluated by cross-flow permeation under an isobaric and isosmotic environment, reveal that both the membranes follow a distinctive rejection trend of Na2SO4 > MgSO4 > NaCl > MgCl2 (Fig. 7a).30–32 For Na2SO4, a maximum solute rejection of 76.1(±1.4) and 86.7(±1.5)% was achieved for TFC-NFA and TFC-NFW, respectively. In comparison to this, the rejection of NaCl was found to be quite low, i.e., 22.9(±1.6) and 25.6(±1.2)% for TFC-NFA and TFC-NFW, respectively. Considering the fact that there is no distinct localized charged site and the membranes carry markedly low surface charge, the rejection performance of at least one of the membranes, namely TFC-NFW, appears to be higher for Na2SO4 (obviously in contrast to NaCl) and more promising than the other fluorinated polyamide-based NF membranes with distinct charged sites as reported by Y. Li et al.30,31 and R. Zhang et al.32 (Table 3). The high rejection of the bivalent anion in a pair with the univalent cation (hydrated radii of Na+ and SO42−: 0.358 and 0.379 nm, respectively) substantiates the governing effect of rejection as Donnan-exclusion.13,52 This results from a strong electrostatic repulsive interaction between the solvated anions and an ambience of negative surface charge exerted by the fluorine-enriched non-ionogenic polyamide of the membrane skin surfaces. We also observed that the extent of such repulsive interaction even increased with prolonged duration of in situ interfacial polymerization (from 60 to 90 s for the TFC-NFA type, Fig. S3 and Table S1, ESI†), on formation of a more dense FPA structure as the top skin layer, leading to a rejection of 92.6(±1.2)% for Na2SO4. However, in the case of MgSO4, the presence of the bivalent cation, Mg2+ (hydrated radius: 0.428 nm), along with the solvated bivalent anion, SO42−, noticeably reduces the extent of Donnan-exclusion.13,52 The rejections of MgSO4 thus appear as 38.3(±1.6) and 52.9(±1.8)% for TFC-NFA and TFC-NFW, respectively. The higher affinity of Mg2+ over Na+ towards the membrane–solution interface could further be attributed to the strong cation–dipole interaction exerted by the C–F segments of the FPA matrices. The lower value of the solute rejection of MgSO4 than that of Na2SO4, in spite of a bigger size of the hydrated ion of Mg2+ than Na+, also signifies that the Donnan-exclusion effect is more dominant than the size-exclusion process, and hence the former phenomenon more than compensates the latter one. A similar effect is also profoundly noticed for the pair of solutes, NaCl and MgCl2, when a further decline was observed in the solute rejection of MgCl2, i.e., 13.3(±1.5) and 19.4(±1.2)% for both TFC-NFA and TFC-NFW, respectively. This further indicates that the solvated univalent Cl− (hydrated radius: 0.332 nm) having lower negative charge density experiences a much weaker repulsive interaction from FPA.13,52 Additionally, the higher solute permeance for MgCl2 as compared to NaCl again indicates the contributory effect of the cation–dipole interaction on the membranes’ ion affinity. The differential rejection efficiency for Na2SO4/NaCl appears better for TFC-NFW in comparison to TFC-NFA, indicating that the effect and ambience of negative surface charge is stronger in case of the former membrane than the latter. The solute rejection trend is in accordance with the post-polymerization conditioning environments and variations in their electrokinetic features, as discussed before. The differential solute rejection (Na2SO4vs. NaCl) performance of our membranes, particularly that of TFC-NFW, seems to outperform some of the recently reported fluorinated polyamide-based NF membranes.30–32 Interestingly, the differential solute rejection performance of TFC-NFW was achieved even without having distinct fixed surface charge on it, whereas residual ionogenic functionalities contributed in creating the surface charge in the case of the previously reported ones.30–32 | ||
| Fig. 7 Nanofiltration performances of TFC-NF membranes: (a) solute rejection against isosmotic solutions of NaCl, Na2SO4, MgCl2 and MgSO4; and (b) initial and stabilized solvent fluxes of the membranes. | ||
A noteworthy observation is also that prolonged duration (90 s) of the in situ interfacial polymerization process enhances the differential solute rejection performance of the membranes, exerting a rejection of 23.4(±1)% for NaCl, as observed with TFC-NFA (Fig. S3 and Table S1, ESI†).
The Psf base membrane, prepared by a non-solvent induced phase inversion method, exhibits a MWCO of 38k Da and pure water permeability of 94(±4) LMH at 1 bar of transmembrane pressure. The solvent fluxes of the composite membranes, presented in Fig. 7b, reflect the physicochemical variations of the skin surfaces of TFC-NFA and TFC-NFW. The higher stabilized solvent flux for TFC-NFA (70 ± 4 LMH) over TFC-NFW (37 ± 3 LMH) is attributed to the consequences of post-polymerization conditioning in hydrophobic and hydrophilic environments, respectively. In addition to the variation in the surface hydrophilic characters, the membranes’ pore structures also play a crucial role, since they get influenced by the inter-chain spacing and resultant packing of polymer chains. The exposure to a hydrophobic (air) environment during fabrication of TFC-NFA likely impedes the intermolecular interactions within the neighbouring chains of the polyamide polymer, which further influences the compactness of the polymer by making it less dense and more open. Such structural freedom of FPA might further enhance the polymeric free volume by disfavouring charge-transfer complex formation, as discussed for TFC-NFA over TFC-NFW.42 These factors perhaps make the membranes’ pore structure more open and increase the free volume, resulting in higher solvent flow. However, in the case of TFC-NFW, the water has penetrated within the hydrophobic polymeric chain segments when the nascent skin layer experiences hydrophilic conditioning (treated in water) during its formation. When polymerization was allowed for a longer duration (90 s) for the TFC-NFA type membrane, we obtained a more hydrophilic surface with a slightly higher negative ζ value (Table S1, ESI†), but the resulting formation of a thicker and dense skin layer reduces the solvent flux to 54(±2) LMH (Fig. S4, ESI†).
This might have, in all probability, resulted in formation of clusters of segments of polymeric chains (hydrophobic zones) away from the influence of water leading to densification of the polymer with a lesser free volume. In addition to charge-transfer complex formation, the strong C–F⋯H–O dipolar interaction also brings the polymer chains closer, making the skin layer tighter to enhance the rejection and lower the solvent flux.13 Furthermore, it is also known that the structures and properties (e.g., pore size, porosity, thickness and surface hydrophilicity) of the Psf base membrane play crucial roles in deciding the solvent fluxes of any TFC membrane.53,54 These properties are highly dependent on the types of solvent used, the concentration of Psf in the casting solution and the resultant viscosity.53,54 Therefore, we assume that one or some of these factors might have reduced the overall solvent flux of the studied polyamide TFC-NF membranes. It is again noticed that the influence of having higher surface area gets overcompensated for TFC-NFW, despite the trend observed in AFM analysis, wherein the surface area of TFC-NFW appears higher than TFC-NFA. During the initial stabilization period, which actually spanned a few hours, it was noticed that the initial solvent flux was significantly high, i.e., 102(±5) and 58(±4) LMH for TFC-NFA and TFC-NFW, respectively, in comparison to their respective stabilized fluxes. The observed tendency towards high compaction also indicates that there is in general a great deal of free volume in the cast polymer, which can be attributed mostly to the lack of strong cohesive non-covalent interactions, and this seems to be a typical structural feature of the current class of FPA.
4. Conclusions
In summary, a new chemical recipe of monomers, particularly a diacyl chloride monomer having a fluoro-acyclic backbone, was explored for the synthesis of a fluorine-enriched polyamide, targeting nanofiltration applications. The surface topography and morphology of the membranes were affected by the stereoelectronic effect of FPA, i.e., the configurational mobility of polymer chains, electron-withdrawing ability of F and relative structural response of hydrophilic/fluorophilic chains. The non-ionogenic FPA-based TFC-NF membranes carried no fixed surface charge but had strikingly low negative ζ values and exhibited high Donnan-exclusion of ions, with substantial differential rejection between multivalent (Na2SO4: 87–93%) and monovalent anions (NaCl: 23–26%). We therefore hope that the illustrated synthetic potential of FPA will open up a new avenue to engineer high-performance membranous polyamides. Because of having unique structural features, the current class of polyamide with α-fluorocarbonyl segments might resist electrophilic substitution, and thus further studies may investigate its potential as an interesting chlorine-tolerant candidate under an oxidative environment. Further research also needs to be undertaken with the studied class of polyamide as it could be effectively used to develop fouling-resistant membranes, because of the low surface free energy of the FPA surface.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the effort of U. R. More of Membrane Development Section, Bhabha Atomic Research Centre, for his support in membrane making and operation of membrane test skids.References
- A. I. Schafer, A. G. Fane and T. D. Waite, Nanofiltration—Principles and Applications, Elsevier, Oxford, UK, 2002 Search PubMed.
- A. W. Mohammad, Y. H. Teow, W. L. Ang, Y. T. Chung, D. L. Oatley-Radcliffe and N. Hilal, Desalination, 2015, 356, 226–254 CrossRef CAS.
- A. Giacobbo, A. M. Bernardes and M. N. de Pinho, Sep. Sci. Technol., 2013, 48, 2524–2530 CrossRef CAS.
- L. D. Nghiem and A. I. Schäfer, Environ. Sci. Technol., 2005, 39, 7698–7705 CrossRef CAS PubMed.
- B. V. Bruggen and C. Vandecasteele, Environ. Pollut., 2003, 122, 435–445 CrossRef PubMed.
- J. Schaep, B. V. Bruggen, S. Uytterhoeven, R. Croux, C. Vandecasteele, D. Wilms, E. V. Houtte and F. Vanlerberghe, Desalination, 1998, 119, 295–302 CrossRef CAS.
- B. A. M. Al-Rashdi, D. J. Johnson and N. Hilal, Desalination, 2013, 315, 2–17 CrossRef CAS.
- A. Akbari, J. C. Remigy and P. Aptel, Chem. Eng. Process., 2002, 41, 601–609 CrossRef CAS.
- J. Garcia-Aleman and J. M. Dickson, J. Membr. Sci., 2004, 239, 163–172 CrossRef CAS.
- J. Tanninen, M. Mänttäri and M. Nyström, J. Membr. Sci., 2006, 283, 57–64 CrossRef CAS.
- H. Wang, Q. Zhang and S. Zhang, J. Membr. Sci., 2011, 378, 243–249 CrossRef CAS.
- T. K. Dey, R. C. Bindal, S. Prabhakar and P. K. Tewari, Sep. Sci. Technol., 2011, 46, 933–943 CrossRef CAS.
- A. Pal, T. K. Dey and R. C. Bindal, Polymer, 2016, 93, 99–114 CrossRef CAS.
- M. R. Teixeira, M. J. Rosa and M. Nystrom, J. Membr. Sci., 2005, 265, 160–166 CrossRef CAS.
- R. J. Petersen, J. Membr. Sci., 1993, 83, 81–150 CrossRef CAS.
- L. Li, S. B. Zhang, X. S. Zhang and G. D. Zheng, J. Membr. Sci., 2007, 289, 258–267 CrossRef CAS.
- L. Li, S. B. Zhang, X. S. Zhang and G. D. Zheng, J. Membr. Sci., 2008, 315, 20–27 CrossRef CAS.
- L. Li, S. B. Zhang and X. S. Zhang, J. Membr. Sci., 2009, 335, 133–139 CrossRef CAS.
- S. Yu, M. Liu, Z. Lu, Y. Zhou and C. Gao, J. Membr. Sci., 2009, 344, 155–164 CrossRef CAS.
- S. Yu, M. Ma, J. Liu, J. Tao, M. Liu and C. Gao, J. Membr. Sci., 2011, 379, 164–173 CrossRef CAS.
- G. Chen, S. Li, X. Zhang and S. Zhang, J. Membr. Sci., 2008, 310, 102–109 CrossRef CAS.
- Y.-H. La, R. Sooriyakumaran, D. C. Miller, M. Fujiwara, Y. Terui, K. Yamanaka, B. D. McCloskey, B. D. Freeman and R. D. Allen, J. Mater. Chem., 2010, 20, 4615–4620 RSC.
- W. Xie, G. M. Geise, B. D. Freeman, H.-S. Lee, G. Byun and J. E. McGrath, J. Membr. Sci., 2012, 403–404, 152–161 CrossRef CAS.
- G. N. B. Baroña, J. Lim and B. Jung, Desalination, 2012, 291, 69–77 CrossRef.
- A. Bera, J. S. Trivedi, S. B. Kumar, A. K. S. Chandel, S. Halder and S. K. Jewrajka, J. Hazard. Mater., 2018, 343, 86–97 CrossRef CAS PubMed.
- J. S. Trivedi, D. V. Bhalani, G. R. Bhadu and S. K. Jewrajka, J. Mater. Chem. A, 2018, 6, 20242–20253 RSC.
- M. F. Jimenez Solomon, Y. Bhole and A. G. Livingston, J. Membr. Sci., 2013, 434, 193–203 CrossRef CAS.
- X. Lu, S. Romero-Vargas Castrilló, D. L. Shaffer, J. Ma and M. Elimelech, Environ. Sci. Technol., 2013, 47, 12219–12228 CrossRef CAS PubMed.
- J. R. Werber, S. K. Bull and M. Elimelech, J. Membr. Sci., 2017, 535, 357–364 CrossRef CAS.
- Y. Li, Y. Su, X. Zhao, R. Zhang, J. Zhao, X. Fan and Z. Jiang, J. Membr. Sci., 2014, 455, 15–23 CrossRef CAS.
- Y. Li, Y. Su, X. Zhao, R. Zhang, Y. Liu, X. Fan, J. Zhu, Y. Ma, Y. Liu and Z. Jiang, Ind. Eng. Chem. Res., 2015, 54, 8302–8310 CrossRef CAS.
- R. Zhang, Y. Li, Y. Su, X. Zhao, Y. Liu, X. Fan, T. Ma and Z. Jiang, J. Mater. Chem. A, 2016, 4, 7892–7902 RSC.
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS PubMed.
- Y. H. La, R. Sooriyakumaran, D. C. Miller, M. Fujiwara, Y. Terui, K. Yamanaka, B. D. McCloskey, B. D. Freeman and R. D. Allen, J. Mater. Chem., 2010, 20, 4615–4620 RSC.
- S.-M. Xue, C.-H. Ji, Z.-L. Xu, Y.-J. Tang and R.-H. Li, J. Membr. Sci., 2018, 545, 185–195 CrossRef CAS.
- X. Yang, ACS Omega, 2019, 4, 13824–13833 CrossRef CAS PubMed.
- N. Misdan, W. J. Lau and A. F. Ismail, J. Polym. Eng., 2015, 35, 71–78 CAS.
- A. Shockravi, E. A. Lotf, A. Javadi and S. Taheri, Polym. J., 2009, 41, 174–180 CrossRef CAS.
- D. Tunc, H. Bouchekif, B. Améduri, C. Jérôme, P. Desbois, P. Lecomte and S. Carlotti, Eur. Polym. J., 2015, 71, 575–584 CrossRef CAS.
- N. S. Myshakina, Z. Ahmed and S. A. Asher, J. Phys. Chem. B, 2008, 112, 11873–11877 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS.
- A. Choudhary, D. Gandla, G. R. Krow and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 7244–7246 CrossRef CAS PubMed.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley Interscience Publication, New York, 1996 Search PubMed.
- P. R. Olivatoa, S. A. Guerreroa, M. H. Yreijoa, R. Rittnerb and C. F. Tormena, J. Mol. Struct., 2002, 607, 87–99 CrossRef.
- A. Choudhary, C. G. Fry and R. T. Raines, ARKIVOC, 2010, 2010, 251–262 Search PubMed.
- N. Xi, X. Sun, M. Li, M. Sun, M. A. Xi, Z. Zhan, J. Yao, X. Bai, Y. Wu and M. Liao, J. Org. Chem., 2018, 83, 11586–11594 CrossRef CAS PubMed.
- E. Neil and G. Marsh, Acc. Chem. Res., 2014, 47, 2878–2886 CrossRef PubMed.
- J. A. Hodges and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 9262–9263 CrossRef CAS PubMed.
- D. R. Iyengar, S. M. Perutz, C. A. Dai, C. K. Ober and E. J. Kramer, Macromolecules, 1996, 29, 1229–1234 CrossRef CAS.
- N. Bruns and J. C. Tiller, Macromolecules, 2006, 39, 4386–4394 CrossRef CAS.
- F. Hof, D. M. Scofield, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2004, 43, 5056–5059 CrossRef CAS PubMed.
- E. R. Nightingale Jr., Phenomenological theory of ion solvation. Effective radii of hydrated ions, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef.
- Q. Xie, S. Zhang, Z. Hong, H. Ma, B. Zeng, X. Gong, W. Shao and Q. Wang, Chem. Eng., 2019, 368, 186–201 CrossRef CAS.
- Q. Xie, S. Zhang, Z. Hong, H. Ma, C. Liu and W. Shao, Ind. Eng. Chem. Res., 2018, 57, 16464–16475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00107d |
| This journal is © The Royal Society of Chemistry 2020 |